Back to Journals » Research and Reports in Tropical Medicine » Volume 14
State-of-the-Art in the Drug Discovery Pathway for Chagas Disease: A Framework for Drug Development and Target Validation
Authors Gabaldón-Figueira JC ![]() , Martinez-Peinado N, Escabia E, Ros-Lucas A, Chatelain E, Scandale I, Gascon J
, Martinez-Peinado N, Escabia E, Ros-Lucas A, Chatelain E, Scandale I, Gascon J ![]() , Pinazo MJ, Alonso-Padilla J
, Pinazo MJ, Alonso-Padilla J
Received 31 March 2023
Accepted for publication 3 June 2023
Published 14 June 2023 Volume 2023:14 Pages 1—19
DOI https://doi.org/10.2147/RRTM.S415273
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mario Rodríguez-Pérez
Juan Carlos Gabaldón-Figueira,1 Nieves Martinez-Peinado,1 Elisa Escabia,1 Albert Ros-Lucas,1,2 Eric Chatelain,3 Ivan Scandale,3 Joaquim Gascon,1,2 María-Jesús Pinazo,2,3 Julio Alonso-Padilla1,2
1Barcelona Institute for Global Health (ISGlobal), Hospital Clínic—University of Barcelona, Barcelona, Spain; 2CIBER de Enfermedades Infecciosas, Instituto de Salud Carlos III (CIBERINFEC, ISCIII), Madrid, Spain; 3Drugs for Neglected Diseases Initiative (DNDi), Geneva, Switzerland
Correspondence: Julio Alonso-Padilla, Barcelona Institute for Global Health (ISGlobal), Hospital Clínic—University of Barcelona. Carrer Rosselló 149, Barcelona, 08036, Spain, Tel +34 932275400 – ext. 4284, Email [email protected]
Abstract: Chagas disease is the most important protozoan infection in the Americas, and constitutes a significant public health concern throughout the world. Development of new medications against its etiologic agent, Trypanosoma cruzi, has been traditionally slow and difficult, lagging in comparison with diseases caused by other kinetoplastid parasites. Among the factors that explain this are the incompletely understood mechanisms of pathogenesis of T. cruzi infection and its complex set of interactions with the host in the chronic stage of the disease. These demand the performance of a variety of in vitro and in vivo assays as part of any drug development effort. In this review, we discuss recent breakthroughs in the understanding of the parasite’s life cycle and their implications in the search for new chemotherapeutics. For this, we present a framework to guide drug discovery efforts against Chagas disease, considering state-of-the-art preclinical models and recently developed tools for the identification and validation of molecular targets.
Keywords: Chagas disease, Trypanosoma cruzi, drug development, screenings, target, animal models
Introduction
Chagas disease, also known as American trypanosomiasis, is the most important neglected tropical disease (NTD) in the Americas. It is caused by the kinetoplastid parasite Trypanosoma cruzi and transmitted by reduviid bugs from the Triatominae subfamily. The disease affects between 6 and 7 million people, most of them in Latin America, leading to over 9,000 deaths per year and being responsible for the loss of over 280,000 disability-adjusted life years (DALYs).1 Apart from vector transmission, the infection can be acquired orally from parasite-contaminated food or drinks, by blood or organ transplantation, and vertically, from infected mothers to their offspring. The three latter are of relevance in endemic and non-endemic regions, like North America, Europe, Australia, and Japan.2,3
While the disease has an initial, mostly asymptomatic acute stage, the majority of people are diagnosed during the chronic stage, when symptomatology appears, generally decades following infection. Symptomatology is characterized by potentially life-threatening damage to the cardiac and/or digestive tissues and estimated to occur in up to 40% of those chronically infected.2 Common manifestations include changes in cardiac conductivity (typically, right bundle branch block or left anterior fascicular blocks; and less commonly sinus node dysfunction, or ventricular arrhythmias) and motility (left ventricular hypokinesis, reduction of the ejection fraction, and changes in diastolic function).4 Digestive symptomatology is typically characterized by enlargement of the colon and esophagus, associated with enteric neuron damage in these tissues.5 Mixed organ damage can also be observed in some patients developing cardiac and digestive manifestations. Central nervous system involvement is less common and may be secondary to immunosuppression,5 or stroke associated with cardiac complications such as heart damage, heart failure, or arrhythmias.6 Diagnosis during the acute stage can be achieved by parasitological or molecular methods, while it relies mostly on serology during chronic stages.2,5
There are two anti-parasitic drugs currently available: benznidazole and nifurtimox. Both are nitroheterocyclic pro-drugs that are metabolized by parasite nitroreductases (TcNTR), which turn them into intermediate products, with anti-parasitic activity producing reactive oxygen species (ROS).7 While highly effective when administered at the initial stages of the disease, like in the case of vertical infections, their efficacy reduces in the chronic stage. Moreover, both have frequent adverse effects linked to their long administration regimes, which hinder adherence to treatment in around 15–20% of treated patients.8,9
In recent years, there have been remarkable advances in drug research for Chagas disease, but progress on the development of new therapies against it is proving slow and difficult. Usually, the process to develop a new drug can take 10–15 years and have an average cost of $1–2 billion for each newly approved drug.10–12 As for other NTDs, there are few economic incentives for the research of new drugs for Chagas disease,13 which has caused modest coordination of research and development efforts, leading to significant knowledge gaps on key aspects of the parasite biology, and ultimately hindering the identification of therapeutic breakthroughs. While the situation is improving, partly due to an increased engagement of pharmaceutical companies and the creation of public–private partnerships such as the Drugs for Neglected Diseases Initiative (DNDi), the drug discovery landscape of Chagas disease remains scarcely populated.14
This is further complicated by T. cruzi’s antigenic and genetic diversity. The parasite has a diploid genome of between 106 and 110 Mb, organized in a variable number of 19 to 40 chromosomes.15 It is estimated to carry up to 22,000 coding sequences, including large families or surface proteins like trans-sialidases, mucins, and mucin-associated surface proteins (MASPs). These play a major role in parasite invasion and antigenicity, and are often subject to recombination events.16,17 Furthermore, T. cruzi is genetically classified into seven major lineages known as discrete typing units (DTUs: TcI – TcVI and TcBat), characterized by different geographic distributions, and populated by isolates with diverse virulence and resistance to drugs.18,19 In addition, the poorly understood mechanisms of pathogenesis at the molecular level and a heterogeneous set of in vitro and in vivo infection models do not contribute to accelerate progress. In this review, we present a comprehensive framework to guide drug development efforts, with a particular emphasis on phenotypic assays, target identification and validation, and available animal models.
Biological Considerations for the Evaluation of Drugs with Activity Against T. cruzi
The life cycle of T. cruzi involves arthropod vectors and mammalian hosts. Metacyclic trypomastigotes (metacyclics) develop from replicative epimastigotes during their transit through the vector posterior digestive tract, and are ultimately expelled in their feces upon feeding from a warm-blooded animal. Metacyclics do not replicate, and infect a host by either accessing its bloodstream through superficial wounds or mucosal membranes, or via the consumption of contaminated food or drinks. They quickly invade the cytoplasm of a host cell and differentiate into amastigotes, the intracellular replicative life stage of the parasite in mammals.2 Upon successive binary replication cycles, amastigotes transform into motile, non-replicative trypomastigotes, which egress from the host cell to the interstice and bloodstream. They can invade cardiac cells and the nervous ganglia below the digestive mucosa, where chronic infection can persist for decades and eventually lead to severe tissue disruptions.19 Despite its predilection for these sites, T. cruzi can invade a wide variety of host cells and tissues, making it necessary for any potential treatment to have a high bioavailability across different anatomic settings.14 Blood circulating trypomastigotes can be taken up by the vectors, becoming epimastigotes in the initial segments of the digestive tract, completing the infection cycle.2 Remarkably, each life stage of the parasite is phenotypically singular, presenting different susceptibility to drugs, and highlighting the major relevance of using mammalian infective forms in drug discovery biological assays.
From a metabolic point of view, the behavior of the parasite during the chronic stage of the infection is also diverse, which has direct implications for its susceptibility to drugs. For instance, it has been described that a small subset of metabolically quiescent amastigotes develops spontaneously in both in vitro and in vivo models. These “persister” or “quiescent” parasites replicate very slowly and are less susceptible to nitroheterocyclic drugs.20,21 Persistent forms can become metabolically active and replicate again in later stages of the infection; a phenomenon that could potentially explain the failure of existing treatments to permanently clear parasitemia in some patients, and thus the need for long-lasting or intermittent treatment regimens.22
Drug Screening Strategies Used to Identify Potentially Useful Compounds
The development of new compounds for the treatment of Chagas disease is guided by its target product profile (TPP). Different TPPs have been reviewed elsewhere,23 but they share key characteristics. An acceptable therapy should be able to (i) benefit patients in the chronic state of infection; (ii) target the most widely distributed parasite strains; (iii) allow shorter treatment schemes; and (iv) be safer and at least as effective as currently available drugs. Additionally, it should ideally be soluble enough to allow oral dosing, lack clinically significant interactions with other medications, and be safe during pregnancy.23,24
The first step in the development pathway is the identification of hit compounds, whose suitability is furtherly evaluated considering aspects outlined in the TPP. Hit compounds can be identified through either phenotypic or target-based approaches, which ultimately reach a common validation pathway before entering in vivo pre-clinical evaluation (Figure 1).
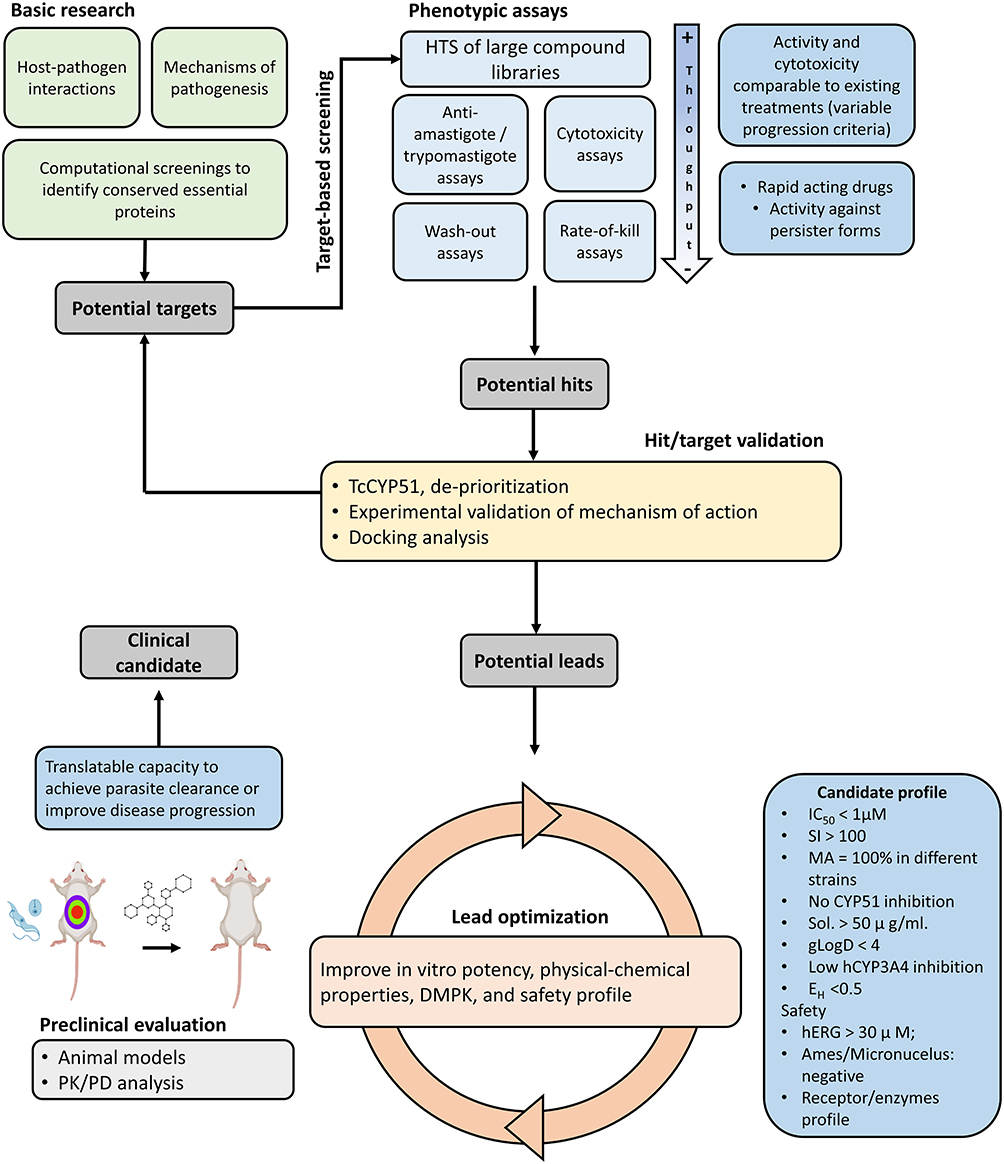 |
Figure 1 General framework for the discovery and validation of novel compounds for the treatment of Chagas disease. Abbreviations: CYP51, 14-α demethylase; DMPK, Drug Metabolism and Pharmacokinetics; gLogD, Distribution coefficient; hCYP3A4, Human cytochrome P450 3A4; hERG, Human ether a-go-go gene; HTS, High throughput screening; IC50, Half maximal inhibitory concentration; PK/PD, Pharmacokinetics and pharmacodynamics; SI, Selectivity Index; MA, Maximal inhibitory activity, Sol, Solubility at physiological pH. |
In target-based screening, molecular biology techniques are used to identify virulence factors likely to be attractive drug targets. For Chagas disease, this approach has been traditionally limited by knowledge gaps that have only recently begun to be addressed, while it has proven useful to identify and de-prioritize targets likely to fail in clinical trials.25 Despite this, a handful of potentially interesting targets have been identified. These have been reviewed elsewhere,26 and include parasite enzymes such as trypanothione-reductase,27,28 enolase,29 cruzain,30 ribose-5-phosphate isomerase (RPI),31 isocitrate dehydrogenase (IDH2),32 dihydrofolate reductase–thymidylate synthase (DHFR-TS),33 pteridine reductase,34 farnesyl diphosphate synthase (FPPS),35 and the parasite sirtuin family.36 More recently, compounds that interfere with the parasite proteasome and mRNA processing machinery have been thoroughly evaluated and gained considerable traction as attractive drug candidates.14 The strategies used to identify and validate these targets are further discussed in the target validation section of this review.
