Back to Journals » OncoTargets and Therapy » Volume 11
STK39 blockage by RNA interference inhibits the proliferation and induces the apoptosis of renal cell carcinoma
Authors Zhao Q, Zhu YJ, Liu L, Wang H, Jiang S, Hu X, Guo J
Received 11 October 2017
Accepted for publication 9 December 2017
Published 16 March 2018 Volume 2018:11 Pages 1511—1519
DOI https://doi.org/10.2147/OTT.S153806
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Qi Zhao,* Yanjun Zhu,* Li Liu, Hang Wang, Shuai Jiang, Xiaoyi Hu, Jianming Guo
Department of Urology, Zhongshan Hospital, Fudan University, Shanghai, China
*These authors contributed equally to this work
Aim: Renal cell carcinoma (RCC), the most frequent type of primary renal malignancies, has a high mortality rate. Serine/threonine kinase 39 (STK39) is associated with various human diseases, including cancers. The current study aimed to investigate the functions of STK39 in RCC.
Materials and methods: STK39 expression levels in RCC and paired normal renal tissue samples were detected by real-time polymerase chain reaction and Western blotting analyses. The biological functions of STK39 were explored in two RCC cell lines with STK39 silence.
Results: STK39 expression was significantly increased in RCC tissues than in normal renal tissues. Suppression of STK39 expression in ACHN and 786-0 cells significantly suppressed cell proliferation and induced cell apoptosis. Consistently, the expression of PCNA and Bcl-2 was remarkably increased, while the expression of Bax was significantly in STK39 knockdown cells compared to control cells. Furthermore, gene set enrichment analysis identified STK39 as an important regulator of p53 and p38 signaling pathways. STK39 knockdown increased p53 expression and inhibited p38 phosphorylation. Moreover, ectopic expression of STK39 in ACHN cells resulted in a reduced p53 expression and increased c-Myc and p-p38 expression. Such effects were suppressed by p38 inhibitor (SB203580).
Conclusion: STK39 may exert its oncogenic function in RCC through p38 signaling. Our data suggest that STK39 may represent a potential therapeutic target against RCC.
Keywords: STK39, renal cell carcinoma, apoptosis, p38
Introduction
Renal cell carcinoma (RCC) is the most frequent type of primary renal neoplasms, accounting for ~90% of cases.1 Various histologic subtypes of RCC have been described, of which clear cell RCC (ccRCC) is the major subtype.2 Metastasis is seen in 20%–30% of RCC patients during diagnosis.3 With the improvement in diagnostic and therapeutic techniques, the 5-year survival rate of RCC has increased to ~55%. Unfortunately, metastatic RCC has a 5-year survival rate of only ~10%.4 The mechanism underlying the development and progression of RCC is far from being completely understood. Thus, there is a strong necessity to identify potential diagnostic markers and pathways or proteins actively engaged in RCC carcinogenesis.
Serine/threonine kinase 39 (STK39, also known as SPAK/PASK/STE20-SPS1 homolog) belongs to the Ste20-like kinase family. It contains an N-terminal proline and alanine repeats (PAPA box), a serine/threonine kinase catalytic domain, a putative nuclear localization signal, and a C-terminal region.5 STK39 interacts with oxidative stress response 1 and several cation chloride cotransporters, KCC3, NKCC1, and NKCC2, thus regulating fluid/ion homeostasis.6 STK39, as a Ser/Thr kinase, is able to activate the mitogen-activated protein kinase p38 signaling pathway, suggesting its involvement in stress responses.5 Recently, increasing evidence has accumulated supporting the roles of STK39 in human diseases. Genetic variants of STK39 are associated with autism,7 hypertension,8–11 and Parkinson’s disease.12,13 Reduced expression of STK39 is correlated with prostate cancer metastasis,14 as well as treatment resistance of breast cancer.15 STK39 is being silenced in B-cell lymphoma and STK39 knockdown protects B cells from DNA double-strand breaks-induced apoptosis through c-Jun NH2-terminal kinase (JNK).16 On the contrary, STK39 expression correlated significantly with the prognosis of non-small-cell-type lung cancer (NSCLC).17 Nevertheless, no reports have described the expression and functions of STK39 in RCC.
In the current study, we analyzed the mRNA and protein expression of STK39 in RCC and paired adjacent normal renal tissues in a cohort of 25 patients. The functions of STK39 on cell proliferation and apoptosis were also investigated. Our results indicated that STK39 may exert anti-apoptotic function in RCC.
Materials and methods
Patients and tissue samples
Ethical approval (B2016-161R) was provided by the Ethics Committee of Zhongshan Hospital (Shanghai, China). Twenty-five patients (14 males and 11 females) with RCC were chosen from Zhongshan Hospital between 2011 and 2013. Written informed consent was collected from all the enrolled patients in accordance to the guidelines of the Ethics Committee. The average age of these patients was 59±9 years. The study cohort consisted of 13 patients at stage I and 12 patients at stage II. During surgical procedures, RCC samples and matched adjacent normal renal samples were collected, fresh-frozen, and stored in −80°C.
