Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Stepwise encapsulation and controlled two-stage release system for cis-Diamminediiodoplatinum
Received 29 January 2014
Accepted for publication 9 April 2014
Published 28 June 2014 Volume 2014:9(1) Pages 3175—3182
DOI https://doi.org/10.2147/IJN.S61570
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Yun Chen,1,* Qian Li,1,2,* Qingsheng Wu1
1Department of Chemistry, Key Laboratory of Yangtze River Water Environment, Ministry of Education, Tongji University, Shanghai; 2Shanghai Institute of Quality Inspection and Technical Research, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Abstract: cis-Diamminediiodoplatinum (cis-DIDP) is a cisplatin-like anticancer drug with higher anticancer activity, but lower stability and price than cisplatin. In this study, a cis-DIDP carrier system based on micro-sized stearic acid was prepared by an emulsion solvent evaporation method. The maximum drug loading capacity of cis-DIDP-loaded solid lipid nanoparticles was 22.03%, and their encapsulation efficiency was 97.24%. In vitro drug release in phosphate-buffered saline (pH =7.4) at 37.5°C exhibited a unique two-stage process, which could prove beneficial for patients with tumors and malignancies. MTT (3-[4,5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide) assay results showed that cis-DIDP released from cis-DIDP-loaded solid lipid nanoparticles had better inhibition activity than cis-DIDP that had not been loaded.
Keywords: stearic acid, emulsion solvent evaporation method, drug delivery, cis-DIDP, in vitro
Introduction
Rosenberg et al’s serendipitous discovery that platinum (Pt) complexes could inhibit cell division sparked great interest in Pt-based antitumor drugs.1 Pt-based complexes, such as cis-diamminedichloroplatinum(II) (cis-[PtCl2(NH3)2]), carboplatin, and oxaliplatin, present good antitumor activity.2 cis-Diamminediiodoplatinum(II), formulated as cis-[(NH3)2PtI2]3 and abbreviated as cis-DIDP, is an intermediate in the synthesis of these Pt anticancer drugs. One common feature of these complexes is that they all contain Pt-N/Pt-I or Pt-N/Pt-Cl coordination bonds with two Pt-N bonds in the cis position. The Pt-I or Pt-Cl bonds in these complexes are chemically much weaker than the Pt-N bond, and are prone to facile hydrolysis under low acidity conditions, yielding charged cis-[Pt(NH3)2(H2O)2]2+ complexes that are highly reactive for deoxyribonucleic acid binding through the N7 atom of either an adenine or guanine base. This binding damages the double-helix structure and interrupts the cell’s transcription and repair mechanisms, killing the cancer cells.4 Enhancing Pt-N bonding not only improves the antitumor activity of the complex and reduces the dose at which 90% of tumors are inhibited, but also decreases drug toxicity, as measured by the median lethal dose, and increases the therapeutic index. The bond energy of Pt-N in cis-DIDP is higher than that in cisplatin.5
Although cis-DIDP is considered to have higher antitumor activity, the drug has not been widely used in clinical practice because of its poor water solubility and low stability. Preparing carriers loaded with cis-DIDP will improve efficacy. Several carriers have been developed. For example, polyamidoamine dendrimers can improve intracellular delivery and reduce undesirable side effects.6 Encapsulation in solid lipid nanoparticles (SLNs), which are drug carriers made from physiological or synthetic lipids, can increase the solubility and stability of pharmaceuticals.7 The present study focuses on designing an SLN carrier system for cis-DIDP. By increasing the stability and the antitumor activity of cis-DIDP, the SLN carrier may increase the performance of cis-DIDP as an anticancer drug.
SLNs can exhibit good drug incorporation and high loading capacity.8,9 Many methods have been used to prepare SLN suspensions, such as high shear homogenization and ultrasound,10 high-pressure melt homogenization,11 supercritical fluids technology,12 salting out,13 and solvent emulsification or evaporation.14,15 In these methods, hydrophobic and hydrophilic surfactants are used to form complexes so that the drug can be loaded into or covered with SLNs. Nanoparticles have been proven to be an effective delivery system, with few side effects, for anticancer drugs.16 Nanometer-sized, controlled-release drug carriers should improve the efficacy and reduce the side effects of cancer drugs, ultimately enhancing patient compliance. Overcoming this challenge in medical scientific research would fulfill an urgent clinical need and advance the pharmaceutical industry.17,18
In this study, cis-DIDP-loaded-SLNs were prepared successfully by emulsification dispersion-ultrasonication using a mixture of the surfactants, polyethylene glycol (PEG-4000) and soy lecithin (SL). This carrier system achieved stepwise encapsulation of cis-DIDP and exhibited two-stage release in vitro. Two-stage release is important for reducing the administration frequency and improving medication compliance. Furthermore, releasing a large quantity of drug within an initial period, followed by slow release, achieves better therapeutic effects.19 The results of this study will provide guidance toward the clinical application of cis-DIDP.
