Back to Journals » Pediatric Health, Medicine and Therapeutics » Volume 8
Steroid-resistant nephrotic syndrome: past and current perspectives
Authors Nourbakhsh N, Mak RH
Received 16 September 2016
Accepted for publication 18 November 2016
Published 11 April 2017 Volume 2017:8 Pages 29—37
DOI https://doi.org/10.2147/PHMT.S100803
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Roosy Aulakh
Noureddin Nourbakhsh, Robert H Mak
Division of Pediatric Nephrology, Rady Children’s Hospital San Diego, University of California, San Diego, La Jolla, CA, USA
Abstract: Patients with steroid-resistant nephrotic syndrome (SRNS) represent a challenging subset of patients with nephrotic syndrome who often fail standard immunosuppression and have a higher likelihood of progressing to end-stage renal disease. Appropriate treatment of SRNS requires an adequate understanding of the historical treatment, renal histopathology, and genetics associated with the disease. The aim of this review is to present a comprehensive appraisal of the history, role of renal biopsy, genetics, and treatment of SRNS.
Keywords: steroid-resistant nephrotic syndrome, focal segmental glomerulosclerosis
Introduction
Nephrotic syndrome is a clinical condition characterized by proteinuria, hypoalbuminemia, edema, and hyperlipidemia. Nephrotic syndrome represents the most common primary glomerular disease in children and affects ~2 per 100,000 children aged <16 years in Europe and North America.1,2 At the time of first presentation, ~80% of children achieve complete remission within 4 weeks of corticosteroid therapy and are classified as having steroid-sensitive nephrotic syndrome (SSNS).3 However, 60%–70% of the patients initially classified as steroid sensitive have >1 relapse.4 Among children who relapse, 30% will be further classified as having frequently relapsing nephrotic syndrome or steroid-dependent nephrotic syndrome.4,5 Steroid resistance, defined as the absence of remission despite 4 weeks of therapy with daily prednisone at a dose of 2 mg/kg/d, encompasses an even smaller proportion of patients. Despite representing a smaller proportion of nephrotic syndrome cases, patients with steroid-resistant nephrotic syndrome (SRNS) have proven more difficult to treat, with 36%–50% progressing to end-stage renal disease within 10 years.6 The aim of this review is to present a comprehensive appraisal of the history, management, genetics, and treatment of SRNS.
Insights from International Society of Kidney Disease in Children (ISKDC)
Between 1967 and 1976, the ISKDC carried out a prospective study of 512 children (from 24 participating clinics) with primary nephrotic syndrome. The participants had a renal biopsy performed at the time of diagnosis before steroid therapy was begun. The prednisone regimen consisted of an initial treatment of 60 mg/m2 (maximum dosage 80 mg/24 h) divided into 2 doses/d for 4 weeks, followed by 40 mg/m2 divided into 2 doses/d on 3 consecutive days/wk for 4 weeks.7 Relapse protocol consisted of 60 mg/m2 divided into 2 doses/d until response (maximum of 4 weeks), followed by 40 mg/m2 divided into 2 doses/d on 3 serial days/wk for 4 weeks.7 As a result of this treatment regimen, the clinical definitions that delineate the various subcategories of nephrotic syndrome surfaced (Table 1).
 | Table 1 Clinical definitions of nephrotic syndrome Abbreviations: d, day; h, hour. |
A significant finding in the context of this review, steroid sensitivity, was defined as the achievement of complete remission within 4 weeks of corticosteroid therapy after initial presentation.7,8 In the ISKDC study, ~80% of children achieved clinical remission within 4 weeks of corticosteroid therapy. Additionally, if renal pathology results were considered, 95% of children with minimal change disease (MCD) on histopathology achieved clinical remission within 8 weeks of corticosteroid therapy after initial presentation.7,8 Beyond the histopathologic findings, other prognostic features in assessing a child’s response to corticosteroid therapy arose: children with nephrotic syndrome younger than 6 years without hypertension, chronic kidney disease (CKD), hypocomplementemia, or signs of systemic disease had an ~85% chance of responding to corticosteroid therapy.7
Length of corticosteroid therapy
In 1988, the German Pediatric Nephrology Working Group, in a study published in Lancet, evaluated the efficacy of the ISKDC standard steroid regimen compared to short-course prednisone therapy.9 In a controlled multicenter study, 61 children with a first episode of idiopathic nephrotic syndrome were randomly assigned to receive short-course prednisone therapy versus standard prednisone therapy. Short-course prednisone therapy consisted of 60 mg/m2 daily until absence of proteinuria for 3 days, followed by 40 mg/m2 every 48 hours until complete remission took place. The standard prednisone therapy was the ISKDC treatment regimen of 60 mg/m2 daily (maximum dosage: 80 mg/24 h) for 4 weeks, followed by 40 mg/m2 every 48 hours for 4 weeks. Patients were also randomly assigned to 1 of 2 of the following relapse therapy treatments: short-course prednisone treatment for relapse (60 mg/m2 daily until proteinuria disappeared for 3 days, followed by 40 mg/m2 every 48 hours until complete remission) versus standard prednisone treatment for relapse (60 mg/m2 daily until response [maximum of 4 weeks], followed by 40 mg/m2 every 48 hours for 4 weeks).
