Back to Journals » Cancer Management and Research » Volume 11
Statins induce cell apoptosis through a modulation of AKT/FOXO1 pathway in prostate cancer cells
Authors Deng JL, Zhang R, Zeng Y, Zhu YS, Wang G ![]()
Received 17 April 2019
Accepted for publication 28 June 2019
Published 31 July 2019 Volume 2019:11 Pages 7231—7242
DOI https://doi.org/10.2147/CMAR.S212643
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chien-Feng Li
Jun-Li Deng,1,2 Rui Zhang,1,2 Ying Zeng,1,2 Yuan-Shan Zhu,3 Guo Wang1,2
1Department of Clinical Pharmacology, Xiangya Hospital, Central South University, Changsha 410008, People’s Republic of China; 2Institute of Clinical Pharmacology, Central South University, Hunan Key Laboratory of Pharmacogenetics, Changsha 410078, People’s Republic of China; 3Department of Medicine, Weill Cornell Medicine, NY, NY 10065, USA
Background: In recent years, statins have been frequently investigated in neoplasms. However, the potential roles of statins on prostate cancer cells and the underlying mechanisms have not been fully elucidated. In current study, we explored the effect and molecular mechanism of statins on cell proliferation and apoptosis in prostate cancer cells.
Methods: Prostate cancer cell were treated with gradient doses of simvastatin and fluvastatin for 24–72 h. Cell proliferation was analyzed by using MTS assay and colony formation. Cell apoptosis was measured by Hoechst staining, flow cytometry and caspase-3 activity. Western blotting was used to evaluate the proteins levels.
Results: Both simvastatin and fluvastatin produced a dose- and time-dependent inhibition of cell viability and colony formation while a promotion of cell apoptosis as evident with increases in caspase-3 activity, cleaved-caspase-3, cleaved-caspase-8 and cleaved-PARP levels in PC3 cells. Similar statin effects were observed in DU145 prostate cancer cells. Furthermore, statins produced a time- and dose-dependent reduction of phosphorylated-AKT and phosphorylated-FOXO1 levels in PC3 cells, and pretreatment of cells with an AKT phosphorylation inhibitor, MK2206, potentiated statins’ effect.
Conclusion: Statins decrease cell proliferation and induce cell apoptosis, probably mediated via a downregulation of AKT/FOXO1 phosphorylation in prostate cancer cells, which may have a potential benefit in prostate cancer prevention and therapy.
Keywords: statins, prostate cancer, apoptosis, AKT/FOXO1, pathway
Introduction
Prostate cancer (PCa) remains to be the most commonly diagnosed noncutaneous malignancy and the second leading cause of cancer-associated mortality among men in Western countries.1 Based on the Cancer Statistics report, there will be about 174,650 new cases and 31,620 deaths in 2019, which represent 20% of all cancer cases and 10% of cancer-related deaths among American men, respectively.2 The treatment of early stage PCa uniquely depends on androgens for proliferation, and the blocking of androgen receptor pathway could greatly produce tumor regression. However, the majority of PCa cells in later stages always inevitably progress to androgen-independent, and no curative therapy is existing for this intractable disease.3 With the advance and improvement of prostate cancer screening approaches, most of the prostate cancer could be diagnosed at an early stage, but it remains as a primary cause of cancer-related death in men of industrialized countries. In particular, there is no curative treatment available in current upon progression to androgen-independent metastatic disease.4,5 Although advanced chemotherapy allows patient outcome greatly improved,4,6 effective mechanism-based therapeutic methods that can obtain long-term improvements in patient outcomes remains lacking.7
The 3-Hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, commonly known as statins, are the most prescribed lipid-lowering drugs in clinic on account of their demonstrated safety and efficacy in prevention and treatment of hyperlipidemia and cardiovascular diseases.8,9 Beyond their potent inhibitory effects on cholesterol biosynthesis, statins appear to have pleiotropic effects in cancer. Previous epidemiologic studies have consistently demonstrated a beneficial role of statin use after diagnosis across the continuum of prostate cancer.10,11,12,13,14 A meta-analysis reported that the use of statins was associated with a 22% decreased risk of metastases (relative risk, 0.78; 95% CI, 0.68–0.87) and a 24% decreased risk of all-cause mortality (relative risk, 0.76; 95% CI, 0.63–0.91) among patients with prostate cancer.15 In a study of 11,772 men with nonmetastatic prostate cancer, Yu et al supported the findings of the meta-analysis and made an important observation that the effect of postdiagnostic statin use on prostate cancer mortality was more pronounced among men who had been taking a statin before diagnosis (HR, 0.55; 95% CI, 0.4–0.74) compared with those who were only taking statins postdiagnosis (HR, 0.76; 95% CI, 0.66–0.88).10 Other studies conducted in men with advanced prostate cancer found that those who were taking a statin at the time of the initiation of androgen-deprivation therapy had a longer time to progression compared with nonusers of statins.11,16 Taking together, all of these studies have demonstrated that statin used after diagnosis may decrease PCa risk and PCa progression. In addition, statins have also been reported to modulate the cell growth, apoptosis, and inflammation.17,18 However, the molecular mechanisms of these statin effects in PCa cells are not fully understood.
The AKT kinase is activated by hormones, growth factors, and chemical drugs, and it regulates the cell proliferation and survival.19–21 The forkhead transcription factor family, FOXO (forkhead box, O class), are downstream targets of AKT and include several subclasses, such as FOXO1, FOXO3, FOXO4, and FOXO6. AKT kinases could phosphorylate FOXO proteins and decrease their transcriptional activity through promoting the process of their redistribution to the cytoplasm.22 FOXO transcription factors play a vital function in cell apoptosis and survival in variety of cell types.23 The AKT/FOXO1 pathway plays an important role in chemoresistance since it is related to cell proliferation, migration, angiogenesis and apoptosis.24 In the present study, we studied the anti-proliferative and pro-apoptotic effects of statins and explored the potential molecular pathway(s) involved in statin actions in prostate cancer cells.
Materials and methods
Chemicals
Simvastatin and fluvastatin were purchased from Sigma-Aldrich (St. Louis, MO, USA). The compounds were dissolved in dimethylsulfoxide (DMSO) and stored at −20 °C until use. The final concentration of DMSO in cell cultures was less than 0.1% (v/v), which did not influence cell growth. MK2206 was obtained from Selleck Chemicals (Houston, TX, USA).