Materials and methods
Materials
PEG-4000 was obtained from Ling Feng Chemical Reagent Co., Ltd., Shanghai, People’s Republic of China. Stearic acid (SA) and SL were purchased from Sinopharm Chemical Reagent Co., Ltd., Shanghai, People’s Republic of China. cis-DIDP was obtained from Kunming Guiyan Pharmaceutical Co., Ltd., Kunming, People’s Republic of China. All chemicals were of analytical grade.
Preparation of an empty SA carrier
Approximately 200 to 350 mg of PEG-4000 was dissolved in 100 ml distilled water; 50 mg SL and 70 mg SA were dissolved in 20 mL trichloromethane (TCM). After complete dissolution, the SL and SA solution was poured into the PEG-4000 solution under vigorous stirring at 30°C–90°C until the TCM evaporated. The resulting precipitate was filtered off, washed five times with distilled water, and then freeze-dried to yield a white product, an empty SA carrier (ESC).
Preparation of cis-DIDP-loaded SLNs
cis-DIDP (0.05 g) and 200 mg to 350 mg PEG-4000 were dissolved in 100 mL distilled water; 50 mg SL and 70 mg SA were dissolved in 20 mL TCM. After complete dissolution, the SL-SA solution was poured into the PEG-4000 solution under vigorous stirring between 30°C–90°C until the TCM had fully evaporated. The precipitate was collected via centrifugation and freeze-dried. The black product, composed of cis-DIDP-loaded SLNs, was soluble in distilled water. To encapsulate adequate amounts of the drug, cis-DIDP was added to the solution before the formation of ESCs.
Characterization of cis-DIDP-loaded SLNs
Morphology and size of cis-DIDP-loaded SLNs were investigated with scanning electron microscopy (SEM; XL30; Philips, Amsterdam, the Netherlands) with energy dispersive spectroscopy (EDS) at an accelerating voltage of 15 kV to identify and measure Pt. Infrared spectra were recorded between 4,000 cm−1 and 400 cm−1 by Fourier transform infrared (FTIR) spectroscopy using a spectrometer equipped with a triglycine sulfate/piezoelectric effect detector and a silicon beam splitter with 1 cm−1 resolution (Nicolet Nexus; Thermo Fisher Scientific, Waltham, MA, USA). The crystal phase and purity of the products were characterized by X-ray powder diffraction (XRD; Focus D8; Bruker Optik GmbH, Ettlingen, Germany) equipped with graphite monochromatized Cu Ka radiation (λ =1.5406 Å), employing a scanning rate of 0.02° s-1. The operating voltage and current were maintained at 40 kV and 100 mA, respectively. The cell lattice constants of the samples were calculated and corrected with MDI Jade (5.0 edition) software (Jade Technologies, Christchurch, New Zealand). The absorbance spectra of ultraviolet (UV) radiation were obtained with a UV-visible spectrometer (Model 8453; Agilent Technologies, Santa Clara, CA, USA). Zeta potential was determined using a Zetasizer Nano (Malvern Instruments, Malvern, UK). Experiments were conducted with particles diluted in phosphate buffered saline (PBS) solution (Sinopharm Chemical Reagent Co., Ltd., Shanghai, People’s Republic of China), and measurements were performed at 25°C.
Evaluation of drug encapsulation and loading capacity
The cis-DIDP-loaded SLNs were dissolved in ethanol. The concentration of DIDP was measured before the SLN dissolution (free DIDP) and after the SLN dissolution (total DIDP), via UV-visible spectroscopy at 359 nm, in quintuplicate. The eleven-point calibration curve was obtained over the concentration range of 0.00255 mg/mL to 1.5 mg/mL cis-DIDP. The quantity of cis-DIDP could be calculated from the calibration curve. The encapsulation efficiency (EE%) and the drug loading capacity (LC%) of the nanoparticles were calculated by Equations 1 and 2, as per Riley et al.20
| (1) |
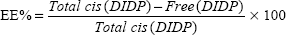
| (2) |
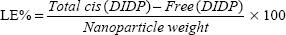
In vitro drug release
The methodology used for this portion of the study was derived from the Chinese Pharmacopoeia (2010 edition), “Plasma determination”.21 cis-DIDP release profiles were determined at 37°C using PBS (pH =7.4) as a release medium in a dissolution cup. In vitro drug release was studied on five nanoparticles obtained at different PEG-4000/SL weight ratios. About 17.5 mg of each nanoparticle was added to 50 mL of the PBS medium. The suspensions were gently stirred at 100 rpm. For each sample, about 5 mL of sample was withdrawn at predetermined time intervals and replaced by an equivalent amount of medium maintained at the same temperature under the same stirring regime. Each sample aliquot was filtered through a microporous membrane (Diameter of pores =5.0 μm). The cis-DIDP content released into the solution was measured by UV spectroscopy as described in the “Characterization of cis-DIDP-loaded SLNs” section at λmax =359 nm. All experiments were performed in the dark, in triplicate. As a control, two release solutions (named “A” and “B”) with the same concentration of SLNs were prepared at the beginning of the release experiment. When 5 mL was withdrawn from A, and replaced with 5 mL of the release solution from B, the drug concentration in the solution remained stable. The limit of detection for cis-DIDP with this method was 0.5 μg/mL, and the method resulted in a linear regression (R2=0.9999) for a range of 3 μg/mL to 300 μg/mL of cis-DIDP.