In the short-course group, urinary remission took place after 14 days of daily prednisone, and complete remission after an additional 16 days of alternate day prednisone.9 However, after 2 years of follow-up, sustained remission was significantly lower after the short-course regimen than after standard treatment (19% versus 41%, p=0.001), and the duration of remission in patients with a relapse was half as long after the short course compared to the standard course (79 days versus 169 days, p=0.004).9 In brief, although complete initial remission of nephrotic syndrome can be obtained with half the standard prednisone dose, the short course is beset with a higher rate of relapses that require additional prednisone administration.
Given the beneficial effect of standard prednisone therapy over short-course prednisone therapy, Ehrich and Brodehl,10 in association with the German Pediatric Nephrology Working Group, conducted a controlled prospective multicenter study of 71 children comparing the efficacy and outcome of long prednisone therapy versus standard prednisone therapy for the initial treatment of childhood nephrotic syndrome. Long prednisone therapy consisted of 60 mg/m2 daily for 6 weeks, followed by alternate day 40 mg/m2 every 48 hours for 6 weeks. The standard prednisone therapy group received the ISKDC treatment of 60 mg/m2 daily for 4 weeks, followed by 40 mg/m2 every 48 hours for 4 weeks. Compared to standard prednisone therapy, sustained remission after 2 years of follow-up was significantly greater after the long course of steroid therapy (49% versus 19%, p=0.0079).10
Thanks to the contributions of the ISKDC and German Pediatric Nephrology Working Group, disparities between the short, standard, and long corticosteroid regimens for nephrotic syndrome were established (Table 2). Long-course prednisone therapy for the initial presentation of SSNS is preferable to the standard regimen and short-course regimen because it leads to the highest percentage of sustained remission rates and reduces the rate of subsequent relapses.10 Of course, these outcomes are accompanied by the higher side effect profile of the long-course prednisone treatment regimen.
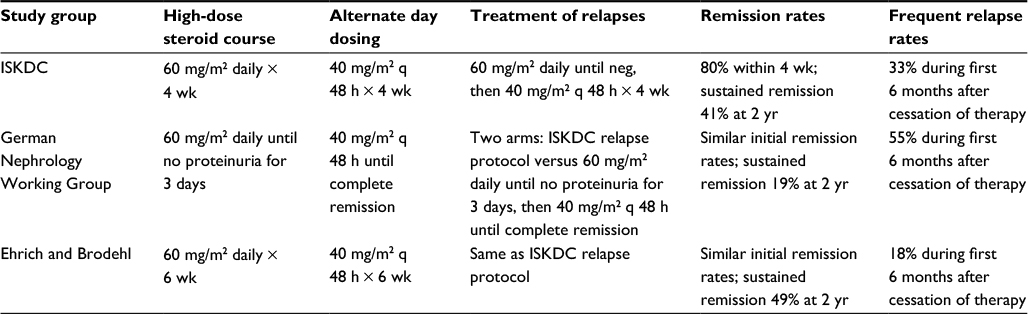 | Table 2 Comparative outcomes of steroid therapy for initial presentation of nephrotic syndrome Abbreviations: h, hour; wk, week; yr, year; ISKDC, International Society of Kidney Disease in Children; q, every; neg, negative. |
Role of renal biopsy
As per ISKDC report, of initial steroid responders, the incidence of focal segmental glomerulosclerosis (FSGS) on renal biopsy was 3%, while 47.5% of initial nonresponders had FSGS on renal biopsy.8 Also, the histopathologic variants of MCD most commonly associated with initial nonresponse were diffuse mesangial hypercellularity, mild mesangial hypercellularity, and focal tubular changes.11 Given the larger proportion of patients with FSGS who progress to end-stage renal disease (ESRD) and the higher likelihood of a relapsing course based on histopathology, many nephrologists face the difficult decision of determining optimal timing of initial biopsy in initial nonresponders with new-onset nephrotic syndrome. ISKDC data suggest that absence of response to steroid therapy at 8 weeks indicates nonresponse; however, given the 6-week high-dose steroid course endorsed by numerous programs, a lack of response at completion of 6 weeks often prompts many nephrologists to pursue renal biopsy.
Genetics
The genetics of SRNS has focused on genes playing a role in cell–cell signaling at the podocyte slit membrane (NPHS1, NPHS2, CD2AP, and PTPRO/GLEPP1), regulation of foot process actin network (ACTN4 and INF2), or foot process–glomerular basement membrane interaction (LAMB2 and ITGA3).12 More recently, podocyte cell migration has been implicated in the pathogenesis of SRNS. Gee et al13 utilized combined homozygosity mapping with whole-exome resequencing in a single family in whom 2 siblings had early-onset SRNS (with renal biopsy finding of diffuse mesangial sclerosis) to identify an ARHGDIA (Rho GDP dissociation inhibitor alpha) mutation that causes SRNS in rats. The investigators demonstrated that ARHGDIA is in complex with RHO GTPases expressed in podocytes of rat glomeruli. These RHO GTPases are responsible for regulation of actin remodeling such that both increased and decreased RHO GTPase signaling interfere with podocyte mobility.13
Additionally, recessive mutations in the kidney ankyrin repeat-containing protein (KANK) family proteins have been found to have essential roles in nephron function via regulation of Rho GTPase activity.14 KANK2 was found to interact with ARHGDIA and knockdown of KANK2 in cultured podocytes increased active GTP-bound RHOA, thus decreasing podocyte migration.14
In terms of the other histopathologic variants of MCD, which portend a steroid-resistant course, genes involved in the pathogenesis of diffuse mesangial sclerosis have been identified.15 Truncating mutations in phospholipase C epsilon 1 (PLCE1) prevent normal glomerular development and lead to diffuse mesangial sclerosis.
