Back to Journals » Journal of Inflammation Research » Volume 19
SPP1+ Macrophages Involved in Juvenile Xanthogranuloma via PPARG Signaling Pathway
Authors Yuan X, Li J, Zhang X, Zhang H, Li P, Liu M, Chen H, Liu R, Zhu S, Feng S, Xiao X, Xing Z, Huang X, Wang C, Tang P
Received 1 March 2026
Accepted for publication 2 May 2026
Published 19 May 2026 Volume 2026:19 603881
DOI https://doi.org/10.2147/JIR.S603881
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Anish R. Maskey
Xiuqing Yuan,1,* Jianhong Li,1,* Xingliang Zhang,2 Huan Zhang,3 Ping Li,1 Mengqi Liu,1 Hefeng Chen,1 Rujing Liu,1 Sihong Zhu,1 Sihang Feng,1 Xing Xiao,1 Zhihao Xing,2,4 Xin Huang,5 Chenyao Wang,6 Pengyue Tang1
1Department of Dermatology, Shenzhen Children’s Hospital of Shantou University Medical College, Shenzhen, Guangdong, People’s Republic of China; 2Institute of Pediatrics, Shenzhen Children’s Hospital of Shantou University Medical College, Shenzhen, Guangdong, People’s Republic of China; 3Department of Pathology, Shenzhen Children’s Hospital of Shantou University Medical College, Shenzhen, Guangdong, People’s Republic of China; 4Department of Clinical Laboratory & Biobank, Shenzhen Children’s Hospital of Shantou University Medical College, Shenzhen, Guangdong, People’s Republic of China; 5Department of Hematology, Guangdong Provincial People’s Hospital (Guangdong Academy of Medical Sciences), Southern Medical University, Guangzhou, Guangdong, People’s Republic of China; 6Department of General Surgery, Shenzhen Children’s Hospital of Shantou University Medical College, Shenzhen, Guangdong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Pengyue Tang, Email [email protected]
Background: Juvenile xanthogranuloma (JXG) is the most common non-Langerhans cell histiocytic disorder in children. However, the pathogenesis remains unclear.
Methods: Lesional and adjacent normal (NOR) skin samples were obtained from patients with histologically confirmed JXG. Each biopsy sample was processed for either bulk or single-cell RNA sequencing (scRNA‑seq). Differentially expressed genes (DEGs) in the bulk transcriptome were identified and subjected to functional enrichment analyses. ScRNA-seq data were analyzed using the Seurat software for cell clustering and annotation. Single-cell regulatory network inference and clustering (SCENIC) and regulon‑module analyses were performed to quantify the differences in regulatory activity between JXG and NOR. Cell–cell communication networks were inferred using the CellChat software. Transcription factor (TF) regulons corresponding to pathway‑focused DEGs were delineated, and their activity and cellular distribution were assessed using the AUCell algorithm. Macrophage developmental trajectories and trajectory-associated genes were inferred using Monocle 3. Immunohistochemistry (IHC) was performed to examine the distribution and expression of SPP1 and related markers in JXG and similar histiocyte-related disorders. Gene set enrichment analysis (GSEA) was used to compare functional differences between SPP1+ and SPP1- macrophage subsets.
Results: A total of 4,656 DEGs were identified, including 2,209 upregulated and 2,447 downregulated genes. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed significant enrichment of the cytokine–cytokine receptor interaction (CCRI) pathway, comprising 158 CCRI-related DEGs. These DEGs revealed a complex immune profile in JXG, characterized by a predominance of pro-inflammatory cytokines despite the presence of M2-like features. Among the immune cell clusters, the M6 regulon module exhibited higher activity in JXG lesions than in NOR lesions. CellChat analysis revealed robust crosstalk between immune and non-immune cells, with SPP1‑mediated signaling being significantly upregulated in JXG. Almost all CCRI-DEG-specific TF regulons were highly and concordantly expressed in the macrophage subsets. Pseudotime analysis resolved two major macrophage branches, and the distribution of the trajectory-associated gene SPP1 closely mirrored that of the CCRI-DEG-specific TF regulons. IHC analysis indicated that SPP1 is an important marker for distinguishing JXG from other similar disorders. GSEA further showed that SPP1+ macrophages displayed activation of the PPARG signaling pathway, suppression of adaptive immune responses, and involvement in lipid homeostasis and functional regulation.
Conclusion: Cytokine-mediated interactions within macrophages play a pivotal role in the pathogenesis of JXG microenvironment, with SPP1+ macrophages showing a close association with pathogenesis, potentially via PPARG-mediated modulation of immune and metabolic homeostasis.
Keywords: JXG, SPP1+ macrophage, cytokine–cytokine receptor interaction pathway, SCENIC, TF regulon
Introduction
Juvenile xanthogranuloma (JXG), the most common non-Langerhans cell histiocytic disorder in infancy and early childhood, typically presents as solitary or multiple cutaneous papules or nodules (cutaneous JXG), and a minority of patients develop systemic involvement (systemic JXG) with potential ocular, pulmonary, hepatic, or central nervous system manifestations that can substantially worsen prognosis and quality of life.1 Although most cutaneous lesions are benign and self‑limited, systemic cases can impose marked clinical, psychological, and economic burdens on the affected families.2
Histopathology and immunophenotyping show that classic JXG lesions are dominated by foamy histiocytes and Touton giant cells that express macrophage-associated markers (CD68⁺, variable factor XIIIa, S100−) but lack Langerhans-cell lineage markers, consistent with a monocyte–macrophage origin and an inflammatory etiology.3 However, a subset of JXG cases presents with atypical histology and non-specific immunophenotypes, making them difficult to distinguish from xanthoma, xanthoma disseminatum (XD), and Blau syndrome by routine H&E staining and conventional immunohistochemical (IHC) staining. Consequently, the disease-specific molecular signatures of JXG remain scarce, underscoring the urgent need to identify robust diagnostic markers.
Transcription factors (TFs) control cell identity, differentiation trajectories, and context‑dependent effector programs in immune cells, including macrophages.4,5 Profiling TF activity and regulatory networks can therefore illuminate the upstream controllers of disease-relevant programs such as macrophage polarization, lipid handling, and cytokine signaling.6 Single-cell regulatory network methods (SCENIC) enable inference of active TF regulons at single-cell resolution and can reveal cell-type-specific regulatory modules that are not apparent from bulk expression alone.7 Applying such approaches to JXG can resolve intralesional cellular heterogeneity, identify TFs that drive lipid-loaded macrophage states, and link TF activity to paracrine cytokine signaling that may sustain lesion formation.
JXG is characterized by the accumulation of foamy histiocytes, which are formed by intracellular lipid deposition within macrophages.3 These lipid-laden cells modulate inflammation, remodel the extracellular matrix, and influence local cellular interactions, resembling foam-cell biology in atherosclerosis and macrophage phenotypes observed in fibrotic and tumor microenvironments.8,9 Theoretically, the pathogenesis of such lesions involves more than passive lipid uptake; it is driven by intricate macrophage-microenvironment interactions. As documented in clinicopathologic surveys of non-Langerhans cell histiocytosis, the spatial organization of histiocytes is orchestrated by localized recruitment signals.10 Furthermore, emerging evidence from signaling inhibition studies suggests that specific molecular mediators—such as receptor tyrosine kinases—regulate the metabolic shift toward lipid sequestration and facilitate the crosstalk between macrophage subsets, thereby sustaining the chronic inflammatory milieu required for Touton giant cell development.11 Secreted phosphoprotein 1 (SPP1, osteopontin) has been implicated in lipid metabolism, phagocytosis, tissue remodeling, and profibrotic signaling in diverse diseases.12 SPP1+ macrophages have been linked to chronic inflammation and coordinated metabolic and structural remodeling in cancer, fibrosis, and metabolic disorders.13–15 Yet, their presence and regulatory architecture in JXG remain undefined.
PPARG signaling is a key regulator of macrophage lipid metabolism and inflammatory responses. In disorders such as atherosclerosis and fatty liver disease, PPARG activation enhances lipid uptake and foam‑cell formation while simultaneously suppressing pro-inflammatory cytokine production and promoting alternative (M2‑like) macrophage polarization.16–18 Beyond these effects, PPARG activity is generally associated with the maintenance of lipid and inflammatory homeostasis. Thus, its activation does not necessarily imply chronic lesion persistence but may instead reflect a compensatory response to a lipid‑rich microenvironment. Although PPARG signaling has been extensively studied in lipid‑metabolic diseases, its relevance to JXG—another condition characterized by lipid-laden macrophages—remains largely unexplored. Given the typically self‑limited nature of cutaneous JXG, PPARG activation in this context may represent a homeostatic mechanism that coordinates lipid handling with the restraint of excessive inflammation. However, to the best of our knowledge, the functional relevance of PPARG signaling in JXG has not been investigated.
It is widely recognized that bulk RNA-seq delineates the altered microenvironment of JXG, whereas single-cell RNA-seq (scRNA-seq) resolves the individual cellular constituents that drive these changes. In this study, we integrated bulk and single-cell transcriptomes to identify candidate cell populations that sculpt into the JXG microenvironment. Rather than enumerating differentially expressed genes (DEGs) in isolation, we first identified the most significantly rewired signaling pathway between JXG and normal tissue, extracted its constituent DEGs, intersected them with the transcriptional regulatory networks that exhibited inter-group differences in the scRNA-seq data, and defined the corresponding TF regulons. Mapping the activity landscape of these regulons allowed us to trace pathway perturbations in specific cellular subsets. To the best of our knowledge, this is the first investigation to dissect JXG pathogenesis at both transcriptomic scales by linking a critical signaling axis to its gene regulatory circuitry and, ultimately, to the responsible cell types and potential pathways. We anticipate that these findings will not only advance our understanding of JXG etiology but also uncover novel biomarkers and therapeutic targets for early intervention and improved clinical outcomes.
Materials And Methods
Sample Collection
Pathological lesional tissues from the JXG and adjacent normal skin were obtained by dermatologic biopsy. Samples were processed and sent to the Beijing Genomics Institute (BGI) for sequencing. After quality control, 11 JXG and 7 NOR samples met the thresholds and were included in bulk RNA‑seq analysis, and 2 JXG and 2 NOR samples were included in the scRNA‑seq analysis. All specimens were collected after written informed consent was obtained from parents or legal guardians of the pediatric patients. The study was approved by the Ethics Committee of Shenzhen Children’s Hospital (approval number: 202309402).