Antitumor activity in vitro
In vitro antitumor activity of cis-DIDP-loaded SLNs was examined by the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay method. The human hepatocellular carcinoma cell line SMMC-7721 (Gibco, Carlsbad, CA, USA) was maintained in Roswell Park Memorial Institute 1640 medium and adjusted to 5×104 cells/mL with 10% fetal calf serum (Gibco, Carlsbad, CA, USA), 25 μg/mL penicillin (Sigma-Aldrich, St Louis, MO, USA), and 25 μg/mL streptomycis (Sigma-Aldrich) under 5% CO2 in a humidified atmosphere at 37°C. The cells were routinely harvested by using trypsin-ethylenediaminetetraacetic acid (trypsin-EDTA) (Gibco) (0.25% trypsin and 0.01% EDTA), and reseeded before reaching confluence. For experiments, 200 μL aliquots of the cell suspension were added into each well of a 96-well plate and incubated overnight, whereupon the medium was changed. The cells were treated for 24 hours with 0.2 mg/mL to 0.5 mg/mL native cis-DIDP or with 0.2 mg/mL to 0.5 mg/mL cis-DIDP-loaded SLNs, in PBS solution. A 20 μL aliquot of MTT solution (5 mg/mL) was then added to each well and incubated for 4 hours. Finally, the solution in the wells was deserted completely and 150 μL of dimethyl sulfoxide was added to each of the wells to dissolve the residue. The optical densities of the dimethyl sulfoxide solutions were determined by a microplate reader (EnVision Multilabel plate reader, PerkinElmer, Waltham, MA, USA) at 490 nm, and cell inhibitions were calculated. The optical density values were averaged for each group. The relative cell viability rate was calculated by dividing the optical density value of the test group by that of the control group.
Results and discussion
Preparation and characterization of ESCs
Selecting optimum ESC morphology
Emulsion temperature and the weight ratio of PEG-4000 to SL (PEG-4000/SL) were found to be important to the morphology of ESCs, because these two variables affected their entrapment efficiency and drug loading content. Figure 1 exhibits SEM images of samples obtained at different temperatures with different PEG-4000/SL weight ratios. Irregular sheets were obtained at low temperature (30°C; Figure 1A). Microspheres with diameters ranging from 5 μm to 10 μm were obtained at 60°C (Figure 1B), which was much more conducive to drug loading. As the temperature increased, spheres began to dissociate and collapse at 80°C (Figure 1C). Therefore, 60°C was identified as the optimal emulsion temperature.
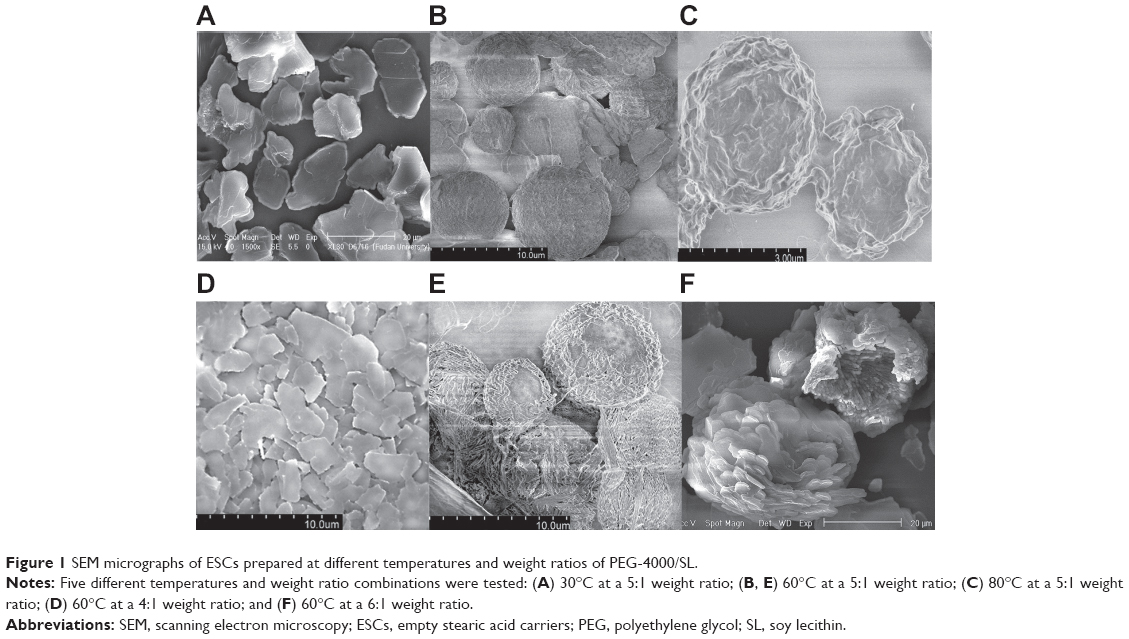 | Figure 1 SEM micrographs of ESCs prepared at different temperatures and weight ratios of PEG-4000/SL. |
The influence of the PEG-4000/SL weight ratio on ESCs at 60°C was also studied. Irregular sheets were obtained at a PEG-4000/SL ratio of 4 (Figure 1D). Upon increasing the PEG-4000/SL ratio to five, ball-shaped structures appeared (Figure 1E). When the PEG-4000/SL ratio reached 6, flat structures appeared and formed pancake-like stacks (Figure 1F).