Cell culture and cell viability assay
The PC3 and DU145 human prostate cancer cell lines were obtained from the American Type Culture Collection (ATCC) (Rockefeller, Maryland, USA). The PC3 prostate cancer cells were cultured in RPMI-1640 medium (BI, Kibbutz Beit Haemek, Israel) supplemented with 10% fetal bovine serum (FBS, BI), 2 mM glutamine (BI), 100 U/ml penicillin (BI) and 100 μg/ml streptomycin (BI) at 37 °C in a 5% CO2 atmosphere. The MTS assay was performed to detect viable cell numbers using a cell proliferation assay Kit from Promega (Madison, WI, USA). For the MTS assay, PC3 and DU145 cells were seeded in 96-well plates at 3×103 cells/wells (six wells per group), cultured for 24 h in the growth media, and then treated with different concentrations of statins (0, 0.02, 0.2, 1, 2, 5, 10 and 20 μM) for 24, 48 and 72 h. The absorbance(A) at 490 nm was detected using a microplate reader.
Cell colony formation assay
Briefly, 1×103 cells/well were initially seeded in a six-well plate. 48 hrs later, the cells were treated with various concentrations of statins as indicated in each experiment and the treatment medium was refreshed every two days. At day 7 of treatment, when the colonies were visible by naked eyes, the cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) for 30 min, stained with 1% crystal violet for 20 min and then counted. The colony numbers were quantified using Image J software. All experiments were repeated three times.
Nuclear staining with Hoechst 33258
Cells were cultured and treated as described above. Briefly, at the end of experiments, cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) for 15 min, stained with 4 μg/ml Hoechst 33,342 (Beyotime Biotechnology, Shanghai, China) for 20 min, and then analyzed under a fluorescent microscope.
Caspase 3 activity assay
The detection of Caspase 3 activity was performed using a Caspase 3 activity assay Kit from Beyotime Biotechnology (Shanghai, China) according to the manufacturer’s instruction. The cells were plated at a density of 2×105 per well in 6-well plates, harvested at the end of statin treatment as indicated in each experiment and lysed in a lysis buffer. The lysis was centrifuged at 16,000× g for 15 min and the supernatant was obtained. For caspase 3 assay, 100 μg of protein lysis was incubated with 100 μl of reaction buffer and 10 μl of colorimetric tetrapeptides and Z-DEVD-pNA at 37 °C for 120 min, and the optical density at 405 nm for liberated p-nitroaniline (pNA) was obtained using a multi-well plate reader.
Cell apoptosis assay by annexin v staining
Annexin V-FITC staining was carried out using an Annexin V-FITC Apoptosis Detection Kit from Beyotime Biotechnology (Shanghai, China) according to the manufacturer’s instruction. Briefly, at the end of experiment, the cells were harvested, washed once in cold PBS, and suspended in 1× binding buffer. The cells were then stained with PI and Annexin V-FITC solution for 15 min in the dark, and analyzed by flow cytometry within 1 h (FC500, Beckman Coulter, Istanbul, Turkey).
Western blot analysis
Western blot analysis was conducted as previously described.25 Briefly, protein extracts from harvested experimental cells were prepared using a lysis buffer from Beyotime Biotechnology (Shanghai, China). Thirty micrograms of protein extracts were loaded to a 12% SDS-PAGE, electrophoretically separated and transferred to polyvinylidene membranes (Beyotime Biotechnology, Shanghai, China). After incubating the membrane with 5% non-fat milk blocking buffer at room temperature for 1 hr, the membrane was incubated with a primary antibody overnight at 4 °C. After the incubation with a horseradish peroxidase-conjugated secondary antibody (1:2500, Promega, Madison, WI, USA) at room temperature for 1 hr, the signal was obtained by using an ECL Western blotting system (Promega, Madison, WI, USA), visualized and quantified using the Bio-Rad ChemiDoc MP system. The primary antibodies against AKT (catalog # 4691p, 1:1000), phospho-AKT(Ser457) (catalog # 4060p, 1:2000), FOXO1 (catalog # 2880p, 1:1000) and phospho-FOXO1 (Ser256) (catalog # 9461p, 1:1000) were obtained from Cell Signaling Technology (Beverly, MA, USA). Antibodies specifically against BAX, BCL-2, caspase-3, caspase-8, poly (ADP-ribose) polymerases (PARP) were obtained from Abcam (Abcam, London, UK). β-actin (Sigma Chemical Co., St. Louis, MO, USA) was used as the loading control.
Statistical analysis
Each experiment was performed at least three times, and data were expressed as mean ± SEM. Statistical analyses were performed using SPSS 23.0 software (SPSS, Inc, Chicago, IL, USA). One-way ANOVA was applied to determine the difference among multiple groups, and the Student’s t-test was adopted to compare the statistical significance between two groups. A p-value less than 0.05 was considered to be statistically significant.
Results
Simvastatin and fluvastatin greatly decrease viable cells in PC3 cells
To assess the effects of statins on cell growth in human prostate cancer PC3 cells, the MTS and colony formation assay were performed. Treatment with either simvastatin or fluvastatin in PC3 cells produced a concentration- and time-dependent decrease in viable cells (Figure 1A, B) and colony formation (Figure 1C-E). The viable cells were decreased noticeably at 1 μM of simvastatin and reached a more than 80% reduction at 20 μM when treated for 72 hrs (Figure 1A). Colony formation was decreased more than 95% at 10 μM simvastatin treatment (Figure 1C, D). Similar effects were obtained when cells were treated with fluvastatin (Figure 1B, C and E). These results indicate that statins have an anti-proliferative or pro-apoptotic effect in PC3 cells.