Differential Expression Analysis
Data processing was performed using R software 4.2.1, and visualization was performed using the ggplot2 package (version 3.4.1). Differential gene expression in the bulk RNA-seq dataset was calculated using the DESeq2 package (version 1.38.3).19 Genes with an adjusted P-value (P-adj) < 0.05, and absolute log fold change (logFC) values > 1 were considered differentially expressed genes (DEGs). The results were visualized using an MAplot.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis of DEGs
GO and KEGG enrichment analyses were conducted for DEGs using the clusterProfiler package (version 4.0) in R.20 The GO enrichment analysis results were divided into three categories: cellular components (CC), biological processes (BP), and molecular functions (MF). Pathways with a P-adj value < 0.05 were considered statistically significant.
Protein–Protein Interaction (PPI) Analysis of Cytokine–Cytokine Receptor Interaction Pathway–Related Differentially Expressed Genes (CCRI-DEGs)
Differentially expressed genes assigned to the “cytokine–cytokine receptor interaction” pathway (ID: hsa04060) in the KEGG enrichment analysis were extracted and defined as CCRI-DEGs. The expression profiles of these genes were visualized using the R package pheatmap (version 1.0.12). To explore potential functional interactions among CCRI-DEGs, a PPI network was constructed using the STRING database (version 11.5; https://string-db.org).21 An interaction confidence score ≥ 0.7 was applied as the cutoff. The resulting network was imported to Cytoscape (version 3.9.1) for topological analysis and visual refinement.22
ScRNAseq Data Processing
ScRNA‑seq was used to identify transcriptionally distinct cell clusters and their defining genes. All analyses were performed using the Seurat R package (version 5.0).23 Raw unique molecular identifier (UMI) counts were log-normalized (NormalizeData), highly variable genes were selected (FindVariableFeatures), and the data were scaled (ScaleData). Principal component analysis (RunPCA) was followed by batch-effect correction with IntegrateLayers, using canonical correlation analysis. The integrated data were embedded in two dimensions with UMAP (RunUMAP), based on the first 30 canonical components, and the cells were clustered using FindNeighbors and FindClusters with a resolution of 0.01. Cluster identities were assigned by comparing the top differentially expressed genes (FindAllMarkers, min; pct = 0.25, log2FC > 0.25) with the established lineage markers. The three most representative marker genes for each cluster were visualized using DimPlot, and cluster‑specific markers were plotted using the scRNAtoolVis package (version 1.0).
Estimation of Cell-Cluster Proportions in JXG versus Normal Tissue
Because only two JXG and two control samples were available for primary scRNA-seq, the cellular composition was inferred using a deconvolution approach. Bulk RNA-seq data from the larger cohort were deconvoluted using BayesPrism2, which applies a Bayesian latent‑variable framework using scRNA‑seq profiles as priors to estimate posterior cell‑type proportions and gene-expression contributions.24 To assess the reliability of the deconvolution, we calculated the posterior Coefficient of Variation (theta.cv) for each estimate. Deconvolution stability was further evaluated via diagnostic plots of theta versus theta.cv. The cell-type proportions derived directly from the scRNA-seq dataset and those inferred via bulk deconvolution were analyzed independently, and visualized using bar charts to verify the consistency of the results.
Inference of Cell-Cell Communication
Cell–cell communication networks within specific clusters were inferred using the CellChat R package, which quantitatively models intercellular signaling based on scRNA-seq data. All analytical procedures and parameter settings followed official CellChat documentation (https://github.com/sqjin/CellChat).
Construction of Gene Regulatory Network (GRN)
Gene regulatory networks in JXG were inferred using SCENIC and regulon‑module analyses to identify modules that reflect transcriptomic differences between JXG and NOR based on scRNA‑seq data.7 Genes within the CCRI‑DEG set intersected with those in the selected regulon module to identify shared components. TFs regulating these intersecting genes were then determined, and a CCRI-DEG–associated GRN was constructed using the igraph package.
Identification of Immune-Cell Subclusters
GRN analysis indicated increased regulon activity in the immune cell compartment of JXG compared to NOR, suggesting a central role of immune subsets in JXG pathogenesis. To identify the specific contributing subpopulations, all immune cells were re-clustered in Seurat (version 5.0) following the workflow described in Method 1.5, using a clustering resolution of 0.1. Cell identities were assigned by matching cluster‑specific differentially expressed genes to canonical markers from the sc‑ImmuCC database.25 Immune subclusters were visualized using DimPlot software.
Investigating the Expression Distribution of CCRI‑DEG–Specific TF Regulons in Macrophages
The M6 regulon module identified in the GRN analysis was used to examine the relationship between CCRI‑DEGs and macrophage-related GRNs. TFs and their downstream targets within this module were retrieved, and CCRI‑DEGs were intersected with these target sets to define the TF-specific regulons composed of each TF and its directly regulated CCRI‑DEGs. Regulon activity in individual macrophages was quantified using AUCell and mapped onto UMAP embeddings to visualize their distribution across macrophage subsets.
Pseudotime Analysis of Macrophage Subpopulations
Since macrophages exhibit distinct functional states along their differentiation trajectory, we sought to determine which developmental stage aligns most closely with the CCRI-DEG–associated TF regulons and may therefore drive JXG pathogenesis. All macrophages were isolated and subjected to pseudotime analysis using Monocle 3 (version 3.0) with the default parameters.26 A single developmental trajectory was reconstructed from the integrated expression matrix, and the top 20 trajectory-associated signature genes were identified.
IHC Was Employed to Evaluate the Diagnostic Utility of Identified JXG-Associated Markers
IHC was performed to assess the diagnostic utility of the JXG-associated markers. Skin sections from patients with JXG and clinically similar disorders (Blau syndrome, xanthoma, and XD) were stained for SPI1, SPP1, and IL‑1B, with CD68 and CD36 included as reference macrophage markers.
Paraffin-embedded tissues from 3 JXG, 3 xanthoma, 2 xanthoma disseminatum, and 3 Blau syndrome cases were processed. Sections (4 μm thick) were deparaffinized, rehydrated, treated with hydrogen peroxide, and subjected to antigen retrieval. Slides were incubated for 30 min with primary antibodies against SPI1 (1:100), SPP1 (1:400), IL‑1B (1:1000), CD68 (1:500), and CD36 (1:300), followed by incubation with peroxidase-conjugated goat anti-rabbit IgG secondary antibody (1:200, 60 min). Staining was visualized using DAB and imaged using bright-field microscopy. The staining intensity was quantified using ImageJ as a percentage of the positive area. Group differences were evaluated using t-tests in GraphPad Prism version 10. Ethical approval was obtained from the Ethics Committee of Shenzhen Children’s Hospital.
Functional Characterization of SPP1+ Macrophages
To investigate the role of SPP1+ macrophages in JXG, SPP1 expression levels were extracted from all macrophage subsets, and the median expression value was used as the threshold to classify cells into SPP1+ and SPP1- groups. Differences in the proportions of SPP1+ macrophages between groups were assessed using the chi-square test. To further explore the functional characteristics of SPP1+ macrophages, DEGs between SPP1+ and SPP1- cells were identified using the FindMarkers function in Seurat and ranked by log-fold change. Gene set enrichment analysis (gseGO and gseKEGG) was performed using the clusterProfiler package, and significantly enriched pathways (p < 0.05) were visualized using the GseaVis package.
Results
Overview of Samples and Study Workflow
Pathological biopsies were obtained from the JXG lesions and adjacent normal skin for bulk RNA-seq and single-cell RNA-seq analyses. The detailed sample information was presented in Table 1. The workflow of this study was shown in Figure 1.
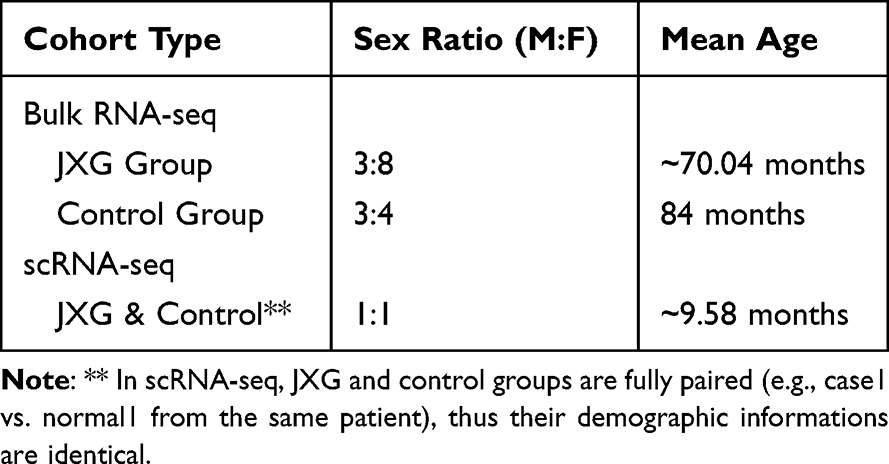 |
Table 1 Clinical Characteristics of Pediatric Patients |
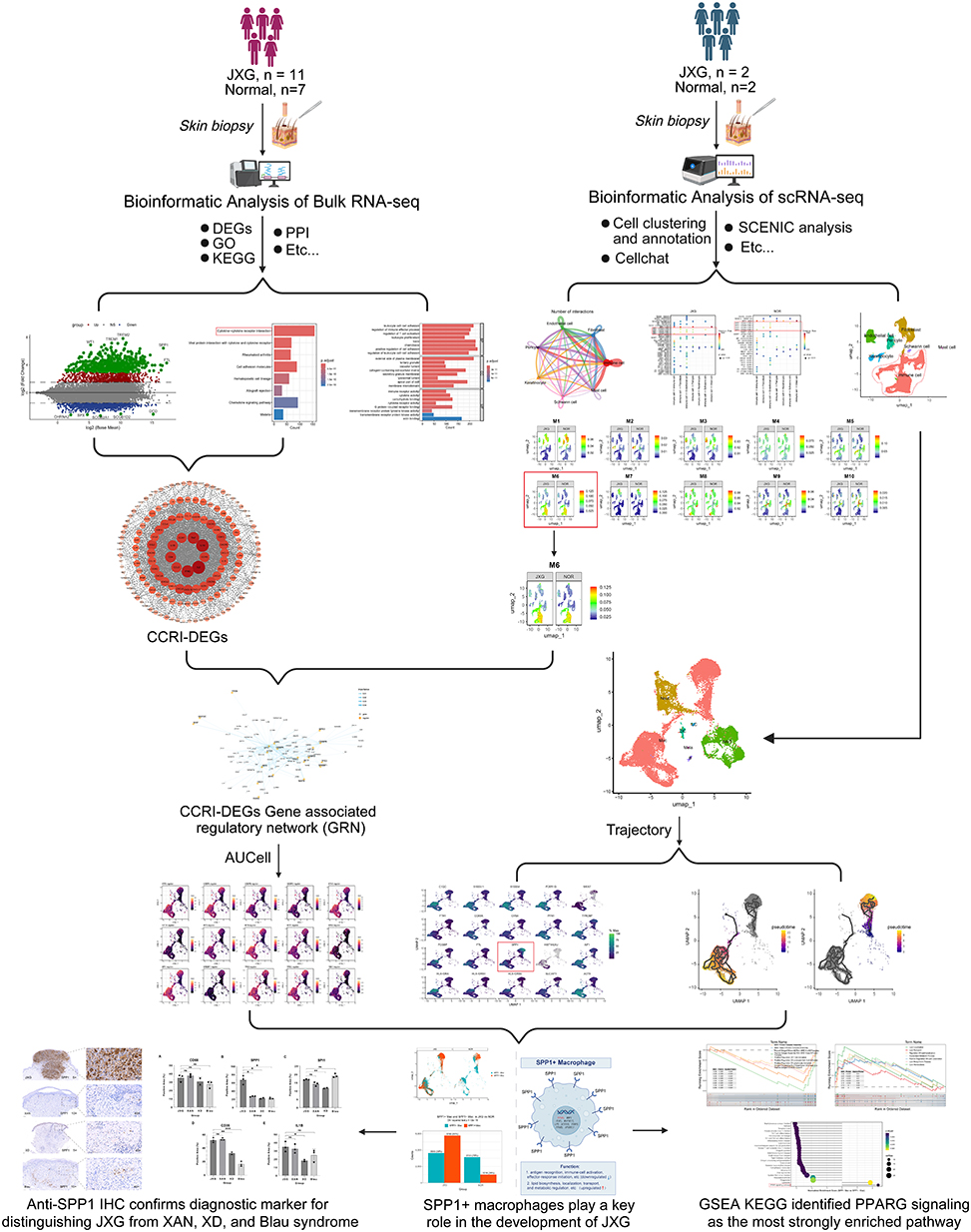 |
Figure 1 An outline of the research workflow. |
Identification of DEGs
A total of 4,656 DEGs were identified according to the criteria set above, including 2,209 upregulated and 2,447 downregulated genes. Upregulated DEGs were shown in red, downregulated DEGs were shown in blue, and those with an absolute logFC greater than 3 were shown in green (Figure 2A).