XRD of ESCs
XRD was used to characterize the phases of ESCs prepared with different weight ratios of PEG-4000/SL at 60°C. The XRD curve of SA (Figure 2A) could be indexed to monoclinic stearic acid (Joint Committee on Powder Diffraction Standards [JCPDS] card number 09-0618) with calculated lattice constants of a =9.51 Å, b =4.95 Å, and c =51.4 Å. The XRD spectrum of ESCs prepared at a weight ratio of 5 PEG-4000/SL (Figure 2C) was similar to that of raw SA. This shows that the ESCs formed from SA maintained the same crystal structure as raw SA, without impurities.
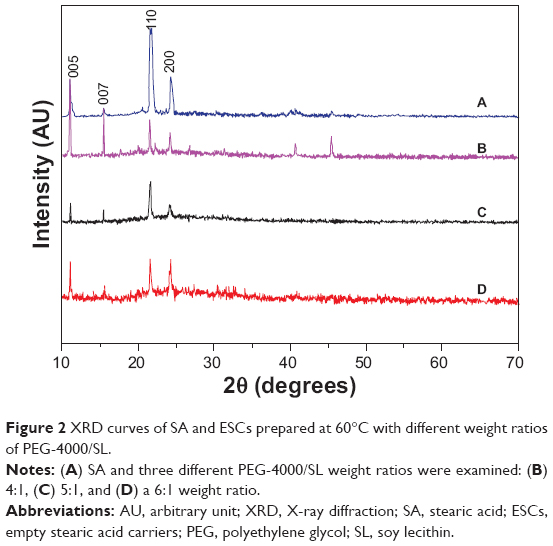 | Figure 2 XRD curves of SA and ESCs prepared at 60°C with different weight ratios of PEG-4000/SL. |
The morphology and phases of ESCs could be tuned successfully by modifying the emulsion temperature and the weight ratio of PEG-4000/SL.
Mechanism of ESC formation
Understanding the process for formation of ESCs was instrumental to drug loading in this study. SEM images indicate that different morphologies can be obtained via controlling the weight ratio of PEG-4000/SL (Figure 3). Initially, the concentration of the emulsifier PEG-4000 was too low for the droplet interface to reach saturation adsorption, and thus was insufficient to form a closely packed interfacial membrane;22 only irregular flakes were obtained (Figures 1D and 3A). Upon increasing the amount of PEG-4000, the emulsifier began to play a role, and the mixture became more stable. The molecular structure of SA has a polar carboxyl head group and a long hydrocarbon chain. Therefore, SA can form hydrogen bonds and can undergo hydrophobic interactions with the emulsifier. This leads to the formation of lamellar sheets which tend to aggregate together, thereby gradually forming self-assembled spheres (Figures 1E and 3B). Compared with the hydroxyl groups in PEG-4000, the size and number of the hydrophobic groups in the spheres are much larger. When the PEG-4000/SL weight ratio increased, the hydrophobic region within the sphere grew in size until the sphere could no longer maintain its integrity, causing it to open into a flower-like structure (Figures 1F and 3C).
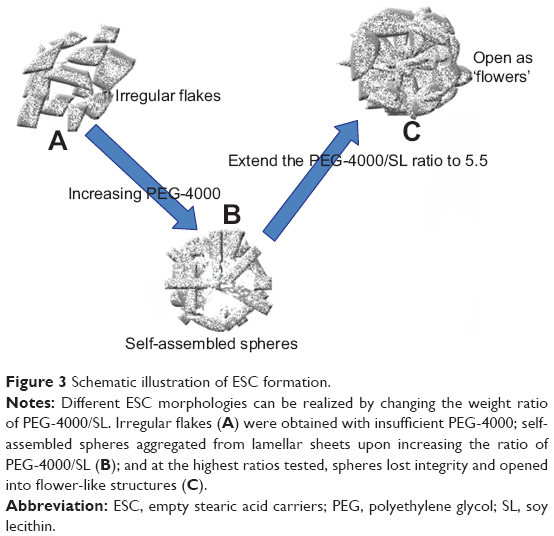 | Figure 3 Schematic illustration of ESC formation. |
It is believed that emulsion stabilizer weight ratios will influence the formation of SLNs.23 The results from a previous study23 indicate that a larger percentage of polyvinyl alcohol can form a stable capsular structure with a consequent increase in encapsulation and actual drug loading. In our current research, uniform spheres formed upon increasing the PEG-4000/SL weight ratio to 5. When the weight ratio of PEG-4000/SL reached 6, the spheres broke. According to the literature, spheres are the preferred structure for high drug encapsulation and loading.23
Our results indicate that ESCs can be used to encapsulate cis-DIDP to increase its stability, decrease its toxicity, and control its release.