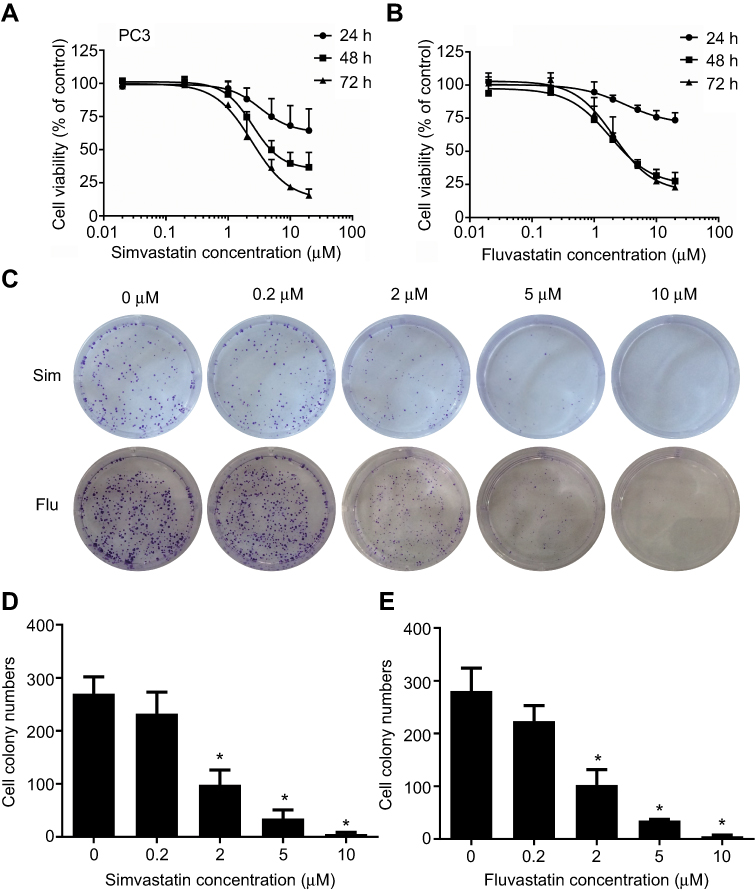 |
Figure 1 Statins, both simvastatin (Sim) and fluvastatin (Flu) produce dose- and time-dependent inhibitions of cell proliferation and colony formation in PC3 cells. (A and B) show the time- and dose-dependent inhibition of cell proliferation by simvastatin (A) and fluvastatin (B). The experiment was performed as described in the Materials and Methods and cell proliferation was measured using MTS assay. Results are expressed as a percentage of vehicle-treated control, and the data are the mean ± SEM (n=3) of three independent experiments. (C) shows representative images of colony formation assay and (D and E) are the mean ± SEM (n=3) of three independent experiments of quantitative colony formation assays following simvastatin (D) and fluvastatin (E) treatment. *p<0.05 compared to corresponding vehicle-treated controls. |
Simvastatin and fluvastatin induce cell apoptosis in PCa cells
To determine whether the growth inhibitory effects of simvastatin and fluvastatin are attributed to the induction of apoptosis in PC3 cells, Hoechst 33258 staining and caspase-3 activity assay were performed. As shown in Figure 2A and B, treatment with statins, simvastatin and fluvastatin, for 48 h produced a concentration-dependent decrease in cell number and obvious morphological changes including chromatin condensation and apoptotic body formation, characteristics of cell apoptosis. Similar time- and dose-dependent inhibition of cell viability and promotion of cell apoptosis were observed in DU145 (Figure 3A, B) and PC3 (Figure 3C, D) PCa cells treated with simvastatin or atorvastatin, respectively, using MTS assay and Hoechst staining.
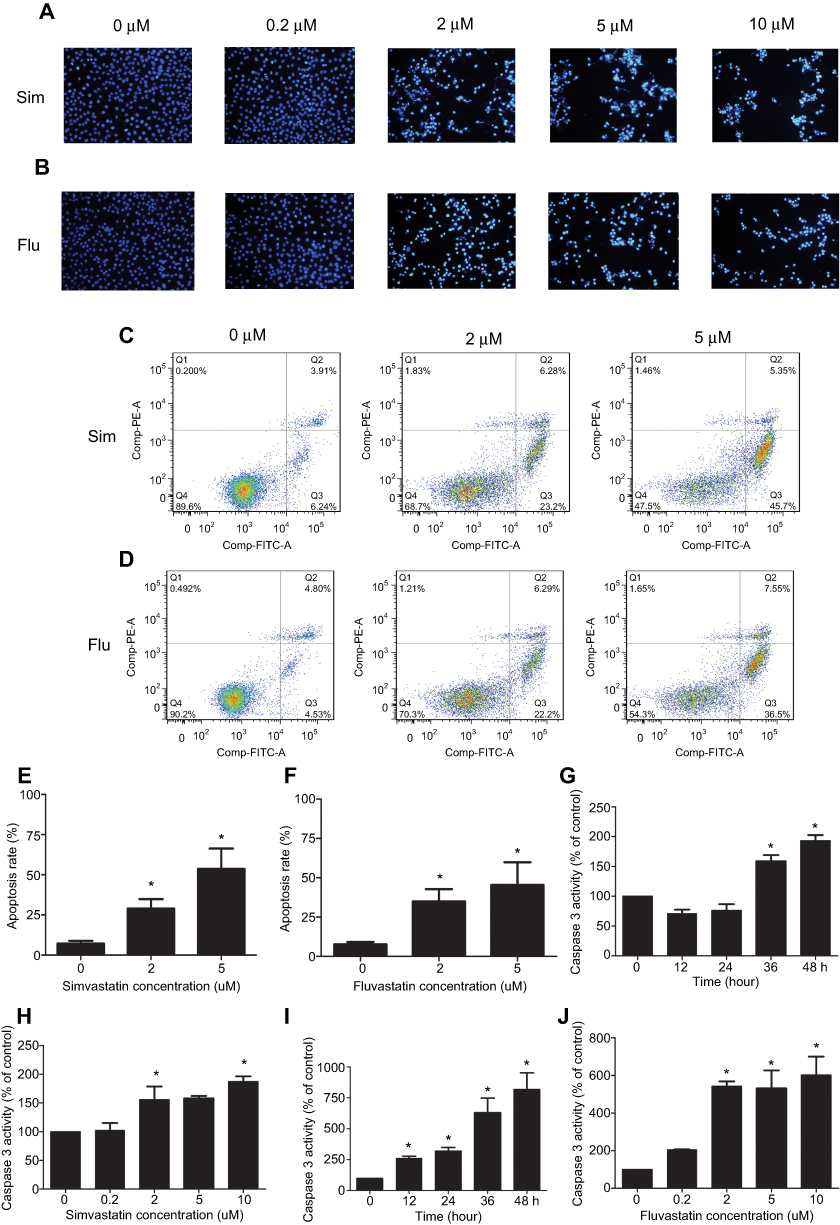 |
Figure 2 Statins, simvastatin (Sim) and fluvastatin (Flu) promote cell apoptosis in PC3 cells. (A and B) show representative images of Hoechst 33358 staining of cells treated with or without simvastatin (A) or fluvastatin (B). PC3 cells were treated for 48 h with the indicated concentrations of simvastatin or fluvastatin, stained with Hoechst 33358, and photographed with a fluorescence microscope using a blue filter at ×200 magnification. (C–F) show representative dot plots (C and D) and quantitation of apoptotic rates (E and F) of Annexin V-FITC apoptotic assay in PC3 cells following the treatment with various doses of simvastatin or fluvastatin, respectively. The apoptotic rate (%) is the sum of late (Q2) and early (Q3) apoptotic cells. The experiment was performed in triplicate, and the data are expressed as the mean ± SEM (n=3) of three independent experiments. Panels G-J show the time (G and I) and dose-dependent (H and J) induction of caspase 3 activity by simvastatin (G and H) or fluvastatin (I and J) treatment in PC3 cells assayed as described in the Methods and materials. The caspase 3 activity was presented as a percentage of corresponding vehicle control and the data are the mean ± SEM (n=3) of three independent experiments. *p<0.05 compared to the corresponding vehicle controls. |