RNA isolation, reverse transcription, and real-time polymerase chain reaction (PCR)
Total RNA was isolated with Trizol reagent according to the manufacturer’s protocol (Invitrogen, Carlsbad, CA, USA). Following treatment with DNase to avoid DNA contamination, NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Rockford, IL, USA) was used to determine the concentrations of RNA samples. Two microgram of total RNA was reverse transcribed by the two-step RT-PCR system (Thermo Fisher Scientific). Subsequently, real-time PCR was performed on an ABI 7300 instrument (Applied Biosystems, Foster City, CA, USA) with the complementary DNA as template by using SYBR Green PCR kit (Thermo Fisher Scientific). The primers for STK39 were 5′-TGCCAGACCAGTATGGATGAA-3′ (forward) and 5′-GGTGTAATAGGTCACTACGTTGG-3′ (reverse), and primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were 5′-CTGGGCTACACTGAGCACC-3′ (forward) and 5′-AAGTGGTCGTTGAGGGCAATG-3′ (reverse). Each sample was run in triplicate. STK39 expression normalized with the internal control GAPDH was calculated using the 2−ΔΔCT method as previously described.18
Protein extraction and Western blotting
Tissue samples and cells were lysed in ice-cold radioimmunoprecipitation assay buffer buffer (Beyotime, Shanghai, China) with fresh-added protease inhibitor cocktail (Sigma-Aldrich, St Louis, MO, USA). Total cell lysates with equal amount of protein for each sample were loaded to sodium dodecyl sulfate-polyacrylamide gel and then transferred to nitrocellulose membranes (Millipore, Bradford, MA, USA). The membranes were incubated with 5% non-fat milk in Tris buffer containing Tween-20 (TBST) for 1 h at room temperature to block non-specific binding. Subsequently, the membranes were probed with primary antibodies (anti-STK39 and anti-c-Myc [Abcam, Cambridge, MA, USA]; anti-Bcl-2 and anti-Bax [Santa Cruz Biotech, Santa Cruz, CA, USA]; anti-p53, anti-p-p38, anti-p38, and anti-GAPDH [Cell Signaling Technology, Danvers, MA, USA]) following the manufacturers’ instructions. After washing three times with TBST, the corresponding horseradish peroxidase-conjugated secondary antibodies (Beyotime) were added. After incubation for 1 h at room temperature and after washing three times with TBST, signals were detected with enhanced chemiluminescence system (Bio-Rad, Richmond, CA, USA). The band density was quantified using ImageJ (http://rsb.info.nih.gov/ij/, Bethesda, MD, USA).
Bioinformatics analysis
A microarray dataset of 72 ccRCC samples and paired non-tumorous samples was downloaded from NCBI Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/). The STK39 expression between the RCC samples and normal samples was compared by Student’s t-test.
To elucidate the possible processes and pathways associated with STK39 expression, Gene set enrichment analysis (GSEA) was performed on the Cancer Genome Atlas (TCGA; https://tcga-data.nci.nih.gov/tcga/) RCC cohort using the GSEA software version 2.0 as previously described.17
Cell culture and treatment
ACHN, 786-0, and HK-2 cells were purchased from the Shanghai Institute of Cell Biology (Shanghai, China). Cells were grown in Dulbecco’s Modified Eagle’s Medium (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA) and penicillin/streptomycin at 37°C with 5% CO2.
siRNA oligonucleotides targeting STK39 (STK39 siRNA) or scrambled control siRNA (Control siRNA) were ordered from Genepharm Technologies (Shanghai, China). siRNA at a concentration of 120 nM was transfected into ACHN and 786-0 cells using Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions. After 48 h, the knockdown efficiency was determined by real-time PCR and Western blotting.
Full-length STK39 cells were cloned into the expression vector pCDNA3.1 (Invitrogen). ACHN cells were transfected with control vector or expression plasmid encoding STK39 and treated with either dimethyl sulfoxide (DMSO) or 10 μM p38 inhibitor, SB203580 (Merck, Darmstadt, Germany). After 48 h, protein samples were extracted and subjected to Western blotting.
Cell proliferation analysis
Cell counting kit-8 (CCK-8; Beyotime) assays were conducted to detect cell proliferation according to the manufacturer’s protocols. In brief, ACHN and 786-0 cells were seeded in 96-well plates. Following culture overnight, the cells were transfected with STK39 siRNA or control siRNA. After incubation for indicated time periods, CCK-8 solution was dispensed to every well. Following culture for 1 h, optical density at a wavelength of 450 nm (OD 450) was determined with a Model 550 microplate reader (BioRad, Richmond, CA, USA).
Apoptosis analysis
ACHN and 786-0 cells were plated in 6-well plates at a density of 3×105 per well. After culturing overnight to allow cell adherence, transfection of STK39 siRNA or control siRNA was carried out as described earlier. At 48 h post-transfection, cell apoptosis was measured by labeling with Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) (eBioscience, San Diego, CA, USA) followed by fluorescence-activated cell sorting (FACScalibur; BD Biosciences, Franklin Lakes, NJ, USA). The percentages of apoptotic cells (lower right quadrant) were estimated from three independent experiments.
Statistical analysis
All experiments were performed three times. Statistical analysis was performed with GraphPad Prism 6.0 (GraphPad Software Inc., La Jolla, CA, USA). Comparisons between two groups and among more than two groups were performed using Student’s t-test and one-way ANOVA followed by Tukey’s post-test, respectively. A P-value less than 0.05 was considered as statistically significant.