In summary, the role of the podocyte is crucial in understanding of the pathogenesis of nephrotic syndrome. The development, migration, basement membrane interaction, and regeneration of the podocyte are all critical processes in the maintenance of foot process integrity. Genetic mutations in any one of these processes can lead to the phenotype of nephrotic syndrome, and more specifically, mutations in genes coding for key podocyte proteins (NPHS2, PLCE1, ACTN4, and TRPC6) cause FSGS, the histopathologic finding most commonly associated with SRNS.12 In all, 24 genes are presently known to be associated with hereditary SRNS (Table 3).16
 | Table 3 Genes associated with SRNS Abbreviations: SRNS, steroid-resistant nephrotic syndrome; FSGS, focal segmental glomerulosclerosis. |
Given the established association of 5 particular gene mutations in nonsyndromic FSGS, these genes will be explored in detail in the following sections, with emphasis being placed on the proposed mechanism of action leading to FSGS. Of note, special emphasis will be placed on the 2 genes (NPHS2 and ACTN4) that are linked not only to FSGS but also to SRNS. Congenital nephrotic syndrome will not be discussed in detail.
NPHS2
Mutations in the NPHS2 gene, which encodes podocin, can cause a recessive form of SRNS.17 Mutations in this gene occur in ~40% of familial and 6%–17% of sporadic SRNS cases.18 Patients typically present from birth to 6 years of age have a steroid-resistant course and reach ESRD before the end of their first decade.19 Although the literature citing the role of nephrin in the podocyte is expansive, little is known about podocin’s role within the podocyte. However, data thus far suggest that podocin binds nephrin either directly or indirectly and thus may augment nephrin-induced stimulation of mitogen-activated protein kinases.20 Also, work on Caenorhabditis elegans species suggests that podocin, an integral membrane protein, is a critical part of membrane-associated proteases, which play a crucial role in protein turnover.21 Dysregulation of protein turnover and the complex interaction between podocin and nephrin is thought to be the core dysfunction triggering the autosomal recessive form of SRNS.
Finally, of particular importance in the genetics of juvenile or adult-onset NS is the p.R229Q variant. This variant, when found in association with one pathogenic NPHS2 mutation, represents the most frequently reported variant in Caucasians, particularly in Europeans with a frequency of 0.03–0.13.22
Alpha-actinin 4
ACTN4, the gene that encodes alpha-actinin 4, is most prominently distributed in podocytes, with some distribution in renal vasculature. Alpha-actinin 4 is responsible for cross-linking bundles of actin filament in the slit diaphragm. Mutations in this protein lead to decreased actin binding, formation of intracellular aggregates, and decreased protein half-life.23 Decreased protein half-life (ie, increased affinity for actin binding) is then thought to alter actin polymerization and thus adversely affect the podocyte cytoskeletal structure.23 Weins et al24 used electron microscopy to show that, compared to wild-type ACTN4, mutant ACTN4 induced formation of a disordered and tangled network of thin filament bundles in vitro. Three different point mutations in ACTN4 were discovered in 3 unrelated families with FSGS: K255E, T259I, and S262P mutations.25 These families showed autosomal dominant inheritance of disease with incomplete penetrance and variable expressivity.25 A typical presentation for patients with ACTN4 mutations is proteinuria in their teenage years or later, with a slow progression to ESRD in their 50s.26
PLCE1
Hinkes et al15 have described a mutation in autosomal recessive nephrotic syndrome type 3, which leads to early-onset nephrotic syndrome. The mutation causes a form of SRNS with the key histologic finding of diffuse mesangial sclerosis.15 The gene product, phospholipase C epsilon (PLCE1), is expressed in the developing kidney in glomerular podocytes and mutations, leading to abnormal protein products that cause an arrest in normal glomerular development. More specifically, given that phospholipase C epsilon catalyzes the hydrolysis of membrane phospholipids to generate inositol 1,4,5-triplephosphate and diacylglycerol, the intracellular pathways of cell growth and differentiation are impaired by mutations in this protein.27 PLCE1 also interacts with IQGAP-1, a podocyte cell junction-associated protein,15 therefore, cell adhesion is affected by mutations in the gene encoding for this protein. Recessive PLCE1 variants were identified in 10/35 (28.6%) families affected by diffuse mesangial sclerosis.15 In another study, recessive PLCE1 pathogenic variants were found in affected individuals from 12 of 68 families (18%) with SRNS.28
TRPC6