In comparison, the phenotypic screening is a serendipitous approach that uses cell-based assays to quantify the effect of a compound on a desired cell line. Phenotypic screenings are followed by the identification and thorough validation of the targets of hit compounds. Notably, the relative scarcity of well-characterized targets for Chagas disease has made of phenotypic screenings the method of choice for identifying new potential therapies.
The Phenotypic Screening Pathway
A phenotypic drug development cascade may start with a high-throughput screening (HTS) campaign using a single concentration of the compounds forming part of a large chemical library whose anti-T. cruzi activity is evaluated on a primary anti-parasitic assay.37–40 The initial hits resulting from this screening are studied again with the same assay, using a dose–response design to calculate the half maximal inhibitory concentrations (IC50), which then allow comparisons between hits and included reference drugs, ie benznidazole or nifurtimox. Noteworthy, this approach relies on the availability of assays with a certain throughput, so that a high number of compounds can be analyzed simultaneously.
To do so, these assays often rely on genetically engineered parasites, modified to express a reporter's activity that facilitates their identification and quantification using spectrophotometric readouts.37,41 Due to its genetic stability and trustworthy performance, together with its simple culture conditions and high production rates, several drug discovery anti-T. cruzi assays have been based on a Tulahuen parasite strain (TcVI) expressing the E. coli β-galactosidase gene.42 An anti-parasitic assay with these parasites and 3T3 mouse embryo fibroblasts was the basis for the development of subsequent HTS-suited assays using different kinds of mammalian host cells.37,43–46 Other techniques allowing a medium to high-throughput detection and quantification of parasite growth rely on the detection of ATP production,47 redox capacity,48 or the use of fluorescent parasites.49
More recently, microscopy-based high-content screening (HCS) approaches have surged as an alternative to obtain quantitative data on the proportion of parasitized cells, or the number of parasites per infected cell. They rely on fluorescent microscopy and image analysis algorithms to identify parasites inside individual cells, allowing the simultaneous processing of several conditions in the host and T. cruzi cells, and a medium throughput of compounds.38,39,50 Remarkably, beyond quantifying parasite growth, these assays can also provide data on the nature and timing of the observed anti-parasitic effects and on the effect of compounds on certain phenotypic characteristics of parasites and cells, including shape, size, presence of vacuoles, or organization of the kinetoplastid DNA (kDNA) and cytoskeleton.51 Additionally, HCS is less likely to be negatively affected by interactions between compounds and reporter enzymes and substrates, or by the effect that colored compounds might have on spectrophotometric readings.51
Regardless of the method used to quantify anti-parasitic effects, it is essential to guarantee that these are specific and not derived from induced host cell damage or death. This can be achieved by also evaluating the cytotoxicity of studied compounds on the same host cell line used in antiparasitic assays and calculating their selectivity index (SI), which is defined as the ratio between its half maximal toxic concentration to the host cells (TC50) and its IC50.45 Importantly, the type of host cell used in anti-parasitic assays must also be taken into consideration when comparing results from different experiments. Parasite infection rates and replication times change from one host cell line to another, modifying the susceptibility observed against certain compounds.52
Specific Complementary Assays
While the evaluation of anti-parasitic effects is easier in epimastigotes and can guide initial drug discovery efforts, the activity of compounds on amastigotes and trypomastigotes should be prioritized, given their central role in the pathogenesis of the disease.
Anti-amastigote assays reflect the potency of compounds on the parasite form typically associated with chronic inflammation and damage and are thus the cornerstone of Chagas disease drug discovery pathways.37,47,49,53 Standardized anti-trypomastigote assays also exist.47,54,55 They are important too, given the resistance of this life-stage to certain compounds and the apparent link to their poor-performance in later stages of in vivo evaluation.47
The capacity of compounds to kill persister, non-replicating forms should also be studied, given their intrinsic resistance to currently available drugs.20,22 This can be evaluated, for instance, using wash-out assays, in which infected cultures are incubated for variable periods of time with a compound, which is washed-out after parasites are no longer detectable Cultures are then maintained for several weeks and monitored daily to record the re-appearance, or not, of trypomastigotes, which would have differentiated from surviving quiescent forms.14,47 Ideally, host cells should be gamma-irradiated prior to these assays to prevent their overgrowth, which can hamper the appropriate quantification of parasite growth.20,56
HCS assays designed to determine whether the parasites simply prevent parasite replication (static effect) or induce a reduction in the number of amastigotes (cidal effect), as well as the time required to produce such effect (rate-of-kill), have also been described and represent increasingly important components of drug discovery cascades.56–58
In addition to addressing the compounds toxicity on the host cells supporting parasite replication, other cell toxicity assays are usually included in the drug discovery cascade. A common model uses the human hepatocarcinoma cell line HepG2, since damage to this line suggests liver toxicity.53,59,60
Importantly, given the wide geographic-phenotypic diversity of T. cruzi, and the fact that any developed drug would be expected to work throughout all endemic regions, complementary in vitro assays should also include evaluating the compounds’ activity against different strains and DTUs.61,62 This seems to be particularly important if potentially slow-acting compounds are being evaluated, as inter-strain differences in susceptibility appear to be more evident following 72 hr of compound exposure.63
Similarly, the de-prioritization of drugs that act on the parasite 14-α-demethylase (TcCYP51), an enzyme involved in the synthesis of sterols for the parasite membrane,56 is also becoming increasingly common. The latter is justified on the failure of azoles posaconazole, ravuconazole, and fosravuconazole in clinical trials.54,64–66 All of these compounds target TcCYP51 and their disappointing clinical results make the case for the importance of understanding parasite biology in detail. Although TcCYP51 inhibitors are commonly identified as promising candidates in screenings, it is now known that endogenous sterol depletion is necessary before cell death occurs, making the activity of these compounds slow and dependent on parasite replication, explaining their lack of anti-parasitic activity on non-replicative trypomastigotes and slow-dividing parasite strains.47,56 These findings have prompted the research community to discard any compounds that act by inhibiting that pathway, regardless of their performance in vitro,56,67 prioritizing fast-acting compounds instead.
Recently, the incorporation of 3D culture systems in drug discovery pathways has gained traction, as these mimic gene transcription and expression more closely in in vivo environments, increasing their translatability. Although 3D cultures have been used to study T. cruzi host–parasite interactions and mechanisms of pathogenesis,68,69 its use in drug discovery remains scarce and mostly restricted to the use of Vero cells and murine primary cardiomyocytes.70,71
It is worth to highlight that as the complexity of assays increases, their throughput decreases, so fewer compounds can move forward in the cascade. While there are no universally accepted pathway progression criteria, an IC50 < 1µM, or at least within <10X that observed for benznidazole, a SI > 10 (ideally >100), and a TC50 > 25 μM in HepG2 cells have been used in the past.23,37,40,53,61,72,73 A summary of other commonly used criteria is presented in Table 1.
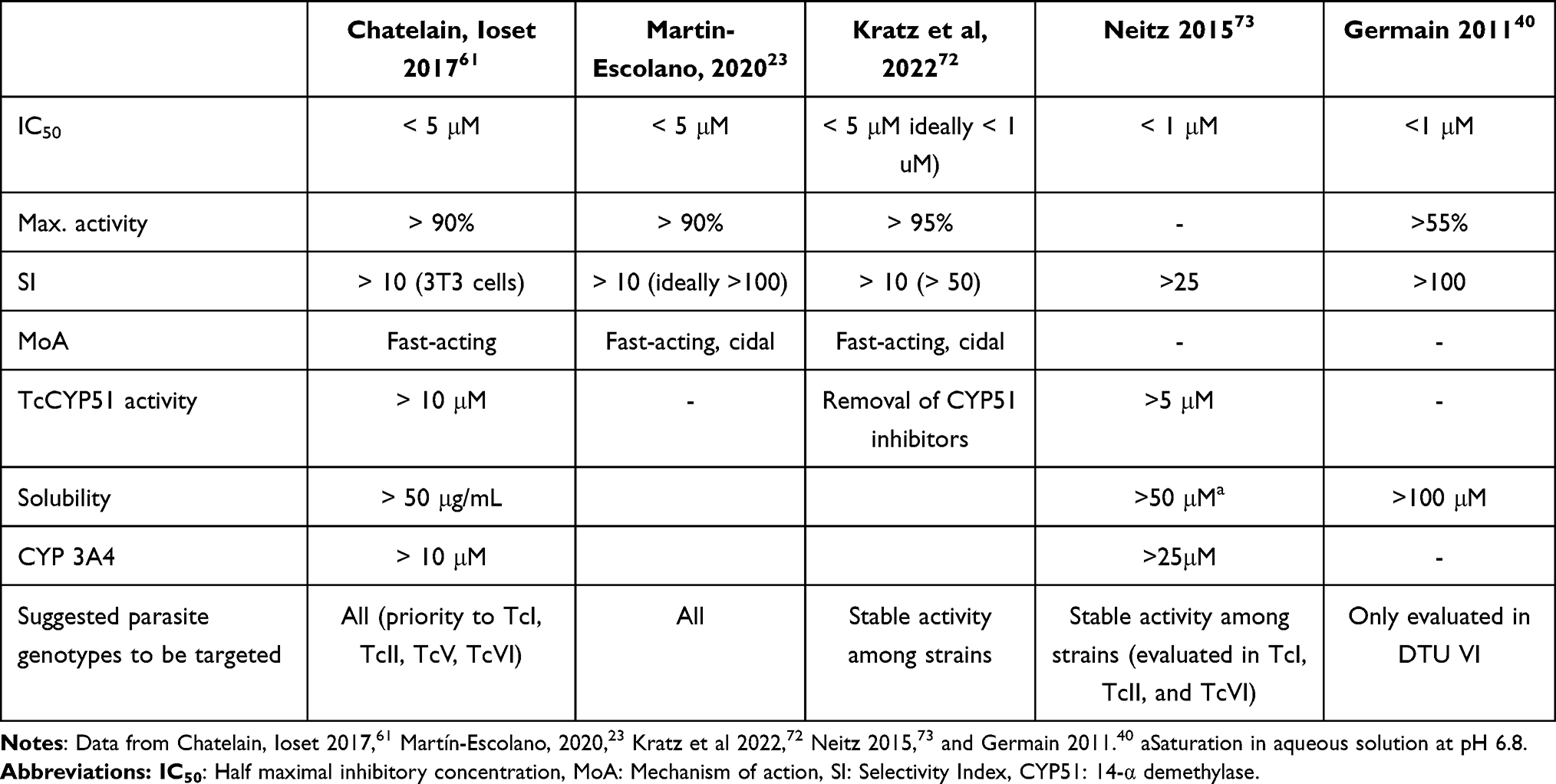 |
Table 1 Commonly Used Criteria to Determine the Progression of a Compound in Phenotypic Development Pathways |
Computer-Assisted Selection of Proteins for Target-Based Screening
An ideal drug target should be essential in pathologically relevant parasite life stages; sufficiently different from related mammalian proteins to be selective; and druggable, ie presenting well-defined domains expected to bind certain types of compounds.74 Large-scale evaluation of these characteristics can greatly reduce the number of potential candidates,75,76 prioritizing proteins that are conserved across different DTUs, but not in mammals,77 and stably expressed in amastigote and trypomastigote stages. Drug targets that are evolutionarily conserved among different parasite strains but differ from the human proteome are expected to have wider clinical efficacy, while minimizing off-target effects. Indeed, some of the most-studied targets in the development of treatments for Chagas disease, such as TcNTR, TcCYP51, or trypanothione-reductase, all lack orthologues in humans.78,79
Other metabolic pathways that differ from the host, such as the purine salvage and folate synthesis pathways, also represent potentially attractive targets.78–80 However, a target should not be discarded solely based on the presence of human orthologues, because highly selective inhibitors of the parasite protein could still be synthesized. Such is the case with compounds inhibiting the cleavage and polyadenylation specificity factor subunit 3 (CPSF3), a protein present in humans but sufficiently different in kinetoplastids so as to be considered a promising target for the development of new treatments against these pathogens.14,81,82 The presence of orthologues in other trypanosomatids can also be incorporated into search strategies. For example, using phenotypic studies against Trypanosoma brucei or Leishmania spp., to identify potential targets in T. cruzi.
