")
Back to Archived Journals » Oncolytic Virotherapy » Volume 5
Spotlight on talimogene laherparepvec for the treatment of melanoma lesions in the skin and lymph nodes
Authors Orloff M
Received 25 June 2016
Accepted for publication 2 September 2016
Published 4 October 2016 Volume 2016:5 Pages 91—98
DOI https://doi.org/10.2147/OV.S99532
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Video abstract presented by Marlana Orloff.
Views: 1382
Marlana Orloff
Department of Medical Oncology, Thomas Jefferson University, Philadelphia, PA, USA
Abstract: On October 27, 2015, talimogene laherparepvec (T-VEC), a first in class intralesional oncolytic virotherapy, was granted the US Food and Drug Administration approval for the treatment of melanoma in the skin and lymph nodes. Its approval has added yet another therapeutic option to the growing list of effective therapies for melanoma. Though the Phase III OPTiM trial has demonstrated its efficacy as a single agent, the target patient population remains narrow. With numerous effective and tolerable treatments available for unresectable and metastatic melanoma, intralesional therapies such as T-VEC are still finding their niche. T-VEC is now widely accepted as option for treatment; however, its combination with various other agents in an effort to expand its use and synergize with other interventions is still being explored. This article will review the pre-clinical and clinical work that eventually led to the Food and Drug Administration approval of this first-in-class agent, as well as address concerns about clinical application and ongoing research.
Keywords: T-VEC, talimogene laherparepvec intralesional, melanoma, oncolytic virus, virotherapy, immunotherapy
Introduction
In 2016, the algorithm for the management of melanoma continues to evolve. Since the US Food and Drug Administration (FDA) approval for the immune checkpoint inhibitor ipilimumab, an anti-CTLA-4 antibody, in 2011, there has been a flood of additional effective therapies for the treatment of now both advanced and high-risk melanoma. In 2011, following ipilimumab came the BRAF targeted agent vemurafenib, followed by two other targeted agents in 2013, dabrafenib, another BRAF inhibitor, and trametinib, a MEK inhibitor. Shortly thereafter two other immune checkpoint inhibitors pembrolizumab and nivolumab, both anti-PD1 antibodies, were also approved starting in 2014. Then, the combination of nivolumab and ipilimumab was approved for metastatic patients on October 1, 2015. All of these agents are given systemically and demonstrate response rates of 15%–70%, but with varied side effect profiles, time to response, and duration of response.1–6 Parallel to the development of the aforementioned agents, a novel intralesional therapy initially called OncoVEXGM-CSF was also being explored. OncoVEXGM-CSF is a oncolytic virus derived from a modified herpes simplex virus type 1 (HSV-1), coupled with the insertion of a gene that encodes for human granulocyte macrophage colony-stimulating factor (GM-CSF), that has come to be known as talimogene laherparepvec (T-VEC).7 On October 27, 2015, T-VEC, a first-in-class intralesional oncolytic virotherapy, was granted FDA approval for the treatment of melanoma in the skin and lymph nodes. The drug is now being used in many US oncology practices and it is further being widely investigated across multiple cancer types and in combination with numerous other agents. This review aims to summarize the data that led up to the development and FDA approval of T-VEC. Further, there will be a comment on the practical considerations of using such an agent in the clinic and a discussion about future directions.
