Back to Journals » Drug Design, Development and Therapy » Volume 11
Spotlight on olaratumab in the treatment of soft-tissue sarcoma: design, development, and place in therapy
Received 9 August 2017
Accepted for publication 22 November 2017
Published 13 December 2017 Volume 2017:11 Pages 3579—3587
DOI https://doi.org/10.2147/DDDT.S121298
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Manfred Ogris
Elizabeth J Davis,1 Rashmi Chugh2
1Department of Internal Medicine, Division of Hematology/Oncology, Vanderbilt University Medical Center, Nashville, TN, 2Department of Internal Medicine, Division of Hematology/Oncology, University of Michigan, Ann Arbor, MI, USA
Abstract: Soft-tissue sarcoma (STS) is a heterogeneous group of tumors that arise from mesenchymal tissue. The prognosis of metastatic STS is poor with a life expectancy of 12–18 months. The mainstay of treatment is chemotherapy with an anthracycline. The addition of other chemotherapeutic agents to an anthracycline has been studied with limited success in improving outcomes for STS patients. Olaratumab is a fully human IgG1 monoclonal antibody that binds to platelet-derived growth factor receptor α (PDGFR-α) preventing binding of its ligands and receptor activation. This drug recently received the US Food and Drug Administration’s accelerated approval for the treatment of advanced STS when combined with doxorubicin. This approval was based upon an improvement in overall survival of patients receiving the combination of doxorubicin and olaratumab compared to those receiving doxorubicin alone. In this review, we have analyzed the available literature on the development of olaratumab, its clinical utility, and its place in therapy. Based on early-phase clinical trials, olaratumab appears to be a promising agent for the treatment of STS.
Keywords: olaratumab, doxorubicin, PDGFR-α, soft-tissue sarcoma
Introduction
Soft-tissue sarcoma (STS) is a rare and heterogeneous group of malignancies. Approximately, 12,000 cases occur annually in the United States, accounting for less than 1% of all cancer diagnoses.1 Within STS, there are more than 50 different histologic subtypes, with variability in their demographics, natural history, and clinical outcomes.2 Treatment for localized disease typically includes surgical resection and radiation therapy, while the role of chemotherapy remains controversial. However, in metastatic STS, systemic treatment is the mainstay of therapy. For the majority of STS histologies, first-line treatment is generally an anthracycline, potentially in combination with another agent.
Until a recent Phase II study evaluating doxorubicin and the platelet-derived growth factor receptor α (PDGFR-α) antibody, olaratumab, numerous drugs have been added to anthracyclines with limited success in improving outcomes for metastatic STS patients. Other cytotoxic agents such as ifosfamide and dacarbazine are often added to improve response rates but at the cost of increased toxicity. A myriad of other agents have been combined with doxorubicin in Phase I, II, and III studies in metastatic STS, such as ifosfamide analogues (palifosfamide,3 evofosfamide4) and targeted agents (bevacizumab,5 cixutumumab6). All studies have shown varying degrees of benefit and toxicity, including prohibitive toxicity, but until recently, no study has demonstrated a significant survival advantage. Thus, the prognosis for metastatic STS has remained poor with overall survival (OS) averaging 12–18 months.7–9 Novel treatment strategies to improve outcomes for patients with STS are desired, and olaratumab appears promising in its early study.
Olaratumab (formerly IMC-3G3) is a fully human IgG1 monoclonal antibody that selectively binds to PDGFR-α. This binding prevents the platelet derived growth factor (PDGF) ligands, PDGF-AA, PDGF-BB, PDGF-AB, and PDGF-CC, from attaching to the receptor and thus prevents receptor activation. It also prevents ligand-induced phosphorylation of downstream signaling molecules, AKT and mitogen-activated protein kinase (MAPK).10 Olaratumab has been studied in the treatment of advanced malignancies and was granted its first global approval in October 2016. Under its Accelerated Approval Program, the US Food and Drug Administration approved olaratumab for the treatment of advanced STS when used in combination with doxorubicin. In November 2016, the European Medicines Agency granted conditional approval for the same indication.11 In this review, we will analyze the design and development of olaratumab and discuss its place in the treatment of STS as well as future directions.