Results
Expression of STK39 is elevated in RCC tissues
To determine whether STK39 expression is upregulated in RCC, 25 pairs of RCC tissues and adjacent non-tumorous normal renal tissues were collected and STK39 mRNA expression was evaluated by real-time PCR. The results showed that STK39 mRNA levels were significantly greater in RCC tissues than in adjacent normal tissues (Figure 1A, P<0.0001). Furthermore, a microarray dataset (accession number: GSE53757) of 72 ccRCC samples and paired non-tumorous sample was downloaded from the NCBI GEO (https://www.ncbi.nlm.nih.gov/geo/). The public available dataset also showed that the expression of STK39 mRNA was increased in human RCC tissues (Figure 1B, P<0.001). We then performed Western blotting to detect STK39 protein expression in 8 paired RCC and normal tissues. The results indicated that STK39 expression was significantly elevated in tumor tissues at translation level (Figure 1C, P<0.05).
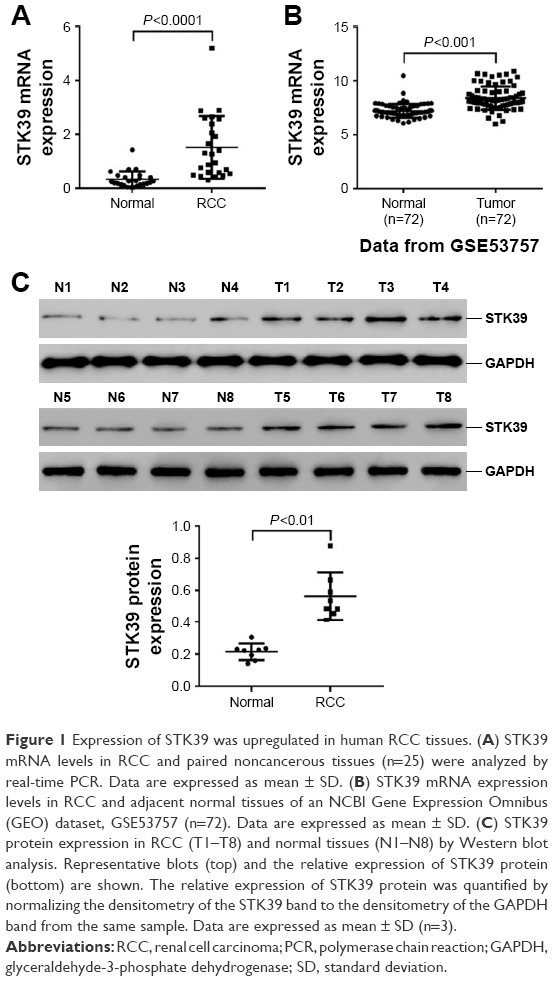 | Figure 1 Expression of STK39 was upregulated in human RCC tissues. (A) STK39 mRNA levels in RCC and paired noncancerous tissues (n=25) were analyzed by real-time PCR. Data are expressed as mean ± SD. (B) STK39 mRNA expression levels in RCC and adjacent normal tissues of an NCBI Gene Expression Omnibus (GEO) dataset, GSE53757 (n=72). Data are expressed as mean ± SD. (C) STK39 protein expression in RCC (T1–T8) and normal tissues (N1–N8) by Western blot analysis. Representative blots (top) and the relative expression of STK39 protein (bottom) are shown. The relative expression of STK39 protein was quantified by normalizing the densitometry of the STK39 band to the densitometry of the GAPDH band from the same sample. Data are expressed as mean ± SD (n=3). |
Silencing of STK39 by RNA interference (RNAi)
Real-time PCR and Western blot analyses were performed to evaluate the mRNA and protein expression of STK39 in two RCC cell lines, ACHN and 786-0, and human renal proximal tubule cell line, HK-2, respectively (Figure 2A). The expression of STK39 was significantly higher in both RCC cell lines than in HK-2 cells.