TRPC6 encodes a calcium channel by the same name, which is located on the podocyte membrane and, in complex with podocin, regulates mechanosensation at the slit diaphragm. In response to phospholipase C activation, activation of TRPC6 leads to increased intracellular calcium concentration. In the kidney, TRPC6 is expressed in renal tubules and glomeruli, with predominance in podocytes. Thus far, 11 different mutations in the TRPC6 gene have been identified in 8 families from different ethnic backgrounds.29,30 Five of these mutations in the TRPC6 gene are gain-of-function mutations that result in intracellular influx of calcium.29,30 While it is unclear exactly how this may lead to FSGS, it is postulated that the increased intracellular calcium may lead to altered podocyte function. Although previously ascribed only as a genetic cause of late-onset autosomal dominant FSGS, a group of Italian investigators have suggested that the TRPC6 variants can also be detected in children with early-onset and sporadic SRNS (4 of 33 patients).31
CD2AP
Another gene which may be involved in the hereditary form of FSGS is the CD2-associated protein (CD2AP) gene. The CD2AP protein is expressed in podocytes and interacts with fyn and synaptopodin, thus maintaining the cytoskeletal architecture of the podocyte. Five different CD2AP heterozygous mutations have been found in pediatric FSGS patients.32 However, the clinical significance of recessive CD2AP mutations has been called into question as a study did not find homozygous CD2AP mutations in a cohort of 42 children with SRNS for whom NPHS1, NPHS2, PLCE1, and WT1 mutations had been previously excluded.33
Syndromic forms of SRNS are far less frequent and may be due to mutations in genes encoding various components involved in glomerular basement integrity. These genes include WT1 and LMX1B (transcriptional factors), LAMB2 and ITGB4 (glomerular basement membrane components), SCARB2 (lysosomal protein), COQ2, PDSS2, and MTTL1 (mitochondrial proteins), and SMARCAL1 (a DNA-nucleosome restructuring mediator). Mutations in NPHS1 are responsible for most cases of congenital nephrotic syndrome.34
Indications for genetic testing
Because there are no clinical findings that correlate with the particular gene defects mentioned, the practitioner commonly must rely on the patient’s response to steroid therapy as a guide to determining steroid resistance. Additionally, there may be some difficulty in distinguishing between true steroid resistance and noncompliance with steroid therapy. Once the classic definition of steroid resistance is met in a young patient (<2 years old), depending on the resources available at the institution, genetic testing may be pursued. Although there are no specific treatment guidelines based on genetic variants of SRNS, recent data suggest that cyclosporine therapy may be more effective in nongenetic forms of SRNS as opposed to genetic forms of SRNS,35 possibly encouraging providers to more frequently request genetic testing in SRNS patients. Additionally, given the poor response to immunosuppressive therapy in genetic forms of SRNS, identification of mutations via genetic screening may assist clinicians in deescalating further immunosuppression options and spare children from significant side effects associated with these medications.
In general, the key features in a patient with nephrotic syndrome, which suggest the need for genetic testing, include age <6 years at diagnosis (especially age <2 years), positive family history of nephrotic syndrome, consanguinity, a steroid-resistant course, and histopathologic findings of FSGS or diffuse mesangial sclerosis on renal biopsy. More specifically, Benoit et al34 have suggested a systematic approach to genetic testing based on age at diagnosis, histopathologic findings, and mode of inheritance (Figure 1).
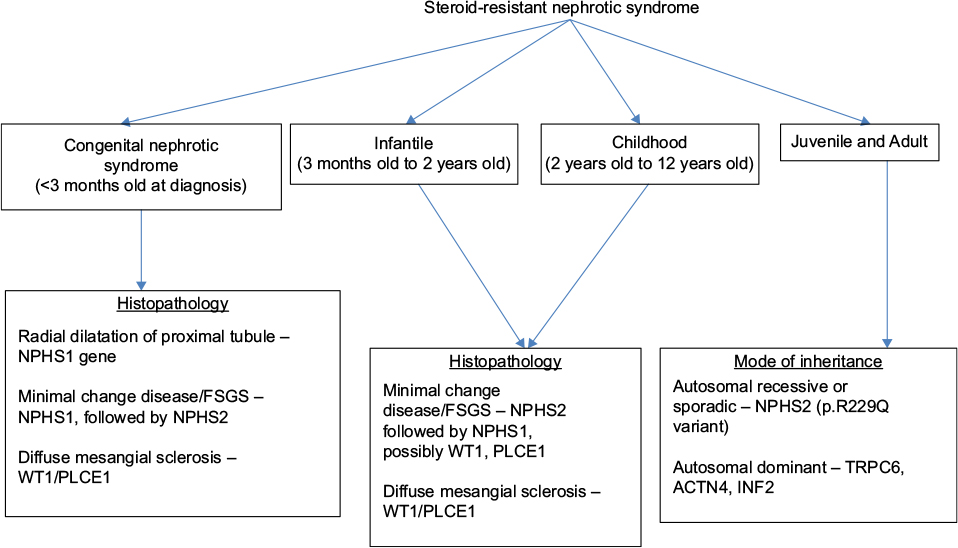 | Figure 1 Systematic approach to genetic testing in children with steroid-resistant nephrotic syndrome. Note: Adapted from Benoit G, Machuca E, Antignac C. Hereditary nephrotic syndrome: a systematic approach for genetic testing and a review of associated podocyte gene mutations. Pediatr Nephrol. 2010;25(9):1621–1632.34 Abbreviations: FSGS, focal segmental glomerulosclerosis; PLCE1, phospholipase C epsilon 1. |
Biomarkers in SRNS
The presence of a circulating serum factor associated with SRNS has long been sought. In a seminal paper by Savin et al,36 the association between a circulating factor and disease activity was evaluated in patients with recurrent FSGS. Serum samples were analyzed in patients with FSGS pre- and postrenal transplant to evaluate if the serum samples of patients with recurrent FSGS increased glomerular-capillary permeability to albumin. A permeability value was assigned to each sample, with the lower permeability values indicating poor glomerular-capillary permeability to albumin and higher permeability values indicating increased glomerular-capillary permeability to albumin. Of the 56 patients with primary FSGS who underwent renal transplantation and were followed for at least 6 months, those without recurrent FSGS had permeability values of 0.14±0.06, while those with recurrent FSGS had permeability values of 0.47±0.06. Patients were then stratified according to their permeability value, with the relative risk of recurrent FSGS being 5 times higher in patients with pretransplantation values of at least 0.50. Of note, this association did not hold true for the 9 children with SSNS who were tested. Plasmapheresis in patients with recurrent FSGS lowered the level of serum activity and decreased proteinuria.