Background on oncolytic viruses
The very thought that a concomitant viral infection during ones cancer course can be beneficial dates back to the 1800s. There have been numerous case reports detailing variable remissions of hematologic malignancies when patients contract infections such as influenza, hepatitis, and measles during their course.8 It was with these signals that the concept of oncolytic virotherapy was born. While early on, the simple parenteral administration of infectious serum was employed, the more modern approach to oncolytic virotherapy involves the administration of a nonpathogenic attenuated virus that preferentially replicates in tumor tissues and causes death of the cancer cell. While direct oncolysis of the tumor due to accumulation of toxic levels of virus within the cancer cell is one mechanism of cell death, there is evidence that there is an additional modulation of the tumor microenvironment, increased antigen presentation, recruitment of cytotoxic T-cells, and the potential to induce a memory immune response.9 Over the past 20 years, there have been numerous viruses explored for their anticancer potential across many tumor types. These include but are not limited to adenovirus, coxsackie virus, HSV, measles, parvovirus, poliovirus, poxvirus, reovirus, Newcastle disease virus, and Seneca Valley virus that have been studied in everything from melanoma to bladder cancer to glioblastoma.9–11
Background on intralesional therapy in melanoma
Similar to the observations of viral infection causing tumor regression, local bacterial infection was witnessed to also have an anticancer effect. In1891, Dr. William B Coley documented that local injections of streptococci and later the combination streptococci and Serratia marcescens into sarcomas, lymphomas, and melanomas had clinical benefit.12–14 Again, akin to the immune responses ignited with oncolytic viruses, local injection of an immunogenic pathogen, or the desired cytokine itself, has the potential to induce both an immediate antitumor response as well as longer lasting immune memory.11 There has been extensive work in the field of intralesional therapy for melanoma. There has been success with the injection of agents such as bacillus Calmette–Guérin, GM-CSF, interleukin 2, rose Bengal, as well as some of the viruses previously listed.11,12 While surgery remains the mainstay of treatment for “resectable” disease in melanoma, and despite all of the advances in systemic therapy, locoregional therapy remains an option for local unresectable disease.
What is T-VEC?
T-VEC is an oncolytic virus that is directly injected into melanoma skin tumors or involved lymph nodes.15 Its development was based on the experience and success of prior work with oncolytic and intralesional therapies in melanoma and gene therapy.7,16 It is derived from a modified HSV-1, coupled with the insertion of a gene that encodes for human GM-CSF. There has been deletion of two nonessential genes, infected cell protein 34.5 (ICP34.5) and ICP47.6,9 The deletion of ICP34.5, a neurovirulence factor, diminishes viral pathogenicity preventing clinical development of herpes sequelae such as fever blisters. The HSV-1-lacking ICP34.5 then may only preferentially replicate in cancer cells and not healthy cells due to the exploitation of the protein kinase R (PKR) activity differential between the two cell types.10 Healthy cells utilize the PKR pathway to halt viral replication, whereas cancer cells inactivate the PKR pathway in an attempt to maintain continuous cell growth; however, this also permits unchecked viral replication. Additionally, the PKR pathway leads to type I IFN signaling to be preserved in healthy cells but absent in tumor cells, again assisting with selective viral replication.10,17 The deletion of ICP47 not only further decreases neurovirulence by augmenting a CD8+ T-cell response, but also enhances antitumor response by blocking ICP47 suppression of tumor antigen presentation.16,18 In addition, the insertion of the gene encoding GM-CSF aims to further enrich the antitumor response by local recruitment of dendritic cells for antigen presentation, increasing T-cell responsiveness, and decreasing both T-regulatory cells and myeloid derived suppressor cells.12,19 The combination of direct oncolysis, controlled virulence, preferential replication, enhanced antigen presentation, augmented antitumor tumor microenvironment, and the potential for both local and systemic antitumor activity led to the enthusiasm and research that would eventually lead to the clinical investigation of T-VEC.
