")
Back to Journals » International Journal of Nanomedicine » Volume 17
Strategy for Avoiding Protein Corona Inhibition of Targeted Drug Delivery by Linking Recombinant Affibody Scaffold to Magnetosomes
Authors Ma S, Gu C, Xu J, He J, Li S, Zheng H, Pang B, Wen Y, Fang Q, Liu W, Tian J
Received 27 September 2021
Accepted for publication 18 January 2022
Published 11 February 2022 Volume 2022:17 Pages 665—680
DOI https://doi.org/10.2147/IJN.S338349
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yan Shen
Shijiao Ma,1 Chenchen Gu,1 Junjie Xu,1 Jinxin He,2 Shuli Li,1 Haolan Zheng,1 Bo Pang,1 Ying Wen,1 Qiaojun Fang,3 Weiquan Liu,1 Jiesheng Tian1
1State Key Laboratory of Agrobiotechnology, College of Biological Sciences, China Agricultural University, Beijing, 100193, People’s Republic of China; 2College of Veterinary Medicine, Shanxi Agriculture University, Taigu, Shanxi, 030801, People’s Republic of China; 3CAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing, 100190, People’s Republic of China
Correspondence: Weiquan Liu; Jiesheng Tian, Tel/Fax +8610-62732676 ; +8610-62731440, Email [email protected]; [email protected]
Purpose: Nanoparticles (NPs) decorated with functional ligands are promising candidates for cancer diagnosis and treatment. However, numerous studies have shown that chemically coupled targeting moieties on NPs lose their targeting capability in the biological milieu because they are shielded or covered by a “protein corona”. Herein, we construct a functional magnetosome that recognizes and targets cancer cells even in the presence of protein corona.
Methods: Magnetosomes (BMPs) were extracted from magnetotactic bacteria, M. gryphiswaldense (MSR-1), and decorated with trastuzumab (TZ) via affibody (RA) and glutaraldehyde (GA). The engineered BMPs are referred to as BMP-RA-TZ and BMP-GA-TZ. Their capacities to combine HER2 were detected by ELISA, the quantity of plasma corona proteins was analyzed using LC-MS. The efficiencies of targeting SK-BR-3 were demonstrated by confocal laser scanning microscopy and flow cytometry.
Results: Both engineered BMPs contain up to ∼ 0.2 mg TZ per mg of BMP, while the quantity of HER2 binding to BMP-RA-TZ is three times higher than that binding to BMP-GA-TZ. After incubation with normal human plasma or IgG-supplemented plasma, GA-TZ-containing BMPs have larger hydrated radii and more surface proteins in comparison with RA-TZ-containing BMPs. The TZ-containing BMPs all can be targeted to and internalized in the HER2-overexpressing breast cancer cell line SK-BR-3; however, their targeting efficiencies vary considerably: 50– 75% for RA-TZ-containing BMPs and 9– 19% for GA-TZ-containing BMPs. BMPs were incubated with plasma (100%) and cancer cells to simulate human in vivo environment. In this milieu, BMP-RA-TZ uptake efficiency of SK-BR-3 reaches nearly 80% (slightly lower than for direct interaction with BMP-RA-TZ), whereas the BMP-GA-TZ uptake efficiency is < 17%.
Conclusion: Application of the RA scaffold promotes and orients the arrangement of targeting ligands and reduces the shielding effect of corona proteins. This strategy improves the targeting capability and drug delivery of NP in a simulated in vivo milieu.
Keywords: affibody, protein corona, magnetosomes, human epidermal growth factor receptor 2, HER2
Introduction
Oncological diseases remain among the most common causes of human mortality worldwide, despite remarkable advances in the medical sciences during recent decades.1 Principal anti-cancer treatment strategies in current use, including radiation therapy, chemotherapy, nucleic acid therapy, and immunotherapy, are frequently associated with severe side-effects related to systemic toxicity due to lack of tumor specificity.2–5 One approach for overcoming this problem is engineering of nanoparticle (NP) surfaces for targeting of tumors through the addition of various synthetic or biological ligands,6 eg small molecules,7 peptides/proteins,8 and antibodies (Abs).9 Targeting with NP-based platforms can enhance efficacy of drug delivery to specific tumor cells with relatively few adverse effects, thus improving diagnosis and therapy. This has been an exciting and fast-progressing research area in recent decades.6
One problematic issue is that host biological conditioning causes immediate adsorption to the NP surface upon blood or tissue contact. A variety of unpredictable and uncontrollable biomolecules, notably proteins, rapidly coat the NP surface and form a protein coating, termed “corona”.10 The presence of a protein corona often results in the reduction or elimination of NP targeting capability by shielding or completely covering relevant functional groups.11–14 Numerous strategies have been tested using surface barrier layers (polymer, protein, or biomimetic coatings) to block the adhesion of corona proteins and prolong blood circulation time of NPs.15,16 However, some of these strategies were found to inhibit the internalization of NPs by cancer cells, resulting in limited therapeutic effectiveness (eg the “PEG dilemma”).17 Some groups introduced various types of cleavable bonds and mechanisms in engineered NPs to prolong the exposure of targeting ligands until they reach target sites.18 However, the above strategies for reducing protein corona effects are generally complicated and not widely used due to the inherent difficulty of in vivo application to diverse types of cancer cells and their external stimuli. It has also been reported that corona compositions strongly depend on the type of disease in human patients.19,20 Therefore, there is a clear need for the development of simple, widely applicable NP systems with a more easily controlled corona for targeted drug delivery.
Herein, we describe a novel, alternative strategy for targeted drug delivery, based on the precoating of functionalized NPs with humanized anti-tumor Abs in an oriented arrangement. We used a type of biogenic NP termed magnetosomes (or bacterial magnetic nanoparticles, BMPs). BMPs are specialized organelles characteristically found in magnetotactic bacteria, composed of membrane-enveloped magnetite (Fe3O4) or greigite (Fe3S4) crystals, with diameters ranging from 30 to 120 nm.21 BMPs act as a compass that can guide bacteria to find the optimum conditions for their growth and survival.22 In addition, the application of BMPs as a nanomaterial is explored owing to their unique crystalline properties, narrow size distribution, and good biocompatibility.23 More importantly, the BMP surface has numerous active chemical groups, particularly primary amino groups, that provide convenient conditions for surface modification.24 We constructed an engineered BMP (termed BMP-RA) by covalent linkage to a recombinant affibody (protein A) scaffold. Affibodies are small molecules (non-Ig proteins) that have high affinity toward specific protein targets. Protein A, as the first applied affibody protein,25 automatically binds Abs and maintains their orientation on the BMP surface.26 Incubation of BMP-RA with Ab results in orientation of the antigen binding site (paratope) of Ab toward the external BMP surface, thus promoting binding to the antigen. Oriented host Abs presumably comprise the majority of protein corona of BMP-RA in the biological milieu. Pre-adsorption of a humanized mAb (trastuzumab, TZ) onto BMP-RA results in the attachment of fewer host compounds to the Ab surface, and BMP-RA-TZ complexes retain their targeting capability even after formation of protein corona.
Materials and Methods
Cell Lines and Materials
The magnetotactic bacterium Magnetospirillum gryphiswaldense MSR-1 is stocked in our laboratory. Cell lines SK-BR-3 (ATCC#HTB-30) and MDA-MB-468 (ATCC#HTB-132) were kindly donated by National Center for Nanoscience and Technology. The anti-HER2 humanized mAb trastuzumab (TZ) was from Roche (Basel, Switzerland). McCoy’s 5A medium, Leibovitz’s L-15 medium, fetal bovine serum (FBS), and rabbit anti-HER2 Ab (cat. MA5-14509) were from Thermo Fisher (Waltham, MA, USA). Human IgG was from Sigma-Aldrich (Germany); 4’,6’-diamidino-2-phenylindole dihydrochloride (DAPI), 1,1-dioctadecyl-3,3,3,3- tetramethylindocarbocyanine perchlorate (DiI), Prussian Blue Iron Stain Kit, and Enhanced BCA Protein Assay Kit were from Solarbio Science & Technology (Beijing, China). Goat anti-rabbit IgG-HRP (cat. Abs20002), rabbit anti-mouse IgG-HRP (Abs.20001), and rabbit anti-goat IgG-HRP (cat. Abs20005) were from Absin Bioscience Inc. (Shanghai, China). Peroxidase-AffiniPure F(ab’)2 fragment rabbit anti-goat IgG (H+L) (cat. 305-036-003) were from Jackson ImmunoResearch Laboratories, Inc. (West Grove, USA).
Human Plasma
Blood samples were taken from 10 healthy male and female volunteer donors aged 20–40 years after obtaining informed consent for blood collection and subsequent analysis, and stored in 10-mL tubes containing K2 EDTA to prevent clotting. Samples were centrifuged (1300 x g, 15 min, 4°C) to remove red and white cells, and supernatants were collected and stored at −80°C for subsequent experiments.
