Back to Journals » Therapeutics and Clinical Risk Management » Volume 16
Statin-Associated Autoimmune Myopathy: Current Perspectives
Authors Tiniakou E ![]()
Received 30 March 2020
Accepted for publication 11 May 2020
Published 27 May 2020 Volume 2020:16 Pages 483—492
DOI https://doi.org/10.2147/TCRM.S197941
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Eleni Tiniakou
Johns Hopkins University School of Medicine, Department of Medicine, Division of Rheumatology, Baltimore, MD, USA
Correspondence: Eleni Tiniakou
Johns Hopkins University School of Medicine, 5200 Eastern Avenue, Mason Lord, Center Tower, Baltimore, MD 21224, USA
Tel +1 410 550-6962
Email [email protected]
Abstract:: Although generally well tolerated, statin users frequently report muscle-related side effects, ranging from self-limiting myalgias to rhabdomyolysis or the rare clinical entity of statin-associated immune-mediated necrotizing myopathy (IMNM). Statin-associated IMNM is based on the development of autoantibodies against 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), the rate-limiting enzyme in cholesterol synthesis and the pharmacologic target of statins, and leads to a necrotizing myopathy requiring immunosuppressive therapy. This review attempts to recapitulate the diverse aspects of anti-HMGCR IMNM, including clinical presentation, diagnostic modalities, genetic risk associations, therapeutic options and potential pathogenetic pathways.
Keywords: stains, myopathy, statin toxicity, statin myopathy, anti-HMGCR
Introduction
Cardiovascular disease (CVD) is the leading cause of death in most developed countries, and a large proportion could be prevented by modifying existing metabolic risk factors, like dyslipidemia.1,2 Therefore, lipid-lowering strategies are one of the cornerstones of primary and secondary prevention of CVD. Statins are the most significant drug of the cholesterol-lowering armamentarium by inhibiting the enzyme hydroxyl-methyl-glutaryl-Co-A reductase (HMGCR), the rate-limiting step in cholesterol synthesis.3
Although generally well tolerated, statin users frequently report muscle-related side effects, ranging from self-limiting myalgias to rhabdomyolysis or the rare entity of statin-associated immune-mediated necrotizing myopathy (IMNM).4 Based on recently published guidelines for the management of cholesterol by the American College of Cardiology and the American Heart Association (ACC-AHA), approximately 9 out of 10 men and more than half of the women age between 60 and 75 years should be on cholesterol lower medications, namely statins.5,6 Therefore, given the projected increase of statin use, we expect similarly an exponential rise even in the rarest side effects.
Statin-associated IMNM is a recently described entity based on the development of autoantibodies against the enzyme HMGCR, and leads to a necrotizing myopathy requiring immunosuppressive therapy.7 In this review, we recapitulate the definition, clinical picture, pathophysiology and therapeutic options of the anti-HMGCR myopathy.
Statin Myopathy
Half of the side effects of statins are related to muscle complaints. The majority of the complaints include nonspecific benign symptoms, and 7–25% will develop a documented muscle event.4,8-10 These include myalgia, myopathy, myositis (including INMN), myonecrosis or rhabdomyolysis.11 While all events could co-exist, it is important to establish a non-ambiguous terminology and note the differences, as the nomenclature will define further management and work up.
Myalgia is muscle aches without the presence of any objective muscle damage, like elevated CPK. Statin-associated myopathy is a broad term and is defined by the presence of muscle weakness. When there is additionally evidence of muscle inflammation (as defined by elevated CPK, muscle edema on MRI imaging studies or muscle biopsy), we can use the term myositis. Lastly, elevation of CPK more than 10 times the upper limit of normal (ULN) can be named myonecrosis and rhabdomyolysis when there is evidence of myoglobulinuria or renal failure.11
Statins can cause myopathy/myositis either as a noninflammatory, toxic effect12 or as a trigger of an autoimmune process have quite similar initial presentation. The difference between the two conditions though is the effect that has the discontinuation of the offending toxic agent (Table 1). While in the first case, the weakness resolves within weeks or months, in the latter case, the statins have triggered a self-sustained inflammatory cycle requiring the addition of immunosuppression in order to reverse the myopathy, as we see in idiopathic inflammatory myopathies (IIM).