Early clinical trials with T-VEC
Phase I
In the Phase I trial of then OncoVEXGM-CSF, 13 patients with varied malignancies and prior treatment were treated with single variable doses, and 17 patients were treated with multiple variable doses.20 Though patient’s primary malignancies varied amongst breast, colorectal, melanoma, and squamous cell carcinoma of the head and neck, all had refractory cutaneous or subcutaneous metastases and were treated with intratumoral injections. In the first cohort, individual patients were treated with single escalating doses of the drug, at 106, 107, and 108 pfu/mL. Of note, only patients who were HSV seropositive received the 108 pfu/mL dose. In the HSV negative group the dose of 107 pfu/mL was determined to be the maximal tolerated dose. The second cohort of patients received multiple doses at varied escalations depending on their seropositivity. In general, the treatment was very well tolerated with minimal side effects including pyrexia, nausea, vomiting, anorexia, and fatigue, but mostly all were ≤ grade 2. Inflammation of injected and uninjected lesions was observed. In addition to safety and tolerability, observations were made specific to patient’s pre- and post-HSV seropositivity. Overall, side effects were more evident in the pretreatment seronegative patients. Patients who were seronegative prior to treatment strongly converted to seropositivity 3 to 4 weeks after their first dose. Immunity to HSV was measured in pretreatment seropositive patients, and while the level of anti-HSV antibody increased during the first few injections, this leveled off and did not seem to have an obvious effect on antitumor response. One patient who had been seropositive developed “fever blisters” during the treatment. The herpetic eruption was swabbed and tested but was not reflective of systemic infection with OncoVEXGM-CSF. There were no complete responses (CR) or partial responses (PR) witnessed. However, a number of patients had “flattening” of their lesions, and stable disease was seen. It was also observed that some lesions appeared to “progress”, getting larger and more inflamed, but ultimately was either called stable disease or tumor necrosis was found on biopsy. The results of this study and the signal that the virus was well tolerated, easily administered, and some evidence of antitumor response, that led the subsequent Phase II trial in patients with melanoma.
Phase II
The Phase II trial of JS1/34.5-/47-/GM-CSF (previously OncoVEXGM-CSF) was conducted among 50 patients with stage IIIc and IV metastatic melanoma.21 Patients were enrolled between January 2006 and February 2008, prior to the FDA approval of novel targeted and systemic immunotherapeutic agents as previously discussed. Prior treatments in this patient population included surgery, dacarbazine, temozolomide, and interleukin 2. Patients were injected first with the 106 pfu/mL dose, as an attempt to seroconvert, based on what was experienced with the high 108 pfu/mL dose in seronegative patients in the Phase I study. Up to 4 mL was injected intratumorally. A second injection of 108 pfu/mL was administered after 3 weeks and then every 2 weeks until disease progression or intolerance. Official disease assessments were made after six injections and then every 12 weeks thereafter. The demographics of the treatment population were such that 10 patients had stage IIIC disease and 40 patients had stage IV (20 patients had M1c disease). A median of six injections were given and 13 objective responses were seen. There were eight CR and five PR, giving an overall response rate (ORR) of 26%. Twelve of the responses were durable for greater than 6 months (7–31 months at the time of the publication). Ultimately, a disease control rate that was maintained for greater than 3 months was noted to be 50%. There were also observations that both local and distant non-injected lesions may respond. This observation is further defined in a post hoc analysis that will be discussed in this article. The adverse events were similar to what were seen in the Phase I study and all were ≤ grade 2. Autoimmune vitiligo was witnessed in three patients. The ORR, tolerability, observation of CR, and antitumor response in distant non-injected lesions commanded the pursuit of a randomized Phase III trial in efforts for FDA approval.
Observations: post hoc analysis
As already noted, during the Phase II study there was the observation that both local and distant uninjected lesions may respond when accessible tumors are injected with JS1/34.5-/47-/GM-CSF. Such a finding was further investigated in the post hoc analysis of the systemic effects of TVEC (previously JS1/34.5-/47-/GM-CSF).22 While the analysis was ultimately published following the randomized Phase III trial and the eventual FDA approval, the results of the analysis are worth noting. When considering the responders in the Phase II trial as already detailed, 11 of the 23 patients (47.8%) with disease control (CR =8, PR =5, and standard deviation =10) had a ≥30% tumor burden reduction in noninjected lesions. Further two of those patients had ≥30% tumor burden reduction in non-injected distant visceral lesions. Notably, the median time to lesion response was the quickest for directly injected lesions at 18.4 weeks than non-injected non-visceral lesions at 23.1 weeks and visceral lesions at 51.3 weeks. These findings were then further evaluated during the subsequent randomized Phase III trial. The expectations of response, nonresponse, and time to response for the varied types of lesions noted in Phase II and additionally in the phase III were important to consider when this agent ultimately made it to clinical practice.