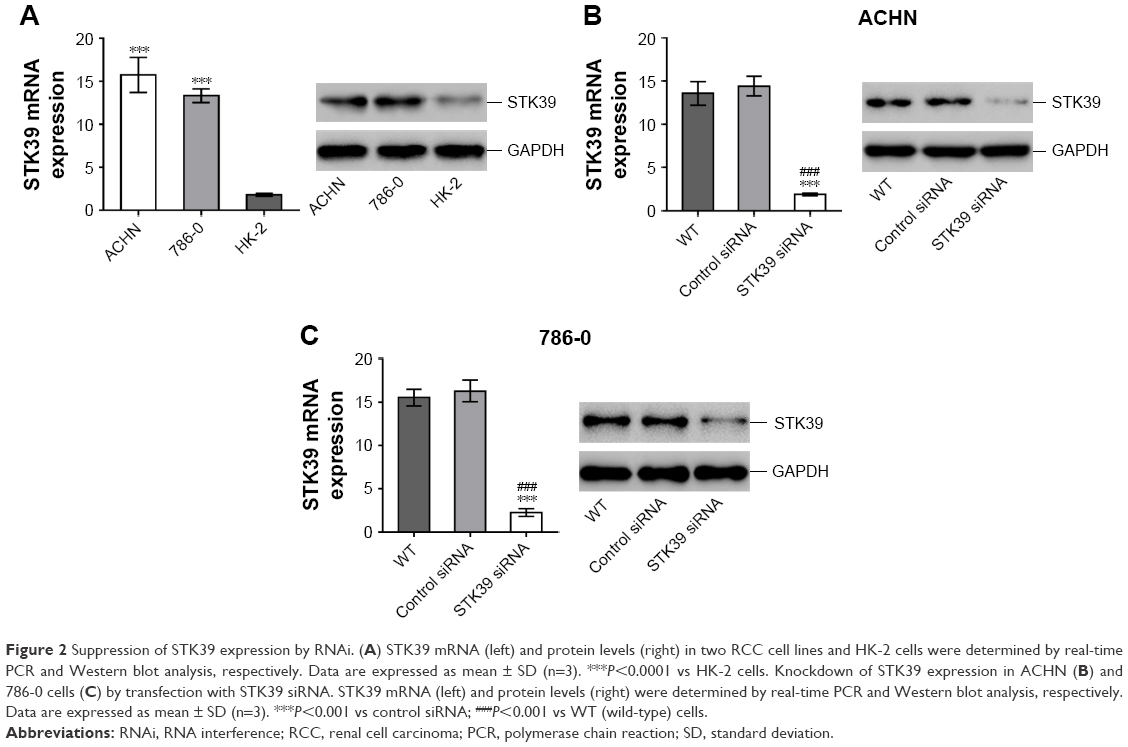 | Figure 2 Suppression of STK39 expression by RNAi. (A) STK39 mRNA (left) and protein levels (right) in two RCC cell lines and HK-2 cells were determined by real-time PCR and Western blot analysis, respectively. Data are expressed as mean ± SD (n=3). ***P<0.0001 vs HK-2 cells. Knockdown of STK39 expression in ACHN (B) and 786-0 cells (C) by transfection with STK39 siRNA. STK39 mRNA (left) and protein levels (right) were determined by real-time PCR and Western blot analysis, respectively. Data are expressed as mean ± SD (n=3). ***P<0.001 vs control siRNA; ###P<0.001 vs WT (wild-type) cells. |
To study the functions of STK39 in RCC cells, firstly, we used a specific siRNA against STK39 to knockdown the expression of STK39 in ACHN and 786-0 cells. Using real-time PCR and Western blotting, we confirmed the successful knockdown of STK39 in both cell lines with a knockdown efficiency of about 85% (Figure 2B and C). Transfection with control siRNA had no effects on the expression of STK39 as compared to cells without any treatment (wild-type, WT).
STK39 knockdown inhibits RCC cell proliferation
CCK-8 assays were then performed to assess the changes to RCC cell proliferation following STK39 knockdown. As demonstrated in Figure 3, the proliferation of ACHN and 786-0 cells was significantly reduced in STK39 siRNA-transfected cells compared with relevant WT and control siRNA-transfected cells. These findings suggested that downregulation of STK39 expression inhibited RCC cell proliferation in vitro.
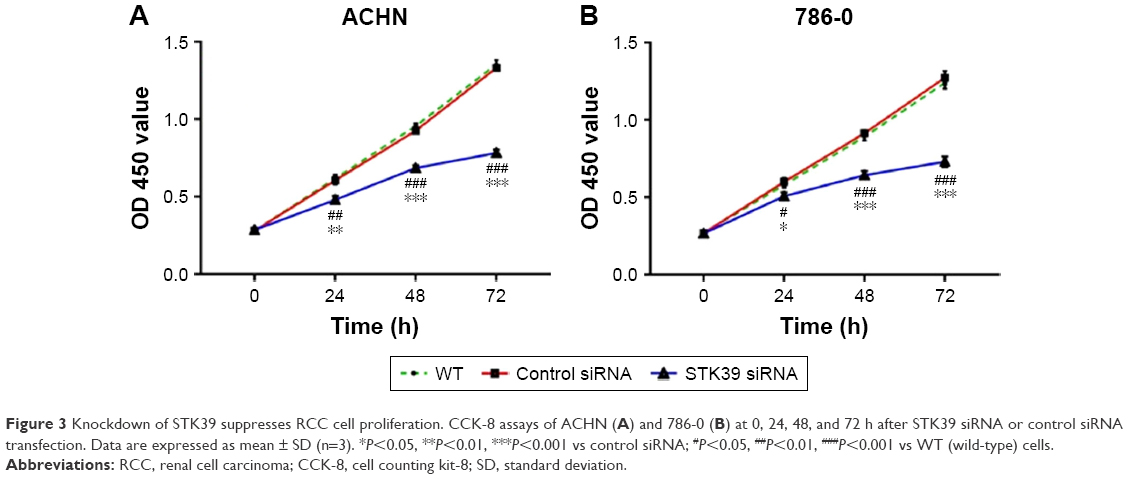 | Figure 3 Knockdown of STK39 suppresses RCC cell proliferation. CCK-8 assays of ACHN (A) and 786-0 (B) at 0, 24, 48, and 72 h after STK39 siRNA or control siRNA transfection. Data are expressed as mean ± SD (n=3). *P<0.05, **P<0.01, ***P<0.001 vs control siRNA; #P<0.05, ##P<0.01, ###P<0.001 vs WT (wild-type) cells. |
STK39 knockdown induces RCC cell apoptosis
We evaluated RCC cell apoptosis after STK39 knockdown by Annexin V-FITC/PI staining and flow cytometry analysis. As shown in Figure 4A and B, knockdown of STK39 in ACHN and 786-0 cells caused a significant increase in the rate of cell apoptosis as compared with cells transfected with control siRNA or without any treatment (WT).
