Back to Journals » Lung Cancer: Targets and Therapy » Volume 12
Spotlight on Amivantamab (JNJ-61186372) for EGFR Exon 20 Insertions Positive Non-Small Cell Lung Cancer
Authors Brazel D, Nagasaka M ![]()
Received 5 September 2021
Accepted for publication 3 November 2021
Published 2 December 2021 Volume 2021:12 Pages 133—138
DOI https://doi.org/10.2147/LCTT.S337861
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sai-Hong Ou
Danielle Brazel,1 Misako Nagasaka1– 3
1Department of Medicine, University of California Irvine School of Medicine, Orange, CA, USA; 2Chao Family Comprehensive Cancer Center, Orange, CA, USA; 3St. Marianna University School of Medicine, Kawasaki, Japan
Correspondence: Misako Nagasaka
Department of Medicine, University of California Irvine School of Medicine, Chao Family Comprehensive Cancer Center, 101 The City Drive South, Orange, CA, 92868, USA
Email [email protected]
Abstract: Non-small cell lung cancer (NSCLC) patients demonstrating sensitizing oncogenic driver mutations have derived clinical benefit from targeted therapy. EGFR mutations constitutively activate the signaling pathway, leading to prosurvival and antiapoptotic signals. Classic sensitizing EGFR mutations, such as exon 19 deletions and exon 21 L858R point mutations, respond well to tyrosine kinase inhibitors (TKIs). On the other hand, EGFR exon 20 in-frame insertions are observed in 4– 12% of EGFR-mutated NSCLC and are resistant to targeted therapy with TKIs. In May 2021, the Federal Drug Administration (FDA) provided accelerated approval to amivantamab (Rybrevant) in adults with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations after treatment with platinum-based chemotherapy. Here, we discuss properties of amivantamab, clinical trial results, and management of patients with EGFR exon 20 insertion mutated NSCLC.
Keywords: amivantamab, epidermal growth factor receptor, mesenchymal-epithelial transition factor, MET, non-small cell lung cancer, tyrosine kinase inhibitors
Introduction
Lung cancer is the leading cause of cancer death in both men and women, accounting for almost 25% of cancer-related deaths.1 The recent development of targeted therapies has provided significant clinical benefit and improved quality of life for non-small cell lung cancer (NSCLC) patients demonstrating sensitizing oncogenic driver mutations. Epidermal Growth Factor Receptor (EGFR) encoded on chromosome 7p11.2 is a transmembrane cell surface receptor implicated in signal transduction pathways that regulate cell proliferation and apoptosis.2 The EGFR has an on and off switch for downstream effects, and many EGFR mutations favor the active state leading to pro-survival and antiapoptotic signals.3,4 EGFR mutations constitutively activate the signaling pathway, even in the absence of a ligand, promoting pro-survival and antiapoptotic signals through phosphatidylinositol 3-kinase (PI3k)/protein kinase B (AKT), extracellular signal-related kinase (ERK)/mitogen-activated protein kinase (MAPK), and signal transducer and activator of transcription (STAT) pathways.2,5,6 Cancer cells with EGFR mutations become dependent on EGFR for survival, making EGFR an attractive target for anticancer therapies.2 Approximately 20% of Caucasians and up to 50% of Asians have EGFR-driven NSCLC.7,8
Classic sensitizing EGFR mutations, including exon 19 deletions and exon 21 L858R point mutations, are more prevalent in women, Asians and never smokers.9,10 These tumors respond well to monotherapy with tyrosine kinase inhibitors (TKIs). Despite the efficacy of TKIs in classic EGFR mutations, resistance commonly develops through subsequent mutations or activation of compensatory pathways (such as cMet).11 In contrast, exon 20 mutations consist of either point mutations, such as S7681, or insertions of 3–21 base pairs.12 As many as 64 different EGFRex20ins mutations were identified by complete genomic profiling.13 Approximately 90% of exon 20 insertion mutations cluster after the C-helix of the tyrosine kinase domain, pushing the C-helix into permanent active confirmation.14 This conformational change shifts the C-helix and phosphate-binding loop into the drug binding pocket.14 Steric hindrance of the drug binding pocket by the C-helix and phosphate-binding loop may explain the resistance to first generation TKIs.15 Second generation TKIs in exon 20 insertion mutations are ineffective due to a dose-limiting toxicity at plasma concentrations below that needed to inhibit signaling pathways.12 The third generation EGFR TKI, osimertinib, although well tolerated has shown limited clinical activity in NSCLC exon 20 insertion mutations with a low overall response rate of 5%.16,17
EGFR exon 20 in-frame insertions are observed in 4–12% of EGFR-mutated NSCLC.18 NSCLC driven by EGFR exon 20 insertion mutations have a worse prognosis and shorter overall survival than more common or “classic” sensitizing EGFR mutations, such as exon 19 deletions and exon 21 L858R point mutations.19,20 Patients with newly diagnosed EGFR exon 20 insertion mutation-driven NSCLC have a median OS of 16.2 months (95% CI: 11.0, 19.4) compared to exon 19 and 21 mutations who have an OS of 25.5 months (95% CI: 24.5, 27.0).21 Until recently, there has been a void of targeted antitumor therapy against NSCLC tumors driven by EGFR exon 20 insertion mutations.
