")
Back to Archived Journals » Orphan Drugs: Research and Reviews » Volume 4
Stiripentol for the treatment of Dravet syndrome
Authors Chiron C
Received 16 January 2014
Accepted for publication 11 February 2014
Published 2 April 2014 Volume 2014:4 Pages 29—38
DOI https://doi.org/10.2147/ODRR.S47619
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Catherine Chiron1–3
1INSERM U1129, Paris, France; 2Paris Descartes University, Paris, France; 3CEA, Gif-sur-Yvette, France
Abstract: Stiripentol (marketed by Biocodex as Diacomit®) is an anticonvulsant drug, structurally unrelated to any other compound, which has recently been approved as adjunctive therapy with clobazam and valproate for Dravet syndrome in Europe, Canada, and Japan. This rare form of early childhood epilepsy is associated with subsequent cognitive impairment, significant risk of death, and high pharmacoresistance. Based on an efficacy signal of stiripentol added to clobazam and valproate in an observational, prospectively conducted, exploratory study including 10% of children with Dravet syndrome, a randomized placebo-controlled trial was specifically dedicated to patients with Dravet syndrome inadequately controlled by clobazam and valproate. Results showed significantly higher responder rates (71% versus 5%; P<0.0001) and decrease in seizure frequency (-69% versus +7%; P<0.002) on stiripentol than on placebo. A second, independently performed, randomized controlled trial confirmed these results 2 years later, and both trials were plotted in a meta-analysis. Efficacy was supported by three subsequent observational studies, with, respectively, 46 (France), 23 (Japan), and 82 (USA) children with Dravet syndrome treated with stiripentol for up to 5 years. Based on the experiences of more than 2,000 patients with Dravet syndrome who were exposed to stiripentol, drowsiness, loss of appetite, and weight loss are the most frequent adverse events and may be reduced by decreasing the dosage of co-medication. The inhibition of stiripentol by the cytochrome P450 complex (CYP2C19, and CYP3A4) leads to clinically significant interactions. Experimental data, both in vitro and in vivo, have definitively established that stiripentol is a GABAergic anticonvulsant and acts on different sites than benzodiazepines. The pharmacodynamic interactions also enhance the anticonvulsant effect of the stiripentol–clobazam combination in patients with Dravet syndrome, irrespective of the GABAergic effect. Stiripentol is currently the only drug specifically indicated in Dravet syndrome.
Keywords: childhood epilepsy, antiepileptic drug trials, orphan drugs, clobazam, GABAergic drugs, CYP450
Introduction
Dravet syndrome (initially known as severe myoclonic epilepsy in infancy) was first described more than 30 years ago.1 It is a rare disease, with an estimated incidence of one in 40,000 children up to the age of 7 years, based on clinical diagnosis.2
Patients with Dravet syndrome present with a fairly homogeneous panel of clinical features, which usually leads to a correct diagnosis when the form is complete (also called typical).2 The first seizures occur in normal infants, before the age of 1 year. These seizures are (tonic–)clonic, generalized, or alternately involving one or the other hemi-body; often provoked by fever; and prolonged as status epilepticus. Typically, from 2 years of age, other seizure types will be associated, including myoclonic, absence, and focal seizures. Cognitive slowing progressively appears, which results in intellectual disability and behavioral disorders. Electroencephalogram is usually normal at onset, after which photosensitivity and nonspecific slow waves and focal spikes may appear. Neuroimaging is negative. Seizures persist into adulthood but become less frequent. Severity of the condition results from the high pharmacoresistance of seizures, the occurrence of slowing cognitive development, and a risk of sudden death of about 10%.
Mutations in SCN1A, a gene of the sodium channel Nav1.1, are the major cause of Dravet syndrome.3 They are found in 75% of patients with Dravet syndrome.4 Of note, similar SCN1A mutations have also been identified in other more or less severe forms of epilepsy.5,6 Besides typical Dravet syndrome, so called atypical or borderline forms have been described (with no myoclonic symptoms or with less severe cognitive outcome, but with the same rate of SCN1A mutations).2 Among girls with Dravet syndrome negative for SCN1A, about 10% were discovered to have mutations in another gene, PCDH19.7
Dravet syndrome is a highly refractory form of epilepsy, and intractability of seizures has even been included in the criteria of diagnosis by the International League Against Epilepsy.8 The relatively numerous treatment options proposed have been disappointing, and not one of them has been associated with a randomized controlled trial except for stiripentol. Among the conventional antiepileptic drugs, valproate and benzodiazepines are the most commonly used first-line compounds, but their efficacy data is incomplete.2 Phenobarbital and phenytoin have been used without success, but beneficial experiences with bromide have been reported in Japanese and German studies.9,10 On the other hand, drugs that produce a worsening effect have been identified, namely: lamotrigine, which may insidiously aggravate seizures in up to 80% of patients;11 carbamazepine and vigabatrin, which exacerbated convulsive seizures in 60% of our patients;12 and rufinamide, which worsened seizures in 30% of patients and was inefficient in another 45%.13 Overdosage of phenobarbital and/or pentothal administered intravenously for prolonged status epilepticus has been occasionally suspected to play a role in the fulminant degradation of neurological status with severe brain atrophy,14 but such an acute encephalopathy has also been reported without any barbiturates during febrile illness in children with Dravet syndrome.15 Among the new antiepileptic drugs, there has been significant experience with topiramate in Dravet syndrome, but only through observational studies. In two of these, patients had not been previously treated with stiripentol, and responder rate was 56% with a complete cessation of seizures in 17%.16,17 In a third study, patients had previously received stiripentol with unsatisfactory results: 78% became responders and 17% stopped having seizures when topiramate was added.18 Levetiracetam was also used in Dravet syndrome in two observational prospective trials: its adjunction resulted in 60% of responders without stiripentol as co-medication,19 but in only 11% of responders in our own series of patients refractory to stiripentol.20 Among 12 retrospectively diagnosed patients with Dravet syndrome having received fenfluramine as long-term add-on therapy, seven remained seizure-free for a long period of time (1–19 years).21 Ketogenic diet, a non-pharmaceutical procedure that is reported more and more in retrospective studies in pediatric epilepsies, including in Dravet syndrome,22–24 was also beneficial in one prospective study that we conducted in Dravet patients, not only in terms of seizures but also with respect to behavior, even in those whose epilepsy had been unsatisfactorily controlled by stiripentol.25
Stiripentol (marketed by Biocodex [Gentilly, France] as Diacomit®) is a new antiepileptic drug structurally unrelated to any other anticonvulsant compound (Figure 1). Stiripentol is currently the only drug for Dravet syndrome that was developed through the methodology of the randomized controlled trial. Positive results led to its approval as adjunctive therapy in Europe (2007), Canada (2012), and Japan (2012). As a result, several recent reviews on stiripentol in Dravet syndrome have been published.26–29 The present review will focus first on our personal experience with stiripentol, probably the most important in the world, and, second, on the most recently reported stiripentol data (Table 1).