While computational target selection still has significant caveats, identifying proteins that comply with the aforementioned criteria is expected to greatly reduce development times and facilitate the design of target-based screenings.83 Furthermore, most of these analyses can be performed quickly using freely available bioinformatic tools, such as those hosted at TriTrypDB (https://tritrypdb.org/tritrypdb/app), part of the VeuPathDB suite, which includes extensive genomic, proteomic, and metabolomic data, as well as information on specific gene expression levels and their involvement in different signaling pathways.84,85 Apart from being essential in refining the number of targets of potential interest, bioinformatic methods are also necessary to study the targets’ interactions with ligands of therapeutic interest. This is a key first step in understanding a drug’s mechanism of action, and highly advisable before progressing any compound into in vivo pre-clinical evaluation.
Evaluation of the Tridimensional Structure of Targets for in silico Experiments
The morphologic study of potential targets in T. cruzi, and therefore, the full pharmacodynamic characterization of compounds likely to bind them has been hindered by the lack of detailed protein models in this species. The first genome sequence of T. cruzi, produced almost 20 years ago, was obtained from the CL Brener strain, a hybrid, polymorphic strain belonging to TcVI that remains the reference for the entire species.86 At present, there are at least 30 genomes from different T. cruzi strains available online, although most remain incomplete and poorly annotated. Only recently, another, less polymorphic TcI strain (Sylvio X10) has been sequenced and described in detail.17,87
The lack of properly annotated sequences, along with the high cost and complexity of crystallographic methods, still limits the number of protein structures available for in silico evaluation. When available, these models are mostly based on CL Brener genes, reducing their generalizability to other DTUs. A partial solution to this issue is the use of computer-generated protein structures. In this regard, AlphaFold, an artificial intelligence (AI) system developed by DeepMind (https://www.deepmind.com/research/highlighted-research/alphafold) represents a valuable tool.88–90 Using deep learning algorithms, the system is capable of predicting accurate protein structures based entirely on their amino acid sequences.91 Moreover, over 200 million predicted structures are already available in the AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/), a virtual repository created in a joint partnership between DeepMind and the European Molecular Biology Laboratory Bioinformatics Institute (EMBL-EBI).89 AlphaFold models of targets of interest can be easily incorporated into drug discovery pipelines by using them, for instance, in in silico docking experiments, where the binding affinity between small molecules and potential targets is predicted, or in inverse virtual screening experiments to determine the mode of action of prospective drugs.75
Nevertheless, before using these models, it is important to evaluate their quality. One option is evaluating the model’s per-residue predicted local distance difference test (pLDDT) score, which scales from 0 to 100. Regions with a pLDDT > 90 are comparable to experimentally generated crystal structures, with amino acid side chains usually set in the correct orientation. This is crucial when modeling active sites in which the orientation of residues is of paramount importance. In addition, the relative positions of a protein’s domains are measured by their predicted aligned error (PAE). A high PAE between two residues indicates that their relative positions are not certain, and their inclusion in docking experiments should be avoided. In particular, considering that a single side chain oriented in the wrong direction can disrupt the results of docking simulations.89 This is a major limitation for the evaluation of targets based on their computationally predicted structures.92
Since AlphaFold relies on multiple sequence alignments, the accuracy of the predictions is also hampered by alignments with low depth. In the context of NTDs, and in particular of Chagas disease, the limited extent of sequence annotation across different T. cruzi strains, and the fact that trypanosomatids are evolutionarily distant from other better-studied organisms entails that many T. cruzi entries in the AlphaFold DB show a comparatively low prediction score.93 In addition, the current release of AlphaFold DB does not contain multimeric structures, greatly hindering the incorporation of some targets into computer-assisted drug discovery.
Even though new iterations of AlphaFold are expected to address some of these issues,89,94,95 the experimental validation of any computationally selected target is still essential before its progression down the development pathway.
Novel Strategies for Experimental Target Validation
Independently of the type of screening strategy used, the appropriate validation of drug targets requires experimental evidence of their predicted role in the parasite. Until recently, the lack of a simple tool for genetic edition in T. cruzi severely limited the capacity to do this. Unlike other trypanosomatid parasites, T. cruzi and some Leishmania species lack the molecular apparatus needed to carry out RNA interference (RNAi) gene knock-out (KO) experiments, widely used to validate the role of potential targets in T. brucei.96 The recent development of different CRISPR-cas9-based editing systems in T. cruzi now offers a quick, relatively simple, and highly versatile way to experimentally validate the role of certain proteins as drug targets in the parasite.97–100 This approach has been used to validate AGC Kinase 1 (AEK1), previously identified as essential in other kinetoplastid parasites using RNAi.101,102 Using a CRISPR-cas9 system, it was demonstrated that the single KO of one of the two copies of the AEK1 gene present in T. cruzi Y strain (TcII) caused an abnormal development of the kinetoplast and the flagella of transfected parasites and that its blockage impaired cytokinesis and cell cycle progression, confirming the role of AEK1 as an attractive drug target.103
A similar approach was used to confirm the role of CPSF3, a protein involved in messenger RNA processing, as the target of AN15368, a new benzoxaborole analog with anti-parasitic activity and apparent low toxicity, and to identify its nature as a prodrug metabolized by a parasite serine carbopeptidase.82 The same method was used to identify the β4/β5 interface in the parasite’s proteasome as the target of a new arylsulfonamide previously shown to present in vitro anti-parasitic activity,37,104 confirming results from previous studies that suggested that inhibition of proteasome activity is an exploitable pathway in the treatment of kinetoplastid infections.105,106 At least five compounds targeting CPSF3 or the kinetoplastid proteasome are currently in development pipelines for the identification of new treatments against kinetoplastid parasites, and three of these are potentially useful in the treatment of Chagas disease (see corresponding section below).14,107
Apart from revealing potentially useful pharmacodynamic (PD) information and possible resistance mechanisms, validating the targets of new compounds is also essential to identify those unlikely to perform well in clinical trials, and discard them early in the development process, as is now regularly done with TcCPY51 inhibitors.14
Lead Generation and Optimization
Hit compounds that show potent specific anti-parasitic properties and certain desired metabolic characteristics are considered leads.24 Lead compounds must have a good PD profile, characterized by a high in vitro activity (killing 100% of parasites), high potency (ideally in the nanomolar range), a high selectivity (SI > 100), a fast mechanism of action, and activity against different T. cruzi strains. Additionally, leads must achieve sterile cure in animal models of chronic infection, while presenting an adequate pharmacokinetic (PK) and safety profile.24
Notably, some of the most pharmacologically relevant physical-chemical characteristics of compounds can be preliminarily evaluated using chemoinformatic servers and in vitro metabolism models, thus providing a rapid assessment of a drug’s absorption, distribution, metabolism, and excretion (ADME) profile.108 Lead candidates should present a high kinetic solubility at physiologically relevant pH (>50 µg/mL), not be unduly hydrophobic at neutral pH (glogD7.4 < 4), show a low inhibition of human cytochrome P450 (CYP3A4) (IC50 > 10 µM), and a low hepatic extraction ratio (EH < 0.5).24
Furthermore, the potential for off-target effects should be addressed through standard toxicology screening assays to study interactions with key enzymes and receptors, such as the human ether a go-go related gene ion channel (hERG or KCNH2), which predicts cardiac toxicity. Similarly, candidate drugs should not be mutagenic (as determined by the Ames or micronucleus assays) and should not cause any significant adverse effects on preliminary animal experiments.14,24 Analogs of lead compounds can then be synthesized, including minor modifications expected to improve their efficacy, or minimize the probability of off-target effects, a process known as hit-to-lead optimization, generally the last step prior to a proof-of-concept evaluation in an animal model of the disease.14,24
Animal Models for in vivo Evaluation
The complex biology of T. cruzi has also hindered the evaluation of new drugs in animal models. The fact that these models had been mostly used to determine the pathogenesis of the different manifestations of chronic disease led to significant diversity, as the parasite strains, animal species, and infection routes used in each one depended on the specific aspects of the disease to be studied.109–111 Such experimental and physiological diversity makes the selection of cure criteria, and the interpretation of results, challenging. Furthermore, a better understanding of the dynamics of Chagas disease has forced the research community to use new endpoints accounting for tissue-specific parasite loads to define therapeutic cure, adding extra complexity to the development and selection of adequate models.112 A variety of animal models of T. cruzi infection have been used to evaluate the performance of promising compounds.109,112 Among those, murine, canine, and primate models are furtherly addressed here, given their importance.
Murine Models
As it happens for other diseases, murine models are the most extensively used in the study of Chagas disease. Until recently, the evaluation of drugs with trypanocide activity had primarily relied on acute models of infection, given their short duration and the high parasitemia observed in this stage, which provides a greater window to detect any trypanocide effect.113 In these models, mice are treated with the test drug during the peak of parasitemia, which usually lasts for 2–4 weeks, depending on the T. cruzi strain used. Traditionally, death (or mortality prevention following drug administration) was the main endpoint assessed; although this has changed recently, due to ethical reasons and the development of new experimental methods, as described in this section.