Loading and controlled release of cis-DIDP
Characterization of cis-DIDP-loaded SLNs
The morphology of cis-DIDP-loaded SLNs and cis-DIDP were observed by SEM-EDS (Figure 4). The carrier maintained its morphology during drug loading (Figure 4A). The amounts of Pt and I in cis-DIDP-loaded SLNs were obtained via EDS (Figure 4B). Zeta-potential measurements show negative values of −33.2±0.917 mV for cis-DIDP-loaded SLNs.
 | Figure 4 The morphology of cis-DIDP-loaded SLNs and cis-DIDP during drug loading. |
FTIR spectrometry was used to find out whether cis-DIDP was encapsulated into SLNs and to characterize the nature of the interaction between cis-DIDP and SLNs. Various physicochemical interactions can alter or broaden the IR absorption peaks. All the cis-DIDP-loaded SLNs were obtained at 60°C with different weight ratios of PEG-4000/SL (Figure 5A–5E; PEG-4000/SL weight ratios of 6, 5.5, 5, 4.5, and 4). The peak at 476 cm−1 was observed in all cis-DIDP-loaded SLNs, and was attributed to the Pt-N bond.24 In addition, the vibrational band at 1600.98 cm−1 represents the primary amino group in cis-DIDP. In the cis-DIDP-loaded SLNs, the amino band was broadened and shifted to 1608.93 cm−1, which indicates an interaction between cis-DIDP and ESCs. FTIR curves of ESCs and cis-DIDP, respectively, are shown in Figure 5F and 5G.
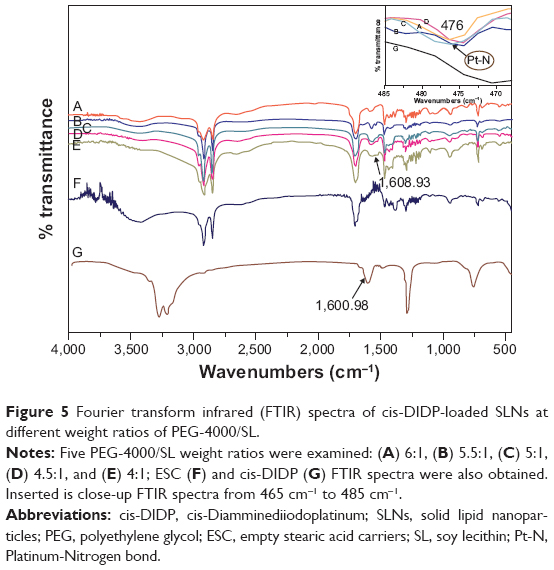 | Figure 5 Fourier transform infrared (FTIR) spectra of cis-DIDP-loaded SLNs at different weight ratios of PEG-4000/SL. |
The UV-visible spectroscopy of cis-DIDP-loaded SLNs was also studied. The peak at 278 nm appeared in the spectra of cis-DIDP-loaded SLNs (Figure 6A–6E), which was shifted by 8 nm from 270 nm of ESCs (Figure 6F). The peak at 359 nm for cis-DIDP (Figure 6G) was also observed, which confirmed that cis-DIDP was successfully loaded into the carrier and sustained stable structure.
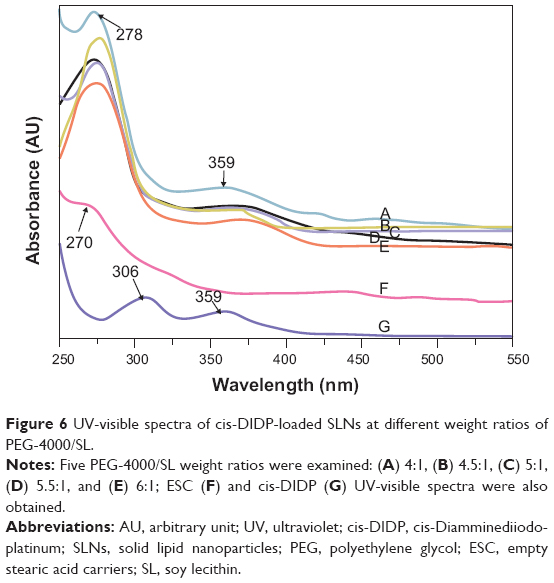 | Figure 6 UV-visible spectra of cis-DIDP-loaded SLNs at different weight ratios of PEG-4000/SL. |
Loading capacity and encapsulation efficiency
The effect of different weight ratios of PEG-4000/SL on the EE and LC of ESCs was determined with the concentration of cis-DIDP-loaded SLNs at 0.35 mg/mL. The maximum EE (97.24%) and LC (22.03%) were achieved at a PEG-4000/SL weight ratio of 5:1 (Figure 7).