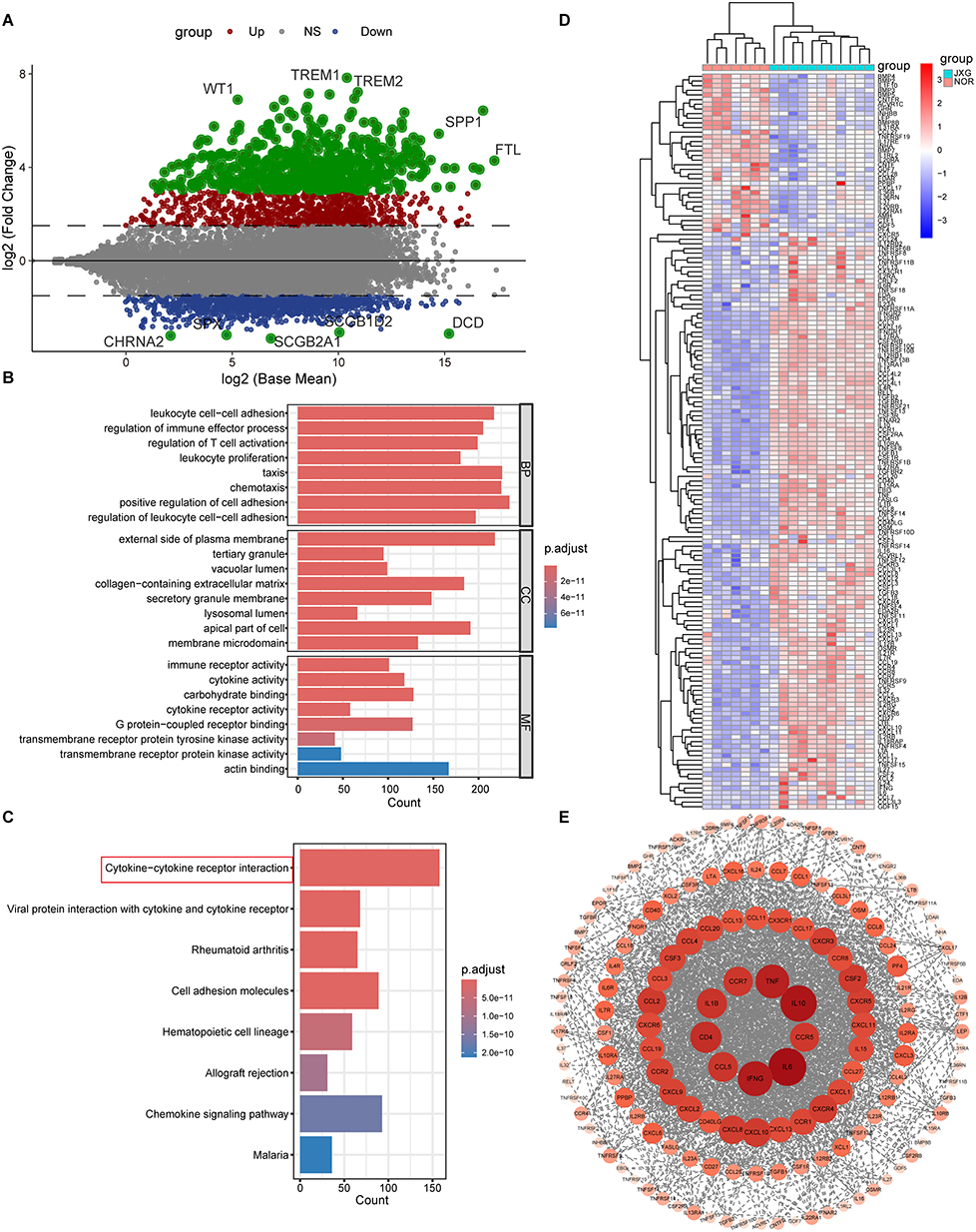 |
Figure 2 Identification of CCRI-DEGs. (A) MAplot displaying DEGs between JXG and NOR groups (|logFC| > 1 and P-adj < 0.05). Blue represents DEGs with lower expression, red with higher expression, and green with a |logFC| value greater than 3. (B) Bar plots presenting the top 8 enriched GO (BP, CC, MF) pathways (P-adj < 0.05). (C) Bar plots illustrating the top 8 enriched KEGG pathways (P-adj < 0.05). (D) pheatmap of CCRI-DEGs. € PPI analysis of CCRI-DEGs. Abbreviations: DEGs, differentially expressed genes; CCRI-DEGs, DEGs belonging to the cytokine-cytokine receptor interaction pathway; PPI, protein-protein interaction. |
Enriched GO and KEGG Pathways
Functional enrichment analysis was performed using all DEGs to explore the biological pathways associated with JXG. A total of 2,453 GO terms (including 2,019 BP, 185 CC, and 249 MF terms) and 138 KEGG pathways were significantly enriched (P-adj < 0.05) (Supplementary Table 1). GO and KEGG analyses showed that DEGs were predominantly enriched in immune-related processes, with cytokine–cytokine receptor interactions representing one of the top KEGG pathways (Figures 2B and C). These findings suggested that JXG is closely associated with immune cell activity and immune-mediated responses.
DEGs Associated with the Cytokine–Cytokine Receptor Interaction Pathway (CCRI‑DEGs)
Based on KEGG enrichment results, we hypothesized that the cytokine–cytokine receptor interaction (CCRI) pathway plays a central role in JXG pathogenesis. Therefore, we extracted all the DEGs annotated in this pathway and identified 158 CCRI‑DEGs, including 124 upregulated and 34 downregulated genes. Unsupervised hierarchical clustering (Figure 2D) clearly separated JXG from normal samples, reflecting the presence of both pro-inflammatory and anti-inflammatory mediators, with pro-inflammatory cytokines being predominant.
Potential Relationships Among CCRI-DEGs
PPI analysis revealed that IL1B, IL6, IL10, CD4, CCL5, CCR5, CCR7, IFNG, and TNF formed the most densely connected hubs among the CCRI‑DEGs (Figure 2E). Therefore, these core nodes are likely to play important roles in the initiation and progression of JXG.
ScRNA-Seq Analysis Revealed a Predominance of Immune Subsets in JXG
Using the Seurat package, we processed the scRNA-seq dataset, identified seven cell clusters based on cluster‑specific markers, and established skin-cell signatures (Figure 3A). Immune cells represented the largest proportion of captured cells, which was consistent with the known histopathology of JXG (Figure 3B). Independent of the scRNA-seq proportions, the deconvolution results of the bulk RNA-seq data corroborated a significantly higher immune cell fraction in JXG than in normal skin (P-adj< 0.001; Figure 3C). Assessment of deconvolution uncertainty revealed that the estimated immune cell fractions in JXG samples exhibited remarkably low variance (mean theta.cv < 0.002), indicating high stability in our posterior estimates (Supplementary Figure 1). The high concordance between the direct scRNA-seq measurements and the independent bulk-scale predictions, supported by robust uncertainty metrics, confirmed the massive infiltration and predominance of immune subsets within the JXG microenvironment.