In May 2021, the Federal Drug Administration (FDA) provided accelerated approval to amivantamab (Rybrevant) in adults with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations whose disease progressed on or after platinum-based chemotherapy. Additionally, the diagnostic test Guardant360® CDx (Guardant Health, Inc.) was approved for diagnosis of these mutations. In this review, we discuss the structure, pharmacodynamics, and pharmacokinetics of amivantamab. We discuss properties of amivantamab in comparison to other EGFR inhibitors, clinical trial results, and management of patients with EGFR exon 20 insertion mutated NSCLC.
Structural Characteristics and Mechanism of Action of Amivantamab
Amivantamab (JNJ-61186372, Rybrevant, Janssen Biotech, Inc) is a bispecific antibody that simultaneously blocks EGF and cMET receptors. It has two arms; one arm of which binds the EGF receptor and the other of which binds the cMet receptor (Figure 1). The hypothesized mechanism is through both ligand blocking and receptor degradation.22 Amivantamab binds to the extracellular domains of EGFR and cMet and blocks the binding of the ligand EGF to EGFR and the binding of the ligand HGF to its receptor, cMet. Amivantamab inhibits ligand-induced receptor activation of both EGFR and cMet, as well as downstream signaling through proteins such as ERK and AKT, which are components of key pro-growth and pro-survival regulatory pathways. It also induces degradation of both receptors in vivo, broadening its impact to include ligand-independent receptor-driven disease.23
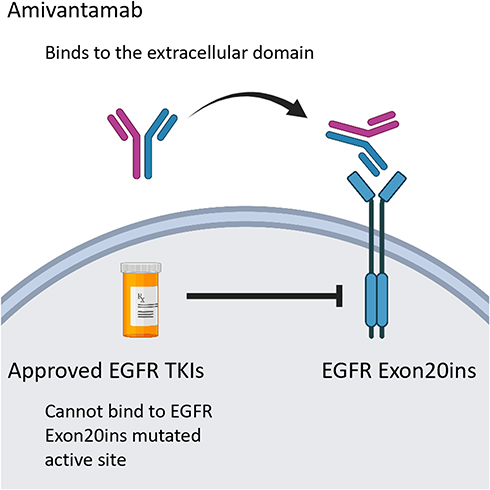 |
Figure 1 Structural characteristics and mechanism of action of amivantamab. |
In contrast to existing targeted antibodies, amivantamab was designed and engineered with a low fructose backbone to enhance binding to FcYRIIIa/CD16a.24 This receptor is found on NK cells, monocytes, and macrophages and triggers stronger antibody-dependent cell-mediated cytotoxicity of NSCLC cells. Due to the reduced fucosylation, amivantamab has an enhanced capacity to engage immune effector cells to eliminate antigen-expressing tumor cells through ADCC.
Pharmacodynamic Properties
The pharmacodynamic response and time to response have not been fully characterized.
In vitro studies show that amivantamab binds the extracellular domain of EGFR and MET receptors with binding affinity (KD) of 1.43 and 0.04 nM, respectively. Amivantamab bound to human EGFR and MET with EC50 values of 0.38 nM and 0.27 nM, respectively.
In vitro studies showed saturation of circulating free EGFR began after a single dose of 350 mg and saturation of MET at a single dose of 140 mg. Complete and durable saturation of both EGFR and MET was reliably achieved at doses ≥700 mg.25
Pharmacokinetics
Data from the first human clinical trial reveal that amivantamab exposure increased proportionally when administered at doses ranging from 350 mg to 1750 mg with a steady state achieved by the 9th infusion. Amivantamab concentration increased rapidly during Cycle 1, gradually decreasing in subsequent cycles to reach a steady state by Cycle 4.25 The half-life of amivantamab is 11.3 (± 4.53) days.
The recommended dose of amivantamab is 1050 mg for patients with baseline body weight <80 kg and 1400 mg for those with a body weight 80 kg and higher. Amivantamab is administered intravenously weekly for four weeks then every two weeks thereafter until disease progression or toxicity. There was no clinically meaningful effect of age, gender, race, creatinine clearance, or mild hepatic impairment on the exposure of amivantamab.