 |
Table 1 Differential Between Self-Limited Statin Myopathy and Statin-Associated Immune-Mediated Necrotizing Myopathy |
Statin-Associated IMNM
IIM are a heterogeneous group of systemic autoimmune syndromes resulting from injury of the skeletal muscles. Patients with IIM exhibit a wide variety of phenotypes and several classification criteria have developed in an effort to divide them in groups with similar patterns. IIM are first defined by Bohan and Peter as polymyositis (PM) or dermatomyositis (DM), based on the presence of symmetric proximal muscle weakness, elevated muscle enzymes, characteristic electromyography (EMG) abnormalities, typical muscle biopsy findings and/or distinguishing skin rash.13,14 However, over the last 40 years, the detection of distinct histological patterns and identification of novel myositis specific antibodies (MSA)15 have exposed the complexity of IIM. Moreover, the different types of IIM can differ based on their muscle biopsy features, defined a new separate category: the immune-mediated necrotizing myopathy (IMNM).16 The diagnosis of IMNM is restricted by the presence of muscle cell necrosis and degeneration, with sparse inflammatory infiltrates. Further subclassification based on MSA has divided IMNM into three homogeneous entities: anti-HMGCR myopathy, anti-SRP myopathy and antibody-negative IMNM.17
Anti-HMGCR Autoantibody
The first report associating statins with the development of an autoimmune myopathy requiring immunosuppressive treatment was published in 1994 and followed by several others.18–26 The first case series suggesting the presence of a statin-associated necrotizing myopathy of an autoimmune nature was published in 2004.27 Later on, Needham et al described eight patients with muscle biopsies consistent with necrotizing myositis and evidence of MHC-I upregulation, while they exhibited improvement with the initiation of immunosuppression, all pointing towards an autoimmune process.28 Additional reports were published in support of this theory,29 but it was not until 2010 when Christopher-Stine et al described a novel autoantibody against 100-kD and 200-kD proteins when screening patients with biopsy proven necrotizing myopathy.30 These patients had proximal muscle weakness, muscle edema on MRI, irritable myopathy on EMG and highly elevated CK levels (mean 10,333 IU/L; range 3052–24,714). Importantly, 83.3% of these patients above the age of 50 years old were on a statin, compared to 25% in DM and 36.8% in PM, implying a correlation with statin exposure. It was subsequently verified that this novel autoantibody was indeed recognizing HMGCR enzyme, which is a 100-kDa protein forming 200-kD dimers as well.31
Anti-HMGCR autoantibodies are highly specific for this type of autoimmune myopathy, and they are not associated per se with hyperlipidemia, self-limiting side effects from statins or genetic muscle disease. Almost 2,000 patients of the community-based Atherosclerosis Risk in Communities (ARIC) Study and 98 French Canadian subjects with familial hypercholesterolemia were screened for the presence of anti-HMGCR antibodies and none of them were positive.32 Subsequently, two different studies of patients with history of statin intolerance (101 patients and 79 patients) were negative for these antibodies, and in all cases the muscle-related side effects were self-limited after cessation of the drug.33,34 Similarly, anti-HMGCR antibodies were not detected in patients with documented muscle dystrophy.35 Therefore, anti-HMGCR antibodies can be used to define anti-HMGCR IMNM and differentiate from the self-limited toxic effect of statins.
Anti-HMGCR autoantibodies can be used not only for diagnosis, but also as a marker of disease activity. The titers of the antibody are strongly associated with CK levels and inversely associated with muscle strength.36 However, since CK is a less expensive test, there is no need to longitudinally monitor the anti-HMGCR levels unless for research purposes. Interestingly, the anti-HMGCR titers always remain positive even when the CPK normalizes and the disease is quiescent.36
Detection Methods of Anti-HMGCR Autoantibody
Detection of the anti-HMGCR autoantibodies in the clinical setting is based on a commercially available ELISA. The sensitivity and specificity of the ELISA are 94.4% and 99.3%, respectively.32 While this ELISA has very high negative predictive value, meaning that a negative result makes it unlikely that the patient does have anti-HMGCR IMNM, the opposite is not true. The positive predictive value of the HMGCR ELISA would be only 0.910 in a specialty clinic, where the pre-test probability is higher, and 0.001 in an unselected population, with a false-positive rate of 10.5%.32 The gold standard for anti-HMGCR detection remains immunoprecipitation, which is only feasible in a research laboratory. Therefore, testing should be reserved only for cases with severe myopathy (highly elevated CPK and/or severe muscle weakness) persisting more than 6 weeks after cessation of statins.7