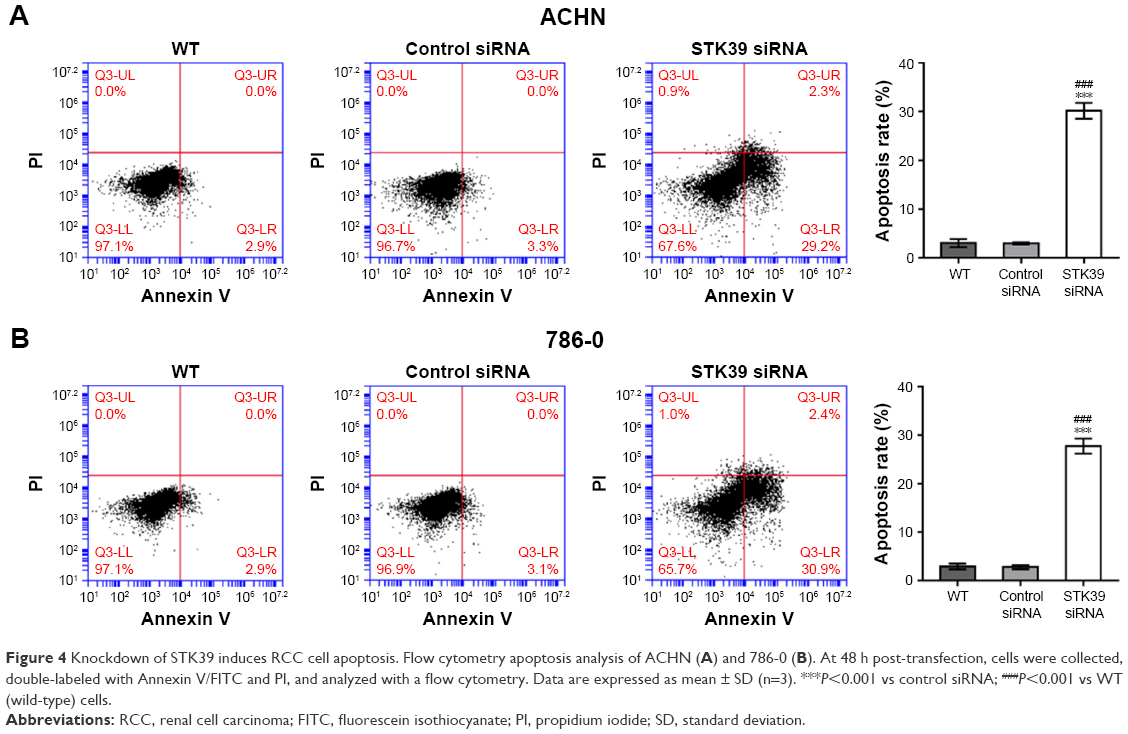 | Figure 4 Knockdown of STK39 induces RCC cell apoptosis. Flow cytometry apoptosis analysis of ACHN (A) and 786-0 (B). At 48 h post-transfection, cells were collected, double-labeled with Annexin V/FITC and PI, and analyzed with a flow cytometry. Data are expressed as mean ± SD (n=3). ***P<0.001 vs control siRNA; ###P<0.001 vs WT (wild-type) cells. |
STK39 knockdown affects the expression of PCNA, Bcl-2, and Bax
In addition, we compared the expression level of proliferation marker, PCNA,19 and apoptosis-regulating proteins, Bcl-2 and Bax,20 by Western blotting. We found that PCNA and the anti-apoptosis protein Bcl-2 were reduced in STK39 siRNA-transfected RCC cells compared to WT and control siRNA-transfected cells, while the apoptosis-promoting protein Bax was increased (Figure 5). These findings confirmed the results of CCK-8 and flow cytometry.