Design
PDGF/PDGFR pathway in cancer
PDGF is a serum growth factor for fibroblasts, smooth muscle cells, and glial cells. The PDGF ligands include four inactive monomeric polypeptide chains (A, B, C, and D) that become active after linkage via disulfide bonds. When dimerized, the five ligands, PDGF-AA, PDGF-BB, PDGF-CC, PDGF-DD, and PDGF-AB, activate two structurally related tyrosine kinase receptors, PDGFR-α and PDGFR-β. Activation of the PDGF/PDGFR system has a very important role in embryogenesis. However, in adulthood, this system is primarily involved in tissue repair and wound healing.
Overactivity of PDGF/PDGFR is associated with many pathologic states including fibrotic and vascular diseases as well as cancer.12–14 Specifically, PDGFR-α has direct tumor as well as stromal effect. Mutations and overexpression of PDGFR-α have been described in numerous malignancies, but the implications of these molecular abnormalities on treatment strategies are not well defined. For example, osteosarcoma, chondrosarcoma, Ewing’s sarcoma, leiomyosarcoma, liposarcoma, undifferentiated pleomorphic sarcoma, prostate, breast, ovarian, and lung cancers have all demonstrated increased expression of PDGFR-α.10,15 Importantly, this overexpression has been associated with worse disease-related outcomes in osteosarcoma, ovarian, breast, and prostate cancers.16–19 Despite PDGFR overexpression, targeting PDGFR with the tyrosine kinase inhibitor, imatinib, has been an unsuccessful strategy in two Phase II studies in patients with PDGFR-positive sarcomas. In contrast, activating mutations in PDGFR-α in a subset of patients with gastrointestinal stromal tumors (GISTs) can be successfully targeted with imatinib.20,21 Occasionally, these PDGFR-mutant GISTs can exhibit primary resistance to tyrosine kinase inhibitors, but affected patients have demonstrated clinical benefit with olaratumab which will be discussed in greater detail subsequently.22,23 These findings suggest that targeting PDGFR as a therapeutic strategy is complex and needs further investigation.24,25
The PDGF/PDGFR-α signaling pathway in cancer involves both autocrine and paracrine growth. The surrounding stromal fibroblasts, blood vessels, and tumor cells express PDGFR-α. Coexpression of PDGF and PDGFR-α in multiple cancer types as well as gene amplification and activation of PDGFR-α provides evidence for the presence of autocrine growth. The role of paracrine growth in the PDGF/PDGFR pathway is supported by data obtained from melanoma xenograft models where PDGF-driven paracrine signaling was necessary for malignant stromal development.26 A second study demonstrated that inducing the PDGF-CC ligand in melanoma cells led to activation of PDGFR-α on stromal fibroblasts causing increased tumor growth.27 These findings provide additional rationale as to how disruption of the PDGF/PDGFR pathway may be a reasonable anti-cancer strategy.
Preclinical studies of olaratumab
Given the preclinical evidence supporting the targeting of the PDGF/PDGFR-α pathway, Loizos et al28 developed five novel antibodies inhibiting PDGFR-α and subjected them to vigorous in vitro evaluation. While all five antibodies demonstrated binding to cell surface receptors, olaratumab was one of two antibodies with the highest binding affinity, ED50 =0.04 nmol/L. Additionally, olaratumab demonstrated strong blocking activity inhibiting receptor binding with IC50s of 0.24 nmol/L and 0.43 nmol/L, when exposed to PDGF-AA and PDGF-BB, respectively.
All antibodies were evaluated for downstream effects, inhibiting ligand-induced receptor phosphorylation. Olaratumab demonstrated the strongest inhibition in response to PDGF-BB. Inhibition of 50% of PDGFR-α phosphorylation occurred at 0.25 nmol/L. Olaratumab also demonstrated the greatest blocking of PDGF-AA and PDGF-BB mitogenesis with EC50s of 8.3 nmol/L and 1.25 nmol/L, respectively. Comparatively, for the other antibodies, blocking of PDGF-AA mitogenesis occurred at EC50s ranging from 14–26 nmol/L.
PDGFR-α antibodies were evaluated for their ability to affect downstream phosphorylation of the key cancer cell survival kinases, Akt and MAPK, in PDGFR-A expressing cell lines, including leiomyosarcoma (SKLMS-1) and gliobastoma (U118). The strongest inhibition was observed with olaratumab, 100% of Akt and 80% of MAPK. In vivo evaluations were also performed in SKLMS-1 and U118 mice xenografts. In both leiomyosarcoma and glioblastoma, olaratumab significantly inhibited tumor growth at 6, 20, and 60 mg/kg doses but was more effective in U118 xenografts. The Cmin for efficacy varied from 155 to 258 μg/mL.28 Collectively, these data suggest a potential role for olaratumab in tumors that express PDGFR-α.