Chronic models, on the other hand, mimic the indeterminate stage of the disease, when parasitemia is low. In this case, mice are treated and monitored over a variable period, typically from 1 week to 2 months. The main efficacy outcome in these studies is the capacity of the compound to achieve sterile cure (parasite clearance in all tissues). Animals are then immunosuppressed to induce the relapse of any remaining parasites and observe a possible reactivation of the infection.114 Distinct combinations of mouse and parasite strains can be used to reflect different aspects of the pathogenesis of human infection. BALB/c, CH3, or C57BL6 mice infected with different parasite strains, including Sylvio X10/4 (TcI), CL Brener (TcVI), Tulahuen (TcVI), Y (TcII), and Brazil (TcI), have been shown to develop cardiac disease with the presence of lymphoid infiltration, fibrosis, and electrocardiographic alterations, similar to those observed in humans.115–118 These findings were reported following both intraperitoneal infection using blood-derived trypomastigotes and oral ingestion of metacyclic trypomastigotes.108
The incorporation of parasites genetically modified to express bioluminescent and fluorescent markers has allowed a detailed monitoring of the dynamics of infection, identifying the persistence of T. cruzi in immune-privileged tissues or following-up the elimination of parasites in other anatomical settings.97,119 These bioluminescence imaging (BLI) models are based on the incorporation of a reporter luciferase gene in the parasites, thereby detecting the light emitted after being exposed to luciferin, the enzyme’s substrate.97,119 BLI models also allow longitudinal, non-invasive evaluation of the drugs´ efficacy to reduce or clear the parasite burden. This, in turn, involves the use of fewer animals, complying with current ethical recommendations for animal testing.120 Bioluminescence and fluorescence can also be observed ex vivo, following necropsy, facilitating the identification of residual parasite nests.
BLI models are currently considered the gold standard for the in vivo evaluation of drugs with trypanocide action, in addition, they can provide valuable insight on the mechanism of action of compounds.121 For instance, BLI in models of chronic infection demonstrated that posaconazole, a TcCYP51 inhibitor, fails to clear parasites from certain infected tissues, leading to increases in parasitemia after immune-suppression, matching results from in vitro studies that explained its lack of clinical benefits.114 These results contrast those obtained with earlier infection models, in which posaconazole was interpreted to be an effective therapy.122 Similar studies have identified serologic profiles associated with parasite clearance, potentially useful for the longitudinal monitoring of treated patients.123
Additionally, murine models of Chagas disease have been developed to study specific characteristics of human infection: Swiss Webster and C57/BL6 mice, respectively, infected with T. cruzi Y and Brazil strains developed significant increases in intestinal transit times and intestinal dilation, which are common manifestations of chronic human digestive Chagas disease.124,125 Although the appearance of those symptoms during the acute infection stage limited their applicability. More recently, the use of BLI/fluorescent models with CH3 mice and parasites from the JR strain (TcI), mimicked human digestive disease more closely, with the delayed transit phenotype persisting and worsening in chronic stages of infection, despite significant reductions in systemic parasitemia.126
Other models for the study of congenital transmission and immune control of infection also exist.127 The latter are particularly relevant as differences in the immune response to infection with T. cruzi of humans and mice can limit the generalizability of studies evaluating the performance of drugs in these animals.14,112 Recently, a genetically modified murine model of Chagas disease that, like humans, lacks the α-1,3-galactosyltransferase (α-GalT) gene has been developed.128 Considering that lytic anti-α-Gal antibodies are an important component of IgG responses in patients with Chagas disease,129,130 such model would resemble the human humoral response against the parasite more closely than other mice that express the enzyme and are therefore tolerant to α-Gal epitopes.14 The use of humanized mice expressing human tissues also translates into more clinically translatable results, but remain prohibitively costly.112
Mice are also used in tolerability studies that precede the evaluation of in vivo efficacy and have the objective of studying the safety and potential adverse effects of experimental drugs, as well as determining their maximum tolerated dose (MTD). This is an important parameter that helps in defining the appropriate dose for subsequent efficacy and safety studies in larger models and in humans.
Canine Models
Dogs are natural hosts of T. cruzi and have been traditionally considered adequate models of Chagas disease, with an electrocardiographic and myocardial pathology that reflects human disease.109,131–133 In particular, dogs can develop a form of the disease that resembles the chronic phase of Chagas disease more closely, with changes in response outcomes following treatment with benznidazole in these animals also matching those observed in human clinical trials.109,133,134 Nonetheless, differences in the half-life of certain drugs in dogs and humans can limit the translational potential of results obtained in this species.112 Furthermore, ethical issues and the slow progression of infection in these animals entail high costs and significant logistical challenges that limit their applicability.
Non-Human Primates
Non-human primates (NHPs) are the most physiologically similar model to human infection and were traditionally considered an important part of the drug development pathway. However, the cost and logistical difficulties of managing these animals have made their use relatively rare.135,136
Most of the studies assessing compounds in NHP models of Chagas disease have involved colonies of naturally infected animals.137,138 A particularly good example of this approach is the recent use of rhesus monkeys (Macaca mulatta) naturally exposed to T. cruzi in the southern United States to evaluate the performance of benzoxaborole AN15368.82
In general, the performance of drugs in NHP models provides valuable clues about their expected efficacy in humans. For instance, the use of posaconazole in primates at plasma levels equivalent to those observed in humans failed to clear parasitaemia, in agreement with results of clinical trials evaluating the drug.139 Nonetheless, infection of NHP with different strains of T. cruzi can lead to varying degrees of disease that do not always correlate with findings observed in human infection.140 The ethical and economic considerations of handling primates in captivity, together with the availability of novel, translatable, and robust murine models, makes the latter a more suitable alternative.112
Discussions on the relevance and translational value of one species over another also tend to miss the fact that current outcomes used in these models are not directly translatable to clinical trials. For instance, the determination of parasitological sterilizing cure, understood as the elimination of parasites from both the blood and tissues, while feasible in animal models, is not possible in human patients. Further improvement on the development of animal models for the disease should, therefore, include the identification of new biomarkers or outcomes that are translatable to a clinical context.
Targeting the Pathogenesis of Chronic Damage as a Complementary Strategy for Drug Development
Work based on murine models suggests that the scarce yet constant presence of parasites in immune-privileged tissues, like in the digestive tract of infected individuals, is necessary for the development of the chronic manifestations of the disease, as these sites are likely to allow the episodic seeding of parasites to other, less permissive organs, like the heart.19 Therefore, a treatment with sterilizing capacity in these anatomical settings remains the main aim of any drug discovery strategy. However, the use of compounds able to target some of the key pathways involved in the development of chronic disease, without necessarily killing the parasites might as well be a promising host-focused complementary strategy. Similar approaches are already used for the treatment of other protozoan infections, such as malaria or leishmaniasis.141,142
Symptomatology of chronic infection with T. cruzi is mostly associated with the development of chronic chagasic cardiomyopathy (CCC), and/or diverse gastrointestinal problems characterized by the dilation of segments of the digestive tract. The clinical and pathological characteristics of CCC are well defined, but the mechanisms through which the condition evolves remain poorly understood, limiting the development of new drugs that prevent it.143
It is accepted that cardiac damage in T. cruzi infection is multifactorial, involving a direct lytic effect on parasitized cells, autonomic dysfunction, and probably the occurrence of immune dysregulation, all ultimately triggered by the presence of parasites in the cardiac parenchyma.143,144 Myocardial fibrosis is also a staple pathological finding,143,145 and might in turn be explained by changes in the coronary microcirculation and the appearance of ischemic areas.144 Thrombotic events, caused both by a direct cytolytic effect of parasitism of endothelial cells, and by the synthesis and secretion of pro-inflammatory and pro-aggregative products by parasites and infected cells, most likely play a role in this.
A lipid inflammatory mediator produced in T. cruzi infection, lysophosphatidylcholine (LPC) C18:1 has been associated with that process.146 LPCs are known to have chemotactic effects on phagocytic cells, while favoring parasite survival upon invasion, making them important virulence factors for several protozoan parasites.147 In addition to this, C18:1-LPC is also a structural analog of the mammal platelet activating factor (PAF), sharing its pro-aggregating effect on platelets.146,148 Inhibiting the synthesis of this and other parasite mediators, or their interaction with their corresponding receptors, could therefore limit the development of organ damage associated with thrombotic effects in the cardiac and systemic microvasculatures. A structural analysis of this molecule suggests that it is synthesized by an enzyme with phospholipase A2 activity (PLA2), although it remains unclear whether this derives from the parasite, its host cell, or both.146 While PLA2-putative genes exist in the genome of T. cruzi,149 their expression and activity on mammalian life stages remains to be confirmed, making further research on this metabolic pathway necessary to determine its role as a potential drug target.
Another route of potential therapeutic interest is that of tumoral growth factor beta (TGF-β), an immune-regulatory cytokine involved in parasite development and progression to chronicity.150,151 TGF-β plays a key role in proliferation and differentiation of tissues, as well as in the remodeling of their extracellular matrix.152 It has also been described to be involved in preventing the development of uncontrolled, potentially life-threatening, inflammatory responses during the acute phases of infection.19 TGF-β polymorphisms have been associated with increased risk of developing CCC and its expression is greatly increased in the early and chronic stages of T. cruzi infection.151,152 Similarly, TGF-β-producing cells are also abundant in the heart of animals with chronic infection, which is also more responsive to the cytokine, showing over-expression of its receptors and mediators, like the connective tissue growth factor (CTGF) and fibronectin, directly associated with collagen deposition and the development of fibrosis.153
Furthermore, treatment with inhibitors of the TGF-β receptor kinase activity reversed collagen deposition and fibrosis in mice infected with the highly virulent Colombiana strain (TcI) of T. cruzi, without reducing the heart parasite load. Such treatment also improved electro and echocardiographic parameters and atrioventricular conduction, suggesting that blocking the TGF-β pathway might lead to improved cardiovascular function in chronic T. cruzi infection, regardless of whether parasite clearance is achieved or not.154
Preventing the development of arrhythmias in infected patients might also be an approach to reduce the burden of the disease. In this context, amiodarone, a widely used class III anti-arrhythmic agent, has also been proposed as a potential complementary treatment for Chagas disease. Apart from its anti-arrhythmic effect, amiodarone also induces the release of Ca+2 from the parasite mitochondrion and acidocalcisomes and kinetoplast, which has been suggested to give it additional anti-parasitic effects.155 Its combination with benznidazole in mouse models of infection also improved electrocardiographic markers of cardiac damage and reduced levels of pro-inflammatory markers and collagen deposition.156 Its use in naturally infected dogs led to improved survival.157 However, a recent meta-analysis of patients with chronic Chagas disease who received amiodarone confirmed its antiarrhythmic benefits, but did not find any evidence of reductions in mortality or hospitalizations.158
In terms of digestive symptomatology, extracts from Lycopodium clavatum have been described to reduce the progression of gastrointestinal disease in rodent models of Chagas disease, probably through modulation of IL-10 production and an enhanced CD4/CD8 T cell response.159,160 Other host-directed therapies can also mediate anti-microbial effects, as it has been demonstrated with the anti-leishmanial effect of ibrutinib, a monoclonal antibody against Bruton’s tyrosine-kinase (BTK), a key modulator of B-cell maturation.142
In any case, it is important to bear in mind that the early clearance of parasites in infected patients remains the only intervention proven to prevent the development of chronic symptomatology, and targeting its pathogenesis without simultaneously killing the parasites is risky and might have potentially dangerous consequences, particularly if it involves the use of immune-modulatory therapies.
Compounds Under Clinical and Preclinical Evaluation
Despite significant progress in the development of new techniques to understand the complex biology of T. cruzi, the number of compounds reaching clinical stage remains low (Table 2).