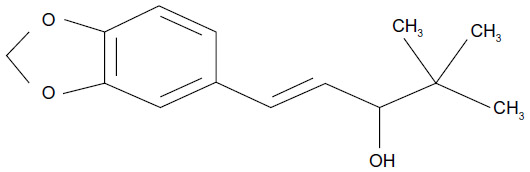 | Figure 1 Structure of stiripentol. |
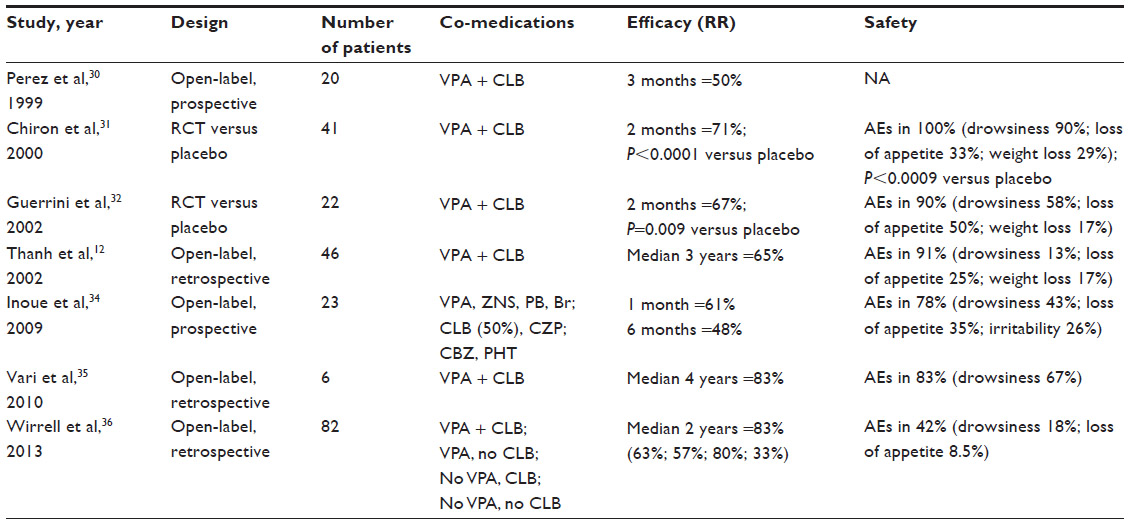 | Table 1 Clinical studies of stiripentol in Dravet syndrome |
Clinical studies: efficacy
Pilot study
The first attempt to use stiripentol in Dravet syndrome was performed in a large, open-label study in which stiripentol was prospectively given as adjunctive therapy at the dose of 60 mg/kg/day for 1 month and 90 mg/kg/day for 2 additional months.30 A large population of 212 children and adolescents, aged from 1 month to 20 years and presenting with various types of refractory epilepsies, was included. At 1 and 3 months of stiripentol treatment, the overall median seizure frequency was significantly lower than at baseline (P<0.0001), with respective responder rates of 49% and 40% that were not statistically different (P=0.08). The two syndromes with the highest responder rates were focal epilepsy (66%) and, unexpectedly, severe myoclonic epilepsy in infancy (Dravet syndrome): 20 children with Dravet syndrome received stiripentol in association with valproate and clobazam (CLB) and ten experienced a decrease in seizure frequency over 50% compared to baseline. Based on these results, two controlled trials were designed to study stiripentol specifically in Dravet syndrome.
Randomized controlled trials
Our first randomized controlled trial was performed in France. This was a multicenter, double-blind, randomized, placebo-controlled trial in children over 3 years of age with typical Dravet syndrome and resistant to valproate and CLB.31 After a 1-month baseline period, stiripentol or placebo was randomly allocated to 21 and 20 patients, respectively, as adjunctive therapy to valproate and CLB, and at a dosage of 50 mg/kg/day for 2 months. The primary endpoint was the percentage of responders in each arm defined as having more than 50% reduction in the frequency of clonic (or tonic–clonic) seizures during the second month of the double-blind period compared to baseline. There were 15 (71%) responders on stiripentol versus only one (5%) on placebo (P<0.0001) (none were seizure-free) (95% confidence interval [CI]: 52–91 [stiripentol] versus 0–15 [placebo]). In addition, nine patients achieved seizure freedom on stiripentol compared to none on placebo. The changes in absolute numbers of seizures from baseline were also more marked on stiripentol (−69%) than on placebo (+7%) (P<0.002).