Mechanism of action
Olaratumab selectively binds human PDGFR-α with high affinity. This prevents binding of PDGF ligands, including PDGF-AA, PDGF-BB, PDGF-AB, and PDGF-CC. Without ligand binding, receptor activation and the downstream signaling cascade does not occur (Figure 1).
 | Figure 1 Olaratumab mechanism of action. |
Pharmacokinetics
Initial pharmacokinetic data for olaratumab were generated from a Phase I study of 19 patients with a variety of solid tumors. Blood was collected up to 168 hours after the first dose and 336 hours after the fourth dose in the 4, 8, and 16 mg/kg weekly dosing cohorts. In the 15 and 20 mg/kg biweekly cohorts, blood was obtained up to 336 hours post-infusion after the first and third doses. Serum olaratumab was measured by a validated ELISA. After first and subsequent doses, the half-life of olaratumab varied from 3.08 to 7.79 and 3.69 to 11.3 days, respectively. After repeated doses of olaratumab, serum concentrations of olaratumab were higher than after a single dose consistent with drug accumulation. The geometric mean Cmin for both 16 mg/kg weekly and 20 mg/kg biweekly was >155 μg/mL, which was above the efficacious concentration in xenograft models as noted previously.29
Pharmacokinetic data from a second Phase I study conducted in Japanese patients with advanced solid tumors demonstrated comparable results. There were three cohorts for this study: olaratumab dosed at 10 and 15 mg/kg on days 1 and 8 of a 21-day cycle and 20 mg/kg given every 2 weeks. Data were available for 15 patients. Serum olaratumab concentrations again increased with repeated dosing, confirming accumulation. Olaratumab 15 mg/kg every 3 weeks resulted in pre-dose blood concentrations >155 μg/mL. They also compared steady-state clearance and exposure to the aforementioned study and found no differences in these parameters, suggesting similarities between Asian and non-Asian patients. However, the small sample size restricted the power for a statistical analysis, so limited conclusions can be drawn. Individual terminal elimination half-life (T1/2) values measured after first and repeated doses were 4.42–9.38 and 4.06–8.83 days, respectively. However, given the short pharamacokinetic sampling time, T1/2 required further validation.30
The largest dataset of pharmacokinetic data was reported from the Phase Ib/II study of olaratumab in STS. Olaratumab 15 mg/kg IV was given on days 1 and 8 of a 21-day cycle. Serum olaratumab concentrations were available for 92 patients. After the first and second doses of olaratumab, mean maximum serum concentration (Cmax) was 284 μg/mL and 293 μg/mL, respectively. At the completion of the cycle, olaratumab decreased to mean trough concentration (Cmin) of 66.5 μg/mL. The steady state was reached during cycle 3 with Cmax ranging from 419 to 487 μg/mL and Cmin ranging from 123 to 156 μg/mL. During cycle 3, T1/2 estimates were obtained and ranged from 6.67 to 14.4 days, slightly longer than was noted during the previous study.31
Clinical development
Phase I studies in advanced solid tumors
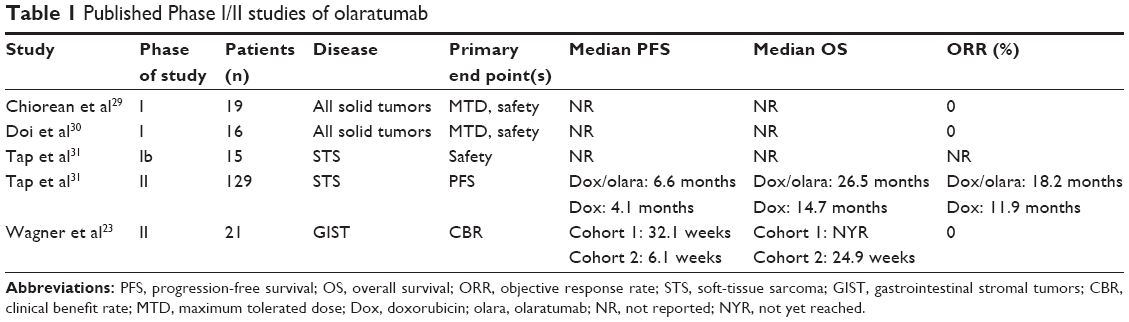
Two Phase I dose escalation studies of olaratumab were conducted in patients with advanced solid tumors (Table 1).29,30 The first was a multicenter, open-label study conducted in adults with good performance status and organ function with the primary objectives to establish the maximum tolerated dose (MTD) and safety profile of olaratumab. Secondary objectives included evaluation of pharmacokinetic and antitumor activity. Olaratumab was administered intravenously either weekly or biweekly. Dose cohorts were selected based upon the pharmacokinetic parameters for olaratumab from the preclinical models in glioblastoma and sarcoma as described previously.28 Olaratumab was dosed intravenously over 60–90 minutes in cohorts with initial dosing of 4 mg/kg IV, 8 mg/kg, and 16 mg/kg weekly ×4 doses followed by a 2-week observation period. Additional cohorts of 15 mg/kg and 20 