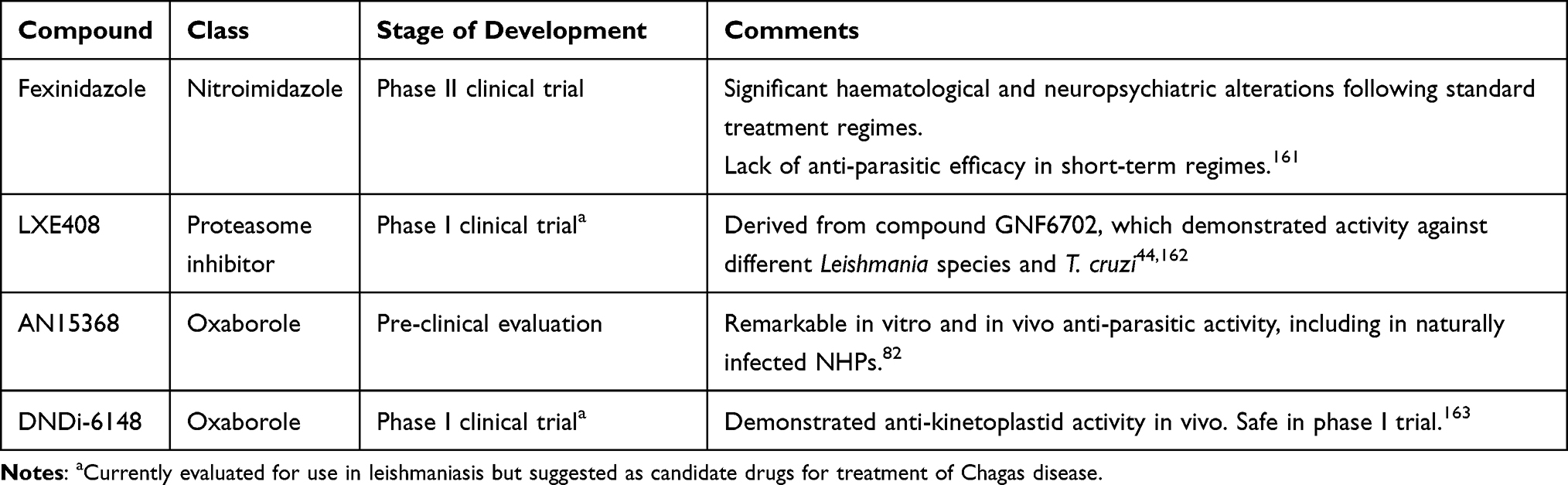 |
Table 2 Compounds Under Evaluation for Their Use in Chagas Disease |
Fexinidazole
Fexinidazole is a nitroimidazole recently approved for its usage in the treatment of human African trypanosomiasis,164 with proven in vivo activity against T. cruzi.165 Fexinidazole is activated by a parasite nitroreductase to a reactive intermediate, which then likely acts against multiple targets within the parasite.166 In a phase II study in Bolivia, the drug revealed a quick and sustained anti-parasitic effect in patients with chronic infection, but the trial was stopped following the development of neutropenia in 20% of the treated patients (clinicaltrials.gov id: NCT02498782). Other alterations, including neuropsychiatric disorders and hepatic toxicity were also observed, and linked to a higher exposure to the drug.161 More recently, data from a multi-center study carried out in Spain (NCT03587766) did not show efficacy with shorter, low-dose regimes of fexinidazole after 12 months of follow-up.167
Other drugs undergoing preclinical evaluation and likely to progress down the clinical pathway include the following:
Proteasome Inhibitors
A kinetoplastid proteasome inhibitor, GNF6702 has demonstrated remarkable in vitro and in vivo activity against T. cruzi, T. brucei, Leishmania donovani, and Leishmania major.44 A new derived molecule with improved solubility, LXE408,162 is currently undergoing phase I clinical trials for its use in visceral leishmaniasis (NCT05593666) but has not been tested against T. cruzi.
Oxaboroles
A benzoxaborole targeting the kinetoplastid mRNA processing factor CPSF3, named AN15368, has demonstrated substantial sterilizing in vitro and in vivo activity against different strains of T. cruzi. The compound also cleared the parasitemia in 19 treated, previously PCR-positive NHPs naturally infected with T. cruzi, as well as its presence in the tissues of euthanized animals. Importantly, the remaining animals showed declining anti-T. cruzi antibody titers in the next 42 months of follow-up, meeting current criteria used to assess treatment efficacy in humans.82
DNDI-6148, another benzoxaborole, is currently in development for the treatment of visceral leishmaniasis.165 It has been recently nominated as a clinical candidate for the treatment of T. cruzi infection.163 While its mechanism of action in T. cruzi has not been described, it is likely to involve, like AN15368, CPSF3 inhibition. An initial study with a single oral ascending dose in healthy volunteers has been completed (EudraCT: 2018–004023-37), and a phase I multiple ascending dose trial is expected to begin in the coming months.
Conclusions
In the last few years, drug discovery efforts focused on kinetoplastid parasites have produced significant advances in the treatment of human African trypanosomiasis and visceral leishmaniasis. In comparison, the drug development landscape for Chagas disease remains scarcely populated and mostly unchanged. The silent clinical progression of the disease, the complex biology of T. cruzi, the poorly understood host–parasite interactions of the long-lasting chronic phase of the infection, as well as the parasite’s remarkable genetic and antigenic diversity pose a challenge to change that reality.
Despite this, the availability of an increasingly large volume of data derived from host-parasite omics studies, together with novel bioinformatic tools, gene editing strategies, and improved animal models better reflecting the human chronic infection phase, have all led to the optimization of existing drug development pathways, and the incorporation of further steps that increase the translatability of in vitro and in vivo experiments to pre-clinical and clinical settings.
This is already leading to the identification and validation of previously unexplored molecular pathways as attractive targets for the development of new treatments. Pending further clinical evaluation of promising compounds, the future treatment of Chagas disease will require the development of drugs with an improved anti-parasitic efficacy, and of complementary therapies that prevent or reverse the organ damage associated with chronic infection.
Acknowledgments
We appreciate the support of the Generalitat of Catalonia Universities and Research Department, Spain (AGAUR: 2021 SGR 01562). The work of N.M.-P., J.C.G.-F., and J.G. was supported by the ISCIII project PI18/01054. This research was supported by CIBER - Consorcio Centro de. Investigación Biomédica en Red - (CB 2021), Instituto de Salud Carlos III, Ministerio de Ciencia e. Innovación and Unión Europea – NextGenerationEU. We also acknowledge the support from the Spanish Ministry of Science, Innovation, and Universities through the “Centro de Excelencia Severo Ochoa 2019–2023′′ Program (CEX2018-000806-S funded by MCIN/AEI/ 10.13039/501100011033) and from the Generalitat of Catalonia through the “CERCA Program.” J.C.G.-F. received support through a fellowship from “la Caixa” Foundation (ID 100010434, fellowship code: LCF/BQ/DI21/11860037). The Drugs for Neglected Diseases initiative (DNDi) is grateful to its donors, public and private, who have provided funding to DNDi since its inception in 2003. A full list of DNDi’s donors can be found at http://www.dndi.org/donors/donors/.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Vos T, Lim SS, Abbafati C, et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the global burden of disease study 2019. Lancet. 2020;396:1204–1222.
2. Pérez-Molina JA, Molina I. Chagas disease. Lancet. 2018;391(10115):82–94. doi:10.1016/S0140-6736(17)31612-4
3. Gascon J, Bern C, Pinazo MJ. Chagas disease in Spain, the United States and other non-endemic countries. Acta Trop. 2010;115(1–2):22–27. doi:10.1016/j.actatropica.2009.07.019
4. Echavarría NG, Echeverría LE, Stewart M, Gallego C, Saldarriaga C. Chagas Disease: chronic Chagas Cardiomyopathy. Curr Probl Cardiol. 2021;46(3):100507. doi:10.1016/j.cpcardiol.2019.100507
5. Echeverria LE, Morillo CA. American Trypanosomiasis (Chagas disease). Infect Dis Clin North Am. 2019;33(1):119–134. doi:10.1016/j.idc.2018.10.015
6. Carod-Artal FJ, Gascon J. Chagas disease and stroke. Lancet Neurol. 2010;9(5):533–542. doi:10.1016/S1474-4422(10)70042-9
7. Francisco AF, Jayawardhana S, Olmo F, et al. Challenges in Chagas disease drug development. Molecules. 2020;25(12):2799. doi:10.3390/molecules25122799
8. Vázquez C, García-Vázquez E, Carrilero B, et al. Tolerance and adherence of patients with chronic Chagas disease treated with benznidazole. J Braz Soc Trop Med. 2023;56. doi:10.1590/0037-8682-0384-2022
9. Tornheim JA, Lozano Beltran DF, Gilman RH, et al. Improved completion rates and characterization of drug reactions with an intensive Chagas disease treatment program in rural Bolivia. PLoS Negl Trop Dis. 2013;7(9):e2407. doi:10.1371/journal.pntd.0002407
10. Wouters OJ, McKee M, Luyten J. Estimated research and development investment needed to bring a new medicine to market, 2009–2018. JAMA. 2020;323(9):844–853. doi:10.1001/jama.2020.1166
11. DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ. 2016;47:20–33. doi:10.1016/j.jhealeco.2016.01.012
12. Hinkson IV, Madej B, Stahlberg EA. Accelerating therapeutics for opportunities in medicine: a paradigm shift in drug discovery. Front Pharmacol. 2020;11. doi:10.3389/fphar.2020.00770
13. Weng H-B, Chen H-X, Wang M-W. Innovation in neglected tropical disease drug discovery and development. Infect Dis Poverty. 2018;7(1). doi:10.1186/s40249-018-0444-1
14. De Rycker M, Wyllie S, Horn D, Read KD, Gilbert IH. Anti-trypanosomatid drug discovery: progress and challenges. Nat Rev Microbiol. 2022;21(1):35–50. doi:10.1038/s41579-022-00777-y
15. Herreros-Cabello A, Callejas-Hernández F, Gironès N, Fresno M. Trypanosoma Cruzi Genome: organization, multi-gene families, transcription, and biological implications. Genes. 2020;11(10):1–26. doi:10.3390/genes11101196
16. El-Sayed NM, Myler PJ, Blandin G, et al. Comparative genomics of trypanosomatid parasitic protozoa. Science. 2005;309:404–409.