A similarly designed placebo-controlled trial was subsequently performed in Italy, and involved eleven patients with typical Dravet syndrome in each arm.32 Results were in the same range as those of the French trial: 67% of responders on stiripentol versus 9% on placebo (P=0.009).
Interestingly both trials were strongly positive in spite of the small numbers of patients included, probably due to the homogeneity of Dravet syndrome combined with an important size-of-treatment effect.
Meta-analyses
Based on these two similarly designed trials, a meta-analysis was performed on their 64 included as intent-to treat patients with Dravet syndrome aged from 3 to 20 years.33 Analysis did not detect any difference in the treatment effect within both studies. Responder rates were significantly higher when stiripentol was added to valproate and CLB compared to when placebo was added to the same combination (odds ratio 32, 95% CI: 6.2–161).
A recently published Cochrane Review did not find any other randomized controlled trials than the two previously described trials evaluating the use of stiripentol in Dravet syndrome.29 Calculation of risk ratio (RR) based on an independent meta-analysis of these trials confirmed that stiripentol was better than placebo with regards to responder rate (22/33 versus 2/31; RR 10.4, 95% CI: 2.6–40.9) and seizure freedom (12/33 versus 1/31; RR 7.9, 95% CI: 1.5–41.2).
Long-term data and supportive observational studies
In the extension period of the French randomized controlled trial, 40 of 41 patients received open stiripentol for a median duration of 25 months (range 10–36 months). Compared to the response during the double-blind period, efficacy was maintained at last visit in 50% of cases.31
In a retrospective study performed in our center (Saint Vincent De Paul Hospital, Paris), we followed 46 successive children with Dravet syndrome receiving stiripentol combined with valproate and CLB for a median of 2.9 years (range 2 months to 5.5 years).12 Initial efficacy was maintained long term in 61% of cases. At last visit, compared to the 3-month period prior to starting stiripentol, monthly seizure frequency was reduced from nine to three (P<0.001). Median seizure duration was also reduced, from 7.5 to 1.5 minutes (P<0.002), and the number of patients with convulsive status epilepticus dropped from 91% to 44% (P<0.001). Efficacy of stiripentol on status epilepticus was particularly notable in the youngest children (<2 years), suggesting that stiripentol as adjunctive therapy can be considered very early in the course of Dravet syndrome.
A prospective, open-label, multicenter Japanese study in 23 patients aged from 1 to 22 years with Dravet syndrome also reported short- and long-term seizure decrease with stiripentol.34 Stiripentol at 50 mg/kg/day was added to patients’ current antiepileptic drug treatments (most commonly valproate, but with or without CLB). At 1 month, following a brief period to stabilize the co-medication dosage, responder rate had reached 61% with two patients seizure-free; seizure duration had shortened in ten patients; and the number of status epilepticus had decreased in six patients. Then, the stiripentol dosage could be modified (maximum 100 mg/kg/day and 4,000 mg/day). Long term (up to 14 months, mean 6 months), responder rate slightly declined to 48%, with one patient remaining seizure-free.
In Italy, six of six children with Dravet syndrome (aged 3–9 years) also experienced more than 75% seizure reduction when stiripentol was added to valproate and CLB, and efficacy was still maintained in five of these children after 3- to 9-year follow-up.35
A retrospective multicenter study recently reported the first experience in the US of stiripentol as adjunctive therapy on 82 children with Dravet syndrome for a median duration of 22 months (10–43 months).36 Global seizure frequency decreased by 63% in the patients who received valproate and CLB as co-medications. The frequency of prolonged seizures and therefore the use of rescue medication and the number of hospitalizations were also reduced, whatever the co-medication (CLB or not, valproate or not).
Tolerability
In the French randomized controlled trial, drug-related adverse effects were noticed in all 21 (100%) patients on stiripentol versus only five (25%) of the patients on placebo (P=0.0009).31 The most frequently reported adverse events in the stiripentol arm were drowsiness (19/21 patients) and loss of appetite (7/21 patients), with loss of weight in six of the latter seven cases. Because some adverse reactions may be associated with elevated plasma concentrations of concomitant anticonvulsants, as stiripentol can inhibit the metabolism of drugs that are substrates for cytochrome P450 isoenzymes (see “Pharmacokinetics”), it had been specified in the protocol that the dosage of co-medication (valproate and/or CLB) could be decreased: this procedure solved the adverse events in 17 of 21 patients. Five patients presented with severe adverse events (three with drowsiness, two with loss of weight) on stiripentol compared to one on placebo (drowsiness); the only one dropout patient for side effects was on placebo.
Adverse events observed in the observational studies with stiripentol in patients with Dravet syndrome were generally similar to those reported in the French randomized controlled trial.31 For example, 42 of 46 patients reported one adverse event with stiripentol, mostly loss of appetite (25%), weight loss (17%), insomnia (13%), somnolence (13%), and ataxia (13%), in the French retrospective long-term study.12 Notice that stiripentol treatment had to be stopped for safety concerns (mostly weight loss) in seven of 46 (15%) patients, who were all over 12 years of age when stiripentol had been introduced.12 In the early phase of the open-label Japanese study in 23 patients, drowsiness (43%), loss of appetite (35%), hyperactivity or irritability (26%), ataxia (22%), and insomnia (9%) were most commonly reported, although many adverse events disappeared after dosage adjustments in the late phase of the study and only one patient discontinued stiripentol for adverse events.34 Adverse events were reported in 42% of patients in the retrospective US study; most common among these were sedation (18%) and reduced appetite (8.5%).36 Four patients (5%) discontinued stiripentol for adverse effects.