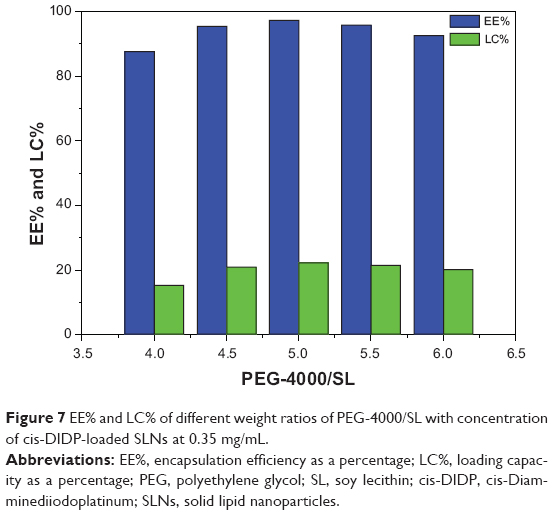 | Figure 7 EE% and LC% of different weight ratios of PEG-4000/SL with concentration of cis-DIDP-loaded SLNs at 0.35 mg/mL. |
In vitro cis-DIDP release
The drug-release behavior of cis-DIDP-loaded SLNs obtained at different weight ratios of PEG-4000/SL was investigated. The percentage of cumulative release was 100% for all formulations after 12 hours. All formulations showed the first drug-release burst at 30 minutes, followed by a steady release which continued for up to 2 hours. A second burst appeared after a period of 3 to 10 hours, followed by another steady-state release (Figure 8). The two-step release could arise from two levels of drug–particle interaction: 1) surface adsorption, which would yield the first release, and 2) matrix interpenetration, which would yield the second release. The release medium can easily penetrate into the wrinkles on the surface of the SLN matrix and dissolve the drug. The dissolved drug rapidly diffused into the release medium and exhibited the first release. The release medium then gradually destroyed the carrier matrix, contributing to the steady-state release. The lifetime of the decomposition matrix differed among the different carrier morphologies obtained by varying the weight ratios of PEG-4000/SL. When the carrier matrix was destroyed completely, another rapid release burst appeared. The release results show that most of the drugs were encapsulated in the carrier. Furthermore, the release results also suggest that cis-DIDP encapsulation is a stepwise process, in which cis-DIDP is wrapped in the carrier during formation of the carrier in the first step, and in the second step cis-DIDP adsorbs onto the carrier after the carrier has formed.
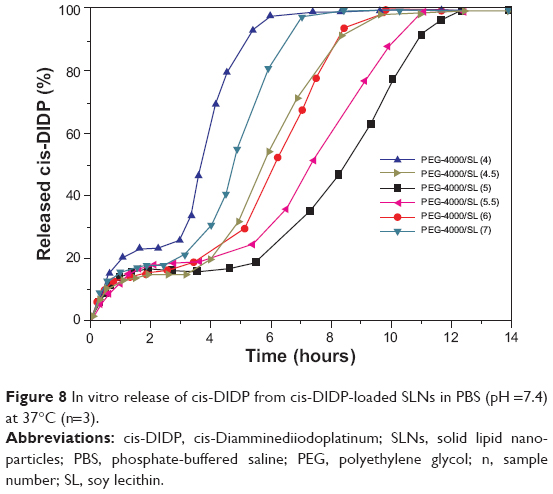 | Figure 8 In vitro release of cis-DIDP from cis-DIDP-loaded SLNs in PBS (pH =7.4) at 37°C (n=3). |
Antitumor activity in vitro
In this study, we conducted different loadings of cis-DIDP-loaded SLNs and used cis-DIDP solution as a positive control. The cis-DIDP-loaded SLNs were added directly to the tumor cells that had been cultured in each well of a well-plate, and the cells were incubated with the drug for 24 hours. The results are shown in Figure 9. The ball-shaped cis-DIDP-loaded SLNs, with actual cis-DIDP contents of 0.088, 0.044, 0.022 and 0.011 mg/mL, achieved cell growth inhibition rates of 68.33%, 50.77%, 33.24%, and 28.42%, respectively. Pure cis-DIDP solutions of 0.2, 0.1, 0.05 and 0.025 mg/mL concentrations achieved cell growth inhibition rates of 75.72%, 64.7%, 45.6%, and 28.16%, respectively (Figure 8). The concentration of cis-DIDP needed to inhibit the growth of SMMC-7721 cells to 50% of control growth rates was 0.0580 mg/mL before loading into the cis-DIDP-loaded SLNs, and 0.0428 mg/mL after loading. The results show that cis-DIDP released from the cis-DIDP-loaded SLNs maintained its inhibition ability. Loading cis-DIDP into SLNs enabled smaller doses to be used, compared to the pure compound, in MTT experiments. In addition, the smaller doses of cis-DIDP delivered via SLNs exhibited higher inhibition of cancer cell growth than the pure compound. The increase in anticancer activity of cis-DIDP when loaded into SLNs can be attributed to an increase in the stability of cis-DIDP in the SLN-loaded system.