17. Talavera-López C, Messenger LA, Lewis MD, et al. Repeat-driven generation of antigenic diversity in a major human pathogen, Trypanosoma cruzi. Front Cell Infect Microbiol. 2021;11:77. doi:10.3389/fcimb.2021.614665
18. Bivona AE, Alberti AS, Cerny N, Trinitario SN, Malchiodi EL. Chagas disease vaccine design: the search for an efficient Trypanosoma cruzi immune-mediated control. Biochim Biophys Acta Mol Basis Dis. 2020;1866(5):165658. doi:10.1016/j.bbadis.2019.165658
19. Pérez-Mazliah D, Ward AI, Lewis MD. Host-parasite dynamics in Chagas disease from systemic to hyper-local scales. Parasite Immunol. 2021;43(2):e12786. doi:10.1111/pim.12786
20. Sánchez-Valdéz FJ, Padilla A, Wang W, Orr D, Tarleton RL. Spontaneous dormancy protects Trypanosoma cruzi during extended drug exposure. Elife. 2018;7. doi:10.7554/eLife.34039
21. Ward AI, Lewis MD, Khan AA, et al. In vivo analysis of Trypanosoma cruzi Persistence foci at single-cell resolution. mBio. 2020;11(4):1–13. doi:10.1128/mBio.01242-20
22. Barrett MP, Kyle DE, Sibley LD, Radke JB, Tarleton RL. Protozoan persister-like cells and drug treatment failure. Nat Rev Microbiol. 2019;17(10):607–620. doi:10.1038/s41579-019-0238-x
23. Martín-Escolano J, Medina-Carmona E, Martín-Escolano R. Chagas disease: current view of an ancient and global chemotherapy challenge. ACS Infect Dis. 2020;6(11):2830–2843. doi:10.1021/acsinfecdis.0c00353
24. Chatelain E. Chagas disease drug discovery: toward a new era. J Biomol Screen. 2015;20(1):22–35. doi:10.1177/1087057114550585
25. Ottilie S, Goldgof GM, Calvet CM, et al. Rapid Chagas disease drug target discovery using directed evolution in drug-sensitive yeast. ACS Chem Biol. 2016;12(2):422–434. doi:10.1021/acschembio.6b01037
26. Kourbeli V, Chontzopoulou E, Moschovou K, et al. An overview on target-based drug design against kinetoplastid protozoan infections: human African trypanosomiasis, Chagas disease and leishmaniases. Molecules. 2021;26(15):4629. doi:10.3390/molecules26154629
27. Fauro R, Presti SL, Bazan C, et al. Use of clomipramine as chemotherapy of the chronic phase of Chagas disease. Parasitology. 2013;140(7):917–927. doi:10.1017/S0031182013000103
28. Villalobos-Rocha JC, Sánchez-Torres L, Nogueda-Torres B, et al. Anti-Trypanosoma cruzi and anti-leishmanial activity by quinoxaline-7-carboxylate 1,4-di-N-oxide derivatives. Parasitol Res. 2014;113(6):2027–2035. doi:10.1007/s00436-014-3850-8
29. Valera-Vera EA, Sayé M, Reigada C, Miranda MR, Pereira CA. In silico repositioning of etidronate as a potential inhibitor of the Trypanosoma cruzi enolase. J Mol Graph Model. 2020;95:107506. doi:10.1016/j.jmgm.2019.107506
30. Bhambra AS, Ruparelia KC, Tan HL, et al. Synthesis and antitrypanosomal activities of novel pyridylchalcones. Eur J Med Chem. 2017;128:213–218. doi:10.1016/j.ejmech.2017.01.027
31. Sinatti V, Baptista LP, Alves-Ferreira M, et al. In silico identification of inhibitors of ribose 5-phosphate isomerase from Trypanosoma cruzi using ligand and structure based approaches. J Mol Graph Model. 2017;77:168–180. doi:10.1016/j.jmgm.2017.08.007
32. Ferreira DD, Mesquita JT, da Costa Silva TA, et al. Efficacy of sertraline against Trypanosoma cruzi: an in vitro and in silico study. J Venom Anim Toxins Incl Trop Dis. 2018;24(1). doi:10.1186/s40409-018-0165-8
33. Juárez-Saldivar A, Schroeder M, Salentin S, et al. Computational drug repositioning for Chagas disease using protein-ligand interaction profiling. Int J Mol Sci. 2020;21(12):4270. doi:10.3390/ijms21124270
34. Mendoza-Martínez C, Correa-Basurto J, Nieto-Meneses R, et al. Design, synthesis and biological evaluation of quinazoline derivatives as anti-trypanosomatid and anti-plasmodial agents. Eur J Med Chem. 2015;96:296–307. doi:10.1016/j.ejmech.2015.04.028
35. Demoro B, Caruso F, Rossi M, et al. Bisphosphonate metal complexes as selective inhibitors of Trypanosoma cruzi farnesyl diphosphate synthase. Dalton Trans. 2012;41(21):6468–6476. doi:10.1039/c2dt12179d
36. Matutino Bastos T, Mannochio Russo H, Silvio Moretti N, et al. Chemical constituents of Anacardium occidentale as inhibitors of Trypanosoma cruzi sirtuins. Molecules. 2019;24(7):1299. doi:10.3390/molecules24071299
37. Peña I, Pilar Manzano M, Cantizani J, et al. New compound sets identified from high throughput phenotypic screening against three kinetoplastid parasites: an open resource. Sci Rep. 2015;5(1). doi:10.1038/srep08771
38. Alonso-Padilla J, Cotillo I, Presa JL, et al. Automated high-content assay for compounds selectively toxic to Trypanosoma cruzi in a myoblastic cell line. PLoS Negl Trop Dis. 2015;9(1):e0003493. doi:10.1371/journal.pntd.0003493
39. Engel JC, Ang KKH, Chen S, et al. Image-based high-throughput drug screening targeting the intracellular stage of Trypanosoma cruzi, the agent of Chagas’ disease. Antimicrob Agents Chemother. 2010;54(8):3326–3334. doi:10.1128/AAC.01777-09
40. Germain AR, Carmody LC, Dockendorff C, et al. Identification of small-molecule inhibitors of Trypanosoma cruzi replication. Bioorg Med Chem Lett. 2011;21(23):7197–7200. doi:10.1016/j.bmcl.2011.09.057
41. Alonso-Padilla J, Rodríguez A, Dumonteil E. High throughput screening for anti–Trypanosoma cruzi drug discovery. PLoS Negl Trop Dis. 2014;8(12):e3259. doi:10.1371/journal.pntd.0003259
42. Buckner FS, Verlinde CLMJ, la Flamme AC, van Voorhis WC. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing beta-galactosidase. Antimicrob Agents Chemother. 1996;40(11):2592–2597. doi:10.1128/AAC.40.11.2592
43. Bettiol E, Samanovic M, Murkin AS, et al. Identification of three classes of heteroaromatic compounds with activity against intracellular Trypanosoma cruzi by chemical library screening. PLoS Negl Trop Dis. 2009;3(2):e384. doi:10.1371/journal.pntd.0000384
44. Khare S, Nagle AS, Biggart A, et al. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature. 2016;537(7619):229–233. doi:10.1038/nature19339
45. Martinez-Peinado N, Cortes-Serra N, Torras-Claveria L, et al. Amaryllidaceae alkaloids with anti-Trypanosoma cruzi activity. Parasit Vectors. 2020;13(1):1–10. doi:10.1186/s13071-020-04171-6
46. Sykes ML, Avery VM. Approaches to protozoan drug discovery: phenotypic screening miniperspectives series on phenotypic screening for antiinfective targets. J Med Chem. 2013;56(20):7727–7740. doi:10.1021/jm4004279
47. MacLean LM, Thomas J, Lewis MD, et al. Development of Trypanosoma cruzi in vitro assays to identify compounds suitable for progression in Chagas’ disease drug discovery. PLoS Negl Trop Dis. 2018;12(7):e0006612. doi:10.1371/journal.pntd.0006612
48. Rolón M, Vega C, Escario JA, Gómez-Barrio A. Development of resazurin microtiter assay for drug sensibility testing of Trypanosoma cruzi epimastigotes. Parasitol Res. 2006;99(2):103–107. doi:10.1007/s00436-006-0126-y
49. Canavaci AMC, Bustamante JM, Padilla AM, et al. In vitro and in vivo high-throughput assays for the testing of anti-Trypanosoma cruzi compounds. PLoS Negl Trop Dis. 2010;4(7):e740. doi:10.1371/journal.pntd.0000740
50. Nohara LL, Lema C, Bader JO, Aguilera RJ, Almeida IC. High-content imaging for automated determination of host-cell infection rate by the intracellular parasite Trypanosoma cruzi. Parasitol Int. 2010;59(4):565–570. doi:10.1016/j.parint.2010.07.007
51. Dantas RF, Dos Santos ECT, Junior FPS. Past and future of trypanosomatids high-throughput phenotypic screening. Mem Inst Oswaldo Cruz. 2022;117. doi:10.1590/0074-02760210402
52. Franco CH, Alcântara LM, Chatelain E, Freitas-Junior L, Moraes CB. Drug discovery for Chagas disease: impact of different host cell lines on assay performance and hit compound selection. Trop Med Infect Dis. 2019;4(2):82. doi:10.3390/tropicalmed4020082
53. Martínez-Peinado N, Cortes-Serra N, Tallini LR, et al. Amaryllidaceae plants: a potential natural resource for the treatment of Chagas disease. Parasit Vectors. 2021;14(1):337. doi:10.1186/s13071-021-04837-9
54. Morillo CA, Waskin H, Sosa-Estani S, et al. Benznidazole and posaconazole in eliminating parasites in asymptomatic T. Cruzi carriers: the STOP-CHAGAS trial. J Am Coll Cardiol. 2017;69(8):939–947. doi:10.1016/j.jacc.2016.12.023
55. Roquero I, Cantizani J, Cotillo I, et al. Novel chemical starting points for drug discovery in leishmaniasis and Chagas disease. Int J Parasitol Drugs Drug Resist. 2019;10:58–68. doi:10.1016/j.ijpddr.2019.05.002
56. de Rycker M, Thomas J, Riley J, et al. Identification of trypanocidal activity for known clinical compounds using a new Trypanosoma cruzi hit-discovery screening cascade. PLoS Negl Trop Dis. 2016;10(4):e0004584. doi:10.1371/journal.pntd.0004584
57. Svensen N, Id SW, Gray Id DW, De M, Id R, Tonelli RR. Live-imaging rate-of-kill compound profiling for Chagas disease drug discovery with a new automated high-content assay. PLoS Negl Trop Dis. 2021;15(10):e0009870. doi:10.1371/journal.pntd.0009870
58. Cantizani J, Gamallo P, Cotillo I, et al. Rate-of-Kill (RoK) assays to triage large compound sets for Chagas disease drug discovery: application to GSK Chagas box. PLoS Negl Trop Dis. 2021;15(7):e0009602. doi:10.1371/journal.pntd.0009602
59. Choi JM, Oh SJ, Lee SY, et al. HepG2 cells as an in vitro model for evaluation of cytochrome P450 induction by xenobiotics. Arch Pharm Res. 2015;38(5):691–704. doi:10.1007/s12272-014-0502-6
60. Martinez-Peinado N, Martori C, Cortes-Serra N, et al. Anti-Trypanosoma cruzi activity of metabolism modifier compounds. Int J Mol Sci. 2021;22(2):688. doi:10.3390/ijms22020688
61. Chatelain E, Ioset JR. Phenotypic screening approaches for Chagas disease drug discovery. Expert Opin Drug Discov. 2017;13(2):141–153. doi:10.1080/17460441.2018.1417380
62. Zingales B, Miles MA, Moraes CB, et al. Drug discovery for Chagas disease should consider Trypanosoma cruzi strain diversity. Mem Inst Oswaldo Cruz. 2014;109(6):828. doi:10.1590/0074-0276140156
63. Sykes ML, Kennedy EK, Avery VM. Impact of laboratory-adapted intracellular Trypanosoma cruzi strains on the activity profiles of compounds with anti-T. cruzi activity. Microorganisms. 2023;11(2):476. doi:10.3390/microorganisms11020476
64. Torrico F, Gascón J, Barreira F, et al. New regimens of benznidazole monotherapy and in combination with fosravuconazole for treatment of Chagas disease (BENDITA): a Phase 2, double-blind, randomised trial. Lancet Infect Dis. 2021;21(8):1129–1140. doi:10.1016/S1473-3099(20)30844-6
65. Torrico F, Gascon J, Ortiz L, et al. Treatment of adult chronic indeterminate Chagas disease with benznidazole and three E1224 dosing regimens: a proof-of-concept, randomised, placebo-controlled trial. Lancet Infect Dis. 2018;18(4):419–430. doi:10.1016/S1473-3099(17)30538-8
66. Molina I, Gómez I Prat J, Salvador F, et al. Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. N Engl J Med. 2014;370(20):1899–1908. doi:10.1056/NEJMoa1313122
67. Riley J, Brand S, Voice M, et al. Development of a fluorescence-based Trypanosoma cruzi CYP51 inhibition assay for effective compound triaging in drug discovery programmes for Chagas disease. PLoS Negl Trop Dis. 2015;9(9):e0004014. doi:10.1371/journal.pntd.0004014
68. Garzoni LR, Adesse D, Soares M, et al. Fibrosis and hypertrophy induced by Trypanosoma cruzi in a three-dimensional cardiomyocyte-culture system. J Infect Dis. 2008;197(6):906–915. doi:10.1086/528373
69. Breyner NM, Hecht M, Nitz N, Rose E, Carvalho JL. In vitro models for investigation of the host-parasite interface - possible applications in acute Chagas disease. Acta Trop. 2020;202:105262. doi:10.1016/j.actatropica.2019.105262
70. Orlando LMR, Lechuga GC, da Silva Lara L, et al. Structural optimization and biological activity of pyrazole derivatives: virtual computational analysis, recovery assay and 3D culture model as potential predictive tools of effectiveness against Trypanosoma cruzi. Molecules. 2021;26(21):6742. doi:10.3390/molecules26216742
71. Nisimura LM, Ferrão PM, da Rocha Nogueira A, et al. Effect of Posaconazole in an in vitro model of cardiac fibrosis induced by Trypanosoma cruzi. Mol Biochem Parasitol. 2020;238:111283. doi:10.1016/j.molbiopara.2020.111283
72. Kratz JM, Gonçalves KR, Romera LM, et al. The translational challenge in Chagas disease drug development. Mem Inst Oswaldo Cruz. 2022;117:e200501. doi:10.1590/0074-02760200501
73. Jeffrey Neitz R, Chen S, Supek F, et al. Lead identification to clinical candidate selection: drugs for Chagas disease. J Biomol Screen. 2015;20(1):101–111. doi:10.1177/1087057114553103
74. Osorio-Méndez JF, Cevallos AM. Discovery and genetic validation of chemotherapeutic targets for Chagas’ disease. Front Cell Infect Microbiol. 2019;9:439. doi:10.3389/fcimb.2018.00439
75. Ros-Lucas A, Martinez-Peinado N, Bastida J, Gascón J, Alonso-Padilla J. The use of alphafold for in silico exploration of drug targets in the Parasite Trypanosoma cruzi. Front Cell Infect Microbiol. 2022;12. doi:10.3389/fcimb.2022.944748
76. Trevisan RO, Santos MM, Desidério CS, et al. In silico identification of new targets for diagnosis, vaccine, and drug candidates against Trypanosoma cruzi. Dis Markers. 2020;2020:1–15. doi:10.1155/2020/9130719
77. Vela A, Coral-Almeida M, Sereno D, et al. In vitro susceptibility of Trypanosoma cruzi discrete typing units (DTUs) to benznidazole: a systematic review and meta-analysis. PLoS Negl Trop Dis. 2021;15(3):e0009269. doi:10.1371/journal.pntd.0009269
78. Pereira CA, Sayé M, Reigada C, et al. Computational approaches for drug discovery against trypanosomatid-caused diseases. Parasitology. 2020;147(6):611–633. doi:10.1017/S0031182020000207
79. Panecka-Hofman J, Poehner I, Wade RC. Anti-trypanosomatid structure-based drug design – lessons learned from targeting the folate pathway. Expert Opin Drug Discov. 2022;17(9):1029–1045. doi:10.1080/17460441.2022.2113776
80. Martinez-Peinado N, Lorente-Macías Á, García-Salguero A, et al. Novel purine chemotypes with activity against Plasmodium falciparum and Trypanosoma cruzi. Pharmaceuticals. 2021;14(7):638. doi:10.3390/ph14070638
81. Kande V, Kalonji WM, Rembry S, et al. Efficacy and safety of acoziborole in patients with human African trypanosomiasis caused by Trypanosoma brucei gambiense: a multicentre, open-label, single-arm, phase 2/3 trial. Lancet Infect Dis. 2022;23:463–470. doi:10.1016/S1473-3099(22)00660-0
82. Padilla AM, Wang W, Jacobs RT, Tarleton RL. Discovery of an orally active benzoxaborole prodrug effective in the treatment of Chagas disease in non-human primates. Nat Microbiol. 2022;7(10):1536–1546. doi:10.1038/s41564-022-01211-y
83. Belew AT, Junqueira C, Rodrigues-Luiz GF, et al. Comparative transcriptome profiling of virulent and non-virulent Trypanosoma cruzi underlines the role of surface proteins during infection. PLoS Pathog. 2017;13(12):e1006767. doi:10.1371/journal.ppat.1006767
84. Aslett M, Aurrecoechea C, Berriman M, et al. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010;38(suppl_1):457–462. doi:10.1093/nar/gkp851
85. Amos B, Aurrecoechea C, Barba M, et al. VEuPathDB: the eukaryotic pathogen, vector and host bioinformatics resource center. Nucleic Acids Res. 2022;50(D1):D898–D911. doi:10.1093/nar/gkab929
86. El-Sayed NM, Myler PJ, Bartholomeu DC, et al. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science. 2005;309:409–415.