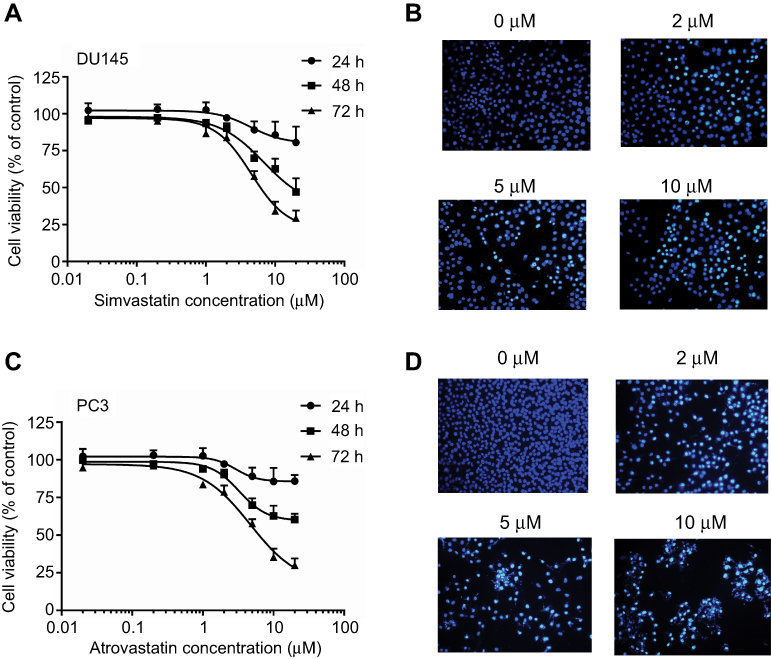 |
Figure 3 Simvastatin or atrovastatin inhibition of cell viability (A and C) and promotion of cell apoptosis (B and D) in DU145 or PC3 cells, respectively. (A) shows a time- and dose-dependent inhibition of cell viability by simvastatin in DU145 cells using MTS assay. The data are the mean ± SEM of three independent experiments (n=3) and presented as a percentage of corresponding vehicle controls. (B) shows representative images of Hoechst 33258 staining of cells treated with or without various doses of simvastatin. (C) shows a time- and dose-dependent inhibition of cell viability by atrovastatin in PC3 cells using MTS assay. (D) shows representative images of Hoechst 33258 staining of cells treated with or without various doses of atrovastatin. The images were taken under fluorescence microscope using a blue filter at ×200 magnification. |
To characterize further the statin-induced cell apoptosis in PC3 cells, flow cytometric analysis with Annexin V-FITC and PI double staining was carried out. Both simvastatin and fluvastatin produced a significant induction of cell apoptosis in PC3 cells in a dose-dependent fashion as shown in Figure 2C-F. Furthermore, treatment with either simvastatin or fluvastatin significantly induced casepase-3 activity (Figure 2G-J) and the cleavage of caspase-3, caspase-8 and PARP (Figure 4A-D) in a dose- and time-dependent manner without significant alterations in BCL-2, BAX (Figure 4E, F) and CHOP (Figure 4G, H) expression in PC3 cells. On the other hand, treatment with simvastatin in PC3 cells produced a marked induction of FAS-L protein expression at doses ranging from 0.2 to 10 μM (Figure 4G, H). These data collectively suggest that statins induced cell apoptosis may involve in death receptor apoptotic pathway but not the ER-stress or mitochondrial apoptotic pathway.
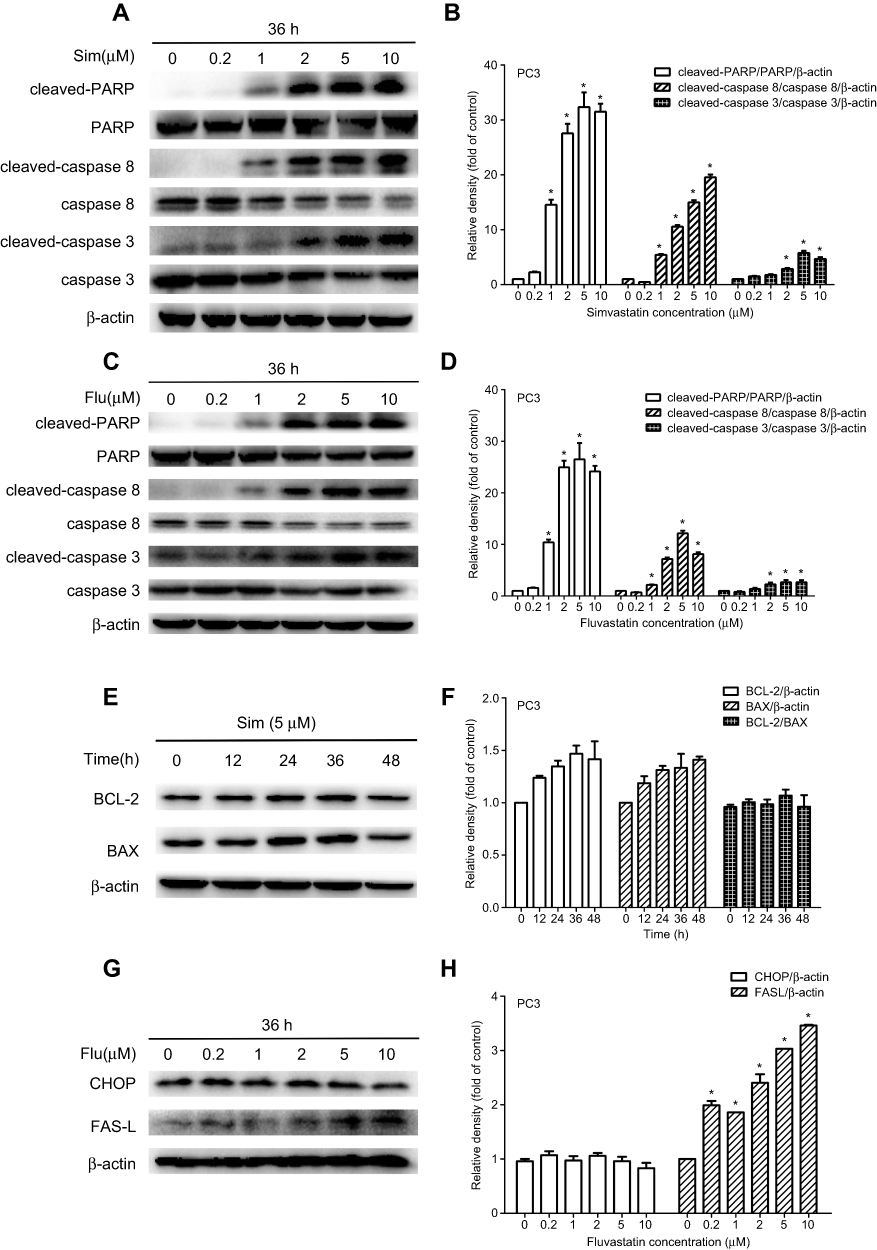 |
Figure 4 Western blot analysis of simvastatin (Sim) and fluvastatin (Flu) promotion of cell apoptosis in PC3 cells. (A and B) and (C and D) show representative Western blot images and quantitative analyses of dose-dependent effects of simvastatin and fluvastatin on the cleavage of PARP, caspase 8 and caspase 3 in PC3 cells, respectively. The quantitative data are the mean ± SEM of three independent experiments and expressed as folds of corresponding control. (E and F) show respectively a representative Western blot image and quantitative analysis of simvastatin effects on BCL-2 and BAX expression in PC3 cells. (G and H) show respectively a representative Western blot image and quantitative analysis of fluvastatin effects on CHOP and FAS-L expression in PC3 cells. *p<0.05 compared to the corresponding control. |