mg/kg were evaluated with every other week dosing. Nineteen patients were treated for the following malignancies: prostate cancer (11), neuroendocrine cancer (5), bladder cancer (1), lung cancer (1), and endometrial cancer (1). The median number of treatments was 9 with median treatment duration of 12 weeks. There were no responses in the study, but 63.2% of patients had stable disease (SD) for a median duration of 3.9 months. The main reason for discontinuing olaratumab was progressive disease. Three patients discontinued olaratumab due to adverse events (AEs). One was fatigue/weight loss related to the study drug, and two were unrelated to drug (spinal compression fracture and arthralgia). No dose-limiting toxicities (DLTs) were observed during the study, so the MTD was not reached. The most common drug-related AEs were fatigue and infusion reactions, occurring in 10.5% of patients (Table 2). There were no grade ≥4 AEs during the study or the 30-day posttreatment follow-up period.29
 | Table 1 Published Phase I/II studies of olaratumab |
 | Table 2 Olaratumab-related adverse events |
A second Phase I study was conducted in a single center and included Japanese adults with advanced primary or recurrent solid tumors with good performance status and organ function (Table 1). Three dosing cohorts were evaluated: olaratumab 10 mg/kg given on days 1 and 8 of a 21-day cycle (cohort 1), olaratumab 20 mg/kg given every 2 weeks (cohort 2), and 15 mg/kg given on days 1 and 8 for every 3 weeks (cohort 3). The primary objectives were safety and pharmacokinetics, and secondary objectives included pharmacodynamics and antitumor activity. Response was assessed every 6 weeks. Sixteen patients were enrolled with the following cancers: colorectal (7), gastrointestinal stromal tumor (4), gastric (2), head and neck (2), and leiomyosarcoma (1). There were no DLTs in the trial, so the MTD was not reached. The best response was SD with a mean duration of 2.8 months in cohorts 1 and 2 and 4.9 months in cohort 3. The patient with leiomyosarcoma treated in cohort 3 had the longest duration of SD in the study, 5.6 months. The median duration of treatment was 13.1, 6, and 7 weeks in cohorts 1, 2, and 3, respectively. There were no AEs that caused treatment discontinuation. The most frequent AEs were pyrexia (25%), proteinuria (25%), constipation (19%), and anorexia (19%); however, only proteinuria was considered to be olaratumab-related AE (Table 2).30
Studies in soft-tissue sarcoma
First-line chemotherapy for advanced or metastatic STS generally includes doxorubicin. However, with this agent alone, survival is poor. Since olaratumab demonstrated antitumor activity in sarcoma xenografts, it was evaluated in combination with doxorubicin to determine if it could improve outcomes (Table 1).29,30 From October 2010 to January 2013, a two-part, open-label Phase Ib and randomized Phase II study of doxorubicin plus olaratumab accrued patients with unresectable or metastatic STS. Fifteen patients were treated on the Phase Ib study, and 133 were randomized on the Phase II study, 66 to combination treatment and 67 to doxorubicin alone. In the Phase Ib portion of the trial, patients received doxorubicin 75 mg/m2 on day 1 and olaratumab 15 mg/kg on days 1 and 8 of a 21-day cycle for up to eight cycles. After cycle 8 and a maximum potential cumulative dose of doxorubicin of 450 mg/m2, patients with stable or responding disease could continue on olaratumab monotherapy. In the Phase II part of the study, patients were randomized to the treatment as described for the Phase Ib study or doxorubicin 75 mg/m2 every 21 days as monotherapy. Patients being treated with doxorubicin alone could cross over to olaratumab monotherapy at disease progression. The primary end point of the Phase Ib study was safety. In the Phase II study, the primary end point was progression-free survival (PFS) and secondary end points were OS, objective response rate (ORR), safety, and pharmacokinetics.
