Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Status epilepticus in patients with genetic (idiopathic) generalized epilepsy
Authors Bosak M ![]() , Pawełczak D, Słowik A
, Pawełczak D, Słowik A ![]()
Received 18 March 2019
Accepted for publication 15 May 2019
Published 20 June 2019 Volume 2019:15 Pages 1585—1592
DOI https://doi.org/10.2147/NDT.S209084
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Magdalena Bosak,1 Dominika Pawełczak,2 Agnieszka Słowik1
1Department of Neurology, Jagiellonian University Medical College, Krakow, Poland; 2Department of Neurology, Jagiellonian University Medical College, Krakow, Poland
Aim of the study: Genetic (idiopathic) generalized epilepsies (GGEs) account for nearly one-third of all epilepsies. The frequency of status epilepticus (SE) in patients with GGEs has been poorly studied. Therefore, this study aimed to evaluate the frequency of different forms of SE in a cohort of patients with GGEs.Materials and methods: Among 153 patients with GGEs treated at the university epilepsy clinic in the period between 1998 and 2018, those with SE were retrospectively identified.Results: Absence SE was diagnosed in 8 patients (13 episodes), while myoclonic SE was found in 2 patients (2 episodes). No cases of tonic–clonic SE were detected in the study cohort. Most SE episodes were found to be provoked by ill-advised antiepileptic drugs or changes in drug regimen. In all the subjects, SE was stopped by intravenous administration of diazepam and/or valproate. Long-term outcome of epilepsy was good, with most patients (70%) being seizure-free.Conclusion: Status epilepticus is not a rare phenomenon in patients with genetic generalized epilepsies, with absence SE being the most common type. Most cases of SE are provoked by ill-advised AEDs or changes in drug regimen. Status epilepticus in GGEs can be easily treated with benozdiazepines and/or valproate. Status epilepticus in GGEs can be easily treated with benozdiazepines and/or valproate.
Keywords: absence status epilepticus, myoclonic status epilepticus, genetic generalized epilepsy
Introduction
Genetic (idiopathic) generalized epilepsies (GGEs) are a well-recognized subgroup of generalized epilepsy with a presumed genetic etiology. They comprise 15–20% of all epilepsies.1,2 GGEs are characterized by different combinations of generalized seizures such as absence, myoclonic, and generalized tonic–clonic and generalized epileptiform discharges on electroencephalography (EEG). Childhood absence epilepsy, juvenile absence epilepsy, juvenile myoclonic epilepsy, and epilepsy with generalized tonic–clonic seizures alone are well-characterized syndromes of GGEs.3 The frequency of status epilepticus (SE) in patients with GGEs has been poorly studied,4 with most studies on SE being case reports or case series. In a recent, population-based study, no cases of typical absence or myoclonic SE were found;5 in a cohort, hospital-based study, absence SE was found to account for <1% of cases, while no episodes of myoclonic SE were identified.6 The aim of this study was to evaluate the frequency of different forms of SE in a cohort of patients with GGEs.
Materials and methods
This was a retrospective cohort study conducted at the Epilepsy Clinic at Jagiellonian University, Krakow, Poland. Patients with GGEs who had been observed and clinically managed in the clinic in the period between 1998 and 2018 were identified from the electronic database and included in the study. The following information was collected from the patients using a structured questionnaire: age, sex, age at the diagnosis of epilepsy, duration of epilepsy, seizure types, family history of epilepsy, and findings of EEG and neuroradiology. Inclusion criteria were as follows: combination of generalized seizure types (absence, myoclonic, and/or generalized tonic–clonic), generalized spike/polyspike and wave discharges with normal background activity on interictal EEG, and normal magnetic resonance imaging. Syndromes of GGEs in patients were classified according to the classification scheme proposed by the International League Against Epilepsy.7 Three patients with Jeavons syndrome were also included in the study. Among the included patients, those with adequately documented episodes of SE were retrospectively identified. In these patients, SE was classified according to a classification system recently proposed by the International League Against Epilepsy.8 The clinical and EEG data of all identified SE episodes were analyzed. The study was conducted in compliance with Declaration of Helsinki and was reviewed and approved by the bioethics committee of the university (122.6120.32.2017 issued 22 Feb 2017). Patients' consent was not required by the bioethics committee as it was a retrospective chart review. Patients' personal data were available only to the senior author who is the treating physician and anonymized before statistical analysis.