Alternate screening methods have been investigated, like addressable laser bead immunoassay (ALBIA), chemiluminescence or immunoblot.37–39 Investigation into the immunofluorescence pattern the anti-HMGCR antibodies are creating, has led to the suggestion of using IF as an initial screening method. Anti-HMGCR autoantibodies exhibit a finely granular cytoplasmic pattern with perinuclear reinforcement in HEp2-cells,38,39 and produce a centrolobular distribution on rat hepatocytes.40 Positivity is observed in 30–61% of anti-HMGCR positive patients for HEp2-cells30,37 and 91% of cases for the rat hepatocytes.40 Albeit the pattern is visible in a small percentage of cells (10%),40 which could reflect a heterogeneous expression of HMGCR in the cell lines.
Association with Statin Exposure
The first description of anti-HMGCR myopathy was based on the Johns Hopkins cohort in 2010 and reported a mean age of patients 55 years (52.4–57.6) and 75% exposure to statins.30 While the use of statins was overwhelming, there were still 25% of patients with similar clinical and laboratory characteristics that were statin naïve. As these patients demonstrated higher levels of CK, more severe muscle weakness and required more intense immunotherapy, it was initially thought that statin exposure was a marker for milder disease.41 Long term follow-up demonstrated that it was actually the age at disease onset that determined the prognosis, with younger patients having more recalcitrant disease regardless of statin use (although they were less likely to have been exposed to statins).36
Since then, different cohorts in diverse geographic locations have reported various percentages of statin use (Table 2). When trying to interpret the different reports however, it is important to note the mean age of the relevant cohort, since statin use is associated with increasing age. Moreover, there is always the question if the patient has been exposed to statins from an alternate, non-documented source, although this has not been proved and remains a hypothesis. A study from the central US described anti-HMGCR IMNM in patients with mean age 50 years old and only 38% reported use of statins (18/47).42 Two cohorts from Australia, with mean age of 70 years old, reported 94–100% of patients exposed to statins.43,44 Similarly, a Czech cohort described 100% statin exposure in their patients with mean age 67 years old,45 and a New Zealand cohort with mean age 76.8 years old reported 75% statin use.46 However, cohorts from France, China, Japan and the USA acknowledged having a much lower statin exposure (44%, 0–15%, 18–37.5% and 38%, respectively).42,47–51 All of them had a mean age around 50 years old (48.9, 50 and 56.4 years old for the French, US and Japanese cohort, while only 5/22 Chinese patients were over 50 years old and the other Chinese cohort had a mean age of 35).47,48,51,52 Lastly, when cohorts from nine different countries, including China and France, were combined, there continues to be a statistically significant percentage of statin users (52 out of 91).38
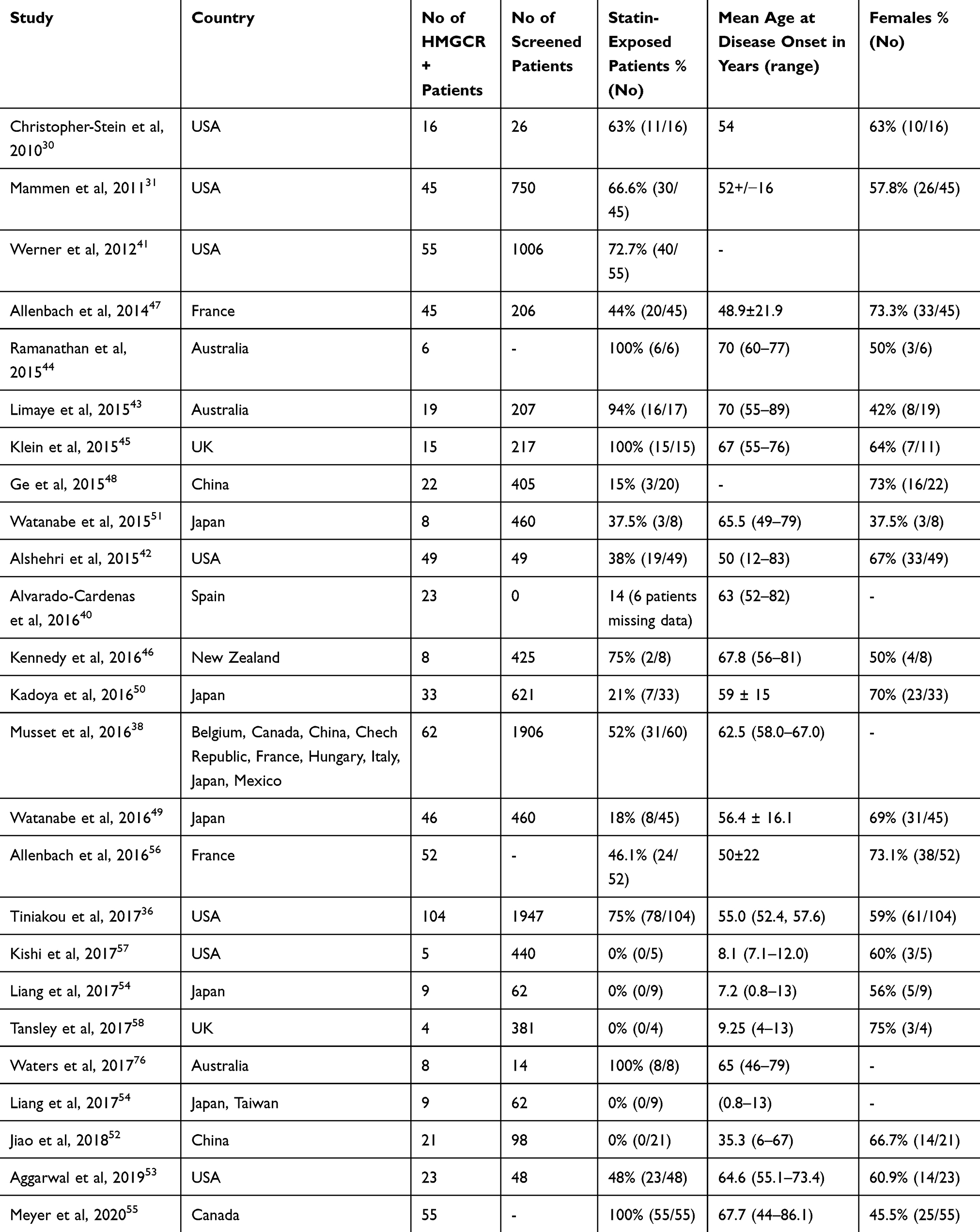 |
Table 2 Demographic Characteristics and Statin Exposure of Patients with Anti-HMGCR+ Myopathy Across Different Studies |
Clinical Presentation
Patients with anti-HMGCR IMNM commonly present with subacute onset, progressive, symmetric proximal muscle weakness with significantly elevated CK levels that persists for months after discontinuation of statins. CK usually ranges in the thousands with values rarely below 1,000. Patients could exhibit myalgias and dysphagia, although this is present in less than one-third of the patients. Extramuscular manifestations, like arthritis or lung involvement, are similarly quite infrequent and if present, they should alert for further work up to rule out other causes of IIM.36,44,45,47,49,53–55 Skin rash, similar to dermatomyositis, can be present in up to 60% in some cohorts,30,36,50,51,54,56–58 although the exact skin pathology is not known. In these cases, the patients are still classified as having anit-HMGCR myositis with a DM-like rash.59
Atypical Clinical Presentation
While the vast majority of patients present with symmetrical, proximal muscle weakness, a small percentage may exhibit atypical features, like asymmetry, scapular winging, disease onset is at a very young age and no report of statin exposure. In these cases, it might be challenging to differentiate from alternate diagnosis, like limb-girdle muscle dystrophy (LGMD), especially given the fact that in 40–60% of LGMD cases no genetic abnormality is detected.60 Anti-HMGCR antibodies have not been detected in cases of genetically confirmed inherited muscle diseases,35 and inversely, patients with recalcitrant anti-HMGCR myopathy undergoing whole-exome sequencing did not reveal any concomitant pathogenic mutation.36 On the other hand though, there have been reported cases of pediatric and young adult patients misdiagnosed as having LGMD, thus avoiding immunosuppression.42,54,57,61–64 Therefore, anti-HMGCR autoantibody is recommended to be used as an evaluation tool in cases of suspected LGMD notwithstanding the lack of statin exposure.