Asymptomatic neutropenia (<1,000/mm3) was observed rarely (in 1%–10% of patients) and resolved after decreasing the stiripentol dosage. According to the EU summary of product characteristics, blood count and liver function should be assessed prior to starting the treatment and checked every 6 months thereafter.37
Based on these published data, a Cochrane Review recommended that further studies be conducted to unequivocally establish the long-term tolerability of stiripentol in the treatment of Dravet syndrome.29 It is notable, however, that most of the pre- and post-approval safety experience included in Biocodex’s stiripentol dossier is not available in the published medical literature: overall, more than 2,000 patients with Dravet syndrome have been exposed to stiripentol, with safety data deriving from compassionate-use approval (France), a Periodic Safety Update Report, and a 5-year postmarketing survey (Europe) completed in 2012. No new stiripentol-related adverse effect was identified within this postmarketing survey.37
Pharmacokinetics
Pharmacokinetic data in healthy adults
In adult volunteers, stiripentol administered alone proved to be rapidly absorbed (with a tmax [time to achieve peak plasma drug concentration (Cmax)] of approximately 1.5 hours) and extensively bound to plasma proteins.38 Both oral formulations in capsules and powder sachets for suspension are bioequivalent for tmax and area under the plasma concentration–time curve, but Cmax is 23% higher using sachets than capsules; medical supervision is therefore recommended if switching between formulations.37 Stiripentol is largely metabolized by the CYP450 complex (CYP1A2, CYP2C19, and CYP3A4), and has shown dose-dependent, nonlinear pharmacokinetics.39,40
Pharmacokinetic data in children
Data are still too limited to establish a clear relationship between dosage, plasma concentration, efficacy, and safety of stiripentol in Dravet syndrome. The mean plasma concentration of stiripentol at steady state was 10 mg/L (95% CI: 8.3–11.7) in the French randomized controlled trial, with a mean stiripentol dosage of 49 mg/kg/day (95% CI: 47–51).31 Preliminary results of a pharmacokinetic study in 35 children with Dravet syndrome (median age 7 years, range 1–17 years) receiving stiripentol (median dosage 45 mg/kg/day, range 27–89) in addition to valproate and CLB showed that apparent volume of distribution, apparent oral clearance, and elimination half-life of stiripentol increased as body weight increased from 10 to 60 kg: from 32 to 192 L, 2.6 to 5.7 L/hour, and 8.5 to 23.5 hours, respectively.37
A retrospective analysis of therapeutic drug monitoring was performed in 75 patients (proportion of Dravet syndrome not given) from three German epilepsy centers.41 Plasma concentrations of stiripentol were 40% lower in children aged 6–12 years and 57.5% lower in children <6 years than in those >12 years. Valproate co-medication did not affect stiripentol plasma concentrations, while CLB increased them by 25% and phenobarbital and phenytoin reduced them by 63%. This study reported an overall “significant nonlinear concentration-to-dose relationship”,41 based on eight patients who were treated with stiripentol dosages up to 40 mg/kg and higher, but note that the nonlinearity was not observed in all eight patients.
Drug interactions