Magnetosome Extraction
M. gryphiswaldense MSR-1 were cultured in a 7.5-L fermentor (BioFlo 110; New Brunswick Scientific, CT, USA). Inoculum was cultured in sodium lactate medium as described in our 2008 report.27 Cells were harvested at 3000 rpm for 30 min at 4 °C. BMPs were extracted as described in our 2019 report.28 In brief, harvested cells were resuspended in 10 mM PBS (10 mL per g bacterial pellet, pH 7.4) and disrupted by ultrasonication (150 W, 30 min) (model JY92-IIN; Scientz; Xiamen, China) on ice. BMPs were collected from solution by magnet at 4 °C overnight, supernatant was discarded, and precipitate was resuspended in PBS. The above steps were repeated until protein in supernatant showed no decrease. BMPs were treated with 1 mg/mL proteinase K for 3 h at 56 °C, electroeluted as described in our 2011 report,29 suspended in PBS at final concentration 1 mg/mL, and sterilized by cobalt-60.
Purification of Recombinant Protein A
Nucleotides of protein A (Z domain) were optimized using a JAVA adaptation tool for soluble expression in E. coli BL21. Recombinant protein containing linker peptides and cysteine was synthesized at N-terminus, cloned into pET28(a+) plasmid with kanamycin-resistant gene, and transformed into E. coli BL21. The protein expressed by recombinant gene was termed RA. E. coli BL21 with plasmid was cultured in LB medium; expression was induced by addition of over 8 h of isopropyl β-D-thiogalactopyranoside (IPTG) at final concentration 1 mM. Cells were collected by centrifugation (10,000 x g, 10 min), resuspended in 10 mM PBS (10 mL per g bacterial pellet, pH 7.4), disrupted by sonication at 30% amplitude (200 W, 30 min) on ice, and centrifuged again (10,000 x g, 30 min). RA protein present in supernatant was purified using HisTrap Fast Flow (FF) Crude Column as per manufacturer’s instructions. Purified RA was freeze-dried and stored at −20 °C for subsequent experiments.
Identification of RA and Its Function
Purified RA was added with protein loading buffer (0.01 (w/v) bromophenol blue, 0.04 M dithiothreitol (DTT), 10% glycerol, 2% (w/v) SDS, 62.5 mM Tris-HCl, pH 6.8), boiled for 10 min at 100 °C, and centrifuged. Supernatants were loaded onto 10% gels and run at 120 V for 1–2 h in a Mini-PROTEAN Tetra Electrophoresis System (Bio-Rad, USA). Gels were stained by Coomassie Brilliant Blue G250 for direct protein imaging, or transferred onto PVDF membrane in TransBlot SD Semi-Dry Electrophoretic Transfer Cell (Bio-Rad) for western blotting. Membranes were blocked with 5% skim milk for 2 h at room temperature (RT), incubated with mouse anti-FLAG tag Ab (dilution 1:2000) for 8 h, washed 3x with PBST (PBS containing 0.05% Tween-20), incubated with HRP-labeled goat anti-mouse Ab for 2 h at RT, and washed 3x with PBST. Proteins on membranes were displayed using Immobilon Western chemiluminescent HRP substrate, and visualized using an imaging system (Fusion FX6, Vilber, France).
Enzyme-Linked Immunosorbent Assay (ELISA) Analysis of Functions of RA and RA-Decorated BMPs
The ability of RA to bind IgG was analyzed by ELISA. 2 μg RA in 100 μL carbonate-bicarbonate buffer (pH 9.6) was incubated on 96-well microtiter plate overnight at 4°C. Plate was washed 3x next day with PBST, added with 300 μL of 1% BSA (diluted in PBST) for 1 h at RT to block nonspecific binding sites, washed 3x with PBST, added with same number of moles of rabbit anti-mouse IgG-HRP and rabbit anti-goat Peroxidase-AffiniPure F(ab’)2 Fragment for 1 h at RT (n=3), washed 3x with PBST, and added with 100 μL of 3,3′,5,5′-tetramethylbenzidine (TMB) solution. After 8 min, reaction was stopped by adding 50 μL of 2 M H2SO4. Absorbance at wavelength 450 nm was read by microtiter plate reader (ELx800, BioTek, USA). Samples without RA input were used as control check.
For BMP-RA, microtiter plate was blocked with 1% BSA overnight, washed 3x, added with 1 ng BMP-RA (n=3), placed over magnet for 5 min to isolate complex, added with rabbit anti-mouse IgG-HRP (1:20000 dilution in PBS) for 1 h at RT, and subsequent steps were the same as for RA. BMP wild-type (BMP-WT) was used as negative control.
For binding of BMP to human epidermal growth factor receptor-2 (HER2), initial processing of microtiter plate was the same as for BMP-RA. Then, plate was added with BMPs (n=3), washed, added for 1 h with HER2 at RT, washed 3x with PBST, added with 100 μL rabbit anti-HER2 Ab, incubated for 1 h with BMPs at RT, washed 3x, added with goat anti-rabbit IgG-HRP for 1 h, and subsequent steps were the same as for BMP-RA.
Preparation of Functional BMPs
TZ functionalized BMPs were prepared using N-succinimidyl 3-(2-pyridyldithio) propionate (SPDP) and glutaraldehyde as cross-linkers, and termed BMP-RA-TZ and BMP-GA-TZ, respectively. For BMP-RA preparation, 1 mg BMP was resuspended in HEPES (10 mM, pH 8.0) containing 1 mM SPDP, the solution was ultrasonicated for 1 min followed by 1-min intervals (this step was repeated 30x), and mixture was washed 3x with HEPES. RA (1 mg/mL) was suspended in 10 mM HEPES, ultrasonicated for 1 min followed by 1-min intervals (repeated 30x), supernatant was removed and washed 3x with HEPES under magnet, and resuspended in HEPES. The BMP-complex was termed BMP-RA. SDS-PAGE was used to confirm RA linkage on magnetosome, 0.2 mg BMP-RA was loaded, and subsequent steps were the same as for RA. For BMP-RA-TZ preparation, TZ solution (500 μg/mL) was suspended with modified BMPs and incubated for 2 h at 37°C with rotation (200 rpm). BMP-RA-TZ was washed 6x with HEPES and stored at 4 °C. BMP-GA-TZ was prepared as described in our 2019 report.30 For confocal laser scanning microscopy (CLSM) and flow cytometry, 1 mg BMP was FITC-labeled by mixing with 300 μL of 1 mg/mL FITC and incubated overnight at 4 °C with shaking.
Plasma–BMP Interaction
BMP samples were incubated with human plasma or IgG-enriched human plasma for 2 h at 37 °C with shaking (200 rpm), and washed 6x with HEPES. For IgG-enriched human plasma experiments, 1 mL human normal plasma was added with 6 mg IgG. Following the last washing step, BMPs were resuspended with 1 mL HEPES in a 1.5-mL Eppendorf tube.
Characterization of BMPs
Transmission electron microscopy (TEM): BMP sample (0.2 mg) was suspended in 1.5-mL Eppendorf tube with 1 mL double-distilled water (ddH2O), ultrasonicated for 1 min, and BMPs were collected under magnet and resuspended in 1 mL ddH2O. These steps were repeated 3x. Then 10 µL of this suspension was dropped onto copper grid and air dried for 10 min. Samples were observed by TEM (model JEM-1230, JEOL, Tokyo, Japan).
Zeta-potential and DLS measurement: BMP sample was resuspended in ddH2O at concentration 0.01 mg/mL and ultrasonicated for 10 min. Samples (n=3) were combined in sample pool and measured using Zetasizer Nano ZS particle size analyzer (Malvern Instruments, Worcestershire, UK) at 25 °C.
SDS-PAGE Analysis
Immediately following the last washing step, defined amount of BMP-corona complex (0.2 mg) was resuspended in protein loading buffer, boiled for 10 min at 100 °C, BMPs were removed by centrifugation (12,000 x g, 20 min), and equal sample volume was loaded on 10% gel. SDS-PAGE was performed at 80 V for 30 min, switched to 120 V for ~90 min until bromophenol blue neared the gel end, and gel was stained with protein silver stain kit.
Quantitative Analysis of TZ on BMPs
TZ supernatant before and after coupling to BMP was stored in advance, and analyzed using Enhanced BCA Protein Assay Kit as per manufacturer’s protocol. Standard curve was constructed based on eight BSA concentration points (0, 0.025, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5 mg/mL). Then 20 μL samples or BSA (control) were placed on 96-well plates, added with 200 μL BCA working solution, mixed thoroughly, incubated for 30 min at 37 °C, and read by microplate spectrophotometer (Power Wave XS2, BioTek) at 562 nm. Sample concentrations (n=3) were calculated based on the standard curve. Amount of TZ coupled to BMP was calculated as TZ amount before coupling -TZ amount TZ after coupling.