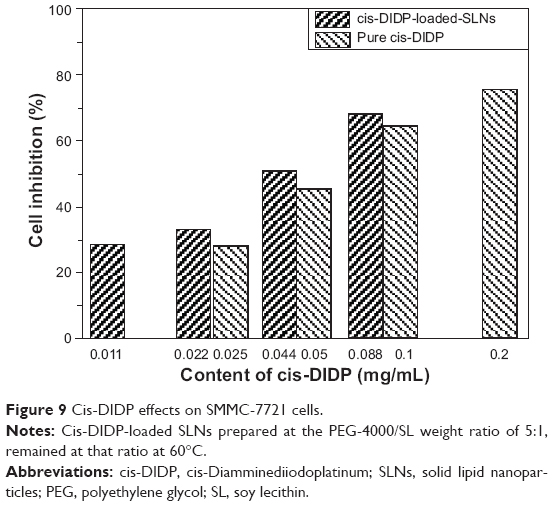 | Figure 9 Cis-DIDP effects on SMMC-7721 cells. |
Our results indicate that cis-DIDP-loaded SLNs can release cis-DIDP continuously from the system when in the amorphous state. Therefore, to a certain extent, encapsulation with SLNs allows for the shortcomings of cis-DIDP in the human body, such as poor water-solubility and instability, to be overcome. Encapsulating cis-DIDP into SLNs by the emulsification dispersion–ultrasonication method is an ideal technique for improving the performance of cis-DIDP.
Surgery is the primary treatment for patients with liver cancer. However, recurrence of liver cancer is very common.25,26 We therefore believe that cis-DIDP-loaded SLNs can be used as an implantable drug delivery system to prevent the recurrence of liver cancer after surgery.
Conclusion
Encapsulation of cis-DIDP into SLNs preserved its bioactivity, and at the same time provided controlled release of the drug. We explored the influences of reaction temperature and PEG-4000/SL weight ratios on the morphology of SLNs. The effects of SLN morphology on drug loading and encapsulation efficiency were also studied. Release curves showed that cis-DIDP-loaded SLNs exhibited two-stage release. Changing the buffer can tune the morphology, and thus the release characteristics of cis-DIDP-loaded SLNs, suggesting a possible future method to fine tune drug release according to the needs of the patient. Cis-DIDP-loaded SLNs are therefore a useful formulation for controlled drug release in cancer therapeutics, particularly because of the favorable biodegradability and biocompatibility of SA. Further investigations of cis-DIDP-loaded SLNs are in progress to improve their drug delivery characteristics. Increasing research efforts devoted to controlled release of cisplatin-like anticancer drugs will continue to lower their cost, decrease side effects, and improve their therapeutic efficacy.
Acknowledgments
The authors are grateful for the financial support of the National Natural Science Foundation (grant numbers 21101118, 91122025, and 21103127) of China; the State Major Research Plan (973) of China (grant number 2011CB932404); the Nano-Foundation of Shanghai, (grant number 11nm0501300); and the Fundamental Research Funds for the Central Universities and the Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials (grant number 2012MCIMKF03).
Disclosure
The authors report no conflicts of interest in this work.