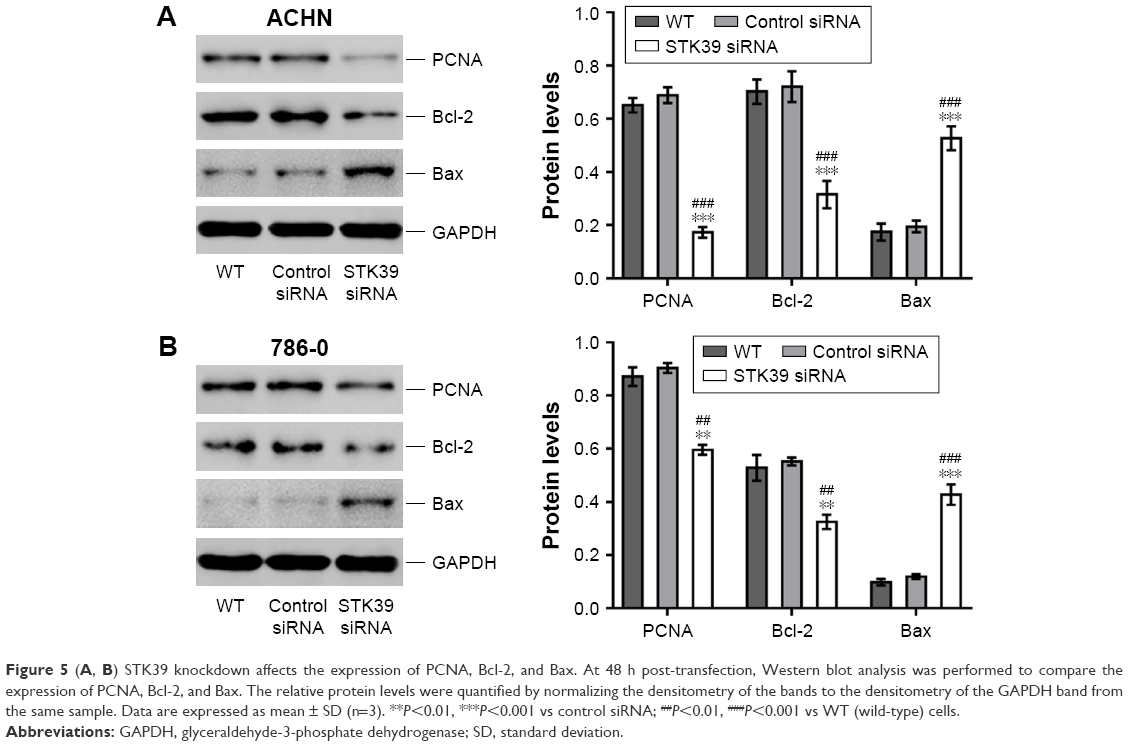 | Figure 5 (A, B) STK39 knockdown affects the expression of PCNA, Bcl-2, and Bax. At 48 h post-transfection, Western blot analysis was performed to compare the expression of PCNA, Bcl-2, and Bax. The relative protein levels were quantified by normalizing the densitometry of the bands to the densitometry of the GAPDH band from the same sample. Data are expressed as mean ± SD (n=3). **P<0.01, ***P<0.001 vs control siRNA; ##P<0.01, ###P<0.001 vs WT (wild-type) cells. |
STK39 is positively correlated with p53 and p38 pathways
Next, we tried to elucidate the pathways that are modulated by STK39 during renal carcinogenesis. By GSEA on RCC cohort from TCGA, we found that genes of p53 and p38 pathways were strongly associated with high expression of STK39 (Figure 6A).
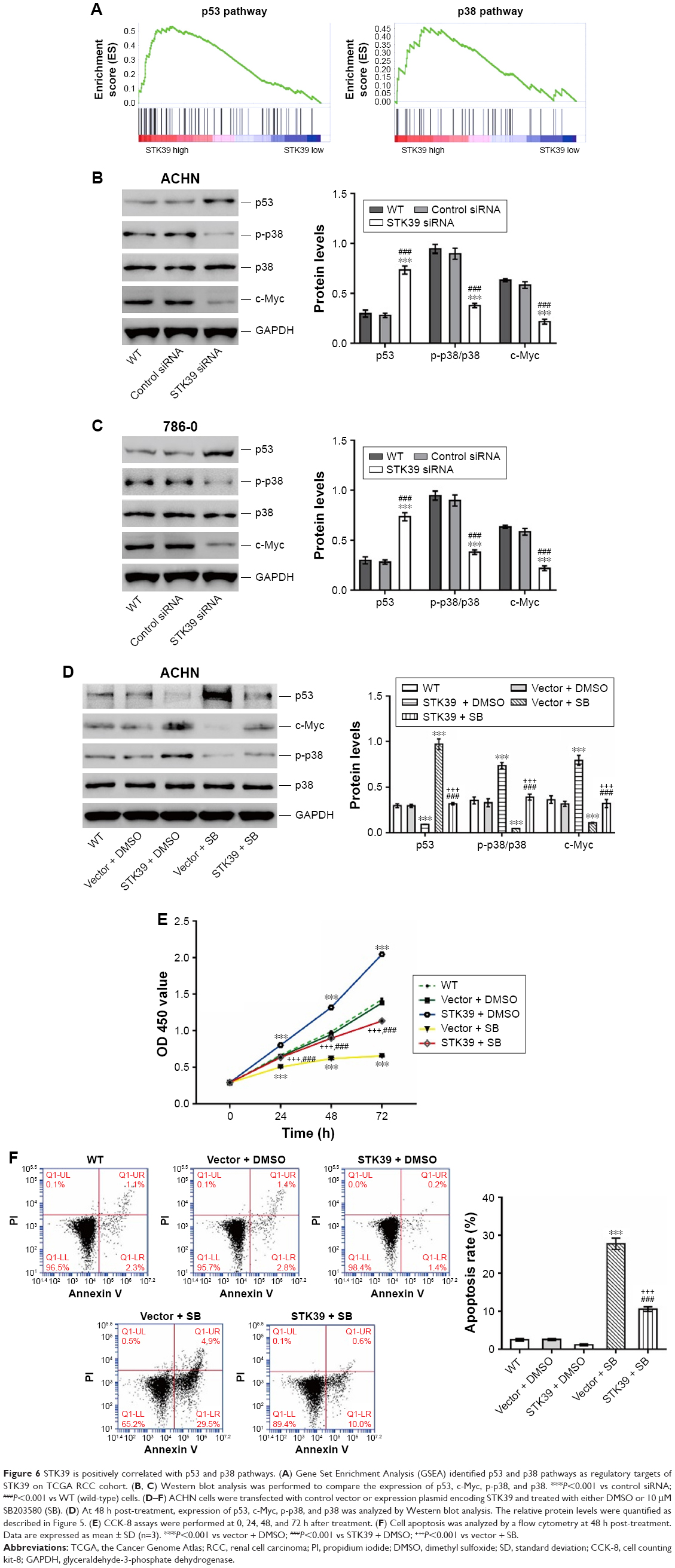 | Figure 6 STK39 is positively correlated with p53 and p38 pathways. (A) Gene Set Enrichment Analysis (GSEA) identified p53 and p38 pathways as regulatory targets of STK39 on TCGA RCC cohort. (B, C) Western blot analysis was performed to compare the expression of p53, c-Myc, p-p38, and p38. ***P<0.001 vs control siRNA; ###P<0.001 vs WT (wild-type) cells. (D–F) ACHN cells were transfected with control vector or expression plasmid encoding STK39 and treated with either DMSO or 10 μM SB203580 (SB). (D) At 48 h post-treatment, expression of p53, c-Myc, p-p38, and p38 was analyzed by Western blot analysis. The relative protein levels were quantified as described in Figure 5. (E) CCK-8 assays were performed at 0, 24, 48, and 72 h after treatment. (F) Cell apoptosis was analyzed by a flow cytometry at 48 h post-treatment. Data are expressed as mean ± SD (n=3). ***P<0.001 vs vector + DMSO; ###P<0.001 vs STK39 + DMSO; +++P<0.001 vs vector + SB. |
To validate the results of GSEA, the levels of p53, p38, phosphorylated p38 (p-p38), and c-Myc were determined by Western blotting (Figure 6B and C). p-p38 and c-Myc were significantly decreased, and p53 was increased after STK39 expression was suppressed compared with the relevant control cells. The levels of p38 remained unchanged after STK39 siRNA transfection.
In order to clarify the involvement of p38 signaling, ACHN cells with STK39 overexpression were exposed to p38 inhibitor (SB203580). The expression of p53 and C-Myc, as well as the phosphorylation of p38, was detected (Figure 6D). STK39 overexpression significantly elevated the levels of C-myc and p-p38, but reduced p53 expression. Such effects were weakened by SB203580 treatment. These findings suggested the regulatory effects of STK39 on p38, p53, and c-Myc. Consistently, the effects of SB203580 treatment that suppressed cell proliferation (Figure 6E) and increased cell apoptosis (Figure 6F) were significantly attenuated by STK39 overexpression. These findings suggested that p38 signaling was involved in the oncogenic function of STK39 in RCC cells.
Discussion