Statins inhibit AKT and FOXO1 phosphorylation in PC3 cells
To investigate the molecular mechanism by which statins affect cell apoptosis, we hypothesized that statin-induced apoptosis in PC3 cells is associated to the AKT/FOXO1 pathway, a vital molecular pathway in cancer development and progression, and investigated the change of molecules related to the AKT/FOXO1 signaling pathway using Western blot analysis. Treatment with statins in PC3 cells significantly decreased the levels of phosphorylated AKT, phosphorylated FOXO1 and the ratios of phosphorylated to total proteins in a time- and dose-dependent manner as shown in Figure 5. When cells were treated with 5 μM of simvastatin or fluvastatin, both phosphorylated AKT and FOXO1 were significantly decreased at 24 h (Figure 5A-D). The levels of both phosphorylated AKT and FOXO1 were significantly decreased when cells were treated for 36 hrs at doses ranging from 2 μM to 10 μM of simvastatin (Figure 5E, F) or fluvastatin (Figure 5G, H). Moreover, as phosphorylation of FOXO1, a transcriptional factor downstream AKT pathway, leads to FOXO1 move out of nucleus, we assessed both the nuclear and cytosolic levels of FOXO1 upon statin treatment. As shown in Figure 6, treatment with statins, either simvastatin or fluvastatin, in PC3 cells resulted in a dose-dependent increase in nuclear FOXO1 while a decrease in cytosolic FOXO1 level, suggesting that a decrease in FOXO1 phosphorylation, presumably due to a reduction in AKT phosphorylation, leads to a nuclear retention of FOXO1.
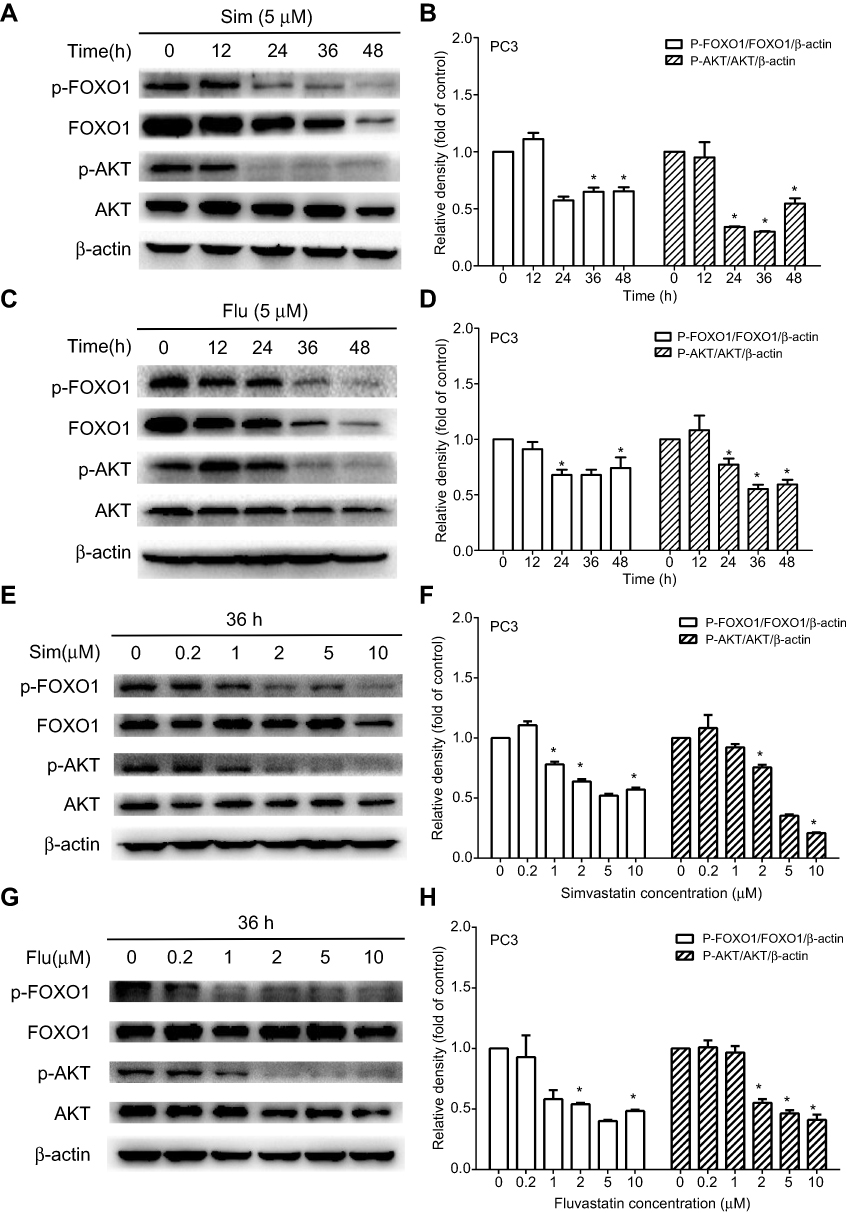 |
Figure 5 Time and dose-dependent inhibition of AKT and FOXO1 phosphorylation by simvastatin (Sim) and fluvastatin (Flu) in PC3 cells. PC3 cells were treated with either 5 μM simvastatin (A) or fluvastatin (C) for 0–48 h or various doses of simvastatin (E) or fluvastatin (G) for 36 h. Total cellular proteins were extracted and analyzed for phosphorylated-AKT (p-AKT, Ser-473), AKT, p-FOXO1 (Ser-256) and FOXO1 by Western blot as described in the Methods. The quantitative data are the mean ± SEM of three independent experiments and expressed as folds of corresponding control (B, D, F, H). *p<0.05 compared to the corresponding control. |
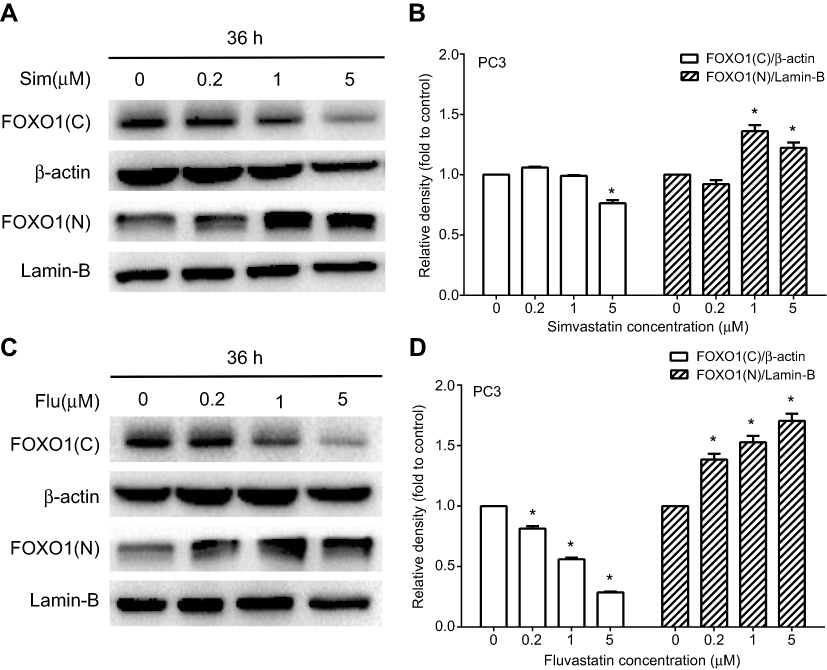 |
Figure 6 A dose-dependent stimulation of FOXO1 nuclear translocation by simvastatin and fluvastatin in PC3 cells. PC3 cells were treated with various doses of either simvastatin (Sim) or fluvastatin (Flu) for 36 h, and the nuclear (N) and cytosolic (C) proteins were isolated and analyzed by Western blotting (A and C). Lamin-B and β-actin were used as a loading control for nuclear and cytosolic proteins, respectively. The quantitative data in (B and D) are the mean ± SEM of three independent experiments and expressed as folds of vehicle control. *p<0.05 compared to the corresponding control. |