In the Phase II study, the median PFS was 6.6 months (95% CI 4.1–8.3) with combination treatment versus 4.1 months (95% CI 2.8–5.4) with doxorubicin alone, p=0.0615. Median OS was 26.5 months (95% CI 20.9–31.7) with doxorubicin plus olaratumab and 14.7 months (95% CI 9.2–17.1) with doxorubicin, p=0.0003. The ORR was 18.2% (95% CI 9.8–29.6) with combination treatment versus 11.9% (95% CI 5.3–22.2) with monotherapy, p=0.3421. Median duration of response was ~8 months in each arm. The most common reason for discontinuing the study treatment in each arm was progressive disease. The most common AE resulting in discontinuation of doxorubicin was a decreased ejection fraction in 7 of the 128 patients (5%) and in discontinuation of olaratumab was an infusion reaction in 2 of the 64 patients (3%). AEs were more common with combination treatment with increased rates of neutropenia (58% vs 35%), mucositis (53% vs 35%), nausea (73% vs 52%), vomiting (45% vs 18%), and diarrhea (34% vs 23%) compared to single-agent doxorubicin. Infusion reactions, including two grade 4 events, occurred in 13% of patients treated with combination therapy and in none of the patients treated with doxorubicin alone31 (Table 2). A Phase III study evaluating doxorubicin with olaratumab has completed accrual, and we are awaiting results to see if this will confirm the promising findings of the Phase II study.
About 5%–10% of GISTs have a mutation in PDGFR-α. Because of the ability of olaratumab to internalize and down-modulate PDGFR expression, olaratumab was evaluated in patients with PDGFR-α-mutant GIST. An open-label, two-stage, multicenter, Phase II study of single-agent olaratumab was performed to evaluate efficacy in two cohorts of patients with previously treated unresectable or metastatic GIST. Thirty patients were enrolled, and 21 patients were treated. Cohort 1 included seven patients with a D842V mutation in PDGFR-α. Cohort 2 included 14 patients without a PDGFR-α mutation, 11 with a KIT mutation, and 3 without mutations in KIT or PDGFR-α. Treatment included olaratumab 20 mg/kg every 2 weeks with radiographic assessment every 6 weeks. The primary end point was clinical benefit rate (CBR) at 12 weeks. Secondary end points included PFS, OS, ORR, and disease control rate.
Results are summarized in Table 1. The CBR at 12 weeks was 50% (95% CI 15.3–84.7) in the PDGFR-α-mutant and 14.3% (95% CI 2.6–38.5) in the PDGFR-α wild-type patients, respectively. When treated with olaratumab, PDGFR-α-mutant patients (all D842V in this study) had a median PFS of 8 months, the median OS had not yet been reached. This compared favorably to the largest historical series of PDGFR-α-mutant GIST patients reporting a median PFS of 2.8 months in patients with D842V mutation versus 28.5 months in patients with all other PDGFR-α mutations. Median OS was 14.7 months in D842V mutants and was not yet reached in all other PDGFR-α mutants.32 Compared to this historical data obtained in the setting of tyrosine kinase inhibitor treatment, olaratumab preliminarily has demonstrated increased efficacy in the D842V-mutant patients. This will need to be confirmed in a larger study. Treatment-related AEs occurred in 76% of patients; however, olaratumab-related AEs ≥ grade 3 occurred in only 10% of patients, with only one serious adverse event due to olaratumab, an episode of syncope. The most common drug-related AEs were fatigue (38%), nausea (19%), headache (19%), infusion reaction (14%), and peripheral edema (14%) (Table 2).23
Place in therapy
Selecting a regimen for a given STS patient requires assessment of both the patient and disease characteristics. Comorbidities, organ function, performance status, extent of disease, burden of symptoms, goals of therapy, and histology affect chemotherapy recommendations. Doxorubicin is often considered first-line treatment for the majority of STS histologies, in patients with metastatic disease, who have not previously received an anthracycline, and with good cardiac function and performance status. Historically, the response rate to doxorubicin alone has varied, ranging from 5% to 27% as has the median OS ranging from 8 to 14.3 months,7,33–36 reflecting the heterogeneity of STS and evolution of response assessment. Other chemotherapy agents, including ifosfamide37–40 and dacarbazine,35 have been added to doxorubicin in an attempt to improve outcomes for STS patients. These combinations can yield higher response rates, particularly in certain sarcoma subtypes, such as synovial sarcoma (ifosfamide) and leiomyosarcoma (dacarbazine); however, these dual-agent cytotoxic regimens also result in increased toxicities (eg, myelosuppression, nausea, fatigue, neurotoxicity) and have not demonstrated an OS benefit.7,41 The addition of targeted agents to doxorubicin such as bevacizumab has yielded other toxicities such as decreased cardiac function.5