Pivotal phase III and FDA approval
As Phase II was completing, the randomized phase III trial (OPTiM) was underway.15 From May 2009 to July 2011, 436 patients across 64 centers in four countries were enrolled. Patients who were in stages IIIB–IV and not surgically resectable were eligible as long as there was at least one cutaneous lesion ≥10 mm in diameter. However, stage IV patients with greater than three visceral metastases, except lung or lymph node, or any visceral metastases >3 cm or uncontrolled liver metastases were excluded. Such an exclusion criterion was likely based on experience gained from the aforementioned post hoc analysis noting only rare and delayed response in non-injected, non-visceral lesions. Higher risk visceral lesions such as those greater than 3 cm, or uncontrolled liver metastases that could result in rapid clinical deterioration if not treated, are unlikely to benefit from single-agent T-VEC and would require systemic therapy. Patients were randomized to receive either T-VEC as given in Phase II or GM-CSF alone injected just subcutaneously at 125 µg/m2 daily for 14/28-day cycle. When considering patient demographics, 57% had stage IIIB, IIIC, and M1a disease and 47% were treatment naïve. Similar to what was seen in Phase II, the ORR in the T-VEC arm was 26.4% versus 5.7% in the GM-CSF arm. Thirty-two patients in the T-VEC arm had a CR (10.8%). Median time to response was again similar to what was seen prior in the T-VEC arm, which was about 4.1 months (1.2–16.7 months). A durable response rate (DRR), which was the primary endpoint and defined as CR + PR lasting ≥6 months, was estimated to be 16.3% in the T-VEC arm versus 2.1% in the GM-CSF arm. In the additional exploratory analyses, it was found that patients getting T-VEC as the first-line treatment did significantly better than patients who were previously treated for their metastatic disease (24% versus 10% DRR). Further, the differences in DRR between T-VEC and GM-CSF group were more significant in the earlier stage IIIB and IIIC, and IVM1a patients (33% versus 0%, 16% versus 2%) as compared to the IVM1b and IVM1c patients (3% versus 4%, 7% versus 3%). Such a contrast is not unexpected given the knowledge about the potential but lower likelihood of visceral non-injected tumor response seen with T-VEC in the Phase II study. The side effects were again similar to what were seen in the Phase I and Phase II studies, though slightly more grade 3/4 events were present. It was generally very well tolerated, though there were six patients with grade 3/4 cellulitis in the T-VEC arm. Ultimately, given the reported outcomes in Phase III, the again witnessed ORR and DRR, tolerability, observation of CR, and also possibility of antitumor response in distant non-injected lesions, the FDA approved T-VEC on October 27, 2015 as the first FDA-approved oncolytic virus therapy for the treatment of melanoma lesions in the skin and lymph nodes.
Recent publication by Andtbacka et al further defines the patterns of clinical response seen in patients treated with T-VEC in the Phase III OPTiM trial.23 Similar to what was seen as part of the post hoc analysis performed in the Phase II trial, after injection with T-VEC, there was a ≥50% decrease in size in 64% of injected lesions. In addition, 34% of uninjected non-visceral and 15% of visceral lesions also had reduction in size. CR was seen in 47% of injected lesions, 22% of non-injected non-visceral lesions, and 9% of visceral lesions. Of the 9% of visceral lesions that responded, 81% were in the lungs, 15% in the liver, and 4% in the thyroid. Median time to response of visceral lesions was 12.3 weeks, which is notably shorter than that previously reported in Phase II where non-injected visceral lesions had a median time to response of 51.3 weeks.