More recently, the soluble urokinase-type plasminogen activator receptor (suPAR) has been suggested as a potential circulating factor in FSGS.37
Reiser et al38 reported that circulating suPAR could activate B3 integrin on podocytes as a means of inducing proteinuria. suPAR concentrations correlated directly with the activity of podocyte B3 integrin. Conversely, inhibition of suPAR by antibodies against suPAR and/or plasmapheresis could lower B3 integrin activity.38 Serum suPAR concentrations were significantly elevated in persons with FSGS, compared to those with MCD, membranous nephropathy, and healthy subjects. In summary, Reiser et al were able to establish and identify the important role of suPAR in the pathogenesis of FSGS.
Therapy
Following failure of standard steroid therapy for nephrotic syndrome, the Tune-Mendoza protocol may serve as a useful adjunct to treatment of SRNS. It is a treatment utilizing high-dose methylprednisolone pulse therapy and oral prednisolone administered for a total of 82 weeks. When this protocol was used to treat patients with SRNS, ~65% went into complete remission and only 25% progressed to CKD.39 However, significant side effects including obesity, growth impairment, hypertension, cataracts, osteoporosis, diabetes, immune suppression, psychosis, abdominal striae, cushingoid facies, and hirsutism can result from prolonged steroid therapy.
ACE-Is/ARBs have long been utilized and found to be effective as antiproteinuric agents in the treatment of adult nephropathies.40 In terms of assessing the renoprotective effect of ACE-Is in children, the ESCAPE trial41 offers data in the largest trial to date. In this trial, 397 children (ages 3–18 years) with CKD (eGFR 11–80 mL/min/1.73 m2) and elevated or high-normal BP received ramipril (6 mg/m2) following a 6-month run-in period, including a 2-month washout of any previous ACE inhibitors. Urinary protein excretion was reduced by 50% on average, with a similar efficacy independent of etiology of renal disease (renal dysplasia or glomerulopathy). Urinary protein excretion was dependent on baseline proteinuria (p<0.0001) and was correlated with antihypertensive efficacy of the drug (p<0.001). Other smaller trials, utilizing ACE-Is and ARBs have found similar results in pediatric patients with proteinuria.42–44 Given the significant renoprotective effects demonstrated in these trials, ACE-I/ARB treatment should be strongly considered in the treatment of hypertensive patients with SRNS.
Oral cyclophosphamide therapy over a 12-week time course has been used in combination with steroids for the treatment of steroid-dependent nephrotic syndrome.45 However, data are lacking for this approach in the treatment of SRNS. Although there are distinct views regarding efficacy of intravenous (IV) cyclophosphamide therapy for treatment of SRNS, some providers have found success with monthly IV cyclophosphamide therapy (500 mg/m2/dose) for a 6-month course.46
Calcineurin inhibitors have long been used in the treatment of SRNS, with the preponderance of data existing for therapy with cyclosporine. Studies have shown that cyclosporine in combination with steroids is effective in retaining remission and reducing relapse rates in patients with SRNS.47,48 Ehrich et al48 have shown that prolonged combination therapy with steroids, cyclosporine and high-dose methylprednisolone maintained the remission rate at 84% for patients with SRNS. Additionally, long-term treatment with cyclosporine in children with SRNS reduced the progression to CKD.49 The question of efficacy of cyclosporine in genetic versus nongenetic causes of SRNS was recently evaluated by Buscher et al.35 Collection of data from 231 patients with SRNS treated at 8 pediatric centers over an observation period of 113 months revealed that 82% of nongenetic SRNS patients responded within 6 months of Csa therapy. Also, 98% of patients with non-genetic SRNS and Csa-induced complete remission maintained normal renal function.35 By contrast, only 3% of patients with genetic SRNS experienced a complete remission, while 16% experienced a partial remission. There was a high rate of progression to ESRD (66% of patients) in patients with genetic SRNS.35 Notable side effects of therapy with cyclosporine include gum hypertrophy, hypertension, hypertrichosis, and nephrotoxicity.
Tacrolimus serves as an alternative to cyclosporine with a slightly more attractive side effect profile as there is less hypertrichosis and gum hypertrophy. However, other side effects, including tremor, hypertension, and diabetes, have been reported. At a dose of 0.1–0.2 mg/kg/d divided in 2 doses, a complete remission rate of 81% was achieved in a study of pediatric patients with SRNS by Jahan et al.50 A target trough level of 5–7 ng/mL is recommended.51
The role of mycophenolate mofetil (MMF) for the treatment of SRNS is uncertain. Data are largely retrospective52 or from single-center experience with a low number of patients.53 In a retrospective study, complete and partial remission for MMF in SRNS was 67%.52 It may seldom be used as an alternative agent in patients with SRNS who have experienced significant side effects due to calcineurin inhibitors. Adverse effects of MMF include abdominal pain, diarrhea, infection, metabolic acidosis, and hyperlipidemia. MMF should be administered at a dose of 1200 mg/m2/d in 2 divided doses for the indication of nephrotic syndrome. Of note, RAS blockade used in combination with MMF has been shown to be effective in patients with steroid-resistant FSGS.54 However, in the study by Montane et al, prior to the initiation of MMF-angiotensin blockade, patients were pretreated with weekly IV methylprednisolone (15 mg/kg/wk) for 4–8 weeks. MMF was given at a dose of 250–500 mg/m2/d.54 In another trial of pediatric patients with SRNS, fosinopril significantly reduced proteinuria.55 Notable side effects of ACE-I/ARB therapy include hyperkalemia, reduction in glomerular filtration rate, and teratogenicity. Teratogenicity is, of course, of particularly high risk with combination MMF and ACE-I/ARB therapy.