Protein Identification and Classification
BMP-corona complexes were obtained by the above methods, and proteins from SDS-PAGE 10% gels were excised and in-gel trypsin digested by the method of Shevchenko et al.31 In brief, 100 µg protein was dissolved in 50 mM ammonium bicarbonate (ABC) solution, reduced with DTT for 45 min at 56 °C, alkylated with iodoacetamide (IAM) for 30 min at RT in the dark, and the solution was transferred to 10K ultrafiltration tube (Vivacon 500, Sartorius, USA) and centrifuged (14000 x g, 20 min). Proteins were washed with 50 mM ABC, added with 2 μg trypsin in 50 μL ABC, and incubated overnight at 37 °C. The ultrafiltration tube was centrifuged (14000 x g, 20 min), digested peptides were transferred to a collection tube, and ABC was added to ultrafiltration tube to wash digested peptides into collection tube. Collected solution was diluted with 0.1% formic acid (FA) for nano-LC-MS analysis. NanoAcquity nano-HPLC (Waters Corp., Milford, MA, USA) was used for nano-LC separation. The trap column was Thermo Acclaim PepMap 100 (75 μm × 2 mm, C18, 3 μm), and the custom-made analytical column was a fused silica capillary (I.D. 100 μm; Polymicro Technologies) filled with 20 cm stationary phase (Aqua 3-μm C18 125A; Phenomenex). Gradient elution program: mobile phase increased linearly from 1% B to 35% B in 65 min. Mobile phase A was 0.1% FA in water; mobile phase B was 0.1% FA in acetonitrile. Raw data were imported into Progenesis QI for Proteomics (build 2.0, Nonlinear Dynamics, Newcastle, UK) for peak alignment and peak picking. The obtained MGF files were submitted to the Mascot search engine (version 2.5.0, Matrix Science, London, UK) and searched against the UniProt human database. Trypsin was selected as the specific enzyme with maximum of two missed cleavages. The mass tolerance was 10 ppm and the MS/MS mass tolerance was 0.02Da. Search results were set to a maximum of 1% false discovery rate with two unique peptides required for positive identification. The data were also preprocessed with Mascot Distiller 2.4 for peak picking. The resulted peak lists were searched against human database using Mascot 2.5 search engine. The exponentially modified protein abundance index (emPAI) was used to estimate the relative concentration of proteins in the sample, as reported previously, to normalize for emPAI difference between runs; the protein content (mol %) of individual proteins in the sample was then calculated by dividing the individual emPAI values by the sum of all emPAI values within the sample and then multiplying it by 100.32
Targeted Cell Uptake Evaluation of BMPs by Flow Cytometry and CLSM
To evaluate uptake of BMP-RA-TZ or BMP-GA-TZ by SK-BR-3 following incubation with human plasma or IgG-enriched human plasma, ~1×105 cells in 1 mL McCoy’s 5A medium with 10% FBS were seeded in a 12-well plate, cultured for 24 h under standard conditions, and then incubated with various samples (BMP-RA-TZ, BMP-RA-TZ-plasma, BMP-RA-TZ-IgG plasma, BMP-GA-TZ, BMP-GA-TZ-plasma, BMP-GA-TZ-IgG plasma) of BMPs (10 μg/mL) in McCoy’s 5A (0% FBS) for 2 h. For analysis of BMP uptake at various plasma concentrations, 1×105 cells were placed in McCoy’s 5A with 0%, 10%, or 100% human plasma, incubated with BMPs (n=3) at indicated concentrations at 37°C for 2 h, and washed 2x with 10 mM PBS. For flow cytometric measurements, cells were treated with 0.5% trypsin-EDTA, collected by centrifugation, washed 3x with PBS (10 mM), resuspended in 400 μL PBS, and FITC fluorescence was measured with a flow cytometer (Calibur, BD Biosciences, San Jose, CA, USA). Cells without BMP were used as control check, and percentage of FITC-positive cells was calculated relative to negative control value. For CLSM, cells were seeded in a 24-well plate with cell slide, incubated with BMPs, washed 2x with 10 mM PBS, stained with 200 μL of 10 μM 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate for 10 min at 37 °C, fixed with 4% polyoxymethylene (POM) for 10 min at RT, and stained with 200 μL of 10 μg/mL 4,6-diamidino-2-phenylindole dihydrochloride for 10 min at 37 °C. Images showing cellular uptake and distribution of BMPs with FITC-fluorescence signals were obtained using an inverted CLSM instrument (model A1, Nikon, Japan). Uptake efficiency of MDA-MB-468 was evaluated by an analogous series of steps.
HER2 Expression in SK-BR-3 and MDA-MB-468 Cells
HER2 expression on SK-BR-3 and MDA-MB-468 cell surfaces was evaluated by CLSM and western blotting. For CLSM, cells were seeded in 24-well plate containing coverslips, incubated for 24 h, fixed with 4% POM for 10 min at RT, incubated with normal goat serum (Solarbio) for 1 h at RT, incubated with rabbit anti-HER2 Ab (1:500 v/v, in 0.01 M PBS; Invitrogen, USA) at 4 °C overnight, incubated with Alexa Fluor 488-conjugated goat-anti-rabbit IgG (1:1000 v/v, in 0.01 M PBS) for 1 h in the dark, and stained with 10 μg/mL DAPI in the dark.
For western blotting, cells were collected following incubation on 6-well plate, washed with PBS, and added with 250 μL native lysis buffer (Solarbio) on ice for 10 min. Supernatant was mixed with SDS-PAGE loading buffer, boiled at 100 °C for 10 min, centrifuged, loaded on 10% gel, transferred to PVDF membrane, and blocked with 5% BSA. Membrane was incubated with rabbit anti-HER2 Ab (1:1000 v/v, in 0.01 M PBS) overnight at 4 °C, washed 3x with TBST (PBS containing 0.05% v/v Tween 20), incubated with HRP-labeled goat anti-rabbit (1:20000 v/v, in 0.01 M PBS) for 2 h at RT, and imaged using an imaging system (Fusion FX6, Vilber).
Prussian Blue Staining
Cells were seeded in 24-well plate containing coverslips, cultured for 24 h under standard condition, incubated with 10 μg/mL BMPs for 2 h, washed 3x, fixed with 4% POM for 10 min at RT, and the iron was detected by Prussian blue staining; ie, equal volumes of Perls stain A and B were mixed, added to fixed cells and tissue sections, and incubated for 30 min at 37 °C. Samples were then washed 3x with Millipore water, immersed in Nuclear Fast Red solution for 2 min, cell slide was washed 3x with Millipore water, and cellular uptake and distribution of BMPs were imaged using a Zeiss microscope (Axio Cam MR3, Germany).
Statistical Analysis
Statistical analysis was performed using Student’s t-test for two groups, and one-way ANOVA for multiple groups. All data are given as mean ± standard deviation (SD). Differences are considered statistically significant at *P <0.05, **P <0.01, ***P <0.001, and ****P <0.0001. Differences are labelled NS for not significant.
Results
Decoration of BMP with a Scaffold Fusion Protein
To maintain targeting capability, surface ligands on a functionalized NP must retain a highly directional orientation. This requires that the BMP be decorated with a protein scaffold. We employ the Z domain (an affibody) as the scaffold. The Z domain is derived from the B domain of Staphylococcal Protein A (SpA), which binds Abs at their fragment crystallizable (Fc) region. Therefore, BMPs decorated with the Z domain (termed BMP-Zs) also bind to the Fc moiety of Abs. Abs have a basic structure that resembles the letter Y. The lower part of the Y represents the Fc moiety, while the top part, the Ab fragment (Fab), has an antigen binding site termed the “paratope”.20 When targeting Abs attach to BMP-Zs, their Fab moieties are oriented toward the outside of resulting complexes to maintain targeting ability.
We constructed a gene fragment of a fusion protein, composed of a FLAG tag, cysteine residue (TGC), and two repeat Z domain units in tandem. The linker sequences in front of the Z domain genes are GSGS and GSGSGS (Figure 1A). The fragment was cloned into the pET28(a+) plasmid and expressed in E. coli BL21(DE3). The resulting fusion protein had moieties of two Z domain units belonging to a recombinant affibody, and is referred to hereafter as “RA“. The FLAG tag was used as a label in the western blotting of RA (Figure 1B). ELISA analysis shows that purified RA can be adsorbed by intact rabbit anti-goat IgG-HRP, but not by the Fab moiety of rabbit IgG (Figure 1C), indicating that the two Z domain units in RA retained activity. The cysteine residue had a free sulfhydryl group, which facilitated the linking of RA to primary amino groups of BMPs using the bifunctional reagent SPDP. WT BMPs were isolated from M. gryphiswaldense MSR-1 and digested with proteinase K (Figure 1D) to remove bacterial proteins on BMP surface and minimize experimental “noise” and other possible adverse effects. BMP-WT and RA were linked by SPDP (Figure 1E); the resulting NP complex is hereafter referred to as ”BMP-RA”. BMP-RA actively adsorbed rabbit anti-mouse IgG-HRP, indicating that the Z domain on BMP-RA retains Fc-binding activity (Figure 1F).