Results
Cohort characteristics
The study population consisted of 153 patients (including 106 women; 69.3%); 13.5% of 1132 patients were treated in the clinic during the study period. The mean age of the patients at the time of inclusion in the study was 31.6 years (SD ±9.8), and the mean age at the onset of epilepsy was 15.6 years (SD ±5.6). The mean duration of follow-up of the cohort members was 9.3 years (SD ±5.2, range 1–20 years). Juvenile myoclonic epilepsy was the most common syndrome observed among patients (74; 48.4%), followed by epilepsy with generalized tonic–clonic seizures alone (43; 28.1%) and juvenile absence epilepsy (29; 18.9%). Childhood absence epilepsy was diagnosed in 4 (2.7%) patients and Jeavons syndrome in 3 (1.9%) patients.
Status epilepticus
A total of 10 (8 female) patients (6,5% of the cohort) with 15 episodes of SE were identified. Absence SE was diagnosed in 8 patients (13 episodes) and myoclonic SE in 2 patients (2 episodes). Tonic–clonic SE was not found in any patient. All patients developed only one type of SE, either absence or myoclonic. The clinical characteristics of the subjects and the episodes of SE are summarized in Table 1.
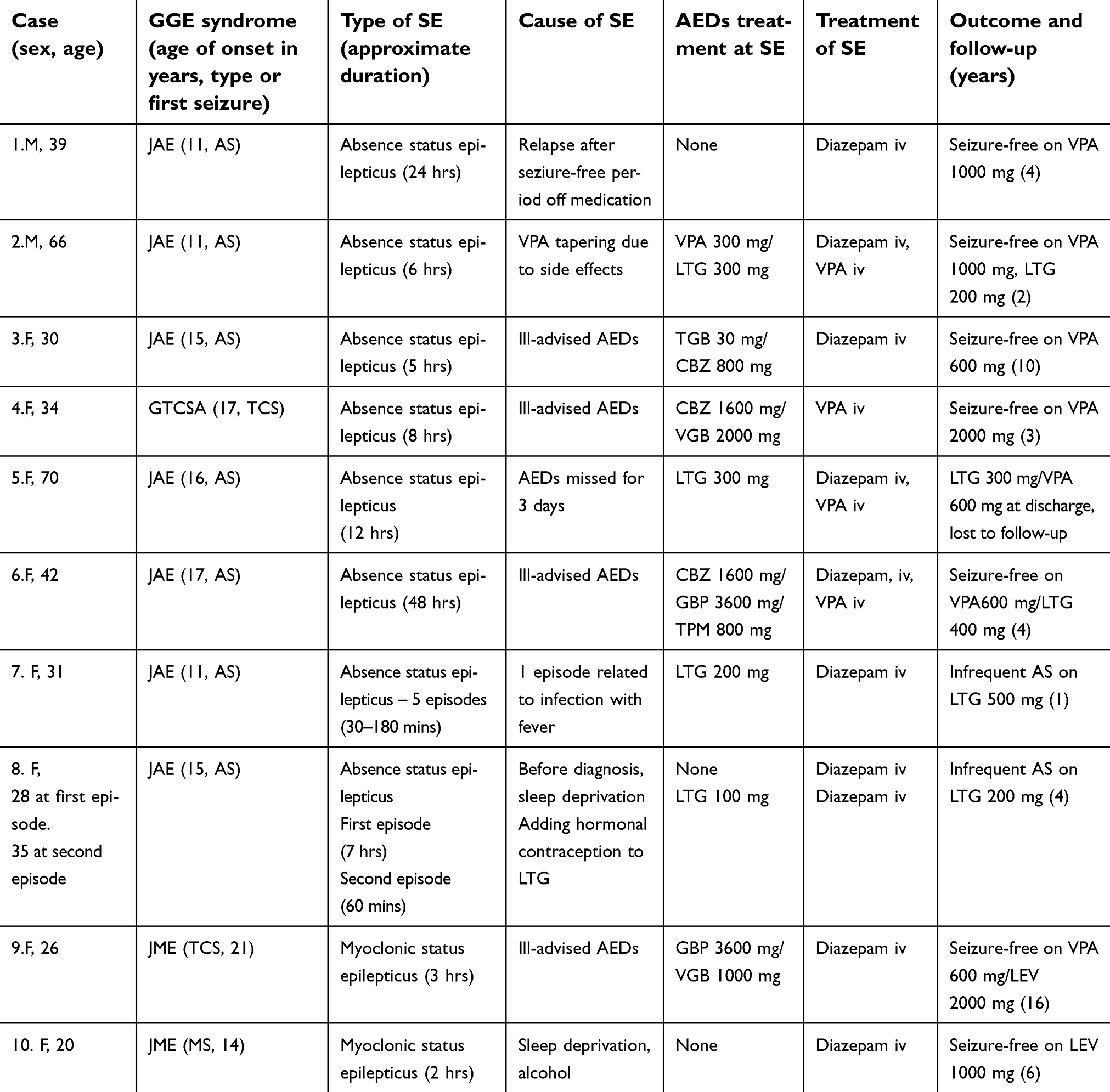 | Table 1 Clinical characteristics of patients and the episodes of SE |
The cardinal clinical features of absence SE were confusion (to different extent), slowing, and difficulties in performing daily activities. Patient 7 had simple gestural automatisms. Patients with myoclonic SE presented with repetitive myoclonic jerks of the upper limbs and slight disorientation. The episode of SE preceded diagnosis of epilepsy in 2 patients (Patients 