Rituximab is a monoclonal antibody directed against the cell surface antigen CD20 expressed on B lymphocytes. It has gained acceptance as a treatment for refractory cases of childhood nephrotic syndrome and, in recent years, is gaining recognition as a first-line steroid-sparing agent in the treatment of children with steroid-dependent nephrotic syndrome.56 Data for rituximab therapy in treatment of SRNS are less robust. An investigation into the efficacy of rituximab for SRNS by the International Pediatric Nephrology Association (IPNA) found that the perceived response rate of rituximab was 44% in children with SRNS compared with 82% for those with steroid-dependent nephrotic syndrome.57 This investigation was based on retrospective assessment of each IPNA associate’s single-center experience. More recently, various investigators have published retrospective data evaluating the efficacy of rituximab used in combination with other immunosuppressive therapies in the treatment of SRNS.58–60 The data are promising; however, there has not yet been a prospective, randomized trial evaluating the efficacy of rituximab in SRNS.
Role of renal transplant in SRNS
Kidney transplantation is the most ideal renal replacement therapy for children with SRNS. Children with SRNS run the risk of recurrence of nephrotic syndrome following transplantation. Recurrence of nephrotic syndrome, if not controlled, can cause delayed graft function, acute rejection, and diminished allograft survival. The majority of information regarding posttransplant outcomes comes from pediatric patients with FSGS, the most common histopathologic finding in patients with SRNS. The risk of recurrence of FSGS posttransplant is ~30%.19 Risk factors for recurrence of FSGS posttransplant include childhood-onset SRNS, rapid progression to ESRD, and recurrence following a previous allograft.61 The concept of a circulating permeability factor as the pathogenic mechanism of SRNS arose following the frequent observation of SRNS recurrence following renal transplantation.62 Some investigators have found that initiation of plasmapheresis may induce remission in 50%–90% of recurrent cases.63,64 The favorable outcomes reported with plasmapheresis in patients with recurrent SRNS have resulted in the use of this treatment in primary SRNS pretransplant.64 Although the preponderance of data currently supports posttransplant use of plasmapheresis for SRNS recurrence, the effectiveness of preoperative plasmapheresis in the prevention of SRNS recurrence following renal transplantation has not yet been firmly established. Dependent on the local practices of renal transplantation programs, plasmapheresis may be part of the pre- or postoperative protocol in patients with SRNS.
Conclusion
While comprising a small minority of the patients presenting with nephrotic syndrome (8%–15%), patients with SRNS prove a particularly challenging set of patients to manage and treat. Many advances have been made in the study of this particular set of patients previously destined to rapidly progress to ESRD. In particular, the genetic profile of such patients has proved critical in unraveling the pathophysiologic mechanisms of disease and in guiding therapeutic options. With further research in the field, it is likely that new mechanisms will be implicated in the pathophysiology of SRNS and further treatment options can be elucidated to optimize treatment of this disease.
Acknowledgment
RHM is supported by NIH grants U01 DK-03012 and R24HD050837.
Disclosure
The authors report no conflicts of interest in this work.
References
El Bakkali L, Rodrigues Pereira R, Kuik DJ, Ket JCF, van Wijk JAE. Nephrotic syndrome in the Netherlands: a population-based cohort study and a review of the literature. Pediatr Nephrol. 2011;26(8):1241–1246. | ||
Report of the International Study of Kidney Disease in Children. Early identification of frequent relapses among children with minimal change nephrotic syndrome. A report of the International Study of Kidney Disease in Children. J Pediatr. 1982;101(4):514–518. | ||
Koskimies O, Vilska J, Rapola J, Hallman N. Long-term outcome of primary nephrotic syndrome. Arch Dis Child. 1982;57(7):544–548. | ||
Tarshish P, Tobin JN, Berstein J, Edelmann CM Jr. Prognostic significance of the early course of minimal change nephrotic syndrome: report of the International Study of Kidney Disease in Children. J Am Soc Nephrol. 1997;8(5):769–776. | ||
Nakanishi K, Iijima K, Ishikura K, et al. Two-year outcome of the ISKDC regimen and frequent-relapsing risk in children with idiopathic nephrotic syndrome. Clin J Am Soc Nephrol. 2013;8(5):756–762. | ||
Mekahli D, Liutkus A, Ranchin B, et al. Long-term outcome of idiopathic steroid-resistant nephrotic syndrome: a multicenter study. Pediatr Nephrol. 2009;24(8):1525–1532. | ||
International Study of Kidney Disease in Children. The primary nephrotic syndrome in children. Identification of patients with minimal change nephrotic syndrome from initial response to prednisone. J Pediatr. 1981;98(4):561–564. | ||