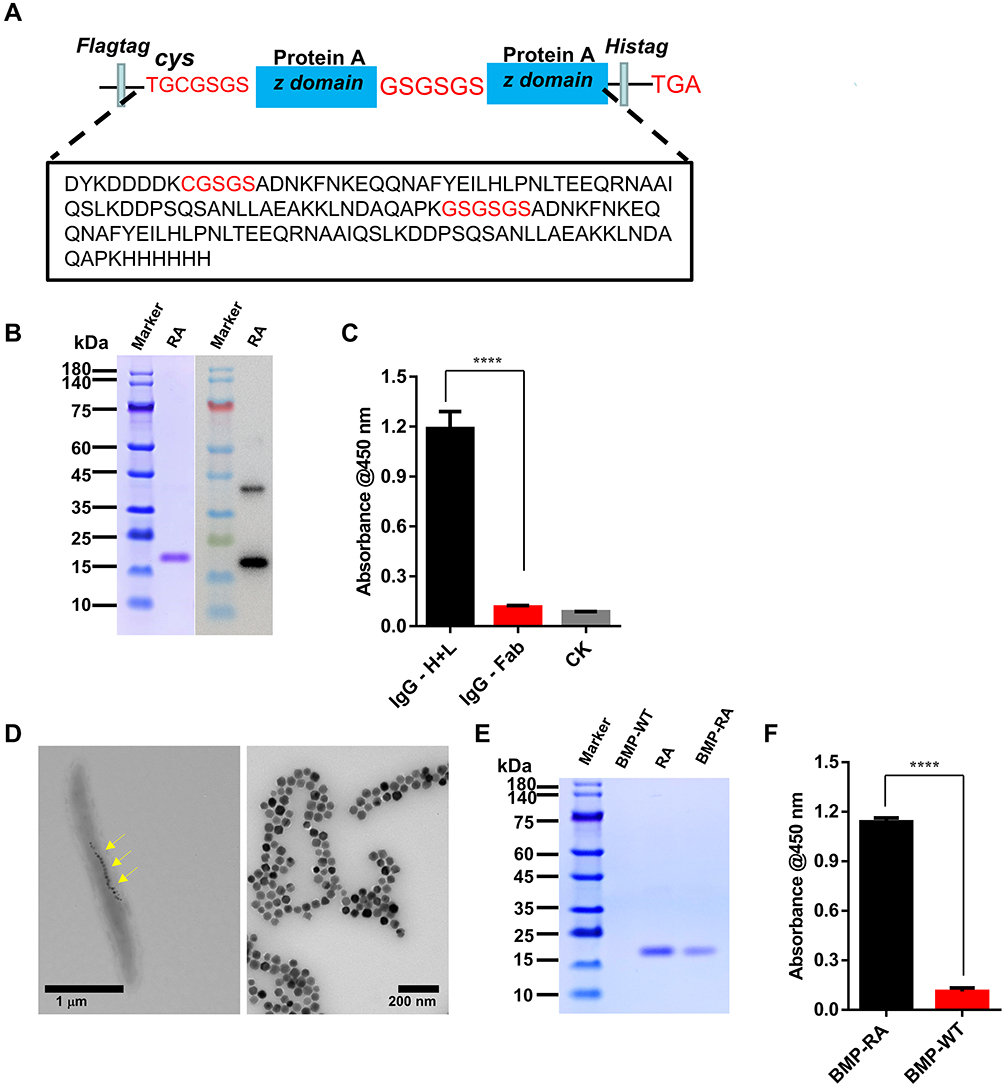 |
Figure 1 Decoration and detection of BMPs with a scaffold fusion protein. (A) Composition (schematic) of recombinant affibody (RA) protein. (B) Purified RAs were separated by SDS-PAGE and analyzed by western blotting with anti-FLAG tag Abs. (C) ELISA with rabbit anti-goat IgG-HRP (IgG-H+L) and rabbit anti-goat Peroxidase-AffiniPure F(ab’)2 Fragment (IgG-Fab) was used to detect the RA binding site of Ab (n=3), CK: control check (no RA added). (D) EM images of M. gryphiswaldense MSR-1 (yellow arrows indicate BMPs) and purified BMPs. (E) SDS-PAGE analysis of RA complexed with BMP membrane (BMP-RA). (F) ELISA analysis of BMP-RA binding to rabbit anti-mouse IgG-HRP (n=3). ****P <0.0001. |
Construction of Targeting BMPs with Humanized Ab
To construct delivery NPs, targeting ligands were linked to BMP-RAs. TZ was used as functional ligand for targeting to HER2-overexpressing cancer cells. TZ is a humanized anti-HER2 mAb used for the treatment of breast and stomach cancer based on Ab-dependent cell-mediated cytotoxicity (ADCC). Humanized Abs have low immunogenicity and avoid the attachment of host molecules to target ligands.34 Incubation of BMP-RAs with TZs led to immobilization of large amounts of TZs on the surface and formation of BMP-RA-TZ complex. SDS-PAGE and protein assay kit were used to measure TZ amount in the complex. TZ directly linked to BMP-WT by glutaraldehyde (BMP-GA-TZ) was used as the control. The engineered BMPs contained similar TZ amounts: 0.2085 ± 0.04861 mg per mg BMP-RA-TZ and 0.1827 ± 0.0522 mg per mg BMP-GA-TZ (Figures 2A and B). TZ also attached to BMP-WT by nonspecific adsorption (referred to as BMP-TZ), but the amount was significantly less than in BMP-RA-TZ and BMP-GA-TZ (Figure S1). Despite similar linkage rates of BMP-RA-TZ and BMP-GA-TZ, the orientation of TZs in these two functionalized NPs was theoretically quite different. TZs on BMP-RA-TZ were presumably highly ordered, with the Fab moiety directed outward,35 whereas TZs on BMP-GA-TZ were oriented in random fashion (Figure 2C). This hypothesis was tested by ELISA analysis of antigen (HER2 protein) amounts on the two engineered BMPs following linkage to HER2. BMP-RA-TZ was found to adsorb >3x more HER2 than BMP-GA-TZ (Figure 2D).
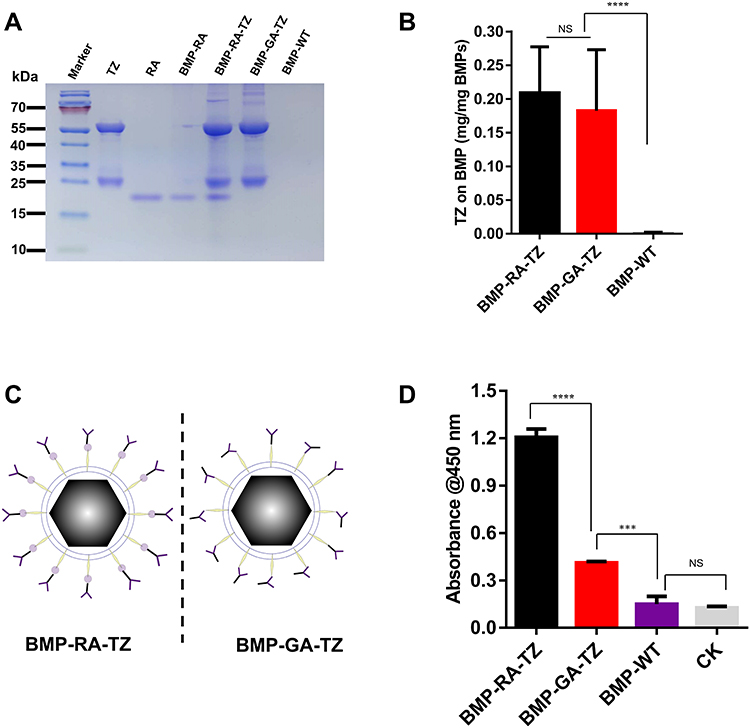 |
Figure 2 Glutaraldehyde (GA) and RA complexes with TZ/BMP (BMP-GA-TZ, BMP-RA-TZ). (A) SDS-PAGE analysis showing presence of TZ on BMP-RA-TZ and BMP-GA-TZ surfaces. (B) TZ quantities, detected by enhanced BCA protein assay kit, similar for BMP-RA-TZ and BMP-GA-TZ (n=3). (C) Orientation of TZ (schematic) on BMP-RA-TZ and BMP-GA-TZ. (D) ELISA analysis of HER2-binding capacity of BMP-RA-TZ and BMP-GA-TZ (n=3), CK: no BMPs added. ***P <0.001; ****P <0.0001. Abbreviation: NS, Not significant. |
Effect of Ab Precoating on BMP Coronas
In organisms, protein coronas may shield or cover active molecules at the NP surface, resulting in mistargeting or unintended scavenging by the liver, kidney, or spleen.36 We hypothesized that Ab binding scaffolds (RAs, in this case) can alter protein components and target ligand orientations in protein corona. We examined and compared protein coronas of our WT and engineered BMPs following incubation with human plasma. Because corona formation on BMPs following plasma incubation resulted in greatly altered surface layer composition (new biological identity),37 we referred to them by adding “-plasma” to the end of the original name; eg “BMP-RA-TZ-plasma” instead of “BMP-RA-TZ”. IgG levels in the blood may increase because of infections, autoimmune processes, inflammation, or malignancy;38 we therefore performed an additional treatment by adding human IgG in normal plasma to simulate the patients’ physiological condition in vitro. To specify this case, the phrase “-IgG plasma” was added to the end of the original name. IgGs are by far the most abundant Abs in the plasma; our subsequent experiments therefore focused only on IgGs rather than all Abs.