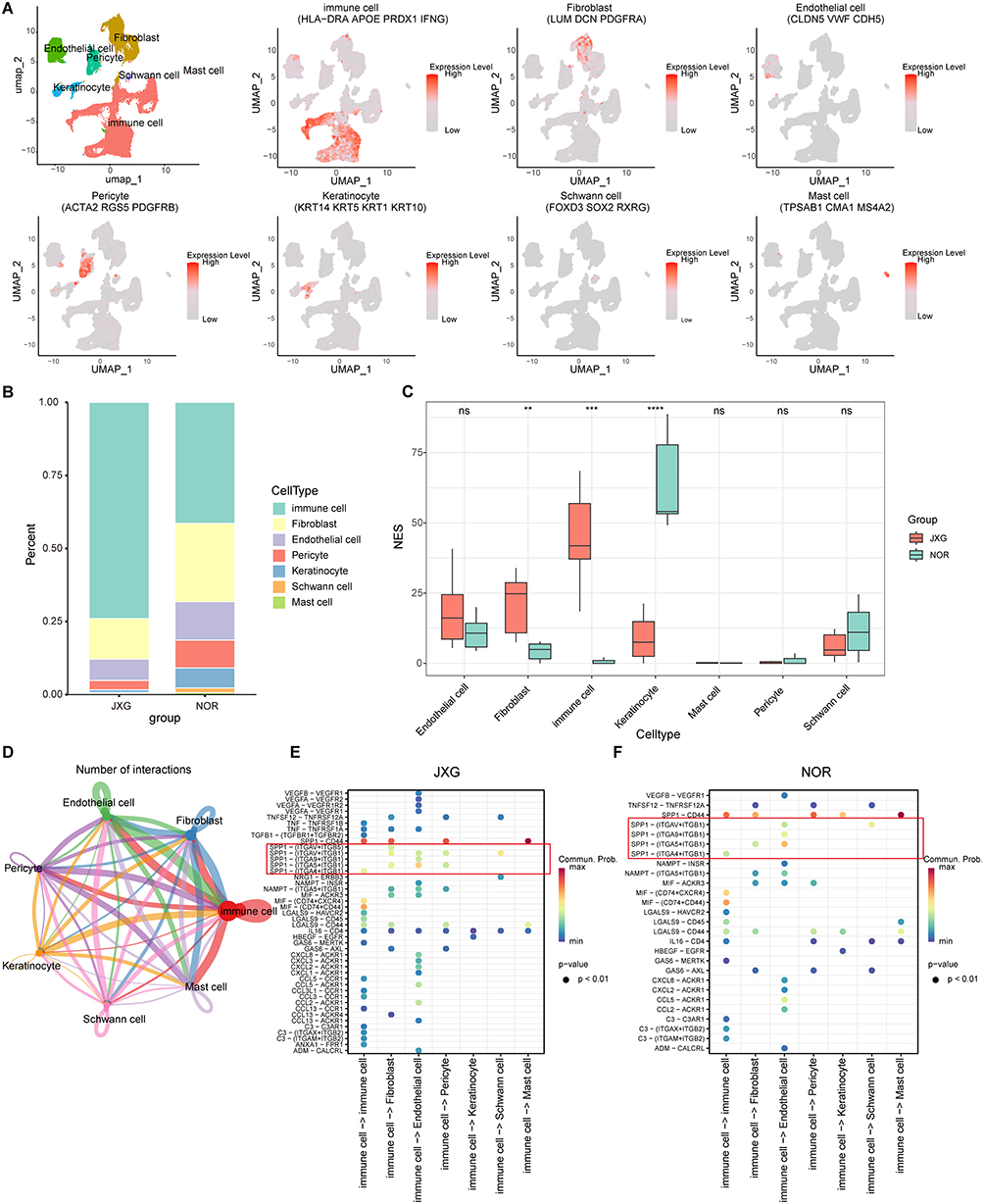 |
Figure 3 Cell cluster annotation, composition, and cell-cell communications in JXG. (A) Annotation of the 7 cell clusters in JXG and NOR groups and related markers of cell clusters. (B) Differentially expressed marker genes of cell clusters. (C) Comparison of cellular compositions between groups after deconvolution of bulk RNA-seq data. (D) Inferred interaction among each cluster. (E and F) SPP1-driven immune–fibroblast crosstalk differed between groups. |
SPP1-Mediated Cellular Communication Differed Between Groups
CellChat analysis revealed extensive and robust interactions among immune cells, fibroblasts, and other cell populations (Figure 3D). Notably, SPP1‑mediated ligand–receptor pairs were markedly enriched in JXG compared to those in the NOR group (Figures 3E and F), suggesting that SPP1‑mediated communication may contribute to JXG pathogenesis.
Elevated M6 Regulon Activity in Immune‑cell Clusters of JXG
Using the SCENIC pipeline, we identified distinct regulon activity patterns between JXG and NOR samples. The M6 module, enriched in immune cell clusters, showed elevated activity in JXG, indicating enhanced immune-related regulatory programs in the lesions. By contrast, the fibroblast-associated M1 module exhibited reduced activity (Figure 4A).
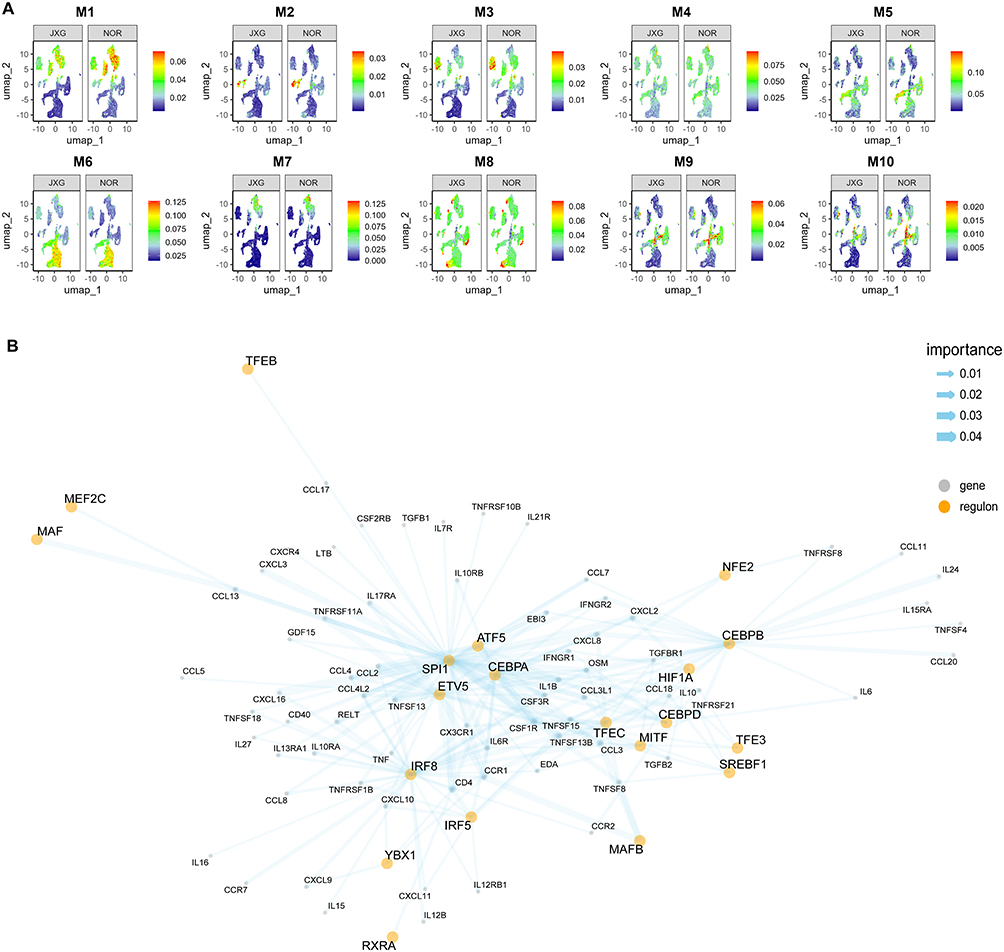 |
Figure 4 Differentiation of regulon activity in JXG and NOR groups. (A) Noticeable regulon activity scores observed in the M1 and M6 regulon modules between groups. (B) Composition of CCRI-DEGs specific gene regulatory network. Yellow dots represent TFs, while grey dots represent target genes. The thickness of the lines indicates the importance of the association between TFs and target genes. |
Discovery of a CCRI-DEGs-Specific GRN
By intersecting CCRI-DEGs with genes from the M6 regulon module, we identified a GRN specific to CCRI-DEGs (Figure 4B). GRN included several TFs, including SPI1, ATF5, ETV5, and CEBPA, each of which directly regulates several downstream genes. Additional TFs, such as NFE2, TFE3, SREBF1, and MAFB, regulate smaller gene sets. These TFs were interconnected through multiple co-regulated targets. In contrast, some TFs such as CEBPB, regulate individual genes, including CCL20, IL24, CCL11, and IL15RA. Together, these findings highlighted the involvement of specific TFs in immune cell clusters and illustrated the integration of CCRI‑DEGs into the regulatory landscape.
Macrophages Constitute a Dominant Fraction of the Immune-Cell Cluster
To determine which immune subsets predominated within the JXG, we re-clustered all immune cells using the Seurat package, yielding six distinct subpopulations: macrophages (Mac), neutrophils (Neut), NK/T cells (NK/T), dendritic cells (DC), innate lymphoid cells (ILC), and B cells (B) (Figure 5A). As melanocytes share transcriptional features with immune cells, they were initially grouped within the immune cluster. However, this was resolved using melanocyte-specific markers (Figure 5B). Notably, macrophages were the largest immune subset, consistent with the prominent macrophage infiltrates observed in the histopathology of JXG.
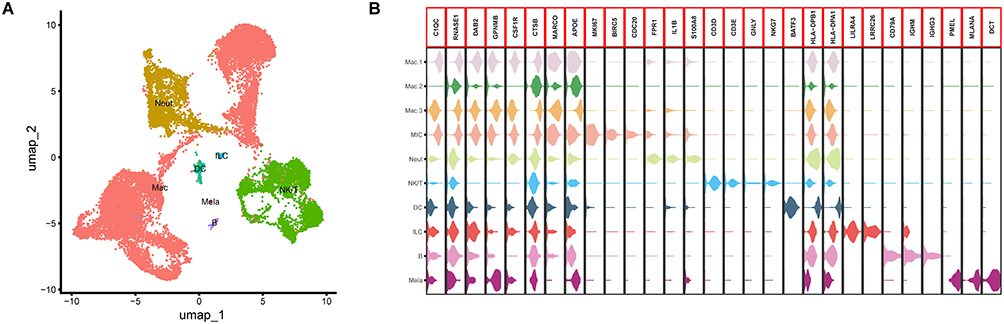 |
Figure 5 Cell cluster annotation of immune cells. (A) Annotation of the different immune cell clusters. (B) Marker genes of different immune cell clusters. |
The CCRI-DEG-Specific TF Regulons Exhibited Highly Concordant Expression Patterns Across Macrophage Subpopulations
As described above, we identified 15 TF regulons specific to CCRI‑DEGs in macrophages and quantified their activity using the AUCell algorithm. Spatial mapping showed that nearly all regulons displayed highly concordant activity patterns across macrophage subpopulations (Figure 6), suggesting that these factors may act synergistically within the same macrophage subsets to shape the transcriptional characteristics of JXG.