T-VEC in the clinic: practical considerations
Patient selection
At diagnosis, about 15% of patients had stage III–IV disease, and about 5% may have what is considered to be injectable disease by definition of what was injected in the aforementioned T-VEC clinical trials.24,25 It has been noted in the discussion of both the phase II and phase III trials, and other commentaries that T-VEC as a single agent should be reserved for earlier stage, limited disease.15,21,26 While this is a narrow patient population, the population does exist and would benefit from a low toxicity treatment with the potential for durable response. When evaluating a new melanoma patient in the clinic, given the increasing number of options for intervention, the option of single-agent T-VEC is discussed carefully. For some patients, an every 2-week injection without the unpredictable immune-related side effects seen with the checkpoint inhibitors, or other systemic side effects seen with targeted BRAF and MEK inhibitors, may be desirable.
Handling and administration
When T-VEC was first made commercially available, there was accompanying guidance on the handling and administration of the agent. It was recommended that since it is a live virus capable of replication, albeit attenuated, there should be appropriate precautions. However, as it was a first-in-class agent, specifics regarding biohazard precautions were not previously known, and there was room for varied interpretation across institutions. As per the package insert, health care providers who are immunocompromised or pregnant should not prepare or administer the agent. Person administering should wear personal protective equipment such as a gown or lab coat, eye protection, and gloves. Any materials that may come in contact with T-VEC should be disposed in accordance with accepted biohazard precautions. In Phase II, 102 swabs were taken from the injection sites in 19 patients at 24–72 hours following the first injection.21 Only one swab tested positive for HSV at <10 pfu/mL, and was negative just prior to the second injection. However, it is still recommended that the exterior of the dressing be wiped with alcohol and the injection site be covered for at least in the first week after each treatment. It is worth noting that the product needs to be stored at –90°C to –70°C and requires about 30–60 minutes for thawing until it can be administered. Further, once thawed the agent needs to be administered immediately, though it can be stored in the refrigerator for 24 (106 pfu/mL) or 48 (108 pfu/mL) hours depending on the concentration.27,28 Such a narrow window for freezing, thawing, and administration requires office and clinic organization to ensure appropriate scheduling and treatment. There is no restriction on the type or level of practitioner allowed to administer T-VEC. With appropriate training, mid-level providers such as nurse practitioners and physician assistants are well suited to administer T-VEC injections in the clinic. Given the aforementioned constraints regarding drug preparation and administration, utilizing staff beyond the physician to ensure timely treatment of the patient is something that should be considered and has been accepted by the mid-level community.28
Financial
Compared with other novel melanoma agents such as ipilimumab, pembrolizumab, nivolumab and the targeted agents vemurafenib, dabrafenib, trametinib, and cobimetinib, which carry a cost of >US $100,000 on average for a course of treatment, the cost of T-VEC was estimated to be around US $65,000. Shortly after FDA approval, there was comment that the drug company behind T-VEC, Amgen, would work with institutions to cap cost per patient per full treatment course at around $65,000. Further, there are a number of assistance programs to help aid patients who may have high co-pays or need free medicine.
Future directions and ongoing clinical trials
As noted previously, patients who will benefit from single-agent T-VEC exist but they form a narrow population. To further expand the utilization of this novel oncolytic virus to other patient populations and to capitalize on its successes, there has been a tremendous effort to further explore its use. See Table 1 for completed T-VEC trials and Table 2 for a summary of ongoing T-VEC trials in melanoma.