How does olaratumab fit into the algorithm for treating STS? The Food and Drug Administration has approved olaratumab for the treatment of STS patients who cannot be cured with surgery or radiation and for whom an anthracycline is appropriate. The Phase II study evaluating the combination doxorubicin with olaratumab suggests relatively minor improvements in ORR and PFS but significant OS improvement for this combination.31
To further evaluate the doxorubicin/olaratumab combination, a Phase III study (NCT02451943) has enrolled ~460 advanced STS patients in a randomized (1:1), placebo-controlled manner with the primary end point of overall survival. Whether the OS benefit will be confirmed in this Phase III study will not be known until at least 2019. For now, adding olaratumab to any patient being considered for single-agent doxorubicin is recommended. The combination is fairly well tolerated, with olaratumab adding relatively little treatment-related toxicity, and potentially confers a survival benefit. It is important to discuss with patients the increased risk for infusion reaction, neutropenia, mucositis, as well as the extra visit needed for a day 8 infusion when receiving combination treatment instead of doxorubicin alone. One caveat to this recommendation is that doxorubicin and ifosfamide is still a combination that could be considered in select cases of advanced STS where the goal is disease response to potentially allow for surgical resection or to ease symptoms related to disease. Although there has not been a direct comparison, the addition of ifosfamide to doxorubicin, as compared to olaratumab, most likely has a higher chance of tumor shrinkage and disease control based on cross-study comparisons, particularly in certain subtypes such as synovial sarcoma and myxoid liposarcoma. However, this potential benefit must be balanced with the increased rates of myelosuppression, nephrotoxicity, and neurotoxicity as well as increased complexity of drug and supportive medication administration. Further evaluation of the efficacy of doxorubicin/olaratumab in synovial sarcoma should be explored as only one patient with this histology received combination treatment in the Phase II study, thus making conclusions about its utility over doxorubicin/ifosfamide challenging.
To confirm the benefit of olaratumab in the metastatic STS population, the results of the Phase III study are necessary. This data may also provide further insight into the Phase II study findings which reported a significant improvement in OS without a significant increase in PFS. While it has been hypothesized that olaratumab may alter the tumor stromal cell environment, and potentially increase the efficacy of subsequent treatments, there are a number of other potential explanations. One possibility is that there was an imbalance in histologies with a greater number of patients with typically more indolent sarcoma subtypes enrolled in the combination treatment arm as compared to the monotherapy arm. This again exemplifies the heterogeneity of STS and the challenges in analyzing data from a diverse group of diseases to formulate conclusions about treatment regimens. Another potential explanation for the discrepancy in OS and PFS is that the posttreatment systemic therapy differed between the study arms. Patients who had received combination treatment were more likely to receive standard sarcoma regimens at the time of progressive disease, whereas patients who had been treated with doxorubicin alone were more likely to receive olaratumab monotherapy, the benefit of which is unknown. Also, olaratumab may alter the tumor stromal cell environment, thus potentially increasing the efficacy of subsequent treatment.