Corona components of engineered BMPs were significantly altered. BMP-RA and BMP-GA, incubated with either normal plasma or IgG plasma, bound considerably more IgG in the corona than BMP-WT (Figure S2). The IgG content of BMP-RA-TZ and BMP-GA-TZ coronas was similar to original TZ content (Figures 3A and S3). Components of these BMP coronas were analyzed by liquid chromatography-mass spectrometry (LC-MS). For each type of corona, the 20 proteins with the highest content based on BMP-WT-plasma are listed in Table S1. Protein species, amounts, and ratios in coronas were strongly affected by the pre-adsorption of TZ and by the type of TZ/BMP linkage (Figures S3 and S4; Table S2). The IgG content was slightly higher for BMP-RA-TZ-IgG plasma than for BMP-RA-TZ-plasma. The protein content was higher for BMP-GA-TZ-plasma than for BMP-RA-TZ-plasma. Out of 114 identified proteins, 61 species had higher content for BMP-GA-TZ-plasma than for BMP-RA-TZ-plasma. The average protein molecular weight (MW) was also higher for BMP-GA-TZ-plasma than for BMP-RA-TZ-plasma (Figures 3B and C; Table S3). Among the proteins with higher content in BMP-GA-TZ-plasma than in BMP-RA-TZ-plasma, 39.3% had MW >60 kDa, and 60.7% had MW ≤60 kDa. Among proteins with higher content in BMP-RA-TZ-plasma than in BMP-GA-TZ-plasma, 75.5% had MW ≤60 kDa. These findings indicate that for BMPs with oriented TZ, fewer and smaller host compounds were incorporated into protein coronas.
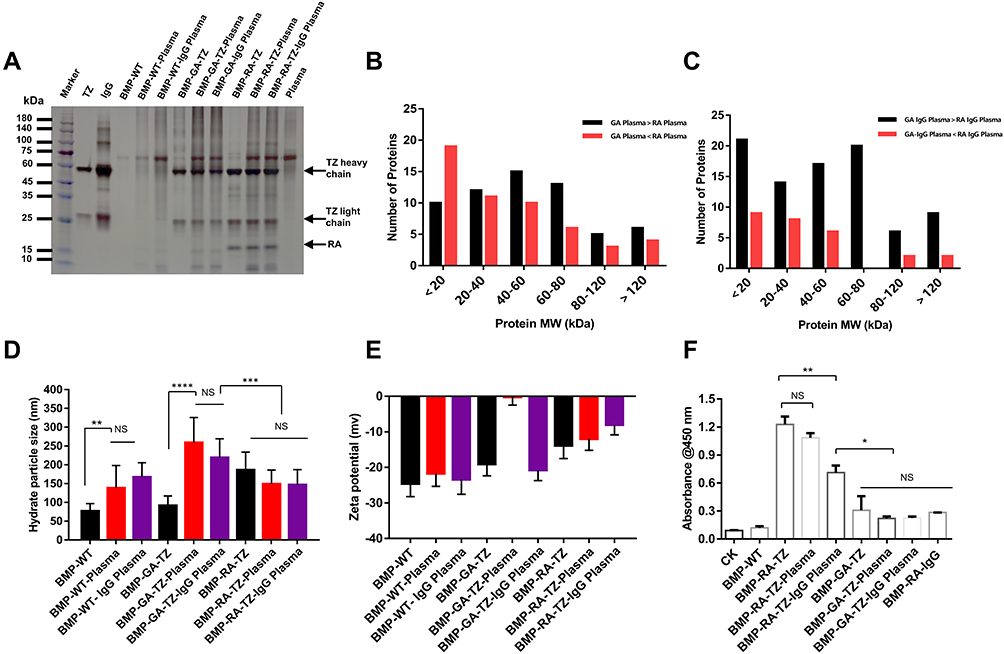 |
Figure 3 Protein corona identification and classification. (A) SDS-PAGE analysis of plasma proteins obtained from BMP-RA-TZ and BMP-GA-TZ following incubation with plasma and IgG plasma. (B and C) Classification based on MW of proteins obtained from BMP-RA and BMP-GA following incubation with plasma and IgG plasma. Average protein MW was higher for BMP-GA-TZ-plasma than for BMP-RA-TZ-plasma. (D) Hydrate particle size analysis of BMPs, BMP-plasma complex, and BMP-IgG plasma complex (n=3). (E) Zeta potential of BMPs, BMP-plasma complex, and BMP-IgG plasma complex (n=3). (F) ELISA analysis of HER2-binding capacity of BMP-RA-TZ and BMP-GA-TZ before and after incubation with plasma and IgG plasma (n=3). *P <0.05; **P <0.01; ***P <0.001; ****P <0.0001. |
TZ pre-adsorption and corona formation affected hydrated radii and zeta potentials of the engineered BMPs. TZ pre-adsorption led to slight increase of hydrated radii and reduced absolute values of zeta potentials. Hydrated radii of GA-TZ-containing BMPs were larger than those of RA-TZ-containing BMPs (Figures 3D and E). The effects of coronas on zeta potentials varied depending on type of BMP. Absolute values of BMP zeta potentials declined to the 10–30 mV range, with the exception of BMP-GA-TZ-plasma. A mixed dispersion system with zeta potential absolute value in this range is considered to have incipient stability;33 thus, it appeared that TZ pre-adsorption and corona formation did not notably affect dispersity of BMPs.
TZ and human IgGs have similar affinities for RA; therefore, some human IgG molecules may competitively bind to BMP-RA-TZ and replace or shield pre-adsorbed TZs. If a large proportion of TZs are repelled or shielded by human IgGs, BMP-RA-TZ-plasma and BMP-RA-TZ-IgG plasma lose their targeting ability. To investigate these possibilities, we used double Ab sandwich ELISA to measure amounts of HER2 bound to BMP-RA-TZ before and after incubation with human plasma. In addition to BMP-GA-TZ, another control (termed BMP-RA-IgG) was constructed by incubating BMP-RA and commercial human IgG. BMP-RA-TZ bound higher amounts of HER2 following incubation with normal plasma than with IgG plasma, suggesting that IgGs could repel or shield some proportion of TZs. In contrast, both BMP-RA-TZ-plasma and BMP-RA-TZ-IgG-plasma bound significantly higher amounts of HER2 than BMP-RA-IgG, BMP-GA-TZ-plasma, and BMP-GA-TZ-IgG plasma, indicating that large amounts of TZ were still present on the BMP-RA-TZ surface (Figure 3F).
Targeting Analysis of TZ Functionalized BMPs
Targeting capacities of the TZ functionalized BMPs were investigated by fluorescence microscopy and flow cytometric analysis. Because TZ is an anti-HER2 mAb, we used two human breast cancer cell lines for evaluation: SK-BR-3 (HER2-overexpressing) and MDA-MB-468 (HER2-negative).39 HER2 expression on the cancer cell surface was determined by the immunofluorescence assay. Anti-HER2 secondary Ab was labeled with Alexa Fluor 488 (bright green fluorescent dye), and cancer cell nuclei were stained by DAPI (blue). CLSM observation revealed surface expression of HER2 on SK-BR-3 but not MDA-MB-468 (Figure S5a). Western blotting analysis yielded similar results (Figure S5b).
Interactions of BMPs with cancer cells were investigated by prestaining BMPs with FITC (green) and cell membranes with DiI (red). Interactions of SK-BR-3 with various BMPs were observed by CLSM. BMPs without RA (BMP-WT, BMP-GA-TZ) were localized mainly bonded to the cell surface. In contrast, BMPs with RA (BMP-RA-TZ, BMP-RA-TZ-plasma, BMP-RA-TZ-IgG plasma) were internalized into the perinuclear region of cancer cells to some degree, in addition to being associated with the cell surface (Figure 4A). These findings suggest that oriented TZ mediated the entry of BMPs into HER2-positive cells. For purposes of flow cytometric analysis (below), cells with engineered BMPs on the surface and cells with some BMPs located internally were not easily distinguishable, and were referred to generally as FITC-positive cells.