7 and 9), whereas in 1 patient (Patient 1) SE occurred after long-term remission off medication. In 3 subjects, absence SE was either followed by (Patients 1 and 5) or interspersed with (Patient 8) a generalized tonic–clonic seizure. In 4 cases, SE was precipitated by contraindicated drugs (carbamazepine, tiagabine, gabapentin, and/or vigabatrin); these patients were referred to the Department of Neurology with the diagnosis of pharmacoresistant focal epilepsy with seizure aggravation. The approximate duration of SE episodes varied from 30 mins to 48 hrs.
EEG data during SE were available for all but 1 patient; in the case of Patients 7 and 8, EEG data were available for 1 episode for each. Video-EEG was available for 4 subjects. In patients who took ill-advised drugs, EEG showed bilateral bursts or continuous discharges of sharp and/or slow waves of 2–4 Hz (Figure 1). In the remaining patients, EEG showed generalized spikes/polyspikes and slow-wave discharges of >2.5 Hz (Figure 2). EEG samples of the remaining patients with absence status epilepticus are presented in supplementary materials (Figures S1–S5).
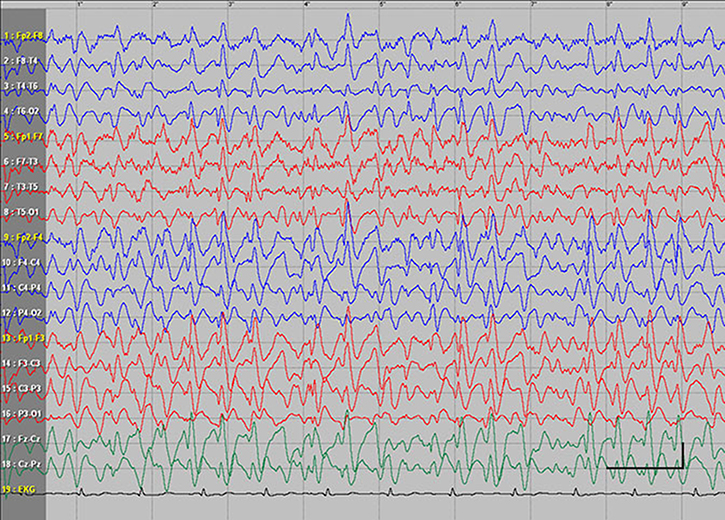 | Figure 1 Absence status epilepticus in patient 6. Vertical bar - 100uV, horizontal bar- 1 sec. |
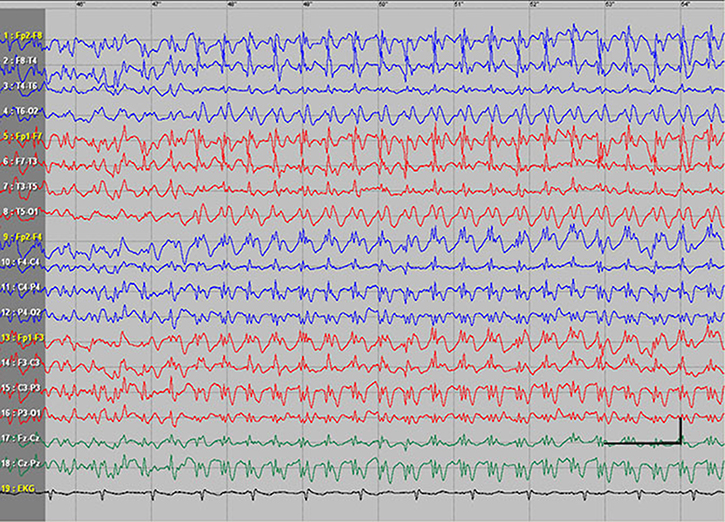 | Figure 2 Absence status epilepticus in patient 1. Vertical bar - 100uV, horizinatl bar - 1 sec. |
In all the cases, SE was stopped by intravenous administration of diazepam and/or valproate. Long-term outcome of epilepsy was found to be good, with most patients (70%) being seizure-free. Long-term treatment included broad-spectrum AEDs: VPA, LTG, and/or LEV.