Preclinical Studies
In preclinical experiments, the low fructose backbone of amivantamab allowed for stronger affinity to F-c dependent mechanisms, enhancing antibody-dependent cellular toxicity.26 Most cytokines upregulated by amivantamab are chemokines which mediates killing by effector cells such as monocytes, macrophages, and neutrophils.22 Trogocytosis is a mechanism where the tumor-targeted antibody mediates the transfer of membrane fragments and ligands from tumor cells to these effector cells. Amivantamab is known to induce receptor degradation through trogocytosis.22,26 Although not as appreciated as phagocytosis, trogocytosis is also an important mechanism that contributes to cell death.
Preclinical studies on Ba/F3 cell lines containing EGFR exon 20 mutations showed amivantamab decreased EGF and cMet receptor expression.23 This study examined five different exon 20 insertions: (V769_D770insASV, D770delinsGY, H773_V774insH, Y764_V765insHH, and D770_N771ins-SVD). A statistically significant and dose-dependent decrease in Ba-F3 cell viability was observed across all five mutations. The proposed mechanism of action is decreased pERK, pAkt, and p-S6 with amivantamab, ultimately leading to inhibited cell proliferation.23 Additionally, amivantamab induced apoptosis via upregulating proapoptotic proteins including BIM and cleaved caspase-3.
Trials in HCC827 cell lines in vivo found the antitumor activity of amivantamab superior to TKI erlotinib and MET inhibitor crizotinib. Tumor growth was inhibited 99.8% (p < 0.05) at day 34 with a durable response noted 8 weeks after amivantamab discontinuation.27
Additionally, in xenograft models, the efficacy of amivantamab was superior to both cetuximab and poziotinib.23
Clinical Trials
Phase I
Accelerated FDA approval was based on CHRYSALIS, a Phase I, multicenter, open label, dose-escalation clinical trial assessing the safety and efficacy of amivantamab (NCT02609776).25 Patients received amivantamab 140 mg IV weekly for four weeks then every two weeks until disease progression or toxicity. The main efficacy outcome measure was overall response rate (ORR) with secondary outcomes of interest including clinical benefit rate, duration of response, progression-free survival, and overall survival. In this platinum-pretreated patient population where the median number of previous therapy was 2 (range 1–7), the ORR was 40% (95% CI: 29%, 51%) with a median response duration of 11.1 months (95% CI: 6.9, not evaluable).28 Complete response was achieved in 4% and partial response was seen in 36% of patients. The clinical benefit rate was 74% (95% CI: 63%, 83%). At most recent data cutoff, 47% of patients remain on treatment. Median progression-free survival was 8.3 months (95% CI: 6.5, 10.9) and median overall survival 22.8 months (95% CI: 14.6, not evaluable).
Safety, Tolerability, and Adverse Events
The most common adverse events include rash (86%), infusion-related reaction (65%), and paronychia (42%). Other less common adverse events include stomatitis, pruritus, hypoalbuminemia, increased ALT, and fatigue. Treatment-related grade 3 or higher adverse events were reported in 16% and adverse events led to drug discontinuation in 4% of patients. 94% of infusion-related reactions occurred with the first infusion.
Discussion/Future Directions
Until recently, targeted therapy against NSCLC tumors with EGFR exon 20 insertion mutations remained an unmet medical need. Preclinical studies show that EGFR exon 20 insertion mutations push the C-helix and phosphate-binding loop into the drug binding pocket, a possible mechanism for the resistance to first generation TKIs.15 Second and third generation EGFR TKIs have also shown disappointing clinical activity against EGFR exon 20 mutations.
Other agents with promising in vitro activity, such as poziotinib and mobocertinib (TAK-788) have shown little clinical efficacy. Poziotinib, an oral pan-HER inhibitor, demonstrated greater anti-tumor activity in PDX models of EGFR exon 20 insertion mutations compared to existing TKIs.14 A Phase II trial (NCT03318939) showed a response rate of only 14.8%.29 Preclinical data shows amivantamab has superior efficacy and tolerability to poziotinib in EGFR exon 20 xenografts.23 Mobocertinib (TAK-788) has shown limited clinical activity in a preliminary phase I/II study (NCT02716116) but treatment was associated with a high incidence of EGFR-driven toxicity.30 This study looked at 22 platinum chemotherapy-failed patients with EGFR20ins without brain metastases, who received mobocertinib 160 mg daily. Results were promising with ORR 30% (95% CI: 19, 35), median DOR of 17.5 months (95% CI: 5.6, not evaluable), and median PFS 7.3 months (95% CI: 5.5, 10.2). Adverse events, most commonly diarrhea and rash, were seen in 99% of patients though grade 3 or higher adverse events occurred in 46% of patients.