Muscle Biopsy Findings
The muscle biopsy findings in anti-HMGCR IMNM are characteristic of a necrotizing myopathy. The predominant feature is necrotic muscle fibers and regenerating fibers with sparse inflammatory infiltrates. Additionally, there can be membrane attack complex deposition on small blood vessels and membrane of few non-necrotic muscle fibers, as well as major histocompatibility complex (MHC) class I upregulation. However, up to one-third of the biopsies can have evidence of inflammatory cell infiltration, 50% of which were identified as scattered T cells (CD4+ and CD8+). These findings are similar regardless of age, statin exposure or geographic location of the patients.42,47,55,65,66
What needs attention is that the self-limited toxic effect of statins can also cause necrosis, regeneration and/or inflammation. However, immune findings like MHC class I upregulation in mature muscle fibers are absent. In cases where these findings are mild, however, it might be hard to interpret the pathologic findings. Therefore, it is preferable to defer the muscle biopsy for at least 4–6 weeks after statin cessation in order to avoid this conundrum, especially in cases when anti-HMGCR testing is not available for diagnosis.
Genetic Risk Factors
HMGCR INMN has one of the strongest associations with HLA class II allele risk factor within autoimmune diseases. The HLA allele DRB1*11:01 has an odds ratio of 24.5 in Caucasians and 56.5 in African Americans.67 The above association has been confirmed by additional cohorts from Australia, Japan and Europe as well.43,68,69 Remarkably, a second HLA class II allele, DRB1*07:01, has been identified as an immunogenetic risk factor in a pediatric cohort, suggesting a different trigger or pathogenetic mechanism.57 Despite this significant association, these alleles are quite frequent in the general population (7–26% of the control population), therefore, not a useful clinical tool for diagnosis.57,67
Disease Prognosis
Based on a longitudinal analysis of the Johns Hopkins cohort including 104 patients followed for approximately 3 years, the age at disease onset was identified as the most important prognostic factor. Every 10 additional years at disease onset was associated with additional 2.2 points in muscle strength. That means that while the majority of patients above 60 years old recovered full strength (85%) within 4 years, this was true for less than half of patients below 52 years old.36 Importantly, statin exposure was not found to be the determining factor for disease progress. Similarly, a Chinese cohort of 21 patients, who were all statin-naïve, verified that the younger patients had a worse prognosis.52 Longitudinal analysis of a Canadian cohort of 55 patients showed that early intervention leads to more efficacious treatment and maintained remission.55 These longitudinal studies emphasize that our efforts should focus on early diagnosis and aggressive treatment especially of the younger population.
Cancer Association
Several studies have investigated the relationship of anti-HMGCR IMNM with malignancy and the results are ambivalent depending on the geographic location. No statistical association with cancer has been found in Australian, Canadian or US cohorts, with the majority of patients reporting statin exposure.36,43,53,55 On the other end, cohorts from France and Japan revealed a significant percentage of patients developing cancer.56,68 Interestingly, 12/33 of anti-HMGCR+ Japanese patients were identified to have synchronous cancer (92% within one year of the myositis diagnosis) and 33% statin exposure, implying that in these cases cancer could have been the trigger for the myositis.50 Therefore, besides the age-appropriate cancer screening, a rigorous work up could potentially be reserved for patients descending from specific regions.
Therapy
The majority of the patients with anti-HMGCR myopathy is statin associated, at least according to the US experience. Rechallenge with statins has led to worsening of the disease,28,44,70 so statins are contraindicated in these patients. It is recommended that it is documented in the patient’s chart of having drug hypersensitivity to statins or including them into their “allergy list”, so future rechallenge will be avoided. Although there is no available data for anti-HMGCR positive patients without history of statin exposure, we would recommend avoidance of the class in these cases as well. Therefore, the first step in management includes discontinuation of statins.
The vast majority of patients with anti-HMGCR myopathy will require aggressive immunosuppression. As there are no controlled studies, the treatment regimen for each patient depends on disease severity and physician preference. Prednisone is regarded commonly as a first-line agent along with the addition of a steroid-sparing agent, like methotrexate, mycophenolate, azathioprine, intravenous immunoglobulin (IVIG) or rituximab. Methotrexate or IVIG seem to be the agent of choice after prednisone for most cohorts,36,44,47,53 and specifically IVIG can be successfully employed for refractory cases. Cases report has also pointed out potential benefit of cyclosporine as well in recalcitrant disease.71 Based on this experience, the 224th European NeuroMuscular Center (ENMC) International Workshop on necrotizing myopathies suggested the concurrent initiation of steroids and methotrexate, adding IVIG in severe cases at disease onset or within 6 months if response has been inadequate. Initial series of 3 patients with anti-HMGCR myositis, who could not tolerate steroids, confirmed efficacy of IVIG monotherapy as initial treatment.17 Especially in cases where symptoms are relatively mild, steroids could be avoided,53,55 and if IVIG is combined with any steroid-sparing agent, remission could be achieved faster.55 Lastly, Rituximab can be reserved as a fourth-line agent.17 Experience with rituximab is limited; there are few reports utilizing it as a rescue therapy for refractory cases,66,72 but with limited benefit. The reason for that could be irreversible widespread muscle atrophy establishing at later stages of the disease. In conclusion, given the association of severity with onset of disease at a young age,36 one could argue that aggressive therapy (IVIG, steroid-sparing agent, with or without steroids) should be considered especially for younger patients and can be tailored for the older patients based on their comorbidities and severity of muscle weakness.