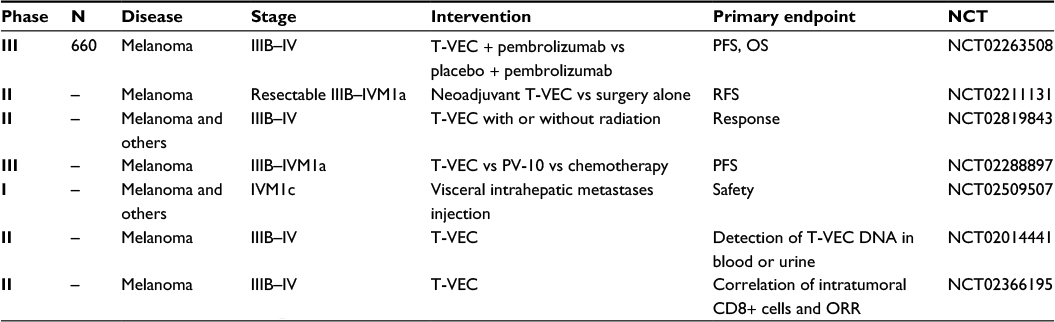 | Table 1 Previous T-VEC trials in melanoma Abbreviations: T-VEC, talimogene laherparepvec; PFS, progression-free survival; OS, overall survival; RFS, recurrence-free survival; ORR, overall response rate. |
 | Table 2 Ongoing T-VEC trials in melanoma Abbreviations: T-VEC, talimogene laherparepvec; PR, partial response; CR, complete response; ORR, overall response rate. |
Combinations
There are now a number of effective therapies for advanced melanoma, and the trend has been that a combination of these agents improves outcomes, which has been shown by the effectiveness of combination checkpoint inhibitors and combination targeted agents.3,6 While most effective combinations thus far have been within a drug class, there is growing enthusiasm for combining therapies between classes and treatment modalities.15 There is already data supporting the combination of T-VEC, an intralesional oncolytic virus, with a number of checkpoint inhibitors. The basic principal supporting the combination is the concept that the introduction of an oncolytic virus may alter an otherwise non-immunogenic tumor microenvironment into one primed for checkpoint inhibitors, in addition to its already direct oncolysis. The possibility of increased antigen presentation and T-cell recruitment due to cell death from oncolysis is attractive to checkpoint inhibitor-stimulated T cells. The results of the Phase Ib trial of ipilimumab with or without T-VEC were reported first at ASCO 2014 meeting prior to T-VEC’s FDA approval (NCT01740297).29 In this Phase Ib, 19 patients were enrolled and 18 were treated with the combination. There was an increase in grade 3/4 side effects at 32%, which was attributed to ipilimumab. The ORR was 41%, with 24% CR and 18% PR, and 35% with stable disease. Median time to response was 2.9 months. Follow-up data then presented at 2015 ASCO again noted an impressive ORR of 56% (33% CR) and DRR of 44%. Survival at 12 and 18 months were 72.2% and 67%, respectively. While the side effect profile resembled that of mostly ipilimumab, the ORR and time to response were much improved over each agent when used alone. Recently updated results of the Phase Ib trial were reported by Puzanov et al.30 After final analysis, ORR was reported at 50% and DRR at 44%. Eighteen-month progression-free survival was 50% and 18-month overall survival was 67%. A Phase 2 was planned and recruited patients; however, with the success and increased tolerability of the anti-PD1 antibodies, combination trials quickly moved to the combination of T-VEC and pembrolizumab. MASTERKEY-265 was a Phase Ib study of T-VEC and pembrolizumab for unresectable stage IIIB-IV melanoma, which has continued and has now moved on to a randomized, placebo-controlled Phase III trial (NCT02263508).31 Results of the efficacy from Phase Ib were recently presented at ASCO 2016 and noted a confirmed/non-confirmed ORR of 48%/57% and CR of 14%/24%.29 Grade 3/4 side effects were reported at 33%, with most common being fatigue, pyrexia, and chills. The side effects seen in the trial were similar to those seen with pembrolizumab, or exaggerated T-VEC related side effects, which are likely more manageable than those seen with ipilimumab. Phase III is underway and set to accrue 660 patients and will randomize the patients in a ratio of 1:1 to pembrolizumab plus T-VEC versus pembrolizumab plus intralesional placebo.
Additional combination trials have been conducted and are ongoing notably in other cancer types that aim to explore T-VEC in combination with chemotherapy and/or radiation in tumors such as breast cancer, head and neck cancer, and sarcoma (NCT02779855, NCT01161498, NCT02453191).