Inhibition of AKT/FOXO1 phosphorylation potentiates statin actions
To provide further evidence that changes in phosphorylation of AKT and/or FOXO1 are associated with statin actions, we studied the influence of a specific AKT phosphorylation inhibitor, MK2206, on statin actions. Pretreatment of PC3 cells with MK2206 (0.2 μM) for 60 min greatly inhibited AKT and FOXO1 phosphorylation (Figure 7A-C), potentiated statin inhibition of AKT and FOXO1 phosphorylation (Figure 7A-C), and caspase 3 and PARP cleavage (Figure 7A, D, E). Moreover, like statins, MK2206 significantly inhibited cell viability and further enhanced statin inhibition of cell viability (Figure 7F). Taken together, this data supports the concept that the effects of statins on cell proliferation and apoptosis in PCa cells is mediated, at least in part, through an AKT/FOXO1 signaling pathway.
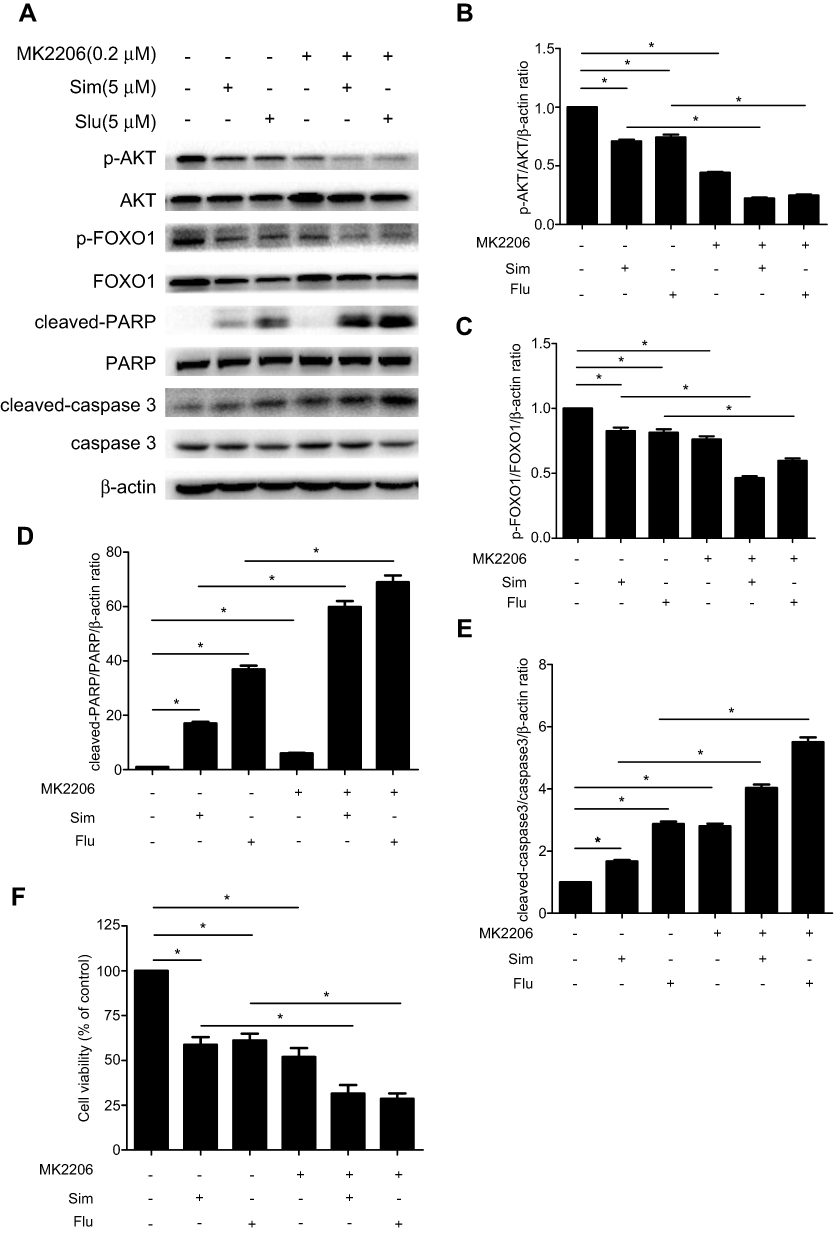 |
Figure 7 Potentiation of simvastatin (Sim) and fluvastatin (Flu) inhibition of AKT and FOXO1 phosphorylation and cell viability while promotion of cell apoptosis by the AKT kinase inhibitor in PC3 cells. PC3 cells were treated with or without 5 μM simvastatin or fluvastatin for 36 h following a pretreatment with or without 0.2 μM MK2206 for 60 min. Total cellular proteins were prepared and immunoblotted for p-AKT(Ser-473), AKT, p-FOXO1 (Ser-256), FOXO1, cleaved-PARP, PARP, cleaved-caspase 3 and caspase 3 (A). β-actin was used as a loading control. The quantitative data in (B-E) are the mean ± SEM of three independent experiments and expressed as folds of vehicle control. (F) shows the alterations of cell viability expressed as percentage of vehicle control in PC3 cells treated with or without MK2206 (0.2 μM), Sim (5 μM) and Flu (5 μM) alone or in combination for 48 h in PC3 cells. *p<0.05 compared to the corresponding control. |
Discussion
In the present study, we have demonstrated that statins produce a dose- and time-dependent induction of cell apoptosis while an inhibition of cell growth in PCa cells. These statin effects involve the modulation of AKT/FOXO1 signaling pathway as evident by a decrease in AKT and FOXO1 phosphorylation and phosphorylated to total AKT and FOXO1 protein ratios. Furthermore, pretreatment with a specific AKT phosphorylation inhibitor potentiated the statin effects on cell viability and cell apoptosis. This data suggests that statins, in addition to being used as anti-hyperlipidemia agents,8 may possess beneficial actions in PCa prevention and treatment.