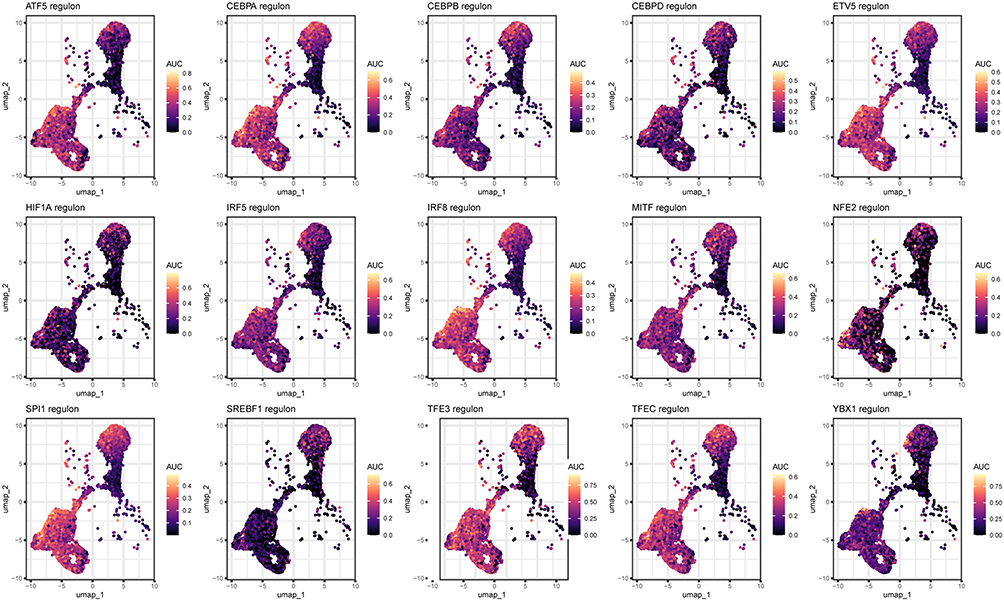 |
Figure 6 Activity scores of different CCRI-DEG-specific TF regulon in macrophages. |
SPP1 as a Trajectory-Associated Marker of Macrophage Developmental Stages
Cell trajectory inference delineated two principal developmental branches within the macrophage population (Figures 7A and B), consistent with known differentiation patterns. Analysis of trajectory-associated genes showed that the spatial distribution of SPP1 expression closely paralleled that of CCRI‑DEG–specific TF regulons (Figure 7C and Figure 6). These findings suggested that SPP1+ macrophages represent a key subpopulation that contributes to the progression of JXG.
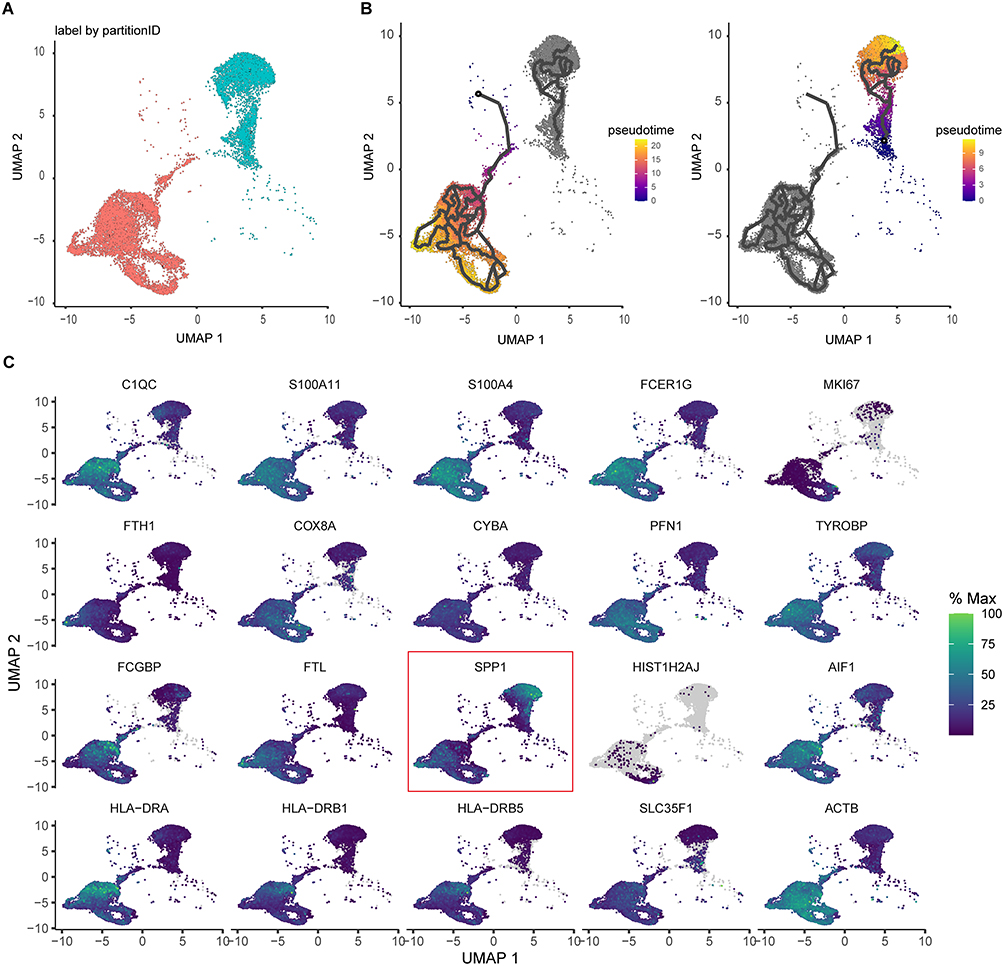 |
Figure 7 Pseudotime trajectory analysis of macrophages. (A) Pseudotime reconstruction reveals two major macrophage differentiation trajectories. (B) Two pseudotime developmental trajectories of macrophage populations. (C) Expression heatmap of the top 20 trajectory-associated genes. |
SPP1 as a Discriminatory Marker Distinguishing JXG From Similar Disorders
IHC of the selected markers (SPI1, SPP1, IL1B, CD36, and CD68) was performed to distinguish JXG from clinically similar disorders, including Blau syndrome, xanthoma, and XD. Representative images were shown in Figure 8 and Supplementary Figures 2–5. Semi-quantitative analysis (Figure 9) revealed no significant differences in CD68 expression between the groups. CD36 expression differed between JXG and XD (p < 0.01) and between JXG and Blau syndrome (p < 0.001). IL1B and SPI1 showed similar patterns with significant differences only between JXG and XD (IL1B, p < 0.05; SPI1, p < 0.01). Notably, SPP1 expression was markedly higher in JXG than in all the other comparison groups (p < 0.05), supporting its utility as a discriminatory marker for differentiating JXG from other histiocytic disorders with overlapping clinical features.
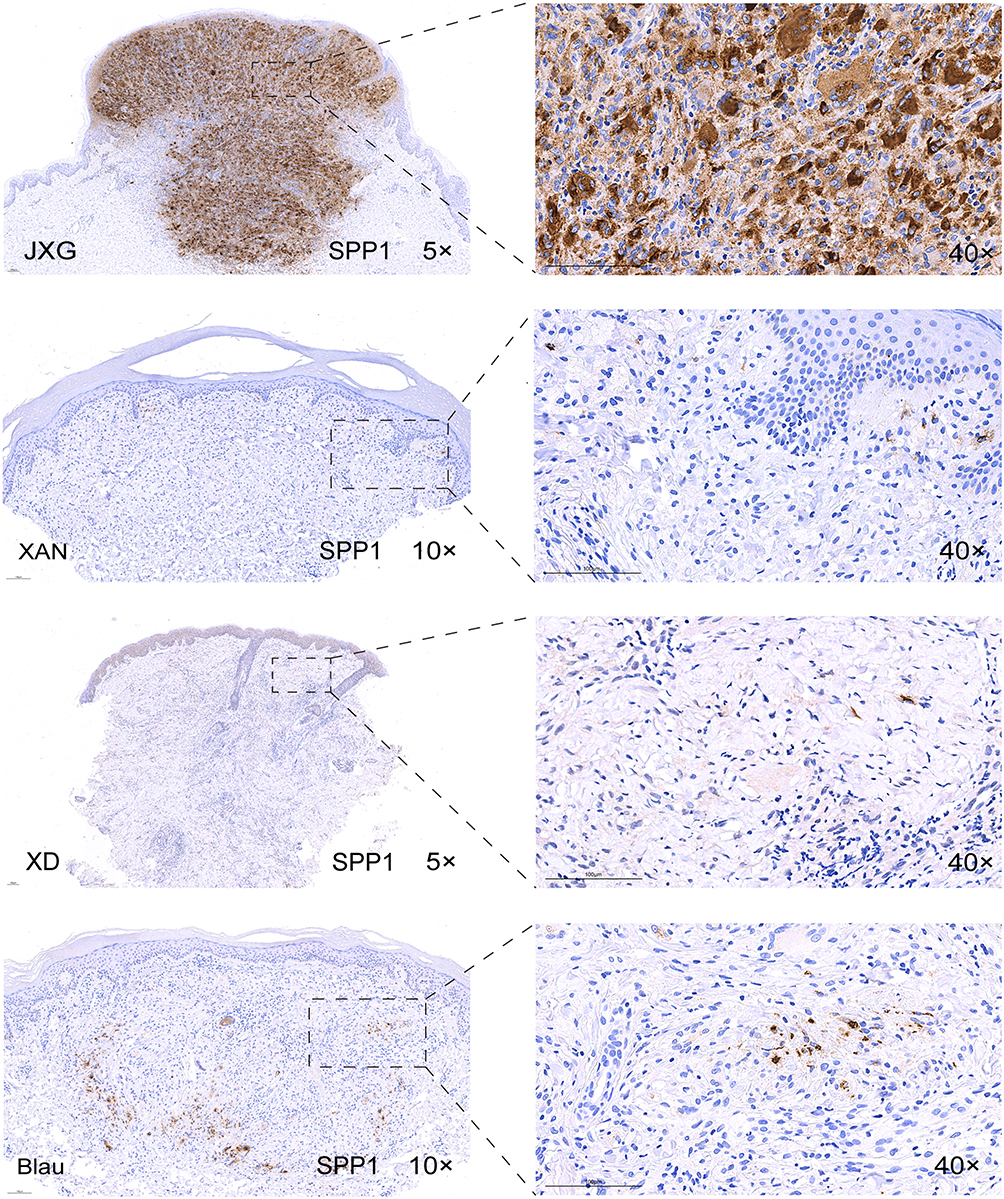 |
Figure 8 IHC staining of SPP1 expression in JXG and other diseases (XAN, XD, Blau). Abbreviations: IHC, immunohistochemistry; XAN, xanthoma; XD, xanthoma disseminatum; Blau, Blau syndrome. |
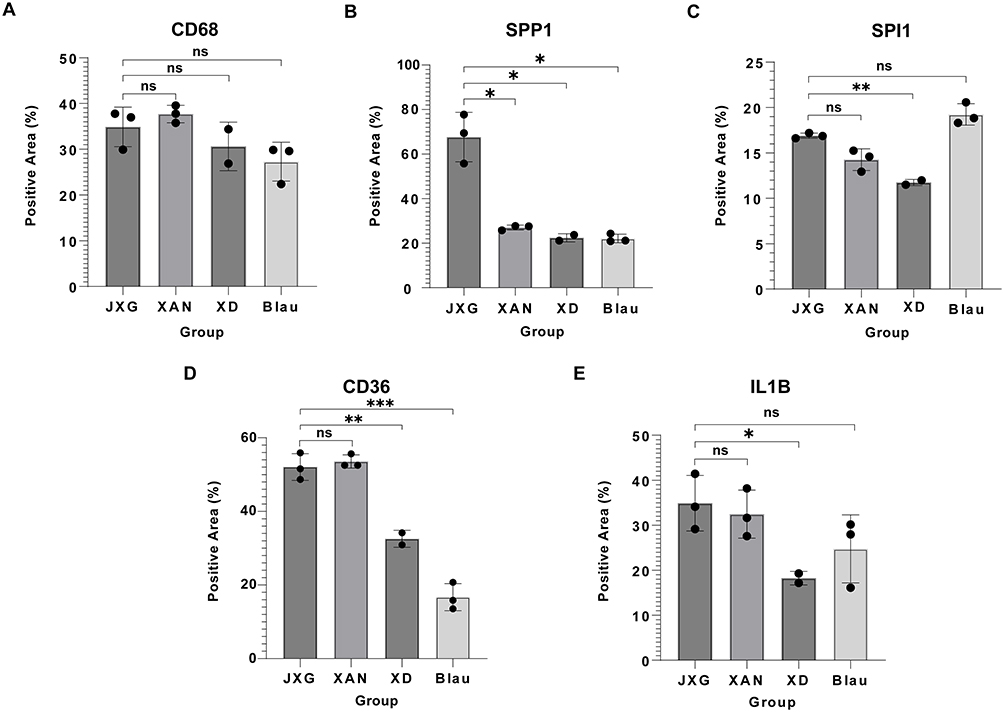 |
Figure 9 Relative expression levels of selected markers based on JXG compared with other disease groups (XAN, XD, Blau). (*p < 0.05; **p < 0.01; ***p < 0.001.) (A) CD68: no significant differences among the groups. (B) SPP1: Significantly higher in JXG than in all the other groups (p < 0.05). (C) SPI1: significantly higher in JXG than in XD (p < 0.01). (D) CD36: significantly higher in JXG than in XD (p < 0.01) and Blau (p < 0.001). (E) IL1B: significantly higher in JXG than in XD (p < 0.05). Abbreviations: XAN, xanthoma; XD, xanthoma disseminatum; Blau, Blau syndrome. |
GSEA Analysis of SPP1+ Macrophages
Quantitative analysis showed that SPP1+ macrophages constituted a larger proportion of macrophages in JXG than in NOR (61% vs. 39% in JXG; 24% vs. 76% in NOR) (Figures 10A and B). A chi-square test confirmed the significant enrichment of SPP1+ macrophages in JXG (p < 0.01) (Figure 10B). GSEA of GO pathways revealed that pathways involved in the initiation and regulation of adaptive immune responses, including antigen recognition, immune cell activation, and effector-response initiation, were significantly downregulated in SPP1+ macrophages. In contrast, pathways associated with lipid homeostasis and functional regulation, such as lipid biosynthesis, localization, transport, and metabolic regulation, exhibited significant positive enrichment (Figures 10C and D). KEGG enrichment analysis further revealed that the PPARG signaling pathway was the most significantly positive enriched signature in SPP1+ macrophages (NES > 2.35, p < 0.01) (Figure 10E).