87. Franzén O, Ochaya S, Sherwood E, et al. Shotgun sequencing analysis of Trypanosoma cruzi I Sylvio X10/1 and comparison with T. cruzi VI CL Brener. PLoS Negl Trop Dis. 2011;5(3):e984. doi:10.1371/journal.pntd.0000984
88. Varadi M, Anyango S, Deshpande M, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2021;50:D439–44.
89. Varadi M, Velankar S. The impact of alphafold protein structure database on the fields of life sciences. Proteomics. 2022. doi:10.1002/PMIC.202200128
90. Bordin N, Dallago C, Heinzinger M, et al. Novel machine learning approaches revolutionize protein knowledge. Trends Biochem Sci. 2022;48(4):345–359. doi:10.1016/j.tibs.2022.11.001
91. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–589. doi:10.1038/s41586-021-03819-2
92. Wong F, Krishnan A, Zheng EJ, et al. Benchmarking AlphaFold-enabled molecular docking predictions for antibiotic discovery. Mol Syst Biol. 2022;18(9). doi:10.15252/msb.202211081
93. Wheeler J, Yurchenko V. A resource for improved predictions of Trypanosoma and Leishmania protein three-dimensional structure. PLoS One. 2021;16(11):e0259871. doi:10.1371/journal.pone.0259871
94. Evans R, O’Neill M, Pritzel A, et al. Protein complex prediction with AlphaFold-Multimer. BioRxiv. 2022:10–2021. doi:10.1101/2021.10.04.463034
95. Mirdita M, Schütze K, Moriwaki Y, et al. ColabFold: making protein folding accessible to all. Nat Methods. 2022;19(6):679–682. doi:10.1038/s41592-022-01488-1
96. Lye LF, Owens K, Shi H, et al. Retention and loss of RNA interference pathways in trypanosomatid protozoans. PLoS Pathog. 2010;6(10):1001161. doi:10.1371/journal.ppat.1001161
97. Cristina Costa F, Francisco AF, Jayawardhana S, et al. Expanding the toolbox for Trypanosoma cruzi: a parasite line incorporating a bioluminescence-fluorescence dual reporter and streamlined CRISPR/Cas9 functionality for rapid in vivo localisation and phenotyping. PLoS Negl Trop Dis. 2018;12(4):e0006388. doi:10.1371/journal.pntd.0006388
98. Lander N, Cruz-Bustos T, Docampo R. A CRISPR/Cas9-riboswitch-based method for downregulation of gene expression in Trypanosoma cruzi. Front Cell Infect Microbiol. 2020;10. doi:10.3389/fcimb.2020.00068
99. Lander N, Chiurillo MA. State-of-The-art CRISPR /Cas9 technology for genome editing in trypanosomatids. J Eukaryot Microbiol. 2019;66(6):981–991. doi:10.1111/jeu.12747
100. Taylor M, Lander N, Yoshida N. Editorial: unravelling T. cruzi Biology. Front Cell Infect Microbiol. 2020;10:382. doi:10.1038/s43018-020-0047-1
101. Jones NG, Thomas EB, Brown E, et al. Regulators of Trypanosoma brucei cell cycle progression and differentiation identified using a Kinome-Wide RNAi screen. PLoS Pathog. 2014;10(1):e1003886. doi:10.1371/journal.ppat.1003886
102. Baker N, Catta-Preta CMC, Neish R, et al. Systematic functional analysis of Leishmania protein kinases identifies regulators of differentiation or survival. Nat Commun. 2021;12(1). doi:10.1038/s41467-021-21360-8
103. Chiurillo MA, Jensen BC, Docampo R, Kumar A. Drug target validation of the protein kinase AEK1, essential for proliferation, host cell invasion, and intracellular replication of the human pathogen Trypanosoma cruzi. Microbiol Spectr. 2021;9(2). doi:10.1128/Spectrum.00738-21
104. Lima ML, Tulloch LB, Corpas-Lopez V, et al. Identification of a proteasome-targeting arylsulfonamide with potential for the treatment of Chagas’ disease. Antimicrob Agents Chemother. 2022;66(1). doi:10.1128/AAC.01535-21
105. Wyllie S, Brand S, Thomas M, et al. Preclinical candidate for the treatment of visceral leishmaniasis that acts through proteasome inhibition. Proc Natl Acad Sci USA. 2019;116(19):9318–9323. doi:10.1073/pnas.1820175116
106. Bijlmakers MJ. Ubiquitination and the proteasome as drug targets in trypanosomatid diseases. Front Chem. 2021;8. doi:10.3389/fchem.2020.630888
107. Vermelho AB, Rodrigues GC, Supuran CT. Why hasn’t there been more progress in new Chagas disease drug discovery? Expert Opin Drug Discov. 2019;15:145–158. doi:10.1080/17460441.2020.1681394
108. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(1):1–13. doi:10.1038/srep42717
109. Chatelain E, Konar N. Translational challenges of animal models in Chagas disease drug development: a review. Drug Des Devel Ther. 2015;9:4807. doi:10.2147/DDDT.S90208
110. Neal RA, van Bueren J. Comparative studies of drug susceptibility of five strains of Trypanosoma cruzi in vivo and in vitro. Trans R Soc Trop Med Hyg. 1988;82(5):709–714. doi:10.1016/0035-9203(88)90208-8
111. Trischmann T, Tanowitz H, Wittner M, Bloom B. Trypanosoma cruzi: role of the immune response in the natural resistance of inbred strains of mice. Exp Parasitol. 1978;45(2):160–168. doi:10.1016/0014-4894(78)90055-3
112. Chatelain E, Scandale I. Animal models of Chagas disease and their translational value to drug development. Expert Opin Drug Discov. 2020;15(12):1381–1402. doi:10.1080/17460441.2020.1806233
113. Romanha AJ, Solange Lisboa de C, Maria de Nazaré Correia S, et al. In vitro and in vivo experimental models for drug screening and development for Chagas disease. Mem Inst Oswaldo Cruz. 2010;105(2):233–238. doi:10.1590/S0074-02762010000200022
114. Francisco AF, Lewis MD, Jayawardhana S, et al. Limited ability of posaconazole to cure both acute and chronic Trypanosoma cruzi infections revealed by highly sensitive in vivo imaging. Antimicrob Agents Chemother. 2015;59(8):4653–4661. doi:10.1128/AAC.00520-15
115. Lewis MD, Francisco AF, Jayawardhana S, et al. Imaging the development of chronic Chagas disease after oral transmission. Sci Rep. 2018;8(1):11292. doi:10.1038/s41598-018-29564-7
116. Eickhoff CS, Lawrence CT, Sagartz JE, et al. ECG detection of murine chagasic cardiomyopathy. J Parasitol. 2010;96(4):758. doi:10.1645/GE-2396.1
117. Mateus J, Guerrero P, Lasso P, et al. An animal model of acute and chronic Chagas disease with the reticulotropic Y strain of Trypanosoma cruzi that depicts the multifunctionality and dysfunctionality of T cells. Front Immunol. 2019;10:918. doi:10.3389/fimmu.2019.00918
118. Francisco AF, Jayawardhana S, Taylor MC, Lewis MD, Kelly JM. Assessing the effectiveness of curative benznidazole treatment in preventing chronic cardiac pathology in experimental models of Chagas disease. Antimicrob Agents Chemother. 2018;62(10). doi:10.1128/AAC.00832-18
119. Lewis MD, Fortes Francisco A, Taylor MC, et al. Bioluminescence imaging of chronic Trypanosoma cruzi infections reveals tissue-specific parasite dynamics and heart disease in the absence of locally persistent infection. Cell Microbiol. 2014;16(9):1285–1300. doi:10.1111/cmi.12297
120. Verderio P, Lecchi M, Ciniselli CM, et al. 3Rs principle and legislative decrees to achieve high standard of animal research. Animals. 2023;13(2):277. doi:10.3390/ani13020277
121. Lewis MD, Francisco AF, Taylor MC, Kelly JM. A new experimental model for assessing drug efficacy against Trypanosoma cruzi infection based on highly sensitive in vivo imaging. J Biomol Screen. 2015;20(1):36–43. doi:10.1177/1087057114552623
122. Cencig S, Coltel N, Truyens C, Carlier Y. Evaluation of benznidazole treatment combined with nifurtimox, posaconazole or AmBisome® in mice infected with Trypanosoma cruzi strains. Int J Antimicrob Agents. 2012;40(6):527–532. doi:10.1016/j.ijantimicag.2012.08.002
123. Francisco AF, Saade U, Jayawardhana S, et al. Comparing in vivo bioluminescence imaging and the Multi-Cruzi immunoassay platform to develop improved Chagas disease diagnostic procedures and biomarkers for monitoring parasitological cure. PLoS Negl Trop Dis. 2022;16(10):e0010827. doi:10.1371/journal.pntd.0010827
124. de Oliveira GM, de Melo Medeiros M, da Silva Batista W, et al. Applicability of the use of charcoal for the evaluation of intestinal motility in a murine model of Trypanosoma cruzi infection. Parasitol Res. 2007;102(4):747–750. doi:10.1007/s00436-007-0829-8
125. Ny L, Holmqvist B, Li H, et al. A magnetic resonance imaging study of intestinal dilation in Trypanosoma cruzi–infected mice deficient in nitric oxide synthase. Am J Trop Med Hyg. 2008;79(5):760. doi:10.4269/ajtmh.2008.79.760
126. Khan AA, Langston HC, Costa FC, et al. Local association of Trypanosoma cruzi chronic infection foci and enteric neuropathic lesions at the tissue micro-domain scale. PLoS Pathog. 2021;17(8):e1009864. doi:10.1371/journal.ppat.1009864
127. Avalos-Borges EE, Rios LE, Jiménez-Coello M, Ortega-Pacheco A, Garg NJ. Animal models of Trypanosoma cruzi congenital transmission. Pathogens. 2022;11(10):1172. doi:10.3390/pathogens11101172
128. Ayala EV, Rodrigues da Cunha G, Azevedo MA, et al. C57BL/6 α-1,3-Galactosyltransferase knockout mouse as an animal model for experimental Chagas disease. ACS Infect Dis. 2020;6(7):1807–1815. doi:10.1021/acsinfecdis.0c00061
129. Almeida IC, Ferguson MAJ, Schenkman S, Travassos LR. Lytic anti- α -galactosyl antibodies from patients with chronic Chagas’ disease recognize novel O -linked oligosaccharides on mucin-like glycosyl-phosphatidylinositol-anchored glycoproteins of Trypanosoma cruzi. Biochem J. 1994;304(3):793. doi:10.1042/bj3040793
130. Milani SR, Travassos LR. Anti-alpha-galactosyl antibodies in chagasic patients. Possible biological significance. Braz J Med Biol Res. 1988;21(6):1275–1286.