To determine the apoptotic pathway(s) involved in statin-induced cell apoptosis, we have analyzed representative biomarkers of intrinsic, extrinsic and ER-related apoptotic pathways, BAX and BCL-2,26,27 caspase-8 and FAS-L,28 and CHOP,29 respectively. Statins significantly increased FAS-L expression level and the cleavage of caspase-8, caspase-3 and PARP without an alteration in BAX, BCL-2 and CHOP (Figure 4), suggesting that the extrinsic death receptor pathway is the major signal pathway involved in statin-induced cell apoptosis in PCa cells, which is in agreement with the previous demonstration.30
To further gain insight of statin actions on cell apoptosis, alterations in AKT/FOXO signaling pathway have been explored. AKT/FOXO signaling pathway plays vital roles in regulating various cellular functions, such as cell growth, apoptosis and survival.31 As a downstream effector of AKT, FOXO1, which is phosphorylated by AKT, may mediate the AKT actions on diverse cellular functions.32,33 FOXO1, a member of the FOXO family, composes of four genes, FOXO1, FOXO3, FOXO4 and FOXO6.34 Phosphorylation of FOXO proteins by AKT decreases the transcriptional activities of FOXO factors apparently through the promotion of translocation of FOXO factors from nucleus to cytosol, and contributes to cell survival, growth and apoptosis.35,36 The FOXO signaling is further transduced to regulating other gene expression and protein functions.23,37 It has been reported that FOXO factors can induce cell apoptosis through stimulating the expression of death receptor ligands, such as tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and FAS ligand, and promote cell cycle arrest through the upregulation of the cell cycle inhibitor p27,kip1 which induces G1 arrest, or GADD45, which causes G2 arrest.38 Overexpression of FOXO1 in LNCaP PCa cells has been revealed to induce cell apoptosis, presumably mediated through the extrinsic apoptotic pathway.39 Dysregulation of FOXO factors have been reported in several tumor types including prostate cancer,40 breast cancer,41 glioblastoma,42 leukemia43 as well as rhabdomyosarcoma.44 In the present study, we have observed for the first time that treatment with statins in PC3 cells significantly decreased AKT and FOXO1 phosphorylation and phosphorylated AKT and FOXO1 to total AKT and FOXO1 protein ratio. Furthermore, inhibition of AKT and FOXO1 phosphorylation using a specific AKT inhibitor potentiated statins’ effects on cell apoptosis and proliferation in PCa cells. This observation is in agreement with a previous report that statins blunted AKT/FOXO signaling in biceps femoris muscle45 and consistent with the concept that the AKT/FOXO1 pathway plays a significant role in cell proliferation and apoptosis.21,23,46 Taken together, these results strongly suggest that AKT/FOXO1 pathway is a potential molecular signal mediating statins-induced cell apoptosis in PCa cells.
Conclusion
In summary, we have demonstrated in the present study that statins promoted cell apoptosis while inhibited cell growth in PCa cells in a dose- and time-dependent manner. These statins effects may be mediated through the extrinsic death receptor pathway and probably involve in the AKT/FOXO1 signaling. This data, together with clinical observations that statins may decrease PCa risk and PCa progression,10,15,47 strongly suggests that statins may be potential agents for prostate cancer prevention and therapy, and warrant further preclinical and clinical investigation.
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China (No. 81673516) and Funding from Central South University of China.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Screening PDQ, Prevention Editorial B. Prostate Cancer Screening (PDQ(R)): health professional version. In: PDQ Cancer Information Summaries. Bethesda (MD): National Cancer Institute (US); 2002.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
3. Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20(13):3001–3015. doi:10.1200/JCO.2002.10.018
4. Pienta KJ, Smith DC. Advances in prostate cancer chemotherapy: a new era begins. CA Cancer J Clin. 2005;55(5):
5. Petrylak DP. New paradigms for advanced prostate cancer. Rev Urol. 2007;9(Suppl 2):S3–s12.
6. Berry WR. The evolving role of chemotherapy in androgen-independent (hormone-refractory) prostate cancer. Urology. 2005;65(6 Suppl):2–7. doi:10.1016/j.urology.2005.03.080
7. LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat. 2008;11(1–2):32–50. doi:10.1016/j.drup.2007.11.003
8. Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet (London, England). 2005;366(9493):1267–1278. doi:10.1016/S0140-6736(05)67394-1
9. Zhao SP, Peng DQ, Yu BL, Huo Y. Rationale and design of China intensive lipid lowering with statins in acute coronary syndrome: the CHILLAS study. Am Heart J. 2009;158(4):509–512.e501. doi:10.1016/j.ahj.2009.07.030
10. Yu O, Eberg M, Benayoun S, et al. Use of statins and the risk of death in patients with prostate cancer. J Clin Oncol. 2014;32(1):5–11. doi:10.1200/JCO.2013.49.4757
11. Harshman LC, Wang X, Nakabayashi M, et al. Statin use at the time of initiation of androgen deprivation therapy and time to progression in patients with hormone-sensitive prostate cancer. JAMA Oncol. 2015;1(4):495–504. doi:10.1001/jamaoncol.2015.0829
12. Platz EA, Leitzmann MF, Visvanathan K, et al. Statin drugs and risk of advanced prostate cancer. J Natl Cancer Inst. 2006;98(24):1819–1825. doi:10.1093/jnci/djj499
13. Mucci LA, Stampfer MJ. Mounting evidence for prediagnostic use of statins in reducing risk of lethal prostate cancer. J Clin Oncol. 2014;32(1):1–2. doi:10.1200/JCO.2013.53.2770
14. Larsen SB, Dehlendorff C, Skriver C, et al. Postdiagnosis statin use and mortality in danish patients with prostate cancer. J Clin Oncol. 2017;35(29):3290–3297. doi:10.1200/JCO.2016.71.8981
15. Raval AD, Thakker D, Negi H, Vyas A, Kaur H, Salkini MW. Association between statins and clinical outcomes among men with prostate cancer: a systematic review and meta-analysis. Prostate Cancer Prostatic Dis. 2016;19(2):151–162. doi:10.1038/pcan.2015.58