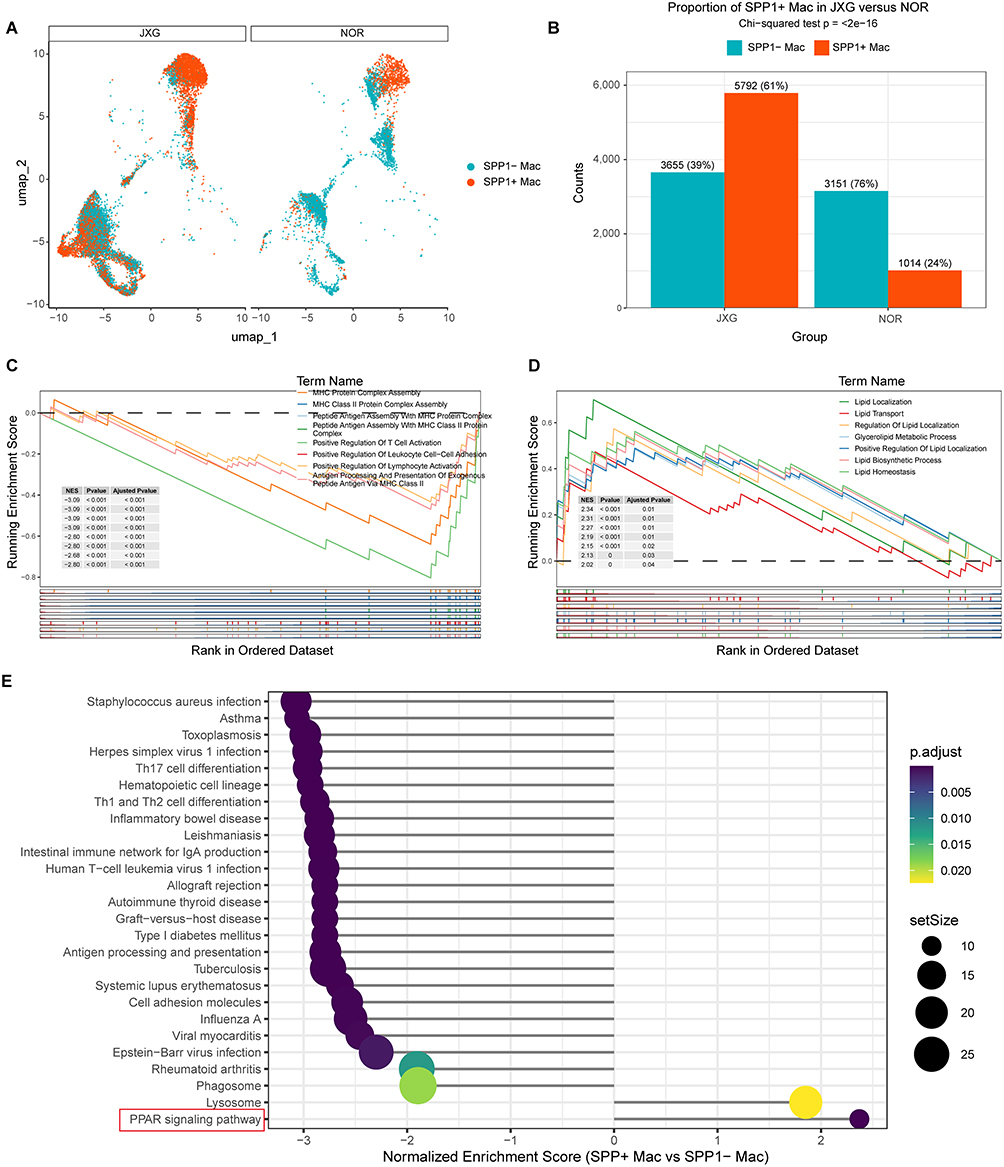 |
Figure 10 Proportion, distribution, and functional characterization of SPP1+ macrophages. (A) Distribution of SPP1+ macrophages across macrophage subclusters. (B) Significantly higher proportion of SPP1⁺ macrophages in JXG than in NOR (p<0.01). (C) GO pathways were significantly negatively enriched in SPP1+ macrophages based on GSEA. (D) GO pathways were significantly positively enriched in SPP1+ macrophages based on GSEA. (E) PPARG signaling was identified as the top positively enriched KEGG pathway based on GSEA. |
Discussion
JXG is the most common non-Langerhans cell histiocytic disorder in children. It typically presents as solitary or multiple yellow-to-red-brown cutaneous nodules or papules on the face, neck, or trunk (cutaneous JXG) and usually follows a benign, self-resolving course.1 However, in a minority of cases, systemic dissemination (systemic JXG) can involve visceral organs and pose life-threatening risks that severely compromise quality of life, imposing heavy psychological and economic burdens on families. Even when confined to the skin, CJXG often exhibits a tendency toward multiplicity and may persist for years, and lesions at critical sites, especially the eye, can lead to irreversible complications such as blindness.27,28
The etiology of JXG remains unclear. It has been proposed as a reactive response to trauma, bacterial or viral infections, physical stimuli, and even an autoimmune process.1 Histologically, JXG is defined by foamy macrophages admixed with a polymorphous inflammatory infiltrate and scattered Touton giant cells,3 underscoring the central role of the inflammatory pathways. Consistent with prior observations, bulk-RNA-seq enrichment analyses of JXG lesions revealed significant activation of immune cell interactions and cytokine–cytokine receptor signaling, with the CCRI pathway emerging as the top KEGG-enriched module. Collectively, these findings suggested that cytokine crosstalk is a pivotal component of the JXG microenvironment. Macrophages are highly plastic innate immune cells that range from pro-inflammatory M1 states to reparative M2 phenotypes. Balanced transitions between these programs are critical for maintaining tissue homeostasis.29,30 JXG is thought to represent a reactive macrophage-mediated process triggered by non-specific insults. However, the immunological mechanisms driving lesion formation remain unclear. Previous studies have indicated that JXG follows a temporal shift from M1‑like macrophages in early lesions to M2‑like populations in more mature or regressing stages.10 However, this phenotypic variability has underscored a major diagnostic challenge: despite the involvement of multiple macrophage subsets, no macrophage-associated marker uniquely identifies JXG, making it difficult to distinguish it from clinically and histologically similar conditions, such as xanthoma and XD.