International Study of Kidney Disease in Children. Prediction of histopathology from clinical and laboratory characteristics at time of diagnosis. Kidney Int. 1978;13(7):159–165. | ||
Short versus standard prednisone therapy for initial treatment of idiopathic nephrotic syndrome in children. Arebeitsgemeinschaft fur Padiatrische Nephrologie. Lancet. 1988;331(8582):380–383. | ||
Ehrich JH, Brodehl J. Long versus standard prednisone therapy for initial treatment of idiopathic nephrotic syndrome in children. Arbeitsgemeinschaft fur Padiatrische Nephrologie. Eur J Pediatr. 1993;152(4):357–361. | ||
International Study of Kidney Disease in Children. Primary nephrotic syndrome in children: clinical significance of histopathologic variants of minimal change and of diffuse mesangial hypercellularity. Kidney Int. 1981;20(6):765–771. | ||
Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular disease. Kidney Int. 2007;71(12):1205–1214. | ||
Gee HY, Saisawat P, Ashraf S, et al. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J Clin Invest. 2013;123(8):3243–3253. | ||
Gee HY, Zhang F, Ashraf S, et al. KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. J Clin Invest. 2015;125(6):2375–2384. | ||
Hinkes B, Wiggins RC, Gbadegesin R, et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet. 2006;38(12):1397–1405. | ||
McCarthy HJ, Bierzynska A, Wherlock M, et al. Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2013;8(4):637–648. | ||
Boute N, Gribouval O, Roselli S, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000;24(4):349–354. | ||
Karle SM, Uetz B, Ronner V, Glaeser L, Hildebrandt F, Fuchshuber A. Novel mutations in NPHS2 detected in both familial and sporadic steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2002;13(2):388–393. | ||
Tejani A, Stablein DH. Recurrence of focal segmental glomerulosclerosis posttransplantation: a special report of the North American Pediatric Renal Transplant Cooperative Study. J Am Soc Nephrol. 1992;2(12 Suppl):S258–S263. | ||
Huber TB, Kottgen M, Schilling B, Walz G, Benzing T. Interaction with podocin facilitates nephrin signaling. J Biol Chem. 2001;276(45):41543–41546. | ||
Tavernarakis N, Driscoll M, Kyrpides NC. The SPFH domain: implicated in regulating targeted protein turnover in stomatins and other membrane-associated proteins. Trends Biochem Sci. 1999;24(11):425–427. | ||
Tsukaguchi H, Sudhakar A, Le TC, et al. NPHS2 mutations in late-onset focal segmental glomerulosclerosis: R229Q is a common disease-associated allele. J Clin Invest. 2002;110(11):1659–1666. | ||
Mukerji N, Damodaran TV, Winn MP. TRPC6 and FSGS: the latest TRP channelopathy. Biochim Biophys Acta. 2007;1772(8):859–868. | ||
Weins A, Schlondorff JS, Nakamura F, et al. Disease-associated mutant alpha-actinin-4 reveals a mechanism for regulating its F-actin-binding affinity. Proc Natl Acad Sci U S A. 2007;104(41):16080–16085. | ||
Feng D, DuMontier C, Pollak MR. The role of alpha-actinin-4 in human kidney disease. Cell Biosci. 2015;5:44. | ||
Kaplan JM, Kim SH, North KN, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. 2000;24(3):251–256. | ||
Wing MR, Bourdon DM, Harden TK. PLC-epsilon: a shared effector protein in Ras-, Rho-, and G alpha beta gamma-mediated signaling. Mol Interv. 2003;3(5):273–280. | ||
Boyer O, Benoit G, Gribouval O, et al. Mutational analysis of the PLCE1 gene in steroid resistant nephrotic syndrome. J Med Genet. 2010;47(7):445–452. | ||
Winn MP, Conlon PJ, Lynn KL, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308(5729):1801–1804. | ||
Zhu B, Chen N, Wang ZH, et al. Identification and functional analysis of a novel TRPC6 mutation associated with late onset familial focal segmental glomerulosclerosis in Chinese patients. Mutat Res. 2009;664(1–2):84–90. | ||
Gigante M, Caridi G, Mentemurno E, et al. TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin J Am Soc Nephrol. 2011;6(7):1626–1634. | ||
Gigante M, Pontrelli P, Montemurno E, et al. CD2AP mutations are associated with sporadic nephrotic syndrome and focal segmental glomerulosclerosis (FSGS). Nephrol Dial Transplant. 2009;24(6):1858–1864. | ||
Benoit G, Machuca E, Nevo F, Gribouval O, Lepage D, Antignac C. Analysis of recessive CD2AP and ACTN4 mutations in steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2010;25(3):445–451. | ||
Benoit G, Machuca E, Antignac C. Hereditary nephrotic syndrome: a systematic approach for genetic testing and a review of associated podocyte gene mutations. Pediatr Nephrol. 2010;25(9):1621–1632. | ||
Buscher AK, Beck BB, Melk A, et al. Rapid response to cyclosporine A and favorable renal outcome in nongenetic versus genetic steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2016;11(2):245–253. | ||
Savin VJ, Sharma R, Sharma M, et al. Circulating factor associated with increased permeability to albumin in recurrent focal segmental glomerulosclerosis. N Engl J Med. 1996;334(14):878–883. | ||