Future directions
Currently, there are several ongoing or recently completed clinical trials evaluating additional roles for olaratumab (Table 3). In addition to the previously mentioned Phase III study, olaratumab is being studied in advanced or metastatic STS in combination with other agents, including pembrolizumab and gemcitabine/docetaxel. A STS neoadjuvant study is examining changes in circulating tumor cells as well as PDGFR-α and PDGFR-β expression with the combination of doxorubicin and olaratumab. Although the role of chemotherapy in the treatment of localized, high-grade STS is controversial, it could be beneficial to evaluate the efficacy of doxorubicin and olaratumab in a neoadjuvant/adjuvant study, potentially in combination with other agents. Ultimately, these trials will allow us to delineate whether the benefit found in the initial randomized Phase II study is confirmed and understand whether there are additional roles for olaratumab in the treatment of STS.
 | Table 3 Summary of ongoing studies with olaratumab in STS |
Author contributions
All authors contributed toward study conception, design, drafting, and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
Rashmi Chugh has received institutional support for research from Lilly. The authors report no other conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. | ||
Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F. World Health Organization Classification of Tumours of Soft Tissue and Bone. Lyon: IARC Press; 2013. | ||
Ryan CW, Merimsky O, Agulnik M, et al. PICASSO III: a Phase III, placebo-controlled study of doxorubicin with or without palifosfamide in patients with metastatic soft tissue sarcoma. J Clin Oncol. Epub 2016 Sep 12. | ||
Tap WD, Papai Z, Van Tine BA, et al. Doxorubicin plus evofosfamide versus doxorubicin alone in locally advanced, unresectable or metastatic soft-tissue sarcoma (TH CR-406/SARC021): an international, multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2017;18(8):1089–1103. | ||
D’Adamo DR, Anderson SE, Albritton K, et al. Phase II study of doxorubicin and bevacizumab for patients with metastatic soft-tissue sarcomas. J Clin Oncol. 2005;23(28):7135–7142. | ||
Chugh R, Griffith KA, Davis EJ, et al. Doxorubicin plus the IGF-1R antibody cixutumumab in soft tissue sarcoma: a phase I study using the TITE-CRM model. Ann Oncol. 2015;26(7):1459–1464. | ||
Judson I, Verweij J, Gelderblom H, et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol. 2014;15(4):415–423. | ||
Schoffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet. 2016;387(10028):1629–1637. | ||
Italiano A, Mathoulin-Pelissier S, Le Cesne A, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer. 2011;117(5):1049–1054. | ||
Shah GD, Loizos N, Youssoufian H, Schwartz JD, Rowinsky EK. Rationale for the development of IMC-3G3, a fully human immunoglobulin G subclass 1 monoclonal antibody targeting the platelet-derived growth factor receptor alpha. Cancer. 2010;116(4 Suppl):1018–1026. | ||
European Medicines Agency. Lartruvo (olaratumab): summary of product characteristics. Science Medicines Health, London, UK. 2016. | ||
Alvarez RH, Kantarjian HM, Cortes JE. Biology of platelet-derived growth factor and its involvement in disease. Mayo Clin Proc. 2006;81(9):1241–1257. | ||
Cao Y. Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol Med. 2013;19(8):460–473. | ||
Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22(10):1276–1312. | ||
Movva S, Wen W, Chen W, et al. Multi-platform profiling of over 2000 sarcomas: identification of biomarkers and novel therapeutic targets. Oncotarget. 2015;6(14):12234–12247. | ||
Chott A, Sun Z, Morganstern D, et al. Tyrosine kinases expressed in vivo by human prostate cancer bone marrow metastases and loss of the type 1 insulin-like growth factor receptor. Am J Pathol. 1999;155(4):1271–1279. | ||
Henriksen R, Funa K, Wilander E, Backstrom T, Ridderheim M, Oberg K. Expression and prognostic significance of platelet-derived growth factor and its receptors in epithelial ovarian neoplasms. Cancer Res. 1993;53(19):4550–4554. | ||
Carvalho I, Milanezi F, Martins A, Reis RM, Schmitt F. Overexpression of platelet-derived growth factor receptor alpha in breast cancer is associated with tumour progression. Breast Cancer Res. 2005;7(5):R788–R795. | ||