Discussion
In this long-term, retrospective study, a total of 15 episodes of SE were identified in 6.5% (10 patients) of the cohort, with 15 episodes per 1428 patient-years. The rate of episodes was much lower than that found in the study of Agathonikou et al (15.4%).9 However, their study comprised patients with syndromes characterized by a high frequency of SE, namely perioral myoclonia with absences and idiopathic generalized epilepsy with phantom absences.10 In contrast, in the present study, two-thirds of the subjects with SE suffered from juvenile absence epilepsy.
It was found that in 40% of patients, SE was provoked by ill-advised antiepileptic drugs (AEDs); the phenomenon of paradoxical aggravation of epilepsy is well known in GGEs.11,12 In this group of patients, ictal EEG findings were inconsistent with GGEs, most probably due to contraindicated treatment. In the opinion of the authors, knowledge and awareness about the subtypes of GGEs should be increased, as the correct classification of syndrome is indispensable for choosing the appropriate treatment. In further 30% of patients, SE episode was caused due to lowering the dose of AEDs.
In Patient 7 with recurrent, mostly unprovoked, episodes of absence SE, absence status epilepsy was suspected.13
This study provided reassuring findings related to the prevalence of generalized convulsive SE in patients with GGEs; no cases of generalized convulsive SE were found in the study cohort.
We acknowledge some limitations of this study. Firstly, due to its retrospective design, some cases of SE could have been missed. Especially episodes of absence SE may have gone unrecognized. Secondly, the number of patients in our cohort was relatively small. Thirdly, we included only adult patients, while the frequency of GGEs is higher in children/adolescents.
Conclusion
SE is not a rare phenomenon in patients with genetic generalized epilepsies, with absence SE being the most common type. This study found that most cases of SE were provoked by ill-advised AEDs or changes in drug regimen. Appropriate treatment of GGEs with broad-spectrum AEDs (VPA, LTG, LEV, TPM) is indispensable for preventing SE episodes. EEG is crucial in the diagnosis of absence SE; however, it may not show classical generalized spikes/polyspikes and slow-wave discharges of >2.5 Hz in patients treated with contraindicated AEDs. Episodes of SE are an easily treatable complication of GGEs.
Acknowledgment
Jagiellonian University Medical College funded the publishing of the manuscript.
Disclosure
MB received honoraria for publications and the participation in advisory meetings from Sanofi; honoraria for lectures, travel expenses and conference fees from Sanofi, Adamed, Teva Pharmaceutical, Neuraxpharm, Glenmark and UCB Pharma. AS received honoraria for lectures from Bayer, Boehringer Ingelheim, Novartis, Polpharma, Bristol-Myers Squibb, Novartis, Biogen, Teva Pharmaceutical, Medtronic and for the participation in advisory meetings from Bayer, Boehringer Ingelheim and Novartis. The authors report no other conflicts of interest in this work.