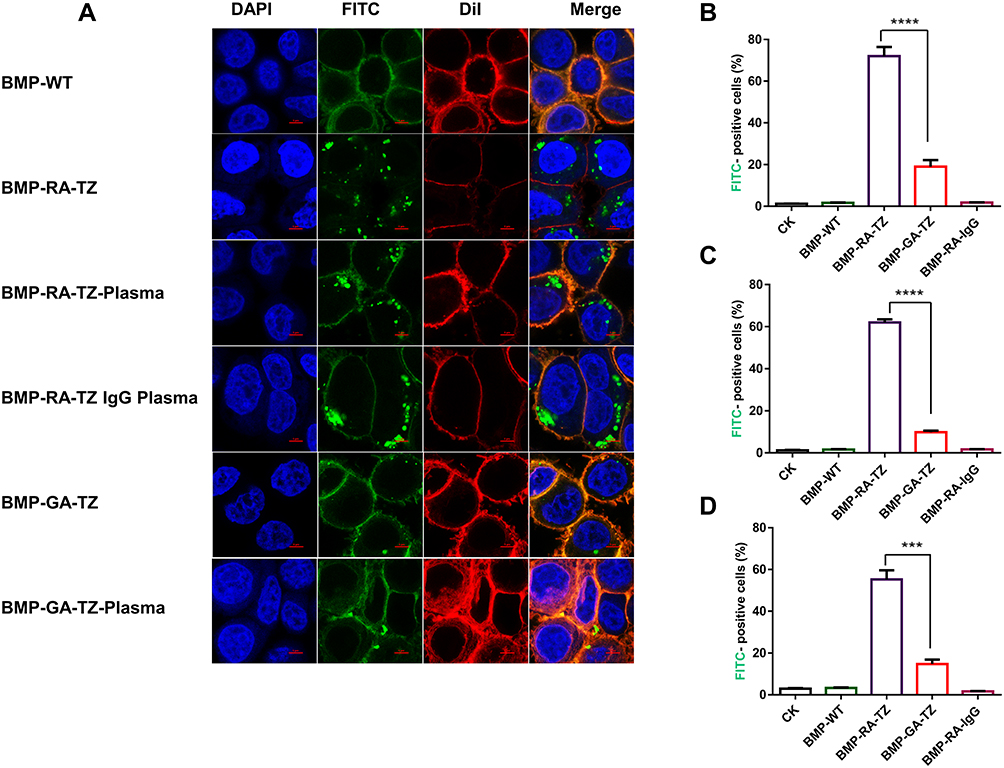 |
Figure 4 Analysis of SK-BR-3 cell uptake of BMPs by CLSM and flow cytometry. (A) CLSM of SK-BR-3 incubated with BMP-RA-TZ and BMP-GA-TZ before and after incubation with plasma and IgG plasma, Scale bar, 5 μm. (B) Percentage (by flow cytometry) of FITC-positive SK-BR-3 after incubation with BMP-RA-TZ and BMP-GA-TZ. (C) Same as (B), using BMP-RA-TZ plasma and BMP-GA-TZ plasma. (D) Same as (B), using BMP-RA-TZ IgG plasma and BMP-GA-TZ IgG plasma. ***P <0.001; ****P <0.0001. |
Percentages of FITC-positive cells were calculated based on flow cytometry. Following incubation with BMP-RA-TZ, BMP-RA-TZ-plasma, and BMP-RA-TZ-IgG plasma, the respective percentages of FITC-positive SK-BR-3 cells were 72%, 61.9%, and 55.2% (Figures 4B–D). FITC-positive percentages were considerably lower for SK-BR-3 incubated with other types of BMPs. BMP-WT, BMP-RA-IgG, BMP-WT-plasma, BMP-RA-IgG-plasma, BMP-WT-IgG plasma, and BMP-RA-IgG-IgG plasma showed essentially no interaction with SK-BR-3. BMP-GA-TZ, BMP-GA-TZ-plasma, and BMP-GA-TZ-IgG plasma interacted to slight degrees with SK-BR-3, but FITC-positive percentages (19%, 9.85%, and 14.8%, respectively) were significantly lower than for RA-TZ-containing BMPs (Figures 4B–D). These findings clearly indicate that targeting capacities of RA-TZ-containing BMPs were significantly stronger than those of GA-TZ-containing BMPs.
Uptake of BMPs by Cancer Cells Mediates TZ Orientation
In view of previous reports that the cellular uptake of nanomaterials is significantly reduced by protein corona,38,40 we evaluated the possibility that TZ/ HER2 interaction mediates internalization of BMPs into breast cancer cells. Although CLSM observation results (preceding section) showed the BMP uptake by some cancer cells, FITC is a dye for proteins; thus, the above results did not rule out the possibility of BMP disintegration when crossing the cell membrane. Prussian blue staining, a method commonly used for detection of iron in biological samples, was applied for microscopic observation.41 By this procedure, BMPs were visualized as blue particles, and cancer cells were dyed red (Figure S6). RA-TZ-containing BMPs were internalized into SK-BR-3, unlike other types of BMPs. These findings confirmed those shown in Figure 4A, and indicate that only oriented TZs can efficiently internalize BMPs into cancer cells.
To simulate human body tumor environment, we incubated BMPs, plasma (100%), and cancer cells together, with the staining procedures described in the preceding section. Treatments with 0% and 10% plasma were performed to evaluate effects of plasma concentration on BMP uptake. CLSM observation showed that BMP-RA-TZ was internalized into SK-BR-3 at each of these three plasma levels, unlike BMP-WT (Figure 5A). Flow cytometric analysis revealed FITC-positive percentages in 0%, 10%, and 100% plasma of 86.5%, 84.6% and 79.4% for BMP-RA-TZ, and 16.4%, 11.7% and 2.61% for BMP-GA-TZ, respectively (Figure 5B). These findings indicate that oriented TZ was required for efficient internalization of BMPs into cancer cells, and that the plasma concentration had negligible effect on the BMP uptake.
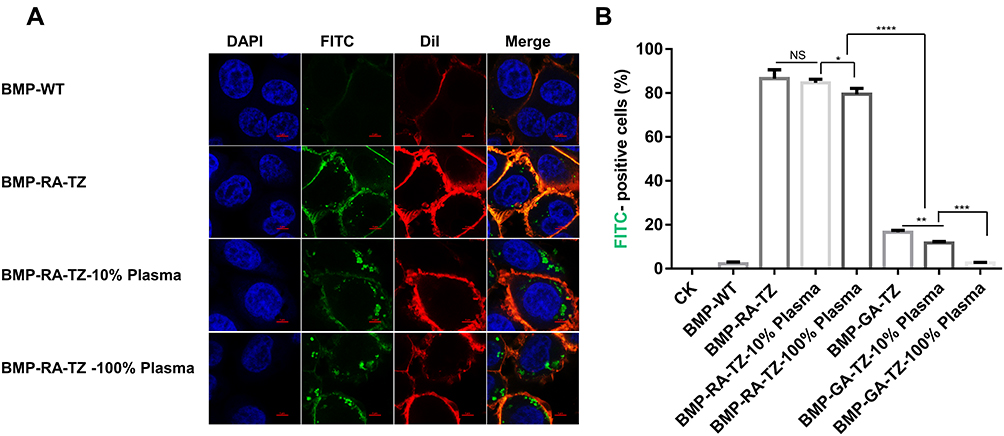 |
Figure 5 Efficiency of BMP-RA-TZ uptake by SK-BR-3 cells at three plasma concentrations. (A) CLSM analysis of SK-BR-3 following incubation with BMP-RA-TZ at three plasma concentrations (0%, 10%, 100%). (B) Percentages (by flow cytometry) of FITC-positive cells for various conditions, scale bars 5 μm. *P <0.05; **P <0.01; ***P <0.001; ****P <0.0001. |
The hypothesis that TZ/HER2 interaction mediates BMP uptake by cancer cells was tested using HER2-negative MDA-MB-468 as negative control. MDA-MB-468 showed no FITC fluorescence upon incubation with WT or engineered BMPs (Figure S7). Flow cytometric analysis in experiments with all BMPs showed that <5% of MDA-MB-468 cells were FITC-positive. These findings indicate that MDA-MB-468 were incapable of the BMP update because of a lack of HER2, and confirm the essential role of oriented TZ in recognition of HER2 and in the BMP uptake by cancer cells.
Discussion
Until the 1980s, the primary category of therapeutic agents was small-molecule drugs (<900 Da, eg chemotherapeutics, antibiotics, steroids). However, only ~10% of the human genome can be targeted by small-molecule drugs.42 New therapeutic functions are available for “new generations” of therapeutic agents developed in recent decades, which include proteins, peptides, mAbs, nucleic acids, and living cells.43 Antibodies, in particular, have great potential as drugs owing to their high target specificity.44 As of 2020, various antibodies were the subjects of >500 ongoing clinical trials and >90 clinical approvals by the US Food and Drug Administration (FDA).45,46 However, the very short half-life of therapeutic Abs means that the application of Ab-based therapy requires multiple administrations and high cost. Ab-based therapy also involves a low degree of tissue penetration. The use of NPs as Ab delivery systems allows the reduction of Ab dosing and increased Ab stability.47 Magnetic NPs (MNPs) are of particular interest as a basis for magnetic drug delivery, as they can be easily conjugated with Abs and delivered to target organs.48 BMPs, as an MNP candidate for drug delivery, have the advantages of single magnetic domain, high crystallinity, strong biocompatibility, and easy surface modification.49 The external boundary of BMPs is a biomembrane, and the large amount of functional groups on surface proteins and lipids facilitates chemical and genetic engineering. In the present study, BMPs adsorbed ~200 μg IgGs per mg, in contrast to 1.5 μg proteins per mg of iron oxide NPs in a previous study.50
An NP-biomolecule (NP-corona) complex is formed following administration of NP into biological fluids, and constitutes the “true identity” of NP in the human body. Mechanisms of corona formation remain poorly understood; however, numerous studies have characterized and identified corona proteins from patient samples.51–53 Ungoverned adsorption of biomolecules onto NPs has multiple effects. These coronas help to decrease the potential cytotoxicity of NPs,54,55 while they also inhibit the accessibility of NP targeting ligands.56 The manipulation and utilization of coronas have potential therapeutic benefits in terms of drug delivery and targeted cancer treatment. Some researchers developed special NP coronas under certain conditions to improve tumoricidal effects,57–60 while others focused on prolonging blood circulation time and enhancing the targeting efficiency of NPs by attempting to minimize or remove NP coronas.14
The coronas of BMPs had not been analyzed until last year.50 Herein, we have developed and described a simple alternative design strategy for directed formation of NP protein corona. Protein scaffolds on BMP surface favor the recruitment of desired proteins (IgG, in this case) in the plasma during corona formation. Full human or humanized Abs (TZ, in this case) can be incorporated (by pre-adsorption) into corona as targeting ligands. The spatial orientation of these Abs can be controlled to enhance their interaction capability with target molecules, and uptake by target cells. Oriented conjugation of Abs to the nanoparticle surface can also be achieved by ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC)/N-hydroxysuccinimide (NHS) chemistry or click chemistry, both of which are necessary to modify the Ab, cumbersome and time consuming.61,62 Our findings indicate that most pre-adsorbed host molecules on protein scaffolds are retained in the corona and maintain their desired functions. In this new design strategy, corona formation is not viewed as a passive obstruction to NP delivery, but rather as a contributor to “NP identity”; ie NP targeting capacity is mediated by specific components (IgG, in this case) in the corona.