Although the bulk of patients requires treatment, there is a small percentage that do not exhibit any weakness. The Johns Hopkins cohort reported approximately 4% (4 out of 104) of their patients did not require treatment due to mild symptoms,36 and 3 out 45 patients of a cohort in France showed improvement of their symptoms after discontinuation of statins.47 However, recently published experience from Canada argues that all patients might benefit from treatment; 22/55 (40%) of their cohort did not have any weakness at diagnosis, but 13 of them eventually developed weakness within a median time of 21.6 months (7.0–95.0) and it was more difficult to control the disease at that stage.55 This is a retrospective study, but demonstrates the need for well-designed clinical trials to answer these questions, especially given our ability to identify patients at an early stage with the use of antibodies.
It still remains the question how to manage hyperlipidemia in these patients, since statins are considered an aggravating factor for the disease. Lipid-lowering medications that avoid the mevalonate pathway, like fenofibrate or ezetimibe, are encouraged starting on a lower and intermittent dosing until it is confirmed they do not cause any side effects. The newer biologic agents proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, which act by increasing the cellular uptake of low-density lipoprotein (LDL), were shown to be safe in patients with anti-HMGCR myopathy and, interestingly enough, allowed 2 patients to decrease their immunosuppression.73 As PCSK9 inhibitors cause downregulation of HMGCR, it is postulated that the spontaneous clinical improvement could be due to decrease of the autoantigen.
Pathogenesis
The pathogenesis of anti-HMGCR myopathy is still under investigation and there are several critical questions that need to be answered. First of all, which is the exact role of statins? Beginning mid-1990s, case reports of autoimmune myopathy following statin use began to emerge.27,28 That led to the hypothesis that statins were associated with this autoimmune process, which was further supported by the finding of a new antibody that was binding actually to HMGCR, the pharmacologic target of statins, and the fact that the majority of patients with this autoantibody were indeed exposed to statins.36,43-46,55 Statins could participate in the initiation of autoimmunity by (1) increasing the expression and availability of the autoantigen HMGCR, and (2) binding to HMGCR causing conformational changes to the protein and alternate processing by antigen presenting cells, leading to immunogenic HMGCR peptides through certain HLA proteins (DRB1*11:01). The counterargument for this hypothesis is that there is a significant percentage of patients never been exposed to statins.47,48,50,51 This could be explained by exposure to statins through diet or it could be that there is a different pathogenic mechanism for these patients. Nevertheless, clinical experience shows worsening of this type of myositis after rechallenging with statins,28,44,70 so the term utilized to describe this entity continues to be statin-associated myopathy.
The second question relates to the role of the anti-HMGCR antibodies. The correlation of the antibody titer with CK and inverse correlation with muscle strength36 was the first indication that these antibodies could be pathogenic. The deposition of membrane attack complex on the surface of muscle fibers, addition of anti-HMGCR antibodies in muscle cultures causing atrophy74 and passive transfer of these antibodies in mice causing muscle weakness support their pathogenic role.75 However, the persistence of anti-HMGCR antibodies even in patients with normal muscle strength, and limited efficiency of plasmapheresis and/or rituximab treatments, continue to be persistent queries in the clinical praxis requiring further studies.
Conclusion
The diagnosis of anti-HMGCR IMNM is based on the detection of antibodies against HMGCR. It is a unique clinical entity in the sense that it is associated with an environmental trigger (statins) and has a strong genetic background (HLA-DRB1*11:01). While investigative work up ought to be reserved only for persistent cases of muscle weakness and/or hyperCKemia after discontinuation of statins, clinical suspicion should be high even in cases of no apparent statin exposure. Increasing data support a pathogenic role of the antibodies, which can lead to the identification of novel therapeutic targets. There are still outstanding questions regarding the epidemiology, pathogenesis and treatment of statin-associated IMNM, which future studies aspire to answer.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Laslett LJ, Alagona P, Clark BA, et al. The worldwide environment of cardiovascular disease: prevalence, diagnosis, therapy, and policy issues: a report from the American College of Cardiology. J Am Coll Cardiol. 2012;60(25 Suppl):S1–49. doi:10.1016/j.jacc.2012.11.002
2. Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364(9438):937–952. doi:10.1016/S0140-6736(04)17018-9
3. Chou R, Dana T, Blazina I, Daeges M, Jeanne TL. Statins for prevention of cardiovascular disease in adults: evidence report and systematic review for the US preventive services task force. JAMA. 2016;316(19):2008–2024. doi:10.1001/jama.2015.15629
4. Rosenson RS, Gandra SR, McKendrick J, et al. Identification and management of statin-associated symptoms in clinical practice: extension of a clinician survey to 12 further countries. Cardiovasc Drugs Ther. 2017;31(2):187–195. doi:10.1007/s10557-017-6727-0
5. Pencina MJ, Navar-Boggan AM, D’Agostino RB, et al. Application of new cholesterol guidelines to a population-based sample. N Engl J Med. 2014;370(15):1422–1431. doi:10.1056/NEJMoa1315665
6. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association task force on practice guidelines. J Am Coll Cardiol. 2014;63(25Pt B):2889–2934. doi:10.1016/j.jacc.2013.11.002
7. Mammen AL, Longo DL. Statin-associated autoimmune myopathy. N Engl J Med. 2016;374(7):664–669. doi:10.1056/NEJMra1515161
8. Hovingh GK, Gandra SR, McKendrick J, et al. Identification and management of patients with statin-associated symptoms in clinical practice: a clinician survey. Atherosclerosis. 2016;245:111–117. doi:10.1016/j.atherosclerosis.2015.12.015
9. Zhang H, Plutzky J, Skentzos S, et al. Discontinuation of statins in routine care settings: a cohort study. Ann Intern Med. 2013;158(7):526–534. doi:10.7326/0003-4819-158-7-201304020-00004
10. El-Salem K, Ababneh B, Rudnicki S, et al. Prevalence and risk factors of muscle complications secondary to statins. Muscle Nerve. 2011;44(6):877–881. doi:10.1002/mus.22205
11. Stroes ES, Thompson PD, Corsini A, et al. Statin-associated muscle symptoms: impact on statin therapy-European Atherosclerosis Society consensus panel statement on assessment, aetiology and management. Eur Heart J. 2015;36(17):1012–1022. doi:10.1093/eurheartj/ehv043
12. Hansen KE, Hildebrand JP, Ferguson EE, Stein JH. Outcomes in 45 patients with statin-associated myopathy. Arch Intern Med. 2005;165(22):2671–2676. doi:10.1001/archinte.165.22.2671
13. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292(7):344–347. doi:10.1056/NEJM197502132920706
14. Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975;292(8):403–407. doi:10.1056/NEJM197502202920807
15. Targoff IN. Laboratory testing in the diagnosis and management of idiopathic inflammatory myopathies. Rheum Dis Clin North Am. 2002;28(4):859–890. doi:10.1016/s0889-857x(02)00032-7.