Previous studies have reported somatic mutations in JXG, including alterations in KRAS, MAP2K1, BRAFV600E, and PIK3CD.31,32 However, recent genomic profiling has indicated that such mutations are uncommon in solitary JXG. Only a small proportion of cases (19%) harbored non-recurrent clonal abnormalities, whereas diffuse or systemic JXG showed more complex genetic changes.33 Despite the low frequency of recurrent driver mutations in classic cases, emerging evidence suggests that genomic instability and specific molecular alterations may critically underpin JXG’s hallmark histopathology. A recent comprehensive survey highlighted that novel somatic mutations in MAPK1 (detected in 27%–74% of cases) and ALK translocations are significant contributors to the molecular landscape of histiocytosis.34,35 These genomic instability mechanisms, particularly those leading to sustained activation of the MAPK/ERK signaling pathway, are thought to drive the characteristic accumulation of foamy histiocytes and the formation of Touton giant cells by disrupting normal myeloid differentiation and lipid processing. Such molecular triggers could induce a state of cellular fusion and metabolic reprogramming, even in the absence of a single, universal driver mutation. These findings suggest that while recurrent genomic alterations are not the sole drivers of classic JXG, the intersection of genomic instability and transcriptional regulatory mechanisms serves as a potential determinant of disease pathogenesis.
TFs occupy upstream positions in signaling networks and coordinate downstream gene expression programs during physiological and pathological processes; therefore, delineating the transcriptional regulatory landscape is critical for understanding the molecular basis of JXG. Using SCENIC, we identified the M6 regulon module as the dominant regulatory program specifically activated in the JXG immune cell clusters. Intersecting M6 targets with CCRI‑DEGs yielded a focused set of CCRI‑related regulons dominated by SPI1, CEBPA, ATF5, and ETV5 TFs, with established roles in myeloid identity, macrophage differentiation, inflammatory tuning, and stress-responsive transcription.36–39 Together, these factors formed an interconnected regulatory axis that may reprogram macrophage states and potentiate cytokine–cytokine receptor interactions, thereby shaping the inflammatory microenvironment characteristics of JXG.
Regulon‑activity mapping demonstrated that these CCRI‑DEG–specific TF regulons exhibited highly concordant activity across macrophage subsets, suggesting coordinated regulation. Pseudotime analysis further revealed that these regulons spanned two major macrophage trajectories, consistent with previous observations that both M1‑ and M2‑like macrophage populations contribute to JXG development. Notably, the expression pattern of SPP1 closely mirrored the activity profiles of CCRI‑DEG–specific TF regulons, indicating that SPP1+ macrophages are a key effector population through which this regulatory network may exert its pathogenic influence.
It is well established that a distinct macrophage subset, characterized by high SPP1 expression, exerts consistent pathophysiological effects across a spectrum of chronic disorders. These SPP1+ macrophages orchestrate inflammatory signaling, extracellular matrix remodeling, fibroblast activation, calcification, and reprogramming of the immune microenvironment, all of which promote disease progression.40 In atherosclerotic plaques, they are associated with calcification, matrix degradation, and plaque instability;41 in the tumor microenvironment, they adopt an immunosuppressive phenotype that correlates with tumor invasion and poor prognosis;13 and in hepatic and pulmonary fibrosis, they secrete profibrotic mediators and modulate cellular adhesion to promote scar formation.14 Similar SPP1-driven microenvironmental alterations and immune dysregulation have been documented in neurodegenerative and autoimmune diseases.15,42 Notably, the contribution of SPP1+ macrophages to JXG pathogenesis has not been investigated.
To assess the role of SPP1 in JXG and determine whether key markers distinguish JXG from clinically similar disorders (xanthoma, XD, and Blau syndrome), we performed IHC on FFPE tissue sections. CD68 showed no significant differences across groups, whereas CD36, SPI1, and IL1B demonstrated variable differential expression. In contrast, SPP1 expression was significantly higher in JXG than in all the other comparison groups, supporting its potential use as a discriminatory marker. Functional analyses further revealed that SPP1+ macrophages suppressed inflammatory and antigen‑presentation pathways, including MHC class II assembly and T‑cell–activating programs, while lipid‑related pathways, such as lipid localization and transport, were significantly upregulated. These features align with previous reports that SPP1+ macrophages often display immunomodulatory profiles and enhanced lipid‑metabolic activity.43–45
Moreover, GSEA‑KEGG analysis suggested significant enrichment of the PPARG signaling pathway in SPP1+ macrophages, consistent with prior evidence that PPARG orchestrates lipid-associated transcriptional programs and shapes the functional phenotype of SPP1‑driven myeloid populations.46,47 Thus, PPARG signaling is known to promote anti-inflammatory, alternatively activated macrophage states, while enhancing lipid uptake and processing.17,48,49 These dual metabolic and immunomodulatory functions are exemplified in atherosclerosis, where PPARG contributes to early foam cell formation and exerts stabilizing anti‑inflammatory effects.50,51 Furthermore, inhibition of PPARG has been shown to reduce lipid uptake in macrophages and mitigate disease progression.52,53 Beyond the metabolic reprogramming of macrophages, direct pharmacological targeting of aberrant signaling pathways has emerged as a promising therapeutic strategy in various macrophage-driven disorders. Recent studies have demonstrated that blocking specific signaling axes—such as the CSF1R pathway or the MAPK/ERK cascade—can effectively modulate macrophage polarization and suppress pathological tissue remodeling.54 In parallel, the development of precision medicine tools, including nanoparticle-mediated delivery systems designed to inhibit intracellular signaling components, has provided new precedents for mitigating inflammatory and fibrotic progression.55 Given these established signaling-blocking mechanisms, there is a compelling rationale for investigating targeted therapies to interrupt the pathogenic macrophage-regulatory axis in systemic or refractory JXG. Taken together, these findings suggested that SPP1+ macrophages may act through PPARG‑associated pathways to integrate lipid metabolism and immune regulation, which were potentially involved in macrophage reprogramming in JXG.
The limitations of this study must be acknowledged. First, we focused exclusively on the CCRI pathway and its associated TF regulons within macrophages, without examining their interactions with other cell populations, such as fibroblasts. Second, due to limited funding, the number of bulk RNA-seq and scRNA-seq samples was relatively small, which may introduce analytical bias; therefore, validation in larger cohorts is necessary. Finally, while our findings suggest an association between PPARG-associated pathways and macrophage reprogramming, this study is limited by the lack of direct experimental perturbation data to establish causality. Therefore, the precise functional contribution of SPP1+ macrophages to JXG pathogenesis, as well as the causal role and downstream effects of the PPARG pathway within these cells, remains to be resolved. Future studies should prioritize experimental dissection of these mechanisms to clarify their roles in disease development and progression, including the use of CRISPR-based genetic manipulation of PPARG or pharmacological interventions with PPARG agonists/antagonists in relevant cellular and animal models.
Conclusions
Our findings demonstrate that cytokine-mediated interactions within macrophages are central to the pathogenesis of JXG. Furthermore, SPP1+ macrophages are closely associated with disease pathogenesis, potentially involving the PPARG signaling pathway as a regulator of immune and metabolic homeostasis.
Data Sharing Statement
The data supporting the findings of this study are available from the corresponding author upon reasonable request. Raw sequencing data were deposited in a public repository (SRA) under accession number PRJNA1402130 (https://www.ncbi.nlm.nih.gov/sra/PRJNA1402130).
Ethics Approval and Consent to Participate
This study was conducted in accordance with the ethical principles outlined in the Declaration of Helsinki. The study protocol, including the collection and analysis of human skin specimens, was reviewed and approved by the Institutional Ethics Committee of Shenzhen Children’s Hospital (Approval No. 202309402). Written informed consent was obtained from parents or legal guardians of the pediatric patients enrolled in the research.
Acknowledgments
We thank Professors Jiaosheng Xu and Bin Zhang (Department of Dermatology, Beijing Children’s Hospital, Capital Medical University, National Center for Children’s Health, Beijing, China) for their expert advice regarding differential diagnosis of JXG. We express our sincere gratitude to Professor Guohong Zhang (Department of Pathology, Provincial Key Laboratory of Infectious Diseases and Molecular Immunopathology, Shantou University Medical College, Shantou, China) for providing technical support for transcriptome sequencing. We also acknowledge Dr. Xiaoning Mao and Dr. Ke Cao (Department of Laboratory Medicine, Shenzhen Children’s Hospital, Shenzhen, China) for their assistance with the skin specimen preservation. Finally, we sincerely thank the patients and their parents for their participation in this study.
Author Contributions
Xiuqing Yuan: Formal analysis (Lead), Writing –original draft (Lead), Conceptualization (Supporting), Writing – review & editing (Lead).
Jianhong Li: Project administration (Lead), Conceptualization (Supporting), Writing – review & editing (Supporting).
Pengyue Tang: Conceptualization (Lead), Funding acquisition (Lead), Investigation (Lead), Writing – review & editing (Lead).
Xingliang Zhang: Investigation (Supporting), Software (Lead), Writing – review & editing (Equal).
Huan Zhang: Methodology (Supporting), Validation (Lead), Writing – review & editing (Supporting).
Ping Li: Project administration (Supporting), Resources (Supporting), Data Curation (Supporting), Writing – review & editing (Supporting).
Mengqi Liu, Hefeng Chen, Rujing Liu, Sihong Zhu, Sihang Feng, Xing Xiao: Resources (Equal), Data Curation (Equal), Writing – review & editing (Equal).
Zhihao Xing, Xin Huang, Chenyao Wang: Supervision (Supporting), Writing – review & editing (Supporting).
All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Guangdong High-level Hospital Construction Fund Clinical Research Project of Shenzhen Children’s Hospital (Grant No. LCYJ2022061).
Disclosure
The authors declare no competing interests.