References
1. Jallon P, Latour P. Epidemiology of idiopathic generalized epilepsies. Epilepsia. 2005;46(Suppl 9):10–14. doi:10.1111/j.1528-1167.2005.00309.x
2. Syvertsen M, Nakken KO, Edland A, Hansen G, Hellum MK, Koht J. Prevalence and etiology of epilepsy in a Norwegian county-A population based study. Epilepsia. 2015;56(5):699–706. doi:10.1111/epi.12972
3. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58:512–521. doi:10.1111/epi.13709
4. Shorvon S, Walker M. Status epilepticus in idiopathic generalized epilepsy. Epilepsia. 2005;46(Suppl 9):73–79. doi:10.1111/j.1528-1167.2005.00316.x
5. Leitinger M, Trinka E, Giovannini G, et al. Epidemiology of status epilepticus in adults: A population-based study on incidence, causes, and outcomes. Epilepsia. 2019;60:53–62. doi:10.1111/epi.14607
6. Rossetti AO, Trinka E, Stähli C, Novy J. New ILAE versus previous clinical status epilepticus semiologic classification: analysis of a hospital-based cohort. Epilepsia. 2016;57:1036–1041. doi:10.1111/epi.13403
7. Engel J. ILAE Commission report. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE task force on classification and terminology. Epilepsia. 2001;42:796–803. doi:10.1046/j.1528-1157.2001.10401.x
8. Trinka E, Cock H, Hesdorffer D, et al. A definition and classification of status epilepticus – Report of the ILAE task force on classification of status epilepticus. Epilepsia. 2015;56:1515–1523. doi:10.1111/epi.13121