16. Hoogendijk JE, Amato AA, Lecky BR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10–12 October 2003, Naarden, The Netherlands. Neuromuscul Disord. 2004;14(5):337–345. doi:10.1016/j.nmd.2004.02.006
17. Allenbach Y, Mammen AL, Benveniste O, Stenzel W. Immune-mediated necrotizing myopathies working group. 224th ENMC international workshop: clinico-sero-pathological classification of immune-mediated necrotizing myopathies Zandvoort, The Netherlands, 14–16 October 2016. Neuromuscul Disord. 2018;28(1):87–99. doi:10.1016/j.nmd.2017.09.016
18. Khattak FH, Morris IM, Branford WA. Simvastatin-associated dermatomyositis. Br J Rheumatol. 1994;33(2):199. doi:10.1093/rheumatology/33.2.199
19. Giordano N, Senesi M, Mattii G, Battisti E, Villanova M, Gennari C. Polymyositis associated with simvastatin. Lancet. 1997;349(9065):1600–1601. doi:10.1016/S0140-6736(05)61628-5
20. Hill C, Zeitz C, Kirkham B. Dermatomyositis with lung involvement in a patient treated with simvastatin. Aust N Z J Med. 1995;25(6):745–746. doi:10.1111/j.1445-5994.1995.tb02870.x
21. Rodriguez-Garcia JL, Serrano Commino M. Lovastatin-associated dermatomyositis. Postgrad Med J. 1996;72(853):694. doi:10.1136/pgmj.72.853.694
22. Noël B, Cerottini JP, Panizzon RG. Atorvastatin-induced dermatomyositis. Am J Med. 2001;110(8):670–671. doi:10.1016/s0002-9343(01)00711-2
23. Riesco-Eizaguirre G, Arpa-Gutiérrez FJ, Gutiérrez M, Toribio E. Severe polymyositis with simvastatin use. Rev Neurol. 2003;37(10):934–936.
24. Takagi A, Shiio Y. Pravastatin-associated polymyositis, a case report. Rinsho Shinkeigaku. 2004;44(1):25–27.
25. Vasconcelos OM, Campbell WW. Dermatomyositis-like syndrome and HMG-CoA reductase inhibitor (statin) intake. Muscle Nerve. 2004;30(6):803–807. doi:10.1002/mus.20127
26. Zuech P, Pauwels C, Duthoit C, et al. Pravastatin-induced dermatomyositis. Rev Med Interne. 2005;26(11):897–902. doi:10.1016/j.revmed.2005.07.005
27. Fauchais A-L, Iba Ba J, Maurage P, et al. Polymyositis induced or associated with lipid-lowering drugs: five cases. Rev Med Interne. 2004;25(4):294–298. doi:10.1016/j.revmed.2003.10.013
28. Needham M, Fabian V, Knezevic W, Panegyres P, Zilko P, Mastaglia FL. Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul Disord. 2007;17(2):194–200. doi:10.1016/j.nmd.2006.10.007
29. Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve. 2010;41(2):185–190. doi:10.1002/mus.21486
30. Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. 2010;62(9):2757–2766. doi:10.1002/art.27572
31. Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum. 2011;63(3):713–721. doi:10.1002/art.30156
32. Mammen AL, Pak K, Williams EK, et al. Rarity of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies in statin users, including those with self-limited musculoskeletal side effects. Arthritis Care Res (Hoboken). 2012;64(2):269–272. doi:10.1002/acr.20662
33. Floyd JS, Brody JA, Tiniakou E, Psaty BM, Mammen A. Absence of anti-HMG-CoA reductase autoantibodies in severe self-limited statin-related myopathy. Muscle Nerve. 2016;54(1):142–144. doi:10.1002/mus.25127
34. Keating P, Young J, George P, Florkowski C, Spellerberg M, Kennedy N. Anti-HMGCR autoantibodies in self-limiting statin-induced myopathy. Int J Rheum Dis. 2017;20(12):2179–2181. doi:10.1111/1756-185X.13025
35. Mammen AL, Casciola-Rosen L, Christopher-Stine L, Lloyd TE, Wagner KR. Myositis-specific autoantibodies are specific for myositis compared to genetic muscle disease. Neurol Neuroimmunol Neuroinflamm. 2015;2(6):e172. doi:10.1212/NXI.0000000000000172
36. Tiniakou E, Pinal-Fernandez I, Lloyd TE, et al. More severe disease and slower recovery in younger patients with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Rheumatology (Oxford). 2017;56(5):787–794. doi:10.1093/rheumatology/kew470