References
1. Hernandez-Martin A, Baselga E, Drolet BA, Esterly NB. Juvenile xanthogranuloma. J Am Acad Dermatol. 1997;36(3):355–20. doi:10.1016/s0190-9622(97)80207-1
2. Samuelov L, Kinori M, Chamlin SL, et al. Risk of intraocular and other extracutaneous involvement in patients with cutaneous juvenile xanthogranuloma. Pediatr Dermatol. 2018;35(3):329–335. doi:10.1111/pde.13437
3. Fraitag S, Emile J-F. Cutaneous histiocytoses in children. Histopathology. 2022;80(1):196–215. doi:10.1111/his.14569
4. Lambert SA, Jolma A, Campitelli LF, et al. The human transcription factors. Cell. 2018;172(4):650–665. doi:10.1016/j.cell.2018.01.029
5. Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. A census of human transcription factors: function, expression and evolution. Nat Rev Genet. 2009;10:252–263. doi:10.1038/nrg2538
6. Locati M, Curtale G, Diversity MA. Mechanisms, and significance of macrophage plasticity. Annu Rev Pathol. 2020;15:123–147. doi:10.1146/annurev-pathmechdis-012418-012718
7. Aibar S, González-Blas CB, Moerman T, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14:1083–1086. doi:10.1038/nmeth.4463
8. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–721. doi:10.1038/nri3520
9. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–462. doi:10.1016/j.immuni.2016.02.015
10. Wegher LSM, Kazmarek LM, Silva ACF, et al. What is the role of different macrophage subsets in the evolution of juvenile xanthogranulomas? Appl Immunohistochem Mol Morphol. 2022:30e54. doi:10.1097/PAI.0000000000001029.
11. Habeeb M, Vasanthan M. Cabozantinib Encapsulated Lipid and polymeric nanoparticles: in-depth exploration of molecular interactions, formulation strategies, and in vitro studies on hepatocellular carcinoma. Biointerface Res. Appl. Chem. 2024;14. doi:10.33263/BRIAC146.143
12. Lang F, Li Y, Yao R, Jiang M. Osteopontin in chronic inflammatory diseases: mechanisms, biomarker potential, and therapeutic strategies. Biology. 2025;14:428. doi:10.3390/biology14040428
13. Bill R, Wirapati P, Messemaker M, et al. CXCL9:SPP1 macrophage polarity identifies a network of cellular programs that control human cancers. Science. 2023;381:515–524. doi:10.1126/science.ade2292
14. Han H, Ge X, Komakula SSB, et al. Macrophage-derived Osteopontin (SPP1) Protects From Nonalcoholic Steatohepatitis. Gastroenterology. 2023;165:201–217. doi:10.1053/j.gastro.2023.03.228
15. Morse C, Tabib T, Sembrat J, et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J. 2019;54:1802441. doi:10.1183/13993003.02441-2018
16. Skoczyńska A, Ołdakowska M, Dobosz A, et al. PPARs in clinical experimental medicine after 35 years of worldwide scientific investigations and medical experiments. Biomolecules. 2024;14:786. doi:10.3390/biom14070786
17. Hernandez-Quiles M, Broekema MF, Kalkhoven E. PPARgamma in metabolism, immunity, and cancer: unified and diverse mechanisms of action. Front Endocrinol. 2021;12:624112. doi:10.3389/fendo.2021.624112
18. Abdalla HB, Napimoga MH, Lopes AH, et al. Activation of PPAR-γ induces macrophage polarization and reduces neutrophil migration mediated by heme oxygenase 1. Int. Immunopharmacol. 2020;84:106565. doi:10.1016/j.intimp.2020.106565
19. Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–138. doi:10.1093/bioinformatics/btp612
20. Wu T, Hu E, Xu S, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2:100141. doi:10.1016/j.xinn.2021.100141
21. Szklarczyk D, Kirsch R, Koutrouli M, et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023:
22. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi:10.1101/gr.1239303
23. Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 2015;33:495–502. doi:10.1038/nbt.3192
24. Chu T, Wang Z, Pe’er D, Danko CG. Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nat Cancer. 2022;3:505–517. doi:10.1038/s43018-022-00356-3
25. Jiang Y, Chen Z, Han N, Shang J, Wu A. sc-ImmuCC: hierarchical annotation for immune cell types in single-cell RNA-seq. Front Immunol. 2023;14:1223471. doi:10.3389/fimmu.2023.1223471
26. Trapnell C, Cacchiarelli D, Grimsby J, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–386. doi:10.1038/nbt.2859
27. Samara WA, Khoo CTL, Say EAT, et al. Juvenile xanthogranuloma involving the eye and ocular adnexa: tumor control, visual outcomes, and globe salvage in 30 patients. Ophthalmology. 2015;122:2130–2138. doi:10.1016/j.ophtha.2015.06.009
28. Smith JL, Ingram RM. Juvenile oculodermal xanthogranuloma. Br J Ophthalmol. 1968;52:696–703. doi:10.1136/bjo.52.9.696
29. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi:10.1016/j.immuni.2010.05.007
30. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi:10.1172/JCI59643
31. Diamond EL, Durham BH, Haroche J, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov. 2016;6:154–165. doi:10.1158/2159-8290.CD-15-0913
32. Chakraborty R, Hampton OA, Shen X, et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood. 2014;124:3007–3015. doi:10.1182/blood-2014-05-577825
33. Paxton CN, O’Malley DP, Bellizzi AM, et al. Genetic evaluation of juvenile xanthogranuloma: genomic abnormalities are uncommon in solitary lesions, advanced cases may show more complexity. Mod Pathol. 2017;30:1234–1240. doi:10.1038/modpathol.2017.50
34. Pai P, Nirmal A, Mathias L, Jain S, Shetty MG, Sundara BK. Molecular mutations in histiocytosis: a comprehensive survey of genetic alterations. Mol Biotechnol. 2025;67:438–455. doi:10.1007/s12033-024-01072-2
35. Chakraborty R, Hampton OA, Abhyankar H, et al. Activating MAPK1 (ERK2) mutation in an aggressive case of disseminated juvenile xanthogranuloma. Oncotarget. 2017;8:46065–46070. doi:10.18632/oncotarget.17521
36. Qi S, Zhang Y, Kong L, et al. SPI1-mediated macrophage polarization aggravates age-related macular degeneration. Front Immunol. 2024;15:1421012. doi:10.3389/fimmu.2024.1421012
37. Zhang -J-J, Shen Y, Chen X-Y, et al. Integrative network-based analysis on multiple Gene Expression Omnibus datasets identifies novel immune molecular markers implicated in non-alcoholic steatohepatitis. Front Endocrinol. 2023;14:1115890. doi:10.3389/fendo.2023.1115890
38. Zhao Y, Zhao C, Guo H, et al. mTORC2 orchestrates monocytic and granulocytic lineage commitment by an ATF5-mediated pathway. iScience. 2023;26:107540. doi:10.1016/j.isci.2023.107540
39. Hu R-D, Zhang W, Li L, et al. Chromatin accessibility analysis identifies the transcription factor ETV5 as a suppressor of adipose tissue macrophage activation in obesity. Cell Death Dis. 2021;12:1023. doi:10.1038/s41419-021-04308-0
40. Liu Y, Xun Z, Ma K, et al. Identification of a tumour immune barrier in the HCC microenvironment that determines the efficacy of immunotherapy. J Hepatol. 2023;78:770–782. doi:10.1016/j.jhep.2023.01.011
41. Zhao Y, Huang Z, Gao L, Ma H, Chang R. Osteopontin/SPP1: a potential mediator between immune cells and vascular calcification. Front Immunol. 2024;15:1395596. doi:10.3389/fimmu.2024.1395596
42. De Schepper S, Ge JZ, Crowley G, et al. Perivascular cells induce microglial phagocytic states and synaptic engulfment via SPP1 in mouse models of Alzheimer’s disease. Nat Neurosci. 2023;26:406–415. doi:10.1038/s41593-023-01257-z
43. Palma A. The landscape of spp1 + macrophages across tissues and diseases: a comprehensive review. Immunology. 2025;176:179–196. doi:10.1111/imm.13952
44. Liu Z, Gao Z, Li B, et al. Lipid-associated macrophages in the tumor-adipose microenvironment facilitate breast cancer progression. Oncoimmunology. 2022;11:2085432. doi:10.1080/2162402X.2022.2085432
45. Jiang Y, Yu W, Hu T, et al. Unveiling macrophage diversity in myocardial ischemia-reperfusion injury: identification of a distinct lipid-associated macrophage subset. Front Immunol. 2024;15:1335333. doi:10.3389/fimmu.2024.1335333
46. Li G, Zhang Y, Jiang H, et al. PPARG/SPP1/CD44 signaling pathway in alveolar macrophages: mechanisms of lipid dysregulation and therapeutic targets in idiopathic pulmonary fibrosis. Heliyon. 2025;11:e41628. doi:10.1016/j.heliyon.2025.e41628
47. Wang H, Dai S, Xie Y, et al. Colorectal cancer cell’s weapon: rnf32 engages spp1+ macrophages to foster liver metastasis, targeted by indole-3-acetic acid. Adv Sci. 2025:e19735. doi:10.1002/advs.202519735.
48. Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;7:48–52. doi:10.1038/83336
49. Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol. 2010;10:365–376. doi:10.1038/nri2748
50. Bouhlel MA, Derudas B, Rigamonti E, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–143. doi:10.1016/j.cmet.2007.06.010
51. Fernandez AZ. Peroxisome proliferator-activated receptors in the modulation of the immune/inflammatory response in atherosclerosis. PPAR Res. 2008;2008:285842. doi:10.1155/2008/285842
52. Wu L, Liu C, Chang D-Y, et al. The attenuation of diabetic nephropathy by annexin a1 via regulation of lipid metabolism through the ampk/pparα/cpt1b pathway. Diabetes. 2021;70:2192–2203. doi:10.2337/db21-0050
53. You L, Wang T, Li W, et al. Xiaozhi formula attenuates non-alcoholic fatty liver disease by regulating lipid metabolism via activation of AMPK and PPAR pathways. J Ethnopharmacol. 2024;329:118165. doi:10.1016/j.jep.2024.118165
54. Chi Y, Jiang H, Yin Y, et al. Macrophage signaling pathways in health and disease: from bench to bedside applications. MedComm. 2020;2025:e70256. doi:10.1002/mco2.70256
55. Thalij KM, You HW, Aher KB, Bhavar GB, Kumbhar ST, Habeeb M. Advances in lipid-based nanomedicine: pathway specific sirna therapy and optimizing delivery for hepatocellular carcinoma. Int J Nanomedicine. 2025;20:10541–10566. doi:10.2147/IJN.S532246
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.