In vitro and in vivo studies have shown that stiripentol is a potent inhibitor of the activity of several CYP enzymes – especially CYP2C19 and CYP3A4.40,42–44 As a result, the plasma concentrations of other antiepileptic drugs metabolized by the same CYP can be increased when they are combined with stiripentol. In patients with Dravet syndrome, stiripentol was approved in combination with valproate and CLB. The trough plasma concentrations of valproate were not significantly increased by adding stiripentol compared to baseline.31 By contrast, those of CLB were approximately doubled (P<0.0001), and those of norclobazam (NCLB), the active metabolite, were multiplied by five to seven (P<0.0001).31 Concomitantly, the concentrations of 4-hydroxynorclobazam, the last and inactive metabolite, were markedly decreased (P<0.0001).31 Such a differing impact of stiripentol on CLB metabolites may be explained by its different inhibitory power on the CYP enzymes (CYP2C19 and CYP3A4) involved in the metabolic chain of CLB (Figure 2).43
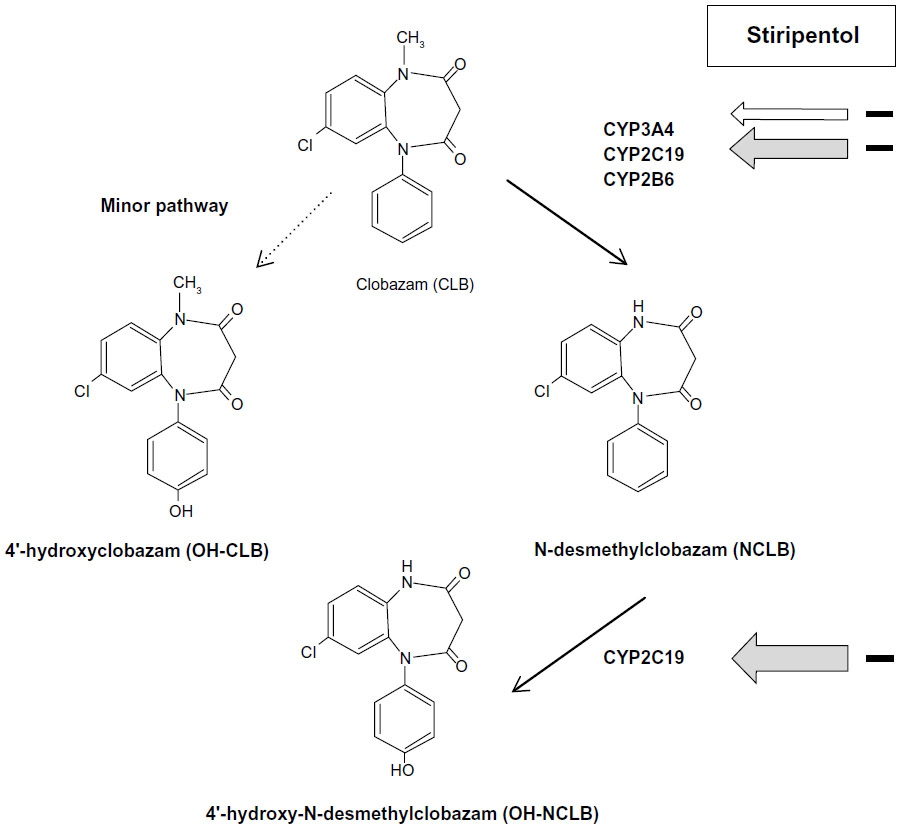 | Figure 2 Metabolic interactions between CLB and stiripentol by the CYP (cytochrome P450) complex. |
Mechanism of action
Based on the overall clinical and preclinical experiences available to date, stiripentol appears to have two types of mechanisms involved in its anticonvulsant effect in Dravet syndrome: the enhancement of γ-aminobutyric acid (GABA) transmission and the interactions with CLB co-medication. There is growing evidence that the first mechanism may sustain the stiripentol efficacy on seizures independently from the second.27,45
Intrinsic GABAergic effects
Intrinsic GABAergic effects were suggested in the past in animal models of seizures induced by electric shock, pentylenetetrazol, and bicuculline.46 Since 2006, the following in vitro and in vivo pharmacodynamic studies have confirmed the GABAergic effects of stiripentol.
When stiripentol was applied to CA3 pyramidal neurons of hippocampus in immature rats, GABAergic postsynaptic currents significantly increased.47 Stiripentol was used at clinically relevant concentrations and the GABA currents increased in a concentration-dependent manner (Figure 3). Stiripentol enhanced the opening duration of the chloride channel (as barbiturates do) rather than its opening frequency (as benzodiazepines do). Therefore, the site of action of stiripentol on the postsynaptic GABAA receptor seemed to be different from that of the benzodiazepine site.
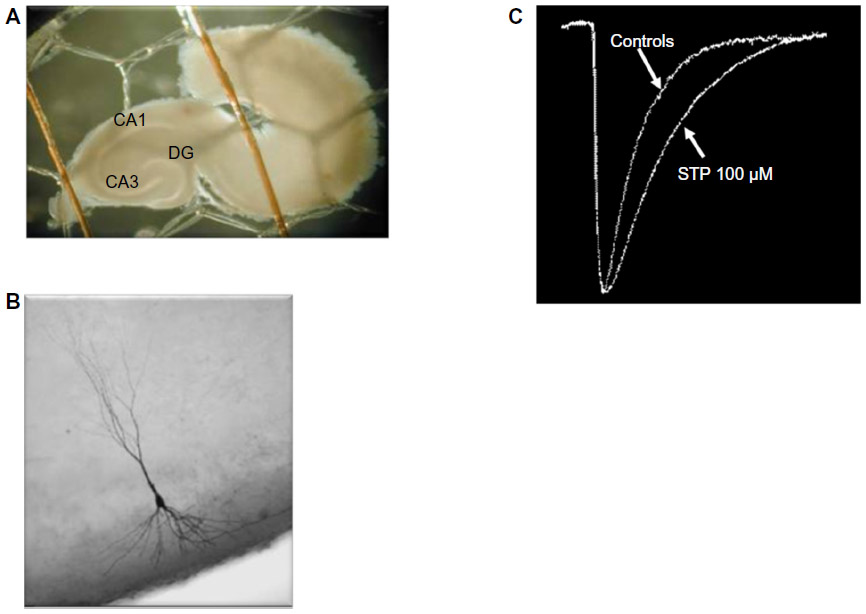 | Figure 3 GABAergic anticonvulsant effect of STP in vitro. |
Analyzing whole-cell currents on recombinant GABAA receptor, Fisher confirmed that stiripentol acts as a direct allosteric modulator of the GABAA receptors.48 Stiripentol disclosed the greatest potentiation on receptors containing α3 and δ subunits, two sites distinct from the γ subunit of benzodiazepines.49 Then, stiripentol and benzodiazepines (diazepam, clonazepam, CLB, and NCLB) were coadministered specifically on α3 recombinant GABAA receptors:50 benzodiazepines were equally effective with or without stiripentol, thus proving that stiripentol and benzodiazepines act independently at GABAA receptors and suggesting that their combination may increase the maximum response beyond that of either drug alone via a pharmacodynamic interaction.