Numerous nano-sized particles, especially within 10–20 nm in diameter, are cytotoxic,63,64 while BMPs exhibit selective cytotoxicity with little toxicity to healthy cells and significantly higher toxicity to cancer cells.65 As the engineered BMPs in this study would probably not be fully covered by the IgGs, we assume that their cytotoxicity is similar to the wild type BMP, and that they could be recognized by the human immune system because of their Fc receptors. A similar strategy that might be more promising is to cover the BMPs with numerous human single chain variable fragments (scFvs) in a designed orientation. Because the scFvs have no Fc moiety like IgG, they lack the macrophage surface receptors. If a BMP is sealed by a scFv coat, theoretically it would be a stealth mode for the human immune system. Besides, as the scFv has low cytotoxicity to healthy cells,66 the BMP-scFv complex is probably also non-cytotoxic.
Conclusion
A novel design strategy for nanoparticle (NP) construction was developed and implemented. The strategy considered the effects of the protein corona, with the NP-corona complex considered as the final identity of the functionalized NP. The Z domain-based recombinant affibody (RA) scaffold was linked to BMPs, which was then pre-adsorbed on anti-HER2 humanized mAb (TZ). Following incubation with human plasma, most of the pre-adsorbed TZ molecules were incorporated into the NP-corona complex and maintained their targeting function. RA maintained TZ molecules in a defined spatial orientation that favored binding to the target antigen (HER2), as demonstrated by the significantly higher efficiency in targeting and cellular uptake of BMP-RA-TZ-plasma and BMP-RA-TZ-IgG plasma complexes, relative to counterpart complexes, where BMP and TZ were directly linked to a chemical bifunctional reagent.
Abbreviations
NP, Nanoparticle; BMP, Magnetosome; TZ, Trastuzumab; RA, Recombinant affibody; GA, Glutaraldehyde; SPDP, N-succinimidyl 3-(2-pyridyldithio) propionate; FBS, Fetal bovine serum; ELISA, Enzyme-linked immunosorbent assay; CLSM, Confocal laser scanning microscopy; HER2, human epidermal growth factor receptor 2.
Ethics Approval and Informed Consent
The volunteers in this study have signed the consent form. This study was approved by the Human Ethics Committee of China Agriculture University (certificate no. CAUHR-2021019). All of the procedures were performed in accordance with the Declaration of Helsinki and relevant policies in China.
Consent for Publication
All authors have provided consent for the manuscript to be published.
Acknowledgments
We thank Beijing Guokerongzhi Bio-Technology Co., Ltd for the help in magnetosome preparation. The authors are also grateful to Dr. S. Anderson and the Charlesworth Group for English editing of the manuscript.
Funding
This study was supported by Key Project of Inter-Governmental International Scientific and Technological Innovation Cooperation (2019YFE0115800), the National Natural Science Foundation of China (No. 31570037, 21577170), and the Project for Extramural Scientists of State Key Laboratory of Agro-biotechnology (2020SKLAB6-6), Beijing Municipal Science and Technology Project (Z201100007920010).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Feigin VL, Nichols E, Alam T, et al. Global, regional, and national burden of neurological disorders, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(5):459–480. doi:10.1016/S1474-4422(18)30499-X
2. Alterio D, Marvaso G, Ferrari A, Volpe S, Orecchia R, Jereczek-Fossa BA. Modern radiotherapy for head and neck cancer. Semin Oncol. 2019;46(3):233–245. doi:10.1053/j.seminoncol.2019.07.002
3. Schirrmacher V. From chemotherapy to biological therapy: a review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int J Oncol. 2019;54(2):407–419. doi:10.3892/ijo.2018.4661
4. Scotte F, Ratta R, Beuzeboc P. Side effects of immunotherapy: a constant challenge for oncologists. Curr Opin Oncol. 2019;31(4):280–285. doi:10.1097/CCO.0000000000000541
5. Stewart MP, Sharei A, Ding X, Sahay G, Langer R, Jensen KF. In vitro and ex vivo strategies for intracellular delivery. Nature. 2016;538(7624):183–192. doi:10.1038/nature19764
6. Salvioni L, Rizzuto MA, Bertolini JA, Pandolfi L, Colombo M, Prosperi D. Thirty years of cancer nanomedicine: success, frustration, and hope. Cancers. 2019;11(12):1855. doi:10.3390/cancers11121855
7. Kue CS, Kamkaew A, Burgess K, Kiew LV, Chung LY, Lee HB. Small molecules for active targeting in cancer. Med Res Rev. 2016;36(3):494–575. doi:10.1002/med.21387
8. Akhavan O, Ghaderi E. Graphene nanomesh promises extremely efficient in vivo photothermal therapy. Small. 2013;9(21):3593–3601. doi:10.1002/smll.201203106
9. McDevitt MR, Chattopadhyay D, Kappel BJ, et al. Tumor targeting with antibody-functionalized, radiolabeled carbon nanotubes. J Nucl Med. 2007;48(7):1180–1189. doi:10.2967/jnumed.106.039131
10. Rampado R, Crotti S, Caliceti P, Pucciarelli S, Agostini M. Recent advances in understanding the protein corona of nanoparticles and in the formulation of ”stealthy” nanomaterials. Front Bioeng Biotechnol. 2020;8:166. doi:10.3389/fbioe.2020.00166
11. Salvati A, Pitek AS, Monopoli MP, et al. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule Corona adsorbs on the surface. Nat Nanotechnol. 2013;8(2):37–43. doi:10.1038/nnano.2012.237
12. Mirshafiee V, Kim R, Park S, Mahmoudi M, Kraft ML. Impact of protein pre-coating on the protein corona composition and nanoparticle cellular uptake. Biomaterials. 2016;75:295–304. doi:10.1016/j.biomaterials.2015.10.019
13. Mirshafiee V, Mahmoudi M, Lou K, Cheng J, Kraftm ML. Protein corona significantly reduces active targeting yield. Chem Commun. 2013;49(25):2557–2559. doi:10.1039/c3cc37307j
14. Dai Q, Yan Y, Guo J, et al. Targeting ability of affibody-functionalized particles is enhanced by albumin but inhibited by serum coronas. ACS Macro Lett. 2015;4(11):1259–1263. doi:10.1021/acsmacrolett.5b00627
15. Fam SY, Chee CF, Yong CY, Ho KL, Mariatulqabtiah AR, Tan WS. Stealth coating of nanoparticles in drug-delivery systems. Nanomaterials. 2020;10(4):787. doi:10.3390/nano10040787
16. Peng Q, Zhang S, Yang Q, et al. Preformed albumin Corona, a protective coating for nanoparticles based drug delivery system. Biomaterials. 2013;34(33):8521–8530. doi:10.1016/j.biomaterials.2013.07.102
17. Zhu Y, Chen C, Cao Z, et al. On-demand PEGylation and dePEGylation of PLA-based nanocarriers via amphiphilic mPEG-TK-Ce6 for nanoenabled cancer chemotherapy. Theranostics. 2019;9(26):8312–8320. doi:10.7150/thno.37128
18. Pinals RL, Chio L, Ledesma F, Landry MP. Engineering at the nano-bio interface: harnessing the protein corona towards nanoparticle design and function. Analyst. 2020;145(15):5090–5112. doi:10.1039/D0AN00633E
19. Hajipour MJ, Raheb J, Akhavan O, et al. Personalized disease-specific protein corona influences the therapeutic impact of graphene oxide. Nanoscale. 2015;7(19):8978–8994. doi:10.1039/C5NR00520E
20. Corbo C, Molinaro R, Tabatabaei M, Farokhzad OC, Mahmoudi M. Personalized protein Corona on nanoparticles and its clinical implications. Biomater Sci. 2017;5(3):378–387. doi:10.1039/C6BM00921B
21. Uebe R, Schuler D. Magnetosome biogenesis in magnetotactic bacteria. Nat Rev Microbiol. 2016;14(10):621–637. doi:10.1038/nrmicro.2016.99
22. Alphandéry E. Applications of magnetosomes synthesized by magnetotactic bacteria in medicine. Front Bioeng Biotechnol. 2014;2:5. doi:10.3389/fbioe.2014.00005
23. Nan X, Teng Y, Tian J, Hu Z, Fang Q. A comprehensive assessment of the biocompatibility of Magnetospirillum gryphiswaldense MSR-1 bacterial magnetosomes in vitro and in vivo. Toxicology. 2021;462:152949. doi:10.1016/j.tox.2021.152949
24. Han L, Li S, Yang Y, Zhao F, Huang J, Chang J. Comparison of magnetite nanocrystal formed by biomineralization and chemosynthesis. J Magn Magn Mater. 2007;313(1):236–242. doi:10.1016/j.jmmm.2007.01.004
25. Nygren PÅ. Alternative binding proteins: affibody binding proteins developed from a small three‐helix bundle scaffold. FEBS J. 2008;275(11):2668–2676. doi:10.1111/j.1742-4658.2008.06438.x
26. Gebauer M, Skerra A. Engineered protein scaffolds as next-generation therapeutics. Annu Rev Pharmacol Toxicol. 2020;60:391–415. doi:10.1146/annurev-pharmtox-010818-021118
27. Liu J, Ding Y, Jiang W, Tian J, Li Y, Li J. A mutation upstream of an ATPase gene significantly increases magnetosome production in Magnetospirillum gryphiswaldense. Appl Microbiol Biotechnol. 2008;81(3):551–558. doi:10.1007/s00253-008-1665-1
28. Xu J, Liu L, He J, et al. Engineered magnetosomes fused to functional molecule (protein A) provide a highly effective alternative to commercial immunomagnetic beads. J Nanobiotechnology. 2019;17(1):37. doi:10.1186/s12951-019-0469-z
29. Guo F, Liu Y, Chen Y, et al. A novel rapid and continuous procedure for large-scale purification of magnetosomes from Magnetospirillum gryphiswaldense. Appl Microbiol Biotechnol. 2011;90(4):1277–1283. doi:10.1007/s00253-011-3189-3
30. Geng Y, Wang J, Wang X, et al. Growth-inhibitory effects of anthracycline-loaded bacterial magnetosomes against hepatic cancer in vitro and in vivo. Nanomedicine. 2019;14(13):1663–1680. doi:10.2217/nnm-2018-0296
31. Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68(5):850–858. doi:10.1021/ac950914h
32. Ishihama Y, Oda Y, Tabata T. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics. 2005;4(9):1265–1272. doi:10.1074/mcp.M500061-MCP200