While there is clear evidence of TVEC’s as a single agent and in combination with other anticancer therapies, there is also an effort to selectively diminish the antiviral response without impairing the antitumor response. Such a concept has been explored and the combination of oncolytic viruses and histone deacetylase inhibitors (HDACIs) may have this effect.32 While HDACIs are known to have varied antitumor effects, they are also found to interfere with the host antiviral immune response by weakening the IFN signaling. A variety of HDACIs including trichostatin A and valproic acid have been explored in combination with HSV oncolytic viruses. Notably, trichostatin A was found to increase viral replication and antineoplastic activity of HSV-1 deficient in ICP34.5 (R849) in oral squamous cell cancer.33 In a review by Forbes et al, there is a summary of a number of other potential pharmacological interventions that when combined with oncolytic viruses may help to enhance antitumor immunity and minimize unwanted antiviral response.34 Such approaches include classic chemotherapy agents like cyclophosphamide and bortezomib, drugs targeting the mTOR pathway like rapamycin and everolimus, DNA demethylating agents like 5-azacytidine, as well as novel viral sensitizers. The strategy of combination therapy to enhance selective viral replication, augment tumor cell oncolytic virus sensitivity, and modulate antitumor and antiviral response may be considered for T-VEC in the future.
Schedule
Though T-VEC was first investigated as a direct intratumoral injection into cutaneous, subcutaneous, and lymph node metastases not amenable to surgical resection, there is a possible role for its use in the neoadjuvant setting. There is an ongoing trial exploring the role of T-VEC administration prior to surgical excision (NCT02211131). While primary outcome is recurrence-free survival, data on rates of R0 surgical resections and pathologic CR, is also being collected.
Visceral injections
Alternative delivery methods to more visceral sites of disease are also being explored. The liver as a site of metastases is at times a difficult disease state to manage. Though there are a number of locoregional therapies for diseases such as hepatocellular carcinoma, liver metastases from colorectal cancer, and liver metastases from uveal melanoma, further therapeutic options are needed. There is an ongoing study evaluating T-VEC injections directly into liver tumors (NCT02509507). For diseases such as uveal melanoma metastatic to the liver, where checkpoint inhibitors have not been successful, potential manipulation of the tumor microenvironment to a more immunogenic environment may be of clinical interest.
Correlative science
In addition to ongoing trials exploring novel combinations, schedules, and delivery methods, there are also ongoing trials investigating patterns of immune response and viral propagation. There is a trial to evaluate the patterns of shedding of T-VEC systemically in the blood and urine (NCT02014441). In addition, there is a trial investigating the correlation of intratumoral CD8+ cell density and response rate (NCT02366195).
Conclusion
T-VEC is now the first FDA-approved oncolytic virus to make it to the clinic. Its development and approval are part of a global cancer immunotherapy renaissance, which has dramatically altered the way in which cancer is approached, most notably melanoma. While there is a focused role for T-VEC’s current on-label use as a single agent, ongoing research on combination therapy and other modifications to dose, administration, and schedule will certainly further expand the drug’s potential in not only melanoma but also in many other cancers.
Disclosure
The authors report no conflicts of interest in this work.