Stiripentol was also shown to be effective in a rodent model of refractory pilocarpine-induced status epilepticus at therapeutically relevant concentrations.51,52 Stiripentol remained effective at terminating seizures that had become resistant to benzodiazepine (diazepam). Whole-cell recording from hippocampal slices confirmed that stiripentol and benzodiazepine potentiate postsynaptic currents at a different site on GABAA receptors. Potentiation by stiripentol persisted during status epilepticus, while potentiation by diazepam was lost, thus suggesting that stiripentol may be efficient on status epilepticus when benzodiazepine is not.
A Nav1.1 zebrafish mutant, which spontaneously exhibits abnormal electrographic activity and convulsive behavior, was recently developed. Stiripentol attenuates mutant seizure activity.53 In addition, screening a large number of US Food and Drug Administration-approved compounds led to identification of a single one that both abolished the behavioral phenotype and electrographic seizures of zebrafish: namely, clemizole. This H1-antihistamine is therefore an unexpected new potential target for Dravet treatment.
Age-related GABAergic effects
Stiripentol has the greatest potentiation on GABAA receptors containing α3 subunit,48 which is a subunit that is highly expressed in the developing brain.54
Interestingly the effect on stiripentol on refractory status epilepticus and the potentiation of GABAergic currents was found to be greater in younger animals than in adults.52 Auvin et al recently observed this age-related efficacy of stiripentol on the pentylenetetrazol model of acute seizures and the lithium–pilocarpine model of status epilepticus comparing young (post-natal age [P]21) and adult (P75) rats.55 Stiripentol prevented seizures from occurring in both models at P21, but not at P75. The authors studied GABAA α3 subunit in both populations: the age-related differences in mRNA expression did not explain the age-related differences in the anticonvulsant effects of stiripentol.55
Stiripentol was recently used in Japan in a model of Dravet syndrome, a heterozygous SCN1A mutant mouse that presents with hyperthermia-induced seizures.56 It was effective in preventing hyperthermia-induced seizures in young (aged 1 month) but not older (aged 5 months) mice. However, this age-related effect has not been demonstrated in humans.
Inhibition of CYP metabolism
In humans, additional evidence that stiripentol may act independently of its pharmacokinetic drug interactions via CYP metabolism has come from pharmacokinetic and pharmacogenetic data. When stiripentol is co-administered with CLB in Dravet syndrome, the main metabolic target is CYP2C19, the inhibition of which results in increased plasma concentrations of CLB and NCLB and of the ratio NCLB/CLB (see “Pharmacokinetics”).43 CYP2C19 has three clinically relevant allelic mutations (*1,*2,*3); mutations *2 and *3 lead to a nonfunctional allele and, therefore, to an absence of pharmacokinetic interaction (no increase of CLB, NCLB, NCLB/CLB) in the case of stiripentol–CLB co-medication.57 Besides the most frequent *1/*1 genotype, the frequency of both other mutations is different around the world: CYP 2C19*2 (present in 13% of Caucasians and 23% of Japanese), CYP 2C19*3 (present in 0% of Caucasians and 10% of Japanese).58
In the observational study performed in Japan on 23 patients with Dravet syndrome, 61% and 48% of patients experienced a more than 50% decrease in seizure frequency in the short and long term, respectively.34 These rates are close to those observed in Europe, although only one-half of the Japanese patients received CLB as co-medication, and 20% of them had a CYP2C19 inactive allele *2 or *3.
We recently performed a post hoc analysis of the pharmacokinetic data from the two aforementioned randomized controlled trials performed in Dravet syndrome.27 Responder rate was similar whether the patients exhibited an increase of CLB, NCLB, or NCLB/CLB on stiripentol or not. For example, 78% of children experienced an increase of NCLB/CLB ratio, but their responder rate (65%) was not different from those who did not (75%). The patients in whom stiripentol could not inhibit CYP2C19 presented with the CYP2C19*1*/2* genotype or had received progabide, a drug likely to be also an inhibitor of CYP2C19, as co-medication.
Altogether, these data show that the antiepileptic effect of stiripentol in Dravet syndrome does not only depend on its pharmacokinetic interaction with CLB.45
Conclusion
Stiripentol was designated as an orphan drug by the European Medicines Agency in 2001 and was approved for Dravet syndrome in Europe in 2007. Fenfluramine also obtained the first-step orphan designation by the European Medicines Agency in 2014, but, to date, stiripentol remains the only drug specifically indicated in Dravet syndrome. Stiripentol is indicated
for use in conjunction with clobazam and valproate as adjunctive therapy of refractory generalized tonic-clonic seizures in patients with severe myoclonic epilepsy in infancy (SMEI, Dravet’s syndrome) whose seizures are not adequately controlled with clobazam and valproate.32
Stiripentol was developed using an exploratory/confirmatory sequence of clinical studies,59 an approach currently recommended by the European Medicines Agency for new drugs in pediatric epilepsy and available on its website (CHMP/EWP/566/98, 22 July 2010). This strategy proved to be successful and ethically beneficial in the identification of rare pediatric epilepsies as potential targets for subsequent focused controlled trials.
Stiripentol has several different mechanisms of action. Experimental data have decisively shown its GABAergic anticonvulsant mechanism of action. Additionally, both pharmacokinetic and pharmacodynamic drug interactions result in a synergistic anticonvulsant effect of the stiripentol–CLB combination, which is therefore considered an example of rational anticonvulsant polytherapy.45,60
Disclosure
The author reports no conflicts of interest in this work.