33. Kapingidza AB, Kowal K, Chruszcz M. Antigen-antibody complexes. Subcell Biochem. 2020;94:465–497.
34. Waldmann H. Human monoclonal antibodies: the benefits of humanization. Methods Mol Biol. 2019;1904:1–10.
35. Tripathi K, Driskell JD. Quantifying bound and active antibodies conjugated to gold nanoparticles: a comprehensive and robust approach to evaluate immobilization chemistry. ACS Omega. 2018;3(7):8253–8259. doi:10.1021/acsomega.8b00591
36. Monopoli MP, Walczyk D, Campbell A, et al. Physical-chemical aspects of protein corona: relevance to in vitro and in vivo biological impacts of nanoparticles. J Am Chem Soc. 2011;133(8):2525–2534. doi:10.1021/ja107583h
37. Nierenberg D, Khaled AR, Flores O. Formation of a protein corona influences the biological identity of nanomaterials. Rep Pract Oncol Radiother. 2018;23(4):300–308. doi:10.1016/j.rpor.2018.05.005
38. Notarangelo LD, Duse M, Ugazio AG. Immunodeficiency with hyper-IgM (HIM). Immunodefic Rev. 1992;3(2):101–121.
39. Mota AL, Evangelista AF, Macedo T, et al. Molecular characterization of breast cancer cell lines by clinical immunohistochemical markers. Oncol Lett. 2017;13(6):4708–4712. doi:10.3892/ol.2017.6093
40. Carter P, Presta L, Gorman CM, et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A. 1992;89(10):4285–4289. doi:10.1073/pnas.89.10.4285
41. Hall AP, Davies W, Stamp K, Clamp I, Bigley A. Comparison of computerized image analysis with traditional semiquantitative scoring of Perls’ Prussian Blue stained hepatic iron deposition. Toxicol Pathol. 2013;41(7):992–1000. doi:10.1177/0192623313476576
42. Milletti F. Cell-penetrating peptides: classes, origin, and current landscape. Drug Discov Today. 2012;17(15–16):850–860. doi:10.1016/j.drudis.2012.03.002
43. Vargason AM, Anselmo AC, Mitragotri S. The evolution of commercial drug delivery technologies. Nat Biomed Eng. 2021;5(9):951–967. doi:10.1038/s41551-021-00698-w
44. Marrocco I, Romaniello D, Yarden Y. Cancer immunotherapy: the Dawn of antibody cocktails. Methods Mol Biol. 2019;1904:11–51.
45. Lu RM, Hwang YC, Liu IJ, et al. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27(1):1. doi:10.1186/s12929-019-0592-z
46. Kaplon H, Reichert JM. Antibodies to watch in 2021. MAbs. 2021;13(1):1860476. doi:10.1080/19420862.2020.1860476
47. Sousa F, Castro P, Fonte P, et al. Nanoparticles for the delivery of therapeutic antibodies: dogma or promising strategy? Expert Opin Drug Deliv. 2017;14(10):1163–1176. doi:10.1080/17425247.2017.1273345
48. Peng XH, Qian X, Mao H, et al. Targeted magnetic iron oxide nanoparticles for tumor imaging and therapy. Int J Nanomed. 2008;3(3):311–321. doi:10.2147/ijn.s2824
49. Alphandery E. Applications of magnetotactic bacteria and magnetosome for cancer treatment: a review emphasizing on practical and mechanistic aspects. Drug Discov Today. 2020;25(8):1444–1452. doi:10.1016/j.drudis.2020.06.010
50. Lai W, Li D, Wang Q, et al. A protein Corona adsorbed to a bacterial magnetosome affects its cellular uptake. Int J Nanomedicine. 2020;15:1481–1498. doi:10.2147/IJN.S220082
51. Kopac T. Protein corona, understanding the nanoparticle-protein interactions and future perspectives: a critical review. Int J Biol Macromol. 2021;169:290–301. doi:10.1016/j.ijbiomac.2020.12.108
52. Di Santo R, Digiacomo L, Quagliarini E, et al. Personalized graphene oxide-protein corona in the human plasma of pancreatic cancer patients. Front Bioeng Biotechnol. 2020;8:491. doi:10.3389/fbioe.2020.00491
53. Caputo D, Papi M, Coppola R, et al. A protein Corona-enabled blood test for early cancer detection. Nanoscale. 2017;9(1):349–354. doi:10.1039/C6NR05609A
54. Hu W, Peng C, Lv M, et al. Protein Corona-mediated mitigation of cytotoxicity of graphene oxide. ACS Nano. 2011;5(5):3693–3700. doi:10.1021/nn200021j
55. Mbeh DA, Akhavan O, Javanbakht T, Mahmoudi M, Yahia L. Cytotoxicity of protein Corona-graphene oxide nanoribbons on human epithelial cells. Appl Surf Sci. 2014;320:596–601. doi:10.1016/j.apsusc.2014.09.155
56. Monopoli MP, Aberg C, Salvati A, Dawson KA. Biomolecular coronas provide the biological identity of nanosized materials. Nat Nanotechnol. 2012;7(12):779–786. doi:10.1038/nnano.2012.207
57. Ye ELL, Joshua U, Cheah J, et al. Exploiting the protein Corona around gold nanorods for low-dose combined photothermal and photodynamic therapy. J Mater Chem B. 2017;5(2):254–268. doi:10.1039/C6TB02743A
58. Hajipour MJ, Akhavan O, Meidanchi A, Laurent S, Mahmoudi M. Hyperthermia-induced protein Corona improves the therapeutic effects of zinc ferrite spinel-graphene sheets against cancer. RSC Adv. 2014;4(107):62557–62565. doi:10.1039/C4RA10862K
59. Assali A, Razzazan S, Akhavan O, Mottaghitalab F, Adeli M, Atyabi F. The bio-interface between functionalized Au NR@ GO nanoplatforms with protein Corona and their impact on delivery and release system. Colloids Surf B Biointerfaces. 2019;173:89–1898.
60. Yeo ELL, Thong PSP, Soo KC, Kah JCY. Protein corona in drug delivery for multimodal cancer therapy in vivo. Nanoscale. 2018;10(5):2461–2472. doi:10.1039/C7NR08509E
61. Haghighi AH, Khorasani MT, Faghih Z, Farjadian F. Effects of different quantities of antibody conjugated with magnetic nanoparticles on cell separation efficiency. Heliyon. 2020;6(4):e03677. doi:10.1016/j.heliyon.2020.e03677
62. Jeong S, Park JY, Cha MG, et al. Highly robust and optimized conjugation of antibodies to nanoparticles using quantitatively validated protocols. Nanoscale. 2017;9(7):2548–2555. doi:10.1039/C6NR04683E
63. Akhavan O, Ghaderi E, Akhavan A. Size-dependent genotoxicity of graphene nanoplatelets in human stem cells. Biomaterials. 2012;33(32):8017–8025. doi:10.1016/j.biomaterials.2012.07.040
64. Yang H, Liu C, Yang D, Zhang H, Xi Z. Comparative study of cytotoxicity, oxidative stress and genotoxicity induced by four typical nanomaterials: the role of particle size, shape and composition. J Appl Toxicol. 2009;29(1):69–78. doi:10.1002/jat.1385
65. Hamdous Y, Chebbi I, Mandawala C, et al. Biocompatible coated magnetosome minerals with various organization and cellular interaction properties induce cytotoxicity towards RG-2 and GL-261 glioma cells in the presence of an alternating magnetic field. J Nanobiotechnology. 2017;15(1):1–18. doi:10.1186/s12951-017-0293-2
66. Ahmad ZA, Yeap SK, Ali AM, Ho WY, Alitheen NBM, Hamid M. scFv antibody: principles and clinical application. Clin Dev Immunol. 2012;2012:1–15. doi:10.1155/2012/980250
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.