37. Drouot L, Allenbach Y, Jouen F, et al. Exploring necrotizing autoimmune myopathies with a novel immunoassay for anti-3-hydroxy-3-methyl-glutaryl-CoA reductase autoantibodies. Arthritis Res Ther. 2014;16(1):R39. doi:10.1186/ar4468
38. Musset L, Allenbach Y, Benveniste O, et al. Anti-HMGCR antibodies as a biomarker for immune-mediated necrotizing myopathies: A history of statins and experience from a large international multi-center study. Autoimmun Rev. 2016;15(10):983–993. doi:10.1016/j.autrev.2016.07.023
39. Shovman O, Gilburd B, Chayat C, et al. Anti-HMGCR antibodies demonstrate high diagnostic value in the diagnosis of immune-mediated necrotizing myopathy following statin exposure. Immunol Res. 2017;65(1):276–281. doi:10.1007/s12026-016-8867-x
40. Alvarado-Cardenas M, Marin-Sánchez A, Martínez MA, et al. Statin-associated autoimmune myopathy: a distinct new IFL pattern can increase the rate of HMGCR antibody detection by clinical laboratories. Autoimmun Rev. 2016;15(12):1161–1166. doi:10.1016/j.autrev.2016.09.005
41. Werner JL, Christopher-Stine L, Ghazarian SR, et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum. 2012;64(12):4087–4093. doi:10.1002/art.34673
42. Alshehri A, Choksi R, Bucelli R, Pestronk A. Myopathy with anti-HMGCR antibodies: perimysium and myofiber pathology. Neurol Neuroimmunol Neuroinflamm. 2015;2(4):e124. doi:10.1212/NXI.0000000000000124
43. Limaye V, Bundell C, Hollingsworth P, et al. Clinical and genetic associations of autoantibodies to 3-hydroxy-3-methyl-glutaryl-coenzyme a reductase in patients with immune-mediated myositis and necrotizing myopathy. Muscle Nerve. 2015;52(2):196–203. doi:10.1002/mus.24541
44. Ramanathan S, Langguth D, Hardy TA, et al. Clinical course and treatment of anti-HMGCR antibody-associated necrotizing autoimmune myopathy. Neurol Neuroimmunol Neuroinflamm. 2015;2(3):e96. doi:10.1212/NXI.0000000000000096
45. Klein M, Mann H, Pleštilová L, et al. Increasing incidence of immune-mediated necrotizing myopathy: single-centre experience. Rheumatology (Oxford). 2015;54(11):2010–2014. doi:10.1093/rheumatology/kev229
46. Kennedy N, Keating P, O’Donnell J. HMGCR-associated myositis: a New Zealand case series and estimate of incidence. Intern Med J. 2016;46(5):622–625. doi:10.1111/imj.13023
47. Allenbach Y, Drouot L, Rigolet A, et al. Anti-HMGCR autoantibodies in European patients with autoimmune necrotizing myopathies: inconstant exposure to statin. Medicine (Baltimore). 2014;93(3):150–157. doi:10.1097/MD.0000000000000028
48. Ge Y, Lu X, Peng Q, Shu X, Wang G, Shu X. Clinical characteristics of anti-3-hydroxy-3-methylglutaryl coenzyme A reductase antibodies in Chinese patients with idiopathic inflammatory myopathies. PLoS One. 2015;10(10):e0141616. doi:10.1371/journal.pone.0141616
49. Watanabe Y, Uruha A, Suzuki S, et al. Clinical features and prognosis in anti-SRP and anti-HMGCR necrotising myopathy. J Neurol Neurosurg Psychiatry. 2016;87(10):1038–1044. doi:10.1136/jnnp-2016-313166
50. Kadoya M, Hida A, Hashimoto Maeda M, et al. Cancer association as a risk factor for anti-HMGCR antibody-positive myopathy. Neurol Neuroimmunol Neuroinflamm. 2016;3(6):e290. doi:10.1212/NXI.0000000000000290
51. Watanabe Y, Suzuki S, Nishimura H, et al. Statins and myotoxic effects associated with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase autoantibodies: an observational study in Japan. Medicine (Baltimore). 2015;94(4):e416. doi:10.1097/MD.0000000000000416
52. Jiao Y, Cai S, Lin J, et al. Statin-naïve anti-HMGCR antibody-mediated necrotizing myopathy in China. J Clin Neurosci. 2018;57:13–19. doi:10.1016/j.jocn.2018.08.010
53. Aggarwal R, Moghadam-Kia S, Lacomis D, et al. Anti-hydroxy-3-methylglutaryl-coenzyme A reductase (anti-HMGCR) antibody in necrotizing myopathy: treatment outcomes, cancer risk, and role of autoantibody level. Scand J Rheumatol. 2019:1–7. doi:10.1080/03009742.2019.1672782.