References
Dravet C. Les epilepsies graves de l’enfant. Vie Med. 1978;8:543–548. French. | |
Dravet C, Oguni H. Dravet syndrome (severe myoclonic epilepsy in infancy). Handb Clin Neurol. 2013;111:627–633. | |
Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68: 1327–1332. | |
Nabbout R, Gennaro E, Dalla Bernardina B, et al. Spectrum of SCN1A mutations in severe myoclonic epilepsy of infancy. Neurology. 2003;60(12):1961–1967. | |
Escayg A, Heils A, MacDonald BT, Haug K, Sander T, Meisler MH. A novel SCN1A mutation associated with generalized epilepsy with febrile seizures plus – and prevalence of variants in patients with epilepsy. Am J Hum Genet. 2001;68(4):866–873. | |
Harkin LA, McMahon JM, Iona X, et al; Infantile Epileptic Encephalopathy Referral Consortium, Sutherland G, Berkovic SF, Mulley JC, Scheffer IE. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain. 2007;130:843–852. | |
Depienne C, Bouteiller D, Keren B, et al. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet. 2009;5(2):e1000381. | |
[No authors listed]. Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia. 1989;30(4):389–399. | |
Tanabe T, Awaya Y, Matsuishi T, et al. Management of and prophylaxis against status epilepticus in children with severe myoclonic epilepsy in infancy (SMEI; Dravet syndrome) – a nationwide questionnaire survey in Japan. Brain Dev. 2008;30(10):629–635. | |
Lotte J, Haberlandt E, Neubauer B, Staudt M, Kluger GJ. Bromide in patients with SCN1A-mutations manifesting as Dravet syndrome. Neuropediatrics. 2012;43(1):17–21. | |
Guerrini R, Dravet C, Genton P, Belmonte A, Kaminska A, Dulac O. Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia. 1998;39:508–512. | |
Thanh TN, Chiron C, Dellatolas G, et al. [Long-term efficacy and tolerance of stiripentol in severe myoclonic epilepsy of infancy (Dravet’s syndrome)]. Arch Pediatr. 2002;9:1120–1127. French. | |
Mueller A, Boor R, Coppola G, et al. Low long-term efficacy and tolerability of add-on rufinamide in patients with Dravet syndrome. Epilepsy Behav. 2011;21(3):282–284. | |
Chipaux M, Villeneuve N, Sabouraud P, et al. Unusual consequences of status epilepticus in Dravet syndrome. Seizure. 2010;19:190–194. | |
Okumura A, Uematsu M, Imataka G, et al. Acute encephalopathy in children with Dravet syndrome. Epilepsia. 2012;53(1):79–86. | |
Nieto-Barrera M, Candau R, Nieto-Jimenez M, Correa A, del Portal LR. Topiramate in the treatment of severe myoclonic epilepsy in infancy. Seizure. 2000;9:590–594. | |
Coppola G, Capovilla G, Montagnini A, et al. Topiramate as add-on drug in severe myoclonic epilepsy in infancy: an Italian multicenter open trial. Epilepsy Res. 2002;49:45–48. | |
Kröll-Seger J, Portilla P, Dulac O, Chiron C. Topiramate in the treatment of highly refractory patients with Dravet syndrome. Neuropediatrics. 2006;37:325–329. | |
Striano P, Coppola G, Pezella M, et al. An open-label trial of levetiracetam in severe myoclonic epilepsy of infancy. Neurology. 2007;69: 250–254. | |
Chhun S, Troude P, Villeneuve N, et al. A prospective open-labeled trial with levetiracetam in pediatric epilepsy syndromes: continuous spikes and waves during sleep is definitely a target. Seizure. 2011;20:320–325. | |
Ceulemans B, Boel M, Leyssens K, et al. Successful use of fenfluramine as an add-on treatment for Dravet syndrome. Epilepsia. 2012;53(7):1131–1139. | |
Laux L, Blackford R. The ketogenic diet in Dravet syndrome. J Child Neurol. 2013;28(8):1041–1044. | |
Caraballo RH. Nonpharmacologic treatments of Dravet syndrome: focus on the ketogenic diet. Epilepsia. 2011;52 Suppl 2:S79–S82. | |
Kang HC, Kim YJ, Kim DW, Kim HD. Efficacy and safety of the ketogenic diet for intractable childhood epilepsy: Korean multicentric experience. Epilepsia. 2005;46(2):272–279. | |
Nabbout R, Copioli C, Chipaux M, et al. Ketogenic diet also benefits Dravet syndrome patients receiving stiripentol: a prospective pilot study. Epilepsia. 2011;52(7):e54–e57. | |
Chiron C, Dulac O. The pharmacologic treatment of Dravet syndrome. Epilepsia. 2011;52(Suppl 2):S72–S75. | |
Nabbout R, Chiron C. Stiripentol: an example of antiepileptic drug development in childhood epilepsies. Eur J Paediatr Neurol. 2012; 16 Suppl 1:S13–S17. | |
Plosker GL. Stiripentol: in severe myoclonic epilepsy of infancy (dravet syndrome). CNS Drugs. 2012;26(11):993–1001. | |
Brigo F, Storti M. Antiepileptic drugs for the treatment of severe myoclonic epilepsy in infancy. Cochrane Database Syst Rev. 2013;19(11):CD010483. | |
Perez J, Chiron C, Musial C, et al. Stiripentol: efficacy and tolerability in children with epilepsy. Epilepsia. 1999;40:1618–1626. | |
Chiron C, Marchand MC, Tran A, et al. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo-controlled syndrome-dedicated trial. STICLO study group. Lancet. 2000;356: 1638–1642. | |