Wei C, El Hindi S, Li J, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med. 2011;17(8):952–960. | ||
Reiser J, Wei C, Tumlin J. Soluble urokinase receptor and focal segmental glomerulosclerosis. Curr Opin Nephrol Hypertens. 2012;21(4):428–432. | ||
Tune BM, Kirpekar R, Sibley RK, Reznik VM, Griswold WR, Mendoza SA. Intravenous methylprednisolone and oral alkylating agent therapy of prednisone-resistant pediatric focal segmental glomeruloscelosis: a long-term follow-up. Clin Nephrol. 1995;43(2):84–88. | ||
MacKinnon M, Shurraw S, Akbari A, Knoll GA, Jaffey J, Clark HD. Combination therapy with an angiotensin receptor blocker and an ACE inhibitor in proteinuric renal disease: a systematic review of the efficacy and safety data. Am J Kidney Dis. 2006;48(1):8–20. | ||
Wuhl E, Mehls O, Schaefer F; ESCAPE Trial Group. Antihypertensive and antiproteinuric efficacy of ramipril in children with chronic renal failure. Kidney Int. 2004;66(2):768–776. | ||
Lama G, Luongo I, Piscitelli A, Salsano ME. Enalapril: antiproteinuric effect in children with nephrotic syndrome. Clin Nephrol. 2000;53(6):432–436. | ||
Ellis D, Vats A, Moritz ML, Reitz S, Grosso MJ, Janosky JE. Long-term antiproteinuric and renoprotective efficacy and safety of losartan in children with proteinuria. J Pediatr. 2003;143(1):89–97. | ||
Wuhl E, Schaefer F. Therapeutic strategies to slow chronic kidney disease progression. Pediatr Nephrol. 2008;23(5):705–716. | ||
Oemar BS, Brodehl J. Cyclophosphamide treatment of steroid dependent nephrotic syndrome: comparison of eight week with 12 week course. Report of Arbeitsgemeinschaft fur Padiatrische Nephrologie. Arch Dis Child. 1987;62(11):1102–1106. | ||
Alshaya HO, Al-Maghrabi JA, Kari JA. Intravenous pulse cyclophosphamide – is it effective in children with steroid-resistant nephrotic syndrome? Pediatr Nephrol. 2003;18(11):1143–1146. | ||
Hamasaki Y, Yoshikawa N, Hattori S, et al. Cyclosporine and steroid therapy in children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2009;24(11):2177–2185. | ||
Ehrich JH, Geerlings C, Zivicnjak M, Franke D, Geerlings H, Gellerman J. Steroid-resistant idiopathic childhood nephrosis: overdiagnosed and undertreated. Nephrol Dial Transplant. 2007;22(8):2183–2193. | ||
Ingulli E, Singh A, Bagi N, Ahmad H, Moazami S, Tejani A. Aggressive, long-term cyclosporine therapy for steroid-resistant focal segmental glomerulosclerosis. J Am Soc Nephrol. 1995;5(10):1820–1825. | ||
Jahan A, Prabha R, Chaturvedi S, Mathew B, Fleming D, Agarwal I. Clinical efficacy and pharmacokinetics of tacrolimus in children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2015;30(11):1961–1967. | ||
Gulati S, Prasad N, Sharma NK, Kumar A, Gupta A, Baburaj VP. Tacrolimus: a new therapy for steroid-resistant nephrotic syndrome in children. Nephrol Dial Transplant. 2008;23(3):910–913. | ||
Kim J, Patnaik N, Chorny N, Frank R, Infante L, Sethna C. Second-line immunosuppressive treatment of childhood nephrotic syndrome: a single center experience. Nephon Extra. 2014;4(1):8–17. | ||
Gargah TT, Lakhoua MR. Mycophenolate mofetil in treatment of childhood steroid-resistant nephrotic syndrome. J Nephrol. 2011;24(2):203–207. | ||
Montane B, Abitbol C, Chandar J, Strauss J, Zilleruelo G. Novel therapy of focal glomerulosclerosis with mycophenolate and angiotensin blockade. Pediatr Nephrol. 2003;18(8):772–777. | ||
Yi Z, Li Z, Wu XC, He QN, Dang XQ, He XJ. Effect of fosinopril in children with steroid-resistant idiopathic nephrotic syndrome. Pediatr Nephrol. 2006;21(7):967–972. | ||
Kemper MJ, Lehnhardt A, Zawischa A, Oh J. Is rituximab effective in childhood nephrotic syndrome? Yes and no. Pediatr Nephrol. 2014;29(8):1305–1311. | ||
Prytula A, Iijima K, Kamei K, et al. Rituximab in refractory nephrotic syndrome. Pediatr Nephrol. 2010;25(3):461–468. | ||
Basu B, Mahapatra TK, Mondal N. Mycophenolate mofetil following rituximab in children with steroid-resistant nephrotic syndrome. Pediatrics. 2015;136(1):e132–e139. | ||
Pescovitz MD, Book BK, Sidner RA. Resolution of recurrent focal segmental glomerulosclerosis proteinuria after rituximab treatment. N Engl J Med. 2006;354(18):1961–1963. | ||
Suyama K, Kawasaki K, Miyazaki K, et al. Rituximab and low-dose cyclosporine combination therapy for steroid-resistant FSGS. Pediatr Int. 2016;58(13):219–223. | ||
Vinai M, Waber P, Seikaly MG. Recurrence of focal segmental glomerulosclerosis in renal allograft: an in-depth review. Pediatr Transplant. 2010;14(3):314–325. | ||
McCarthy ET, Sharma M, Savin VJ. Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2010;5(11):2115–2121. | ||
Cheong HI, Han HW, Park HW, et al. Early recurrent nephrotic syndrome after renal transplantation in children with focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2000;15(1):78–81. | ||
Vécsei AK, Müller T, Schratzberger EC, et al. Plasmapheresis-induced remission in otherwise therapy-resistant FSGS. Pediatr Nephrol. 2001;16(11):898–900. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.