References
Rosenberg B, Vancamp L, Krigas T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature. 1965;205:698–699. | ||
Wong E, Giandomenico CM. Current status of platinum-based antitumor drugs. Chem Rev. 1999;99(9):2451–2466. | ||
Lu JF, Wang K, Sun XZ, et al. Effects of cisplatin and its analogues on the permeability of human erythrocyte membrane. Met Based Drugs. 1995;2(2):73–80. | ||
Xu PS, Van Kirk EA, Murdoch WJ, et al. Anticancer efficacies of cisplatin-releasing pH-responsive nanoparticles. Biomacromolecules. 2006;7(3):829–835. | ||
Sheng LX, Su ZM, Ning L, et al. The electronic structure and biological activity of the type of cis-[Pt(NH3)2X2] (X=F, Cl, Br, I) complexes. J Northeast Normal University. 1993;3:143–145. Available from: http://www.cnki.net/KCMS/detail/detail.aspx?QueryID=16&CurRec=49&dbcode=CJFQ&dbname=CJFD7993&filename=DBSZ199303030&urlid=&yx=&uid=WEEvREcwSlJHSldTTGJhYlJRZTdBc0FGcTBDZG11MkZEQmZRZkpadE1CTHRMaGxtM2FCRzRHaG5jOGFvSXRXRQ==&v=MTI4OTZVTDNOSVMvWWRMS3hGOUxNckk5R1pJUjhlWDFMdXhZUzdEaDFUM3FUcldNMUZyQ1VSTDZmWXVScEZ5cms. Accessed June 11, 2014. Chinese. | ||
Yellepeddi VK, Kumar A, Maher DM, Chauhan SC, Vangara KK, Palakurthi S. Biotinylated PAMAM dendrimers for intracellular delivery of cisplatin to ovarian cancer: role of SMVT. Anticancer Res. 2011; 31(3):897–906. | ||
zur Mühlen A, Schwarz C, Mehnert W. Solid lipid nanoparticles (SLN) for controlled drug delivery – drug release and release mechanism. Eur J Pharm Biopharm. 1998;45(2):149–155. | ||
Gokce EH, Korkmaz E, Dellera E, Sandri G, Bonferoni MC, Ozer O. Resveratrol-loaded solid lipid nanoparticles versus nanostructured lipid carriers: evaluation of antioxidant potential for dermal applications. Int J Nanomedicine. 2012;7:1841–1850. | ||
Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery – a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–177. | ||
Gasco MR, inventor; Gasco MR, assignee. Method for producing solid lipid microspheres having a narrow size distribution. United States patent US 5250236. 1993 Oct 5. | ||
Mehnert W, Mäder K. Solid lipid nanoparticles: production, characterization and applications. Adv Drug Deliv Rev. 2001;47(2–3):165–196. | ||
Mawson S, Johnston KP, Combes JR, DeSimone JM. Formation of poly (1, 1, 2, 2-tetrahydroperfluorodecyl acrylate) submicron fibers and particles from supercritical carbon dioxide solutions. Macromolecules. 1995;28(9):3182–3191. | ||
Allémann E, Leroux JC, Gurny R, Doelker E. In vitro extended-release properties of drug-loaded poly (DL-lactic acid) nanoparticles produced by a salting-out procedure. Pharm Res. 1993;10(12):1732–1737. | ||
Sjöström B, Bergenståhl B. Preparation of submicron drug particles in lecithin-stabilized o/w emulsions 1. Model studies of the precipitation of cholesteryl acetate. Int J Pharm. 1992;88(1):53–62. | ||
Westesen K, Siekmann B. Investigation of the gel formation of phospholipid-stabilized solid lipid nanoparticles. Int J Pharm. 1997; 151(1):35–45. | ||
Cuenca AG, Jiang H, Hochwald SN, Delano N, Cance WG, Grobmyer SR. Emerging implications of nanotechnology on cancer diagnostics and therapeutics. Cancer. 2006;107(3):459–466. | ||
Khiati S, Luvino D, Oumzil K, Chauffert B, Camplo M, Barthélémy P. Nucleoside-lipid-based nanoparticles for cisplatin delivery. ACS Nano. 2011;5(11):8649–8655. | ||
Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7(8):573–584. | ||
Van Bommel Kjeld SC, Stuart Marc CA, Feringa Ben Li, Van Zsch Jan. Two-stage enzyme medicated drug release from LMWG hydrogels. Org Biol Chem. 2005;3916):2917–2920. | ||
Riley T, Govender T, Stolnik S, et al. Colloidal stability and drug incorporation aspects of micellar-like PLA-PEG nanoparticles. Colloids Surface B. 1999;16(1):147–159. | ||
Chinese Pharmacopecia Commission. Chinese pharmacopecia (2010 edition). Chinese Medical Science Press: Beijing. 2010. Chinese. | ||
Trotta M, Cavalli R, Ugazio E, Gasco MR. Phase behavior of microemulsion systems containing lecithin and lysolecithin as surfactants. Int J Pharm. 1996;143(1):67–73. | ||
Dalmoro A, Lamberti G, Titomanlio G, Barba AA, d’Amore M. Enteric microparticles for targeted oral drug delivery. AAPS Pharm Sci Tech. 2010;11(4):1500–1507. | ||
Nakamoto K, McCarthy JP, Fujita J, Condrate RA, Behnke GT. Infrared studies of ligand-ligand interaction in dihalogenodiammineplatinum (II) complexes. Inorg Chem. 1965;4(1):36–43. | ||
Li L, Tang F, Liu H, et al. In vivo delivery of silica nanorattle encapsulated docetaxel for liver cancer therapy with low toxicity and high efficacy. ACS Nano. 2010;4(11):6874–6882. | ||
Kannan A, Hettiarachchy N, Johnson MG, Nannapaneni R. Human colon and liver cancer cell proliferation inhibition by peptide hydrolysates derived from heat-stabilized defatted rice bran. J Agric Food Chem. 2008;56(24):11643–11647. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.