54. Liang W-C, Uruha A, Suzuki S, et al. Pediatric necrotizing myopathy associated with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies. Rheumatology (Oxford). 2017;56(2):287–293. doi:10.1093/rheumatology/kew386
55. Meyer A, Troyanov Y, Drouin J, et al. Statin-induced anti-HMGCR myopathy: successful therapeutic strategies for corticosteroid-free remission in 55 patients. Arthritis Res Ther. 2020;22(1):5. doi:10.1186/s13075-019-2093-6
56. Allenbach Y, Keraen J, Bouvier A-M, et al. High risk of cancer in autoimmune necrotizing myopathies: usefulness of myositis specific antibody. Brain. 2016;139(Pt 8):2131–2135. doi:10.1093/brain/aww054
57. Kishi T, Rider LG, Pak K, et al. Association of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase autoantibodies with DRB1*07:01 and severe myositis in juvenile myositis patients. Arthritis Care Res (Hoboken). 2017;69(7):1088–1094. doi:10.1002/acr.23113
58. Tansley SL, Betteridge ZE, Simou S, et al. Anti-HMGCR autoantibodies in juvenile idiopathic inflammatory myopathies identify a rare but clinically important subset of patients. J Rheumatol. 2017;44(4):488–492. doi:10.3899/jrheum.160871
59. Mammen AL, Allenbach Y, Stenzel W, et al. 239th ENMC international workshop: classification of dermatomyositis, Amsterdam, the Netherlands, 14–16 December 2018. Neuromuscular Disord. 2020;30(1):70–92. doi:10.1016/j.nmd.2019.10.005
60. Thompson R, Straub V. Limb-girdle muscular dystrophies - international collaborations for translational research. Nat Rev Neurol. 2016;12(5):294–309. doi:10.1038/nrneurol.2016.35
61. Mohassel P, Landon-Cardinal O, Foley AR, et al. Anti-HMGCR myopathy may resemble limb-girdle muscular dystrophy. Neurol Neuroimmunol Neuroinflamm. 2019;6(1):e523. doi:10.1212/NXI.0000000000000523
62. Mohassel P, Foley AR, Donkervoort S, et al. Anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase necrotizing myopathy masquerading as a muscular dystrophy in a child. Muscle Nerve. 2017;56(6):1177–1181. doi:10.1002/mus.25567
63. Tard C, Tiffreau V, Jaillette E, et al. Anti-HMGCR antibody-related necrotizing autoimmune myopathy mimicking muscular dystrophy. Neuropediatrics. 2017;48(6):473–476. doi:10.1055/s-0037-1604402
64. Tansley SL, Simou S, Shaddick G, et al. Autoantibodies in juvenile-onset myositis: their diagnostic value and associated clinical phenotype in a large UK cohort. J Autoimmun. 2017;84:55–64. doi:10.1016/j.jaut.2017.06.007
65. Chung T, Christopher-Stine L, Paik JJ, Corse A, Mammen AL. The composition of cellular infiltrates in anti-HMG-CoA reductase-associated myopathy. Muscle Nerve. 2015;52(2):189–195. doi:10.1002/mus.24642
66. Ashton C, Junckerstorff R, Bundell C, Hollingsworth P, Needham M. Treatment and outcomes in necrotising autoimmune myopathy: an Australian perspective. Neuromuscul Disord. 2016;26(11):734–740. doi:10.1016/j.nmd.2016.08.013
67. Mammen AL, Gaudet D, Brisson D, et al. Increased frequency of DRB1*11:01 in anti-hydroxymethylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Care Res (Hoboken). 2012;64(8):1233–1237. doi:10.1002/acr.21671
68. Ohnuki Y, Suzuki S, Shiina T, et al. HLA-DRB1 alleles in immune-mediated necrotizing myopathy. Neurology. 2016;87(18):1954–1955. doi:10.1212/WNL.0000000000003160
69. Rothwell S, Chinoy H, Lamb JA, et al. Focused HLA analysis in caucasians with myositis identifies significant associations with autoantibody subgroups. Ann Rheum Dis. 2019;78(7):996–1002. doi:10.1136/annrheumdis-2019-215046
70. Wu Y, Lach B, Provias JP, Tarnopolsky MA, Baker SK. Statin-associated autoimmune myopathies: a pathophysiologic spectrum. Can J Neurol Sci. 2014;41(5):638–647. doi:10.1017/cjn.2014.22
71. Morishima R, Matsubara S, Sugaya K, et al. Treatment with cyclosporine A for statin-naïve anti-HMGCR antibody-associated necrotizing myopathy. J Clin Rheumatol. 2019;25(5):e72–e73. doi:10.1097/RHU.0000000000000922
72. Landon-Cardinal O, Allenbach Y, Soulages A, et al. Rituximab in the treatment of refractory anti-HMGCR immune-mediated necrotizing myopathy. J Rheumatol. 2019;46(6):623–627. doi:10.3899/jrheum.171495
73. Tiniakou E, Rivera E, Mammen AL, Christopher-Stine L. Use of proprotein convertase subtilisin/kexin type 9 inhibitors in statin-associated immune-mediated necrotizing myopathy: a case series. Arthritis Rheum (Hoboken, NJ). 2019;71(10):1723–1726. doi:10.1002/art.40919
74. Arouche-Delaperche L, Allenbach Y, Amelin D, et al. Pathogenic role of anti-signal recognition protein and anti-3-hydroxy-3-methylglutaryl-CoA reductase antibodies in necrotizing myopathies: myofiber atrophy and impairment of muscle regeneration in necrotizing autoimmune myopathies. Ann Neurol. 2017;81(4):538–548. doi:10.1002/ana.24902
75. Bergua C, Chiavelli H, Allenbach Y, et al. In vivo pathogenicity of IgG from patients with anti-SRP or anti-HMGCR autoantibodies in immune-mediated necrotising myopathy. Ann Rheum Dis. 2019;78(1):131–139. doi:10.1136/annrheumdis-2018-213518
76. Waters MJ and Limaye V. Clinico-serologic features of statin-induced necrotising autoimmune myopathy in a single-centre cohort. Clin Rheumatol. 2018;37:543–547.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.