16. Jung J, Lee C, Lee C, et al. Effects of statin use on the response duration to androgen deprivation therapy in metastatic prostate cancer. Korean J Urol. 2015;56(9):630–636. doi:10.4111/kju.2015.56.9.630
17. Matusewicz L, Meissner J, Toporkiewicz M, Sikorski AF. The effect of statins on cancer cells–review. Tumour Biol. 2015;36(7):4889–4904. doi:10.1007/s13277-015-3551-7
18. Stryjkowska-Gora A, Karczmarek-Borowska B, Gora T, Krawczak K. Statins and cancers. Contemp Oncol (Pozn). 2015;19(3):167–175. doi:10.5114/wo.2014.44294
19. Zheng W, Wang H, Zeng Z, et al. The possible role of the Akt signaling pathway in schizophrenia. Brain Res. 2012;1470:145–158. doi:10.1016/j.brainres.2012.06.032
20. Zhu GC, Yu CY, She L, et al. Metadherin regulation of vascular endothelial growth factor expression is dependent upon the PI3K/Akt pathway in squamous cell carcinoma of the head and neck. Medicine. 2015;94(6):e502. doi:10.1097/MD.0000000000000874
21. Zheng D, Zhu G, Liao S, et al. Dysregulation of the PI3K/Akt signaling pathway affects cell cycle and apoptosis of side population cells in nasopharyngeal carcinoma. Oncol Lett. 2015;10(1):182–188. doi:10.3892/ol.2015.3218
22. Tzivion G, Dobson M, Ramakrishnan G. FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta. 2011;1813(11):1938–1945. doi:10.1016/j.bbamcr.2011.06.002
23. Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta. 2011;1813(11):1978–1986. doi:10.1016/j.bbamcr.2011.03.010
24. Zeng Z, Wang H, Shang F, et al. Lithium ions attenuate serum-deprivation-induced apoptosis in PC12 cells through regulation of the Akt/FoxO1 signaling pathways. Psychopharmacology. 2016;233(5):785–794. doi:10.1007/s00213-015-4168-7
25. Sun H, Wang G, Peng Y, et al. H19 lncRNA mediates 17beta-estradiol-induced cell proliferation in MCF-7 breast cancer cells. Oncol Rep. 2015;33(6):3045–3052. doi:10.3892/or.2015.3899
26. Deegan S, Saveljeva S, Logue SE, et al. Deficiency in the mitochondrial apoptotic pathway reveals the toxic potential of autophagy under ER stress conditions. Autophagy. 2014;10(11):1921–1936. doi:10.4161/15548627.2014.981790
27. Yang J, Ning J, Peng L, He D. Effect of curcumin on Bcl-2 and Bax expression in nude mice prostate cancer. Int J Clin Exp Pathol. 2015;8(8):9272–9278.
28. Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25(34):4798–4811. doi:10.1038/sj.onc.1209608
29. Wang L, Fu P, Zhao Y, et al. Dissociation of NSC606985 induces atypical ER-stress and cell death in prostate cancer cells. Int J Oncol. 2016;49(2):529–538. doi:10.3892/ijo.2016.3555
30. Goc A, Kochuparambil ST, Al-Husein B, Al-Azayzih A, Mohammad S, Somanath PR. Simultaneous modulation of the intrinsic and extrinsic pathways by simvastatin in mediating prostate cancer cell apoptosis. BMC Cancer. 2012;12:409. doi:10.1186/1471-2407-12-409
31. Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9(1):59–71. doi:10.1111/j.1582-4934.2005.tb00337.x
32. Zaballos MA, Santisteban P. FOXO1 controls thyroid cell proliferation in response to TSH and IGF-I and is involved in thyroid tumorigenesis. Mol Endocrinol. 2013;27(1):50–62. doi:10.1210/me.2012-1032
33. Yu JJ, Wu YX, Zhao FJ, Xia SJ. miR-96 promotes cell proliferation and clonogenicity by down-regulating of FOXO1 in prostate cancer cells. Med Oncol. 2014;31(4):910. doi:10.1007/s12032-014-0374-0
34. Tuteja G, Kaestner KH. Forkhead transcription factors II. Cell. 2007;131(1):192. doi:10.1016/j.cell.2007.09.016
35. Lam EW, Francis RE, Petkovic M. FOXO transcription factors: key regulators of cell fate. Biochem Soc Trans. 2006;34(Pt 5):722–726. doi:10.1042/BST0340722
36. Burgering BM, Kops GJ. Cell cycle and death control: long live Forkheads. Trends Biochem Sci. 2002;27(7):352–360.
37. Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117(4):421–426. doi:10.1016/s0092-8674(04)00452-0
38. Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91(2):231–241. doi:10.1016/s0092-8674(00)80405-5
39. Huang H, Muddiman DC, Tindall DJ. Androgens negatively regulate forkhead transcription factor FKHR (FOXO1) through a proteolytic mechanism in prostate cancer cells. J Biol Chem. 2004;279(14):13866–13877. doi:10.1074/jbc.M314143200
40. Modur V, Nagarajan R, Evers BM, Milbrandt J. FOXO proteins regulate tumor necrosis factor-related apoptosis inducing ligand expression. Implications for PTEN mutation in prostate cancer. J Biol Chem. 2002;277(49):47928–47937. doi:10.1074/jbc.M207509200
41. Hu MC, Lee DF, Xia W, et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117(2):225–237. doi:10.1016/s0092-8674(04)00302-2
42. Seoane J, Le HV, Shen L, Anderson SA, Massague J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117(2):211–223. doi:10.1016/s0092-8674(04)00298-3
43. Parry P, Wei Y, Evans G. Cloning and characterization of the t(X;11) breakpoint from a leukemic cell line identify a new member of the forkhead gene family. Genes Chromosomes Cancer. 1994;11(2):79–84. doi:10.1002/gcc.2870110203
44. Galili N, Davis RJ, Fredericks WJ, et al. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993;5(3):230–235. doi:10.1038/ng1193-230
45. Mallinson JE, Constantin-Teodosiu D, Sidaway J, Westwood FR, Greenhaff PL. Blunted Akt/FOXO signalling and activation of genes controlling atrophy and fuel use in statin myopathy. J Physiol. 2009;587(1):219–230. doi:10.1113/jphysiol.2008.164699
46. Palacios F, Prieto D, Abreu C, et al. Dissecting chronic lymphocytic leukemia microenvironment signals in patients with unmutated disease: microRNA-22 regulates phosphatase and tensin homolog/AKT/FOXO1 pathway in proliferative leukemic cells. Leuk Lymphoma. 2015;56(5):1560–1565. doi:10.3109/10428194.2014.990900
47. Allott EH, Howard LE, Cooperberg MR, et al. Postoperative statin use and risk of biochemical recurrence following radical prostatectomy: results from the Shared Equal Access Regional Cancer Hospital (SEARCH) database. BJU Int. 2014;114(5):661–666. doi:10.1111/bju.12720
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.