Guerrini R, Tonnelier S, d’Athis P, Rey E, Vincent J, Pons G. Stiripentol in severe myoclonic epilepsy in infancy (SMEI): a placebo-controlled Italian trial. Epilepsia. 2002;43(Suppl 8):S155. | |
Kassaï B, Chiron C, Augier S, et al. Severe myoclonic epilepsy in infancy: a systematic review and a meta-analysis of individual patient data. Epilepsia. 2008;49:343–348. | |
Inoue Y, Ohtsuka Y, Oguni H, et al. Stiripentol open study in Japanese patients with Dravet syndrome. Epilepsia. 2009;50:2362–2368. | |
Vari MS, Prato G, Siri L, et al. Long term efficacy of stiripentol in Dravet syndrome: an open label trial of 6 cases. Bollettino Lega italiana contro L’Epilessia. 2010;140:215–216. | |
Wirrell EC, Laux L, Franz DN, et al. Stiripentol in Dravet syndrome: results of a retrospective US study. Epilepsia. 2013;54(9):1595–1604. | |
Diacomit: EPAR – Product Information 2013;http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/000664/WC500036519.pdf. | |
Levy RH, Lin HS, Blehaut HM, Tor JA. Pharmacokinetics of stiripentol in normal man: evidence of nonlinearity. J Clin Pharmacol. 1983;23(11–12):523–533. | |
Levy RH, Loiseau P, Guyot M, Blehaut HM, Tor J, Moreland TA. Michaelis-Menten kinetics of stiripentol in normal humans. Epilepsia. 1984;25(4):486–491. | |
Tran A, Rey E, Pons G, et al. Influence of stiripentol on cytochrome P450-mediated metabolic pathways in humans: in vitro and in vivo comparison and calculation of in vivo inhibition constants. Clin Pharmacol Ther. 1997;62(5):490–504. | |
May TW, Boor R, Mayer T, et al. Concentrations of stiripentol in children and adults with epilepsy: the influence of dose, age, and comedication. Ther Drug Monit. 2012;34(4):390–397. | |
Cazali N, Tran A, Treluyer JM, et al. Inhibitory effect of stiripentol on carbamazepine and saquinavir metabolism in human. Br J Clin Pharmacol. 2003;56(5):526–536. | |
Giraud C, Treluyer JM, Rey E, et al. In vitro and in vivo inhibitory effect of stiripentol on clobazam metabolism. Drug Metab Dispos. 2006;34:608–611. | |
Yamamoto Y, Takahashi Y, Imai K, et al. Influence of CYP2C19 polymorphism and concomitant antiepileptic drugs on serum clobazam and N-desmethyl clobazam concentrations in patients with epilepsy. Ther Drug Monit. 2013;35(3):305–312. | |
Mott DD, Grosenbaugh DK, Fisher JL. Polytherapy with stiripentol: consider more than just metabolic interactions. Epilepsy Behav. 2013;29(3):585. | |
Poisson M, Huguet F, Savattier A, Bakri-Logeais F, Narcisse G. A new type of anticonvulsant, stiripentol. Pharmacological profile and neurochemical study. Arzneimittelforschung. 1984;34(2):199–204. | |
Quilichini PP, Chiron C, Ben-Ari Y, Gozlan H. Stiripentol, a putative antiepileptic drug, enhances the duration of opening of GABA-receptor channels. Epilepsia. 2006;47:704–716. | |
Fisher JL. The anti-convulsant stiripentol acts directly on the GABA(A) receptor as a positive allosteric modulator. Neuropharmacology. 2009;56:190–197. | |
Fisher JL. The effects of stiripentol on GABA(A) receptors. Epilepsia. 2011;52 Suppl 2:76–78. | |
Fisher JL. Interactions between modulators of the GABA(A) receptor: stiripentol and benzodiazepines. Eur J Pharmacol. 2011;654: 160–165. | |
Grosenbaugh DK, Mott DD. Stiripentol in refractory status epilepticus. Epilepsia. 2013;54 Suppl 6:103–105. | |
Grosenbaugh DK, Mott DD. Stiripentol is anticonvulsant by potentiating GABAergic transmission in a model of benzodiazepine-refractory status epilepticus. Neuropharmacology. 2013;67:136–143. | |
Baraban SC, Dinday MT, Hortopan GA. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat Commun. 2013;4:2410. | |
Laurie DJ, Wisden W, Seeburg PH. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J Neurosci. 1992;12(11):4151–4172. | |
Auvin S, Lecointe C, Dupuis N, et al. Stiripentol exhibits higher anticonvulsant properties in the immature than in the mature rat brain. Epilepsia. 2013;54(12):2082–2090. | |
Cao D, Ohtani H, Ogiwara I, et al. Efficacy of stiripentol in hyperthermia-induced seizures in a mouse model of Dravet syndrome. Epilepsia. 2012;53(7):1140–1145. | |
Kosaki K, Tamura K, Sato R, Samejima H, Tanigawara Y, Takahashi T. A major influence of CYP2C19 genotype on the steady-state concentration of N-desmethylclobazam. Brain Dev. 2004;26:530–534. | |
Desta Z, Zhao X, Shin JG, Flockhart DA. Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin Pharmacokinet. 2002;41:913–958. | |
Chiron C, Kassai B, Dulac O, Pons G, Nabbout R. A revisited strategy for antiepileptic drug development in children: designing an initial exploratory step. CNS Drugs. 2013;27(3):185–195. | |
Brigo F, Ausserer H, Tezzon F, Nardone R. When one plus one makes three: the quest for rational antiepileptic polytherapy with supraadditive anticonvulsant efficacy. Epilepsy Behav. 2013;27:439–442. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.