Previous studies showed that STK39 is aberrantly expressed in several types of tumors14–17 and its functions in cancer are also analyzed. Hendriksen et al reported that primary prostate cancer patients with lower mRNA levels of STK39 had a higher incidence of metastases following radical prostatectomy.14 In another study, Cleator et al revealed that reduced STK39 expression was correlated with treatment resistance of breast cancer by expression microarray analysis.15 Balatoni et al observed a reduced expression of STK39 in B-cell lymphoma, and they found that STK39 knockdown in B cells suppressed DNA double-strand breaks-induced apoptosis through JNK.16 However, contradictory roles for STK39 have been reported in NSCLC. Li et al found that STK39 expression is upregulated in NSCLC and that knocking down of STK39 expression reduced lung cancer cell proliferation, migration, and invasion.17 These studies indicate the complicated functions of STK39 in different types of cancers. The present study was the first to show the increased expression of STK39 in RCC tissues at both transcriptional and translational levels, suggesting the potential involvement of STK39 in RCC development.
The present study also investigated the functions of STK39 on RCC cell proliferation and apoptosis in vitro. Knocking down of STK39 expression in two RCC cell lines significantly inhibited cell proliferation and induced cell apoptosis, which was in line with the previous findings in NSCLC.17 PCNA is a prognostic marker for RCC.21 Anti-apoptotic effect of Bcl-2 and pro-apoptotic effect of Bax have been widely studied in various cell types.20 Here, STK39 silencing caused a decrease in the protein expression of PCNA and Bcl-2, and an increase in that of Bax, confirming the biological functions of STK39 on cell proliferation and apoptosis.
p53 and p38 signaling pathways, which play crucial roles in the regulation of cell proliferation, transformation, and apoptosis, are important during carcinogenesis.19,22 p38 has been reported to activate p53 in response to ultraviolet radiation.23 C-Myc, a downstream transcription factor of p38,24 acts as an oncogene during renal carcinogenesis.25 Previous studies have demonstrated that STK39 is able to activate the p38 signaling pathway.5,17 The current study identified that STK39 regulated the p53 and p38 signaling pathways by GSEA analysis. This finding was validated by Western blotting on RCC cells with STK39 silenced. More importantly, STK39 overexpression significantly induced p38 phosphorylation and c-Myc expression, and suppressed p53 expression. Such regulatory effects were attenuated by the treatment of p38 inhibitor, SB203580. More importantly, STK39 overexpression significantly attenuated the suppressed cell proliferation and increased cell apoptosis caused by SB203580 treatment. These data suggested that STK39 might exert oncogenic functions by activating p38 pathway.