References
Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11(2):155–164. | ||
Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. | ||
Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–39. | ||
Ribas A, Hamid O, Daud A, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315(15):1600–1609. | ||
Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16(4):375–384. | ||
Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. | ||
Liu BL, Robinson M, Han ZQ, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10(4):292–303. | ||
Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15(4):651–659. | ||
Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14(9):642–662. | ||
Kohlhapp FJ, Kaufman HL. Molecular pathways: mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res. 2016;22(5):1048–1054. | ||
Dharmadhikari N, Mehnert JM, Kaufman HL. Oncolytic virus immunotherapy for melanoma. Curr Treat Options Oncol. 2015;16(3):326. | ||
Sloot S, Rashid OM, Sarnaik AA, Zager JS. Developments in intralesional therapy for metastatic melanoma. Cancer Control. 2016;23(1):12–20. | ||
Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin Orthop Relat Res. 1991;(262):3–11. | ||
Bickels J, Kollender Y, Merinsky O, Meller I. Coley’s toxin: historical perspective. Isr Med Assoc J. 2002;4(6):471–472. | ||
Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–2788. | ||
Harrington KJ, Puzanov I, Hecht JR, et al. Clinical development of talimogene laherparepvec (T-VEC): a modified herpes simplex virus type-1-derived oncolytic immunotherapy. Expert Rev Anticancer Ther. 2015;15(12):1389–1403. | ||
Davis KL, Korom M, Morrison LA. Herpes simplex virus 2 ICP34.5 confers neurovirulence by regulating the type I interferon response. Virology. 2014;468–470:330–339. | ||
Goldsmith K, Chen W, Johnson DC, Hendricks RL. Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J Exp Med. 1998;187(3):341–348. | ||
Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. 2010;17(3):718–730. | ||
Hu JC, Coffin RS, Davis CJ, et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 2006;12(22):6737–6747. | ||
Senzer NN, Kaufman HL, Amatruda T, et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpes virus in patients with unresectable metastatic melanoma. J Clin Oncol. 2009;27(34):5763–5771. | ||
Kaufman HL, Amatruda T, Reid T, et al. Systemic versus local responses in melanoma patients treated with talimogene laherparepvec from a multi-institutional phase II study. J Immunother Cancer. 2016;4:12. | ||
Andtbacka RH, Ross M, Puzanov I, et al. Patterns of clinical response with talimogene laherparepvec (T-VEC) in patients with melanoma treated in the OPTiM Phase III clinical trial. Ann Surg Oncol. Epub 2016 Jun 24. | ||
Balch CM, Gershenwald JE, Soong SJ, Thompson JF. Update on the melanoma staging system: the importance of sentinel node staging and primary tumor mitotic rate. J Surg Oncol. 2011;104(4):379–385. | ||
Beasley G, Tyler D. In-transit melanoma metastases: incidence, prognosis, and the role of lymphadenectomy. Ann Surg Oncol. 2015;22(2):358–360. | ||
Coffin R. Interview with Robert Coffin, inventor of T-VEC: the first oncolytic immunotherapy approved for the treatment of cancer. Immunotherapy. 2016;8(2):103–106. | ||
Imlygic Insert. Available from: http://pi.amgen.com/united_states/imlygic/imlygic_pi.pdf. Accessed June 10, 2016. | ||
Hoffner B, Iodice GM, Gasal E. Administration and handling of talimogene laherparepvec: an intralesional oncolytic immunotherapy for melanoma. Oncol Nurs Forum. 2016;43(2):219–226. | ||
Puzanov I, Milhem MM, Andtbacka RHI, et al. Primary analysis of a phase Ib multicenter trial to evaluate safety and efficacy of talimogene laherparepvec (T-VEC) and ipilimumab (ipi) in previously untreated, unresected stage IIIB-IV melanoma. J Clin Oncol. 2014;32:5s. | ||
Puzanov I, Milhem MM, Minor D, et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol. 2016;34(22):2619–2626. | ||
Long GV, Dummer R, Ribas A, et al. Efficacy analysis of MASTERKEY-265 phase 1b study of talimogene laherparepvec (T-VEC) and pembrolizumab (pembro) for unresectable stage IIIB-IV melanoma. J Clin Oncol. 2016;34(suppl; abstr 9568). | ||
Marchini A, Scott EM, Rommelaere J. Overcoming barriers in oncolytic virotherapy with HDAC inhibitors and immune checkpoint blockade. Viruses. 2016;8(1):9. | ||
Katsura T, Iwai S, Ota Y, Shimizu H, Ikuta K, Yura Y. The effects of trichostatin A on the oncolytic ability of herpes simplex virus for oral squamous cell carcinoma cells. Cancer Gene Ther. 2009;16(3):237–245. | ||
Forbes NE, Krishnan R, Diallo JS. Pharmacological modulation of anti-tumor immunity induced by oncolytic viruses. Front Oncol. 2014;4:191. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.