131. Malcolm EL, Saunders AB, Vitt JP, Boutet BG, Hamer SA. Antiparasitic treatment with itraconazole and amiodarone in 2 dogs with severe, symptomatic Chagas cardiomyopathy. J Vet Intern Med. 2022;36(3):1100–1105. doi:10.1111/jvim.16422
132. de Lana M, Giunchetti RC. Dogs as a model for chemotherapy of Chagas disease and leishmaniasis. Curr Pharm Des. 2020;27(14):1741–1756. doi:10.2174/1381612826666201228142703
133. Santos FM, Lima WG, Gravel AS, et al. Cardiomyopathy prognosis after benznidazole treatment in chronic canine Chagas’ disease. J Antimicrob Chemother. 2012;67(8):1987–1995. doi:10.1093/jac/dks135
134. Guedes PMDM, Veloso VM, Tafuri WL, et al. The dog as model for chemotherapy of the Chagas’ disease. Acta Trop. 2002;84(1):9–17. doi:10.1016/S0001-706X(02)00139-0
135. Espinola Carvalho CM, Bonecini-almeida MA, Xavier SS, et al. Chronic Chagas’ disease in Rhesus monkeys (Macaca mulatta): evaluation of parasitemia, serology, electrocardiography, echocardiography and radiology. Am J Trop Med Hyg. 2003;68(6):683–691. doi:10.4269/ajtmh.2003.68.683
136. Seah SKK, Marsden PD, Voller A, Pettitt LE. Experimental Trypanosoma cruzi infection in rhesus monkeys—The acute phase. Trans R Soc Trop Med Hyg. 1974;68(1):63–69. doi:10.1016/0035-9203(74)90254-5
137. Williams JT, Dick EJ, VandeBerg JL, Hubbard GB. Natural Chagas disease in four baboons. J Med Primatol. 2009;38(2):107–113. doi:10.1111/j.1600-0684.2008.00308.x
138. Vitelli-Avelar DM, Sathler-Avelar R, Mattoso-Barbosa AM, et al. Cynomolgus macaques naturally infected with Trypanosoma cruzi-I exhibit an overall mixed pro-inflammatory/modulated cytokine signature characteristic of human Chagas disease. PLoS Negl Trop Dis. 2017;11(2):e0005233. doi:10.1371/journal.pntd.0005233
139. Cox LA, Olivier M, Spradling-Reeves K, et al. Nonhuman primates and translational research-cardiovascular disease. ILAR J. 2017;58(2):235–250. doi:10.1093/ilar/ilx025
140. de Almeida EA, Navarro MR, Guariento ME, Carvalhal SDS. Infecção experimental de macacos cebus apella sp pelo Trypanosoma cruzi: avaliação clínica, eletrocardiqgráfica e anatomopatológica. Rev Soc Bras Med Trop. 1992;25(1):7–12. doi:10.1590/S0037-86821992000100002
141. Wei L, Adderley J, Leroy D, et al. Host-directed therapy, an untapped opportunity for antimalarial intervention. Cell Rep Med. 2021;2(10):100423. doi:10.1016/j.xcrm.2021.100423
142. Varikuti S, Volpedo G, Saljoughian N, et al. The potent ITK/BTK inhibitor ibrutinib is effective for the treatment of experimental visceral leishmaniasis caused by Leishmania donovani. J Infect Dis. 2019;219(4):599. doi:10.1093/infdis/jiy552
143. Rossi MA, Tanowitz HB, Malvestio LM, et al. Coronary microvascular disease in chronic Chagas cardiomyopathy including an overview on history, pathology, and other proposed pathogenic mechanisms. PLoS Negl Trop Dis. 2010;4(8):e674. doi:10.1371/journal.pntd.0000674
144. Bonney KM, Luthringer DJ, Kim SA, Garg NJ, Engman DM. Pathology and pathogenesis of Chagas heart disease. Annu Rev Pathol. 2020;24:421–447.
145. Nardi Gemme C, Silva TQAC, Martins LC, et al. Diffuse myocardial fibrosis and cardiomyocyte diameter are associated with heart failure symptoms in Chagas cardiomyopathy. Front Cardiovasc Med. 2022;9. doi:10.3389/fcvm.2022.880151
146. Gazos-Lopes F, Oliveira MM, Hoelz LVB, et al. Structural and functional analysis of a platelet-activating Lysophosphatidylcholine of Trypanosoma cruzi. PLoS Negl Trop Dis. 2014;8(8):e3077. doi:10.1371/journal.pntd.0003077
147. Silva-Neto MAC, Lopes AH, Atella GC. Here, there, and everywhere: the ubiquitous distribution of the immunosignaling molecule lysophosphatidylcholine and its role on Chagas disease. Front Immunol. 2016;7. doi:10.3389/fimmu.2016.00062
148. Gomes MT, Monteiro RQ, Grillo LA, et al. Platelet-activating factor-like activity isolated from Trypanosoma cruzi. Int J Parasitol. 2006;36(2):165–173. doi:10.1016/j.ijpara.2005.09.016
149. Belaunzarán ML, Lammel EM, De Isola ELD. Phospholipases A in trypanosomatids. Enzyme Res. 2011;2011:1–10. doi:10.4061/2011/392082
150. Waghabi MC, Keramidas M, Feige JJ, Araujo-Jorge TC, Bailly S. Activation of transforming growth factor β by Trypanosoma cruzi. Cell Microbiol. 2005;7(4):511–517. doi:10.1111/j.1462-5822.2004.00481.x
151. Ferreira RR, Fabiana da Silva M, Alves GF, et al. TGF- β polymorphisms are a risk factor for Chagas disease. Dis Markers. 2018;2018:1–10. doi:10.1155/2018/4579198
152. Ferreira RR, Waghabi MC, Bailly S, et al. The search for biomarkers and treatments in Chagas disease: insights from TGF-beta studies and immunogenetics. Front Cell Infect Microbiol. 2022;11. doi:10.3389/fcimb.2021.767576
153. Ferreira RR, de Souza EM, de Oliveira FL, et al. Proteins involved on TGF-β pathway are up-regulated during the acute phase of experimental Chagas disease. Immunobiology. 2016;221(5):587–594. doi:10.1016/j.imbio.2016.01.009
154. Rodrigues Ferreira R, da Silva Abreu R, Vilar-Pereira G, et al. TGF-β inhibitor therapy decreases fibrosis and stimulates cardiac improvement in a pre-clinical study of chronic Chagas’ heart disease. PLoS Negl Trop Dis. 2019;13:e0007602. doi:10.1371/journal.pntd.0007602
155. Benaim G, Paniz Mondolfi AE. The emerging role of amiodarone and dronedarone in Chagas disease. Nat Rev Cardiol. 2012;9(10):605–609. doi:10.1038/nrcardio.2012.108
156. Barbosa JMC, Pedra Rezende Y, de Melo TG, et al. Experimental combination therapy with amiodarone and low-dose benznidazole in a mouse model of Trypanosoma cruzi acute infection. Microbiol Spectr. 2022;10(1). doi:10.1128/spectrum.01852-21
157. Madigan R, Majoy S, Ritter K, et al. Investigation of a combination of amiodarone and itraconazole for treatment of American trypanosomiasis (Chagas disease) in dogs. J Am Vet Med Assoc. 2019;255(3):317–329. doi:10.2460/javma.255.3.317
158. Stein C, Migliavaca CB, Colpani V, et al. Amiodarone for arrhythmia in patients with Chagas disease: a systematic review and individual patient data meta-analysis. PLoS Negl Trop Dis. 2018;12(8):e0006742. doi:10.1371/journal.pntd.0006742
159. Brustolin Aleixo CF, Ferraz FN, Massini PF, et al. Beneficial immunomodulatory and neuro digestive effect in Trypanosoma cruzi infection after Lycopodium clavatum 13c treatment. Microb Pathog. 2017;112:1–4. doi:10.1016/j.micpath.2017.09.026
160. Varikuti S, Jha BK, Volpedo G, et al. Host-directed drug therapies for neglected tropical diseases caused by protozoan parasites. Front Microbiol. 2018;9:2655. doi:10.3389/fmicb.2018.02655
161. Torrico F, Gascón J, Ortiz L, et al. A phase 2, randomized, multicenter, placebo-controlled, proof-of-concept trial of oral fexinidazole in adults with chronic indeterminate Chagas disease. Clin Infect Dis. 2023;76(3):e1186–e1194. doi:10.1093/cid/ciac579
162. Nagle A, Biggart A, Be C, et al. Discovery and characterization of clinical candidate LXE408 as a kinetoplastid-selective proteasome inhibitor for the treatment of leishmaniases. J Med Chem. 2020;63(19):10773–10781. doi:10.1021/acs.jmedchem.0c00499
163. Mowbray CE, Braillard S, Glossop PA, et al. DNDI-6148: a novel benzoxaborole preclinical candidate for the treatment of visceral leishmaniasis. J Med Chem. 2021;64(21):16159–16176. doi:10.1021/acs.jmedchem.1c01437
164. Bernhard S, Kaiser M, Burri C, Mäser P. Fexinidazole for human African trypanosomiasis, the fruit of a successful public-private partnership. Diseases. 2022;10(4):90. doi:10.3390/diseases10040090
165. Bahia MT, Andrade IM, Martins TA, et al. Fexinidazole: a potential new drug candidate for Chagas disease. PLoS Negl Trop Dis. 2012;6:e1870.
166. Wittlin S, Mäser P. From magic bullet to magic bomb: reductive bioactivation of antiparasitic agents. ACS Infect Dis. 2021;7(10):2777–2786. doi:10.1021/acsinfecdis.1c00118
167. DNDi. Fexinidazole for Chagas. DNDi: Drugs for Neglected Diseases Initiative, Chagas Disease Portfolio; 2023.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Structural Modelling of Platelet Activating Factor Acetyl Hydrolase in Leishmania donovani, Trypanosoma cruzi, and Trypanosoma brucei: Implications on Therapeutics for Leishmaniasis, Chagas Disease, and Sleeping Sickness

Goswami A, Koley T, Rajan MV, Madhuri P, Upadhyay N, Das U, Kumar M, Ethayathulla AS, Hariprasad G
Infection and Drug Resistance 2023, 16:2117-2128
Published Date: 11 April 2023