9. Agathonikou A, Panayiotopoulos CP, Giannakodimos S, Koutroumanidis M. Typical absence status in adults: diagnostic and syndromic considerations. Epilepsia. 1998;39:1265–1276.
10. Panayiotopoulos CP. Syndromes of idiopathic generalized epilepsies not recognized by the International league against epilepsy. Epilepsia. 2005;46(Suppl 9):57–66. doi:10.1111/j.1528-1167.2005.00314.x
11. Benbadis SR, Tatum WO
12. Thomas P, Valton L, Genton P. Absence and myoclonic status epilepticus precipitated by antiepileptic drugs in idiopathic generalized epilepsy. Brain. 2006;129:1281–1292. doi:10.1093/brain/awl047
13. Genton P, Ferlazzo E, Thomas P. Absence status epilepsy: delineation of a distinct idiopathic generalized epilepsy syndrome. Epilepsia. 2008;49:642–649. doi:10.1111/j.1528-1167.2007.01467.x
Supplementary materials
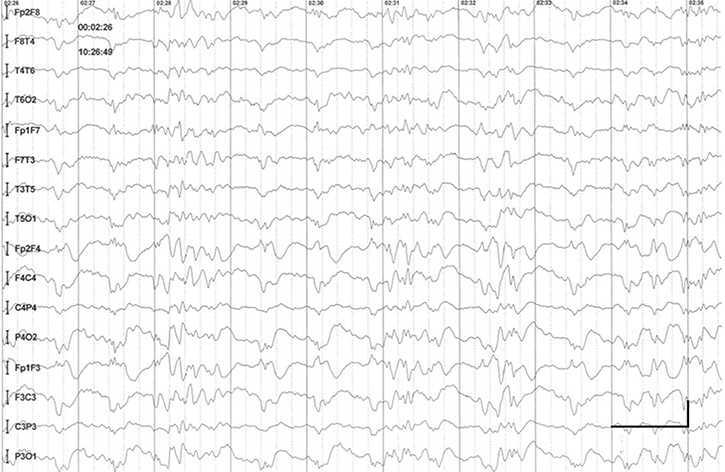 | Figure S1 Absence status epilepticus in patient 2. Vertical bar - 100uV, horizontal bar - 1 sec. |
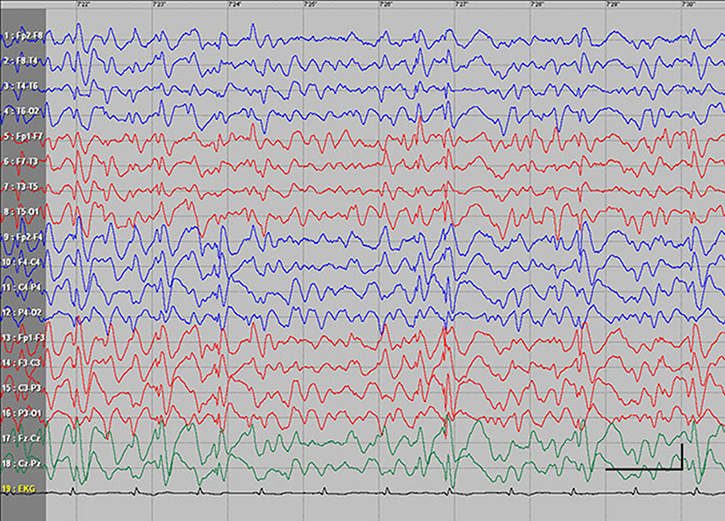 | Figure S2 Absence status epilepticus in patient 3. Vertical bar - 100uV, horizontal bar - 1 sec. |
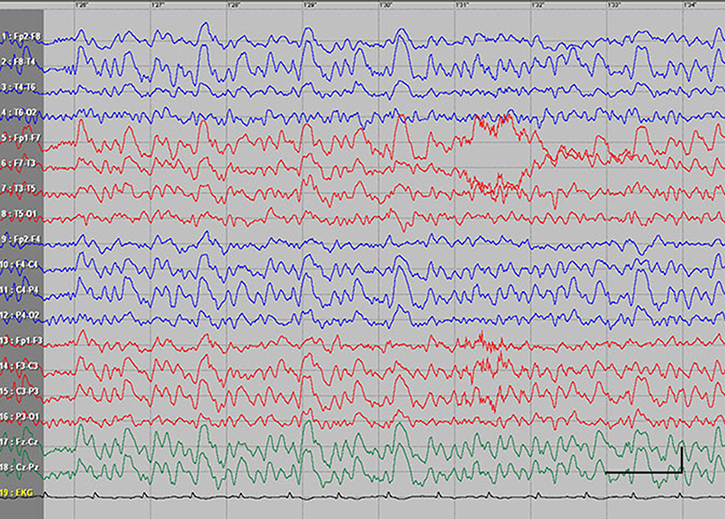 | Figure S3 Absence status epilepticus in patient 4. Vertical bar - 100uV, horozontal bar - 1 sec. |
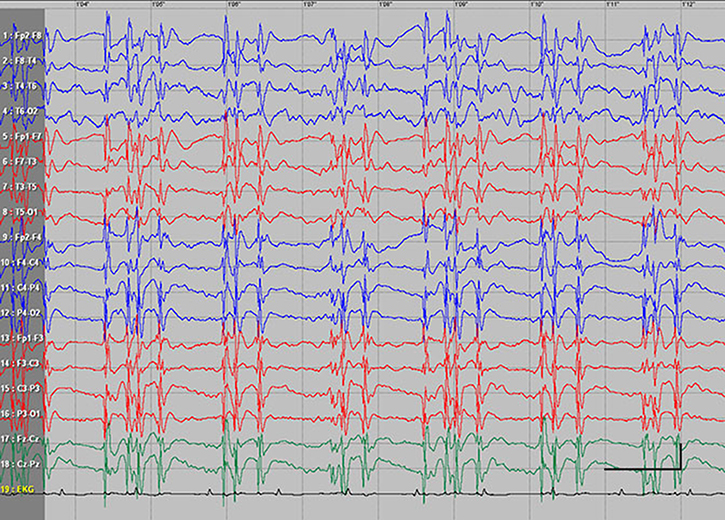 | Figure S4 Absence status epilepticus in patient 5. Vertical bar - 100uV, horizontal bar - 1 sec. |
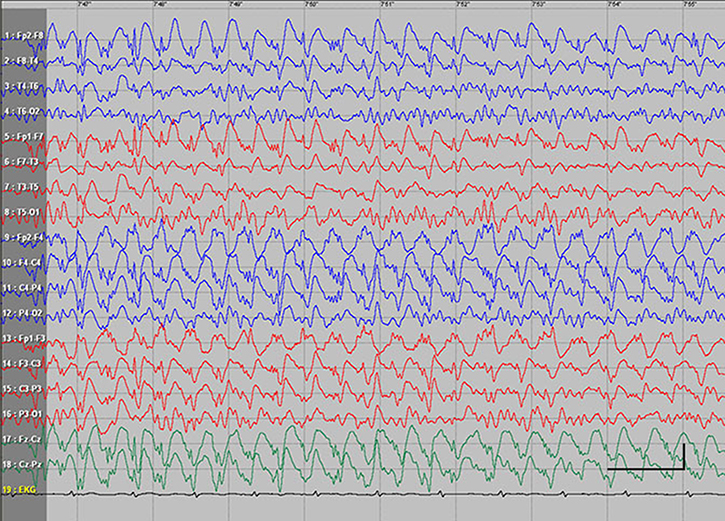 | Figure S5 Absence status epilepticus in patient 8. Vertical bar- 100uV, horizontal bar - 1 sec. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.