Conclusion
In summary, the data presented here suggest that STK39 is overexpressed in RCC tissues. STK39 knockdown in RCC cells inhibits cell proliferation by inducing cell apoptosis via regulating p38 signaling pathway. Thus, STK39 may be a diagnostic biomarker and treatment target for RCC.
Disclosure
The authors report no conflicts of interest in this work.
References
Ljungberg B, Campbell SC, Choi HY, et al. The epidemiology of renal cell carcinoma. Eur Urol. 2011;60(4):615–621. | ||
Cheville JC, Lohse CM, Zincke H, Weaver AL, Blute ML. Comparisons of outcome and prognostic features among histologic subtypes of renal cell carcinoma. Am J Surg Pathol. 2003;27(5):612–624. | ||
Rydzanicz M, Wrzesinski T, Bluyssen HA, Wesoly J. Genomics and epigenomics of clear cell renal cell carcinoma: recent developments and potential applications. Cancer Lett. 2013;341(2):111–126. | ||
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. | ||
Johnston AM, Naselli G, Gonez LJ, Martin RM, Harrison LC, DeAizpurua HJ. SPAK, a STE20/SPS1-related kinase that activates the p38 pathway. Oncogene. 2000;19(37):4290–4297. | ||
Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J Biol Chem. 2002;277(52):50812–50819. | ||
Ramoz N, Cai G, Reichert JG, Silverman JM, Buxbaum JD. An analysis of candidate autism loci on chromosome 2q24–q33: evidence for association to the STK39 gene. Am J Med Genet B Neuropsychiatr Genet. 2008;147(7):1152–1158. | ||
Umedani LV, Chaudhry B, Mehraj V, Ishaq M. Serene threonine kinase 39 gene single nucleotide A-G polymorphism rs35929607 is weakly associated with essential hypertension in population of Tharparkar, Pakistan. J Pak Med Assoc. 2013;63(2):199–205. | ||
Zhao H, Qi Y, Wang Y, et al. Interactive contribution of serine/threonine kinase 39 gene multiple polymorphisms to hypertension among northeastern Han Chinese. Sci Rep. 2014;4:5116. | ||
Chen L-Y, Zhao W-H, Tian W, et al. STK39 is an independent risk factor for male hypertension in Han Chinese. Int J Cardiol. 2012;154(2):122–127. | ||
Wang Y, O’Connell JR, McArdle PF, et al. Whole-genome association study identifies STK39 as a hypertension susceptibility gene. Proc Natl Acad Sci. 2009;106(1):226–231. | ||
Li N-N, Tan E-K, Chang X-L, et al. Genetic association study between STK39 and CCDC62/HIP1R and Parkinson’s disease. PLoS One. 2013;8(11):e79211. | ||
Chang KH, Wu YR, Chen YC, Fung HC, Lee-Chen GJ, Chen CM. STK39, but not BST1, HLA-DQB1, and SPPL2B polymorphism, is associated with Han-Chinese Parkinson’s disease in Taiwan. Medicine. 2015;94(41):e1690. | ||
Hendriksen PJ, Dits NF, Kokame K, et al. Evolution of the androgen receptor pathway during progression of prostate cancer. Cancer Res. 2006;66(10):5012–5020. | ||
Cleator S, Tsimelzon A, Ashworth A, et al. Gene expression patterns for doxorubicin (Adriamycin) and cyclophosphamide (cytoxan) (AC) response and resistance. Breast Cancer Res Treat. 2006;95(3):229–233. | ||
Balatoni CE, Dawson DW, Suh J, et al. Epigenetic silencing of Stk39 in B-cell lymphoma inhibits apoptosis from genotoxic stress. Am J Pathol. 2009;175(4):1653–1661. | ||
Li Z, Zhu W, Xiong L, Yu X, Chen X, Lin Q. Role of high expression levels of STK39 in the growth, migration and invasion of non-small cell type lung cancer cells. Oncotarget. 2016;7(38):61366. | ||
Sánchez-Alonso JA, López-Aparicio P, Recio MN, Pérez-Albarsanz MA. Polychlorinated biphenyl mixtures (Aroclors) induce apoptosis via Bcl-2, Bax and caspase-3 proteins in neuronal cell cultures. Toxicol Lett. 2004;153(3):311–326. | ||
Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8(4):275–283. | ||
Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281(5381):1322–1326. | ||
Morell-Quadreny L, Clar-Blanch F, Fenollosa-Enterna B, Perez-Bacete M, Martinez-Lorente A, Llombart-Bosch A. Proliferating cell nuclear antigen (PCNA) as a prognostic factor in renal cell carcinoma. Anticancer Res. 1997;18(1B):677–682. | ||
Bradham C, McClay DR. p38 MAPK in development and cancer. Cell Cycle. 2006;5(8):824–828. | ||
Bulavin DV, Saito S, Hollander MC, et al. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 1999;18(23):6845–6854. | ||
Stoneley M, Chappell SA, Jopling CL, Dickens M, MacFarlane M, Willis AE. c-Myc protein synthesis is initiated from the internal ribosome entry segment during apoptosis. Mol Cell Biol. 2000;20(4):1162–1169. | ||
Yamada Y, Hidaka H, Seki N, et al. Tumor-suppressive microRNA-135a inhibits cancer cell proliferation by targeting the c-MYC oncogene in renal cell carcinoma. Cancer Sci. 2013;104(3):304–312. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.