Kubo T, Piperdi S, Rosenblum J, et al. Platelet-derived growth factor receptor as a prognostic marker and a therapeutic target for imatinib mesylate therapy in osteosarcoma. Cancer. 2008;112(10):2119–2129. | ||
Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708–710. | ||
Dagher R, Cohen M, Williams G, et al. Approval summary: imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8(10):3034–3038. | ||
Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352–5359. | ||
Wagner AJ, Kindler H, Gelderblom H, et al. A Phase II study of a human anti-PDGFRalpha monoclonal antibody (Olaratumab, IMC-3G3) in previously treated patients with unresectable and/or metastatic gastrointestinal stromal tumors. Ann Oncol. 2017;28(3):541–546. | ||
Grignani G, Palmerini E, Stacchiotti S, et al. A phase 2 trial of imatinib mesylate in patients with recurrent nonresectable chondrosarcomas expressing platelet-derived growth factor receptor-alpha or -beta: an Italian Sarcoma Group study. Cancer. 2011;117(4):826–831. | ||
Sugiura H, Fujiwara Y, Ando M, et al. Multicenter phase II trial assessing effectiveness of imatinib mesylate on relapsed or refractory KIT-positive or PDGFR-positive sarcoma. J Orthop Sci. 2010;15(5):654–660. | ||
Forsberg K, Valyi-Nagy I, Heldin CH, Herlyn M, Westermark B. Platelet-derived growth factor (PDGF) in oncogenesis: development of a vascular connective tissue stroma in xenotransplanted human melanoma producing PDGF-BB. Proc Natl Acad Sci U S A. 1993;90(2):393–397. | ||
Anderberg C, Li H, Fredriksson L, et al. Paracrine signaling by platelet-derived growth factor-CC promotes tumor growth by recruitment of cancer-associated fibroblasts. Cancer Res. 2009;69(1):369–378. | ||
Loizos N, Xu Y, Huber J, et al. Targeting the platelet-derived growth factor receptor alpha with a neutralizing human monoclonal antibody inhibits the growth of tumor xenografts: implications as a potential therapeutic target. Mol Cancer Ther. 2005;4(3):369–379. | ||
Chiorean EG, Sweeney C, Youssoufian H, et al. A phase I study of olaratumab, an anti-platelet-derived growth factor receptor alpha (PDGFRalpha) monoclonal antibody, in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2014;73(3):595–604. | ||
Doi T, Ma Y, Dontabhaktuni A, Nippgen C, Nippgen J, Ohtsu A. Phase I study of olaratumab in Japanese patients with advanced solid tumors. Cancer Sci. 2014;105(7):862–869. | ||
Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488–497. | ||
Cassier PA, Fumagalli E, Rutkowski P, et al. Outcome of patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors in the tyrosine kinase inhibitor era. Clin Cancer Res. 2012;18(16):4458–4464. | ||
Schoenfeld DA, Rosenbaum C, Horton J, Wolter JM, Falkson G, DeConti RC. A comparison of adriamycin versus vincristine and adriamycin, and cyclophosphamide versus vincristine, actinomycin-D, and cyclophosphamide for advanced sarcoma. Cancer. 1982;50(12):2757–2762. | ||
Judson I, Radford JA, Harris M, et al. Randomised phase II trial of pegylated liposomal doxorubicin (DOXIL/CAELYX) versus doxorubicin in the treatment of advanced or metastatic soft tissue sarcoma: a study by the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2001;37(7):870–877. | ||
Borden EC, Amato DA, Rosenbaum C, et al. Randomized comparison of three adriamycin regimens for metastatic soft tissue sarcomas. J Clin Oncol.1987;5(6):840–850. | ||
Chawla SP, Papai Z, Mukhametshina G, et al. First-line aldoxorubicin vs doxorubicin in metastatic or locally advanced unresectable soft-tissue sarcoma: a phase 2b randomized clinical trial. JAMA Oncol. 2015;1(9):1272–1280. | ||
Santoro A, Tursz T, Mouridsen H, et al. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol. 1995;13(7):1537–1545. | ||
Antman KH. Natural history and epidemiology of malignant mesothelioma. Chest. 1993;103(4 Suppl):373S–376S. | ||
Antman K, Crowley J, Balcerzak SP, et al. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol. 1993;11(7):1276–1285. | ||
Edmonson JH, Ryan LM, Blum RH, et al. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol. 1993;11(7):1269–1275. | ||
Verma S, Younus J, Stys-Norman D, Haynes AE, Blackstein M; Members of the Sarcoma Disease Site Group of Cancer Care Ontario’s Program in Evidence-Based Care. Meta-analysis of ifosfamide-based combination chemotherapy in advanced soft tissue sarcoma. Cancer Treat Rev. 2008;34(4):339–347. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.