Promising results from the Phase I CHRYSALIS trial provided the basis for FDA accelerated approval of amivantamab. This study assessed 81 patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations after treatment with platinum-based chemotherapy. The authors found an overall response rate of 40% with median duration of response of 11.1 months. These findings are clinically meaningful considering the population of interest is relapsed metastatic or unresectable NSCLC with a 5-year survival rate of less than 10%.25
For full approval, the FDA will require results of a Phase III trial. The phase III PAPILLON trial (NCT04538664) is currently investigating amivantamab in combination with carboplatin-pemetrexed chemotherapy compared to chemotherapy alone in the first line treatment setting of metastatic NSCLC with exon 20 insertion mutations. The primary outcome of interest is progression-free survival at 18 months with additional outcomes of interest including overall response rate, duration of response, and tolerability (Figure 2). Janssen is investigating the safety and efficacy of amivantamab in combination with lazertinib versus osimertinib in advanced or metastatic NSCLC patients with classic EGFR mutations in the phase III MARIPOSA study (NCT04487080).
 |
Figure 2 Trial schema for the phase 3 PAPILLON study (NCT04538664). |
Future Directions
The CHRYSALIS study included only patients with treated and asymptomatic brain metastases. Given the high propensity for brain metastases in lung cancer, further research is needed into CNS activity of amivantamab against untreated brain lesions. Given that TKIs have better brain penetration than antibody therapies, more data is needed on mobocertinib, a EGFR20ins mutation and HER2 antibody which is currently under review as a potential breakthrough therapy.
Additional studies are also needed to elaborate mechanisms of resistance to amivantamab and investigate potential treatment strategies following resistance.
Conclusion
Amivantamab’s unique mechanism of action as an antibody to both EGFR and cMet, as well as low fructose backbone with high affinity for FcYRIIIa/CD16a may account for increased selectivity and efficacy with decreased toxicity when compared to other targeted therapies for EGFR exon 20 insertion mutation NSCLC.
Abbreviations
AKT, protein kinase B; CI, confidence interval; ERK, extracellular signal-related kinase; EGFR, epidermal growth factor receptor; FDA, federal drug administration; MAPK, mitogen-activated protein kinase; MET, mesenchymal-epithelial transition factor; NSCLC, non-small cell lung cancer; ORR, overall response rate; PI3K, phosphatidylinositol 3 kinase; TKI, tyrosine kinase inhibitor; STAT, signal transducer and activator of transcription.
Data Sharing Statement
Not applicable to this study.
Ethics Approval and Consent to Participate
This report did not meet criteria for IRB approval.
Consent for Publication
The patient has provided consent to publish the above manuscript.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No funding was secured for this report.
Disclosure
Dr Misako Nagasaka reports personal fees from AstraZeneca, Caris Life Sciences, Daiichi Sankyo, Takeda, Novartis, EMD Serono, Blueprint Medicines, Janssen, Pfizer, Lilly, and Genentech, and non-financial support from An Heart, outside the submitted work. The authors declare that they have no other competing interests.
References
1. Key Statistics in Lung Cancer. American Cancer Society. Available from: https://www.cancer.org/cancer/lung-cancer/about/key-statistics.html.
2. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. J Natl Cancer Inst. 2005;97:339–346. doi:10.1093/jnci/dji055
3. Kumar A, Petri ET, Halmos B, Boggon TJ. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J Clin Oncol. 2008;26(10):1742–1751. doi:10.1200/JCO.2007.12.1178
4. Eck MJ, Yun C-H. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim Biophys Acta BBA - Proteins Proteomics. 2010;1804(3):559–566. doi:10.1016/j.bbapap.2009.12.010
5. Nagano T, Tachihara M, Nishimura Y. Mechanism of resistance to epidermal growth factor receptor-tyrosine kinase inhibitors and a potential treatment strategy. Cells. 2018;7(11):212. doi:10.3390/cells7110212
6. Nguyen K-SH, Kobayashi S, Costa DB. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non–small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin Lung Cancer. 2009;10(4):281–289. doi:10.3816/CLC.2009.n.039
7. Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311(19):1998. doi:10.1001/jama.2014.3741
8. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 world health organization classification of lung tumors. J Thorac Oncol. 2015;10(9):1243–1260. doi:10.1097/JTO.0000000000000630
9. Sequist LV, Bell DW, Lynch TJ, Haber DA. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol off J Am Soc Clin Oncol. 2007;25(5):587–595. doi:10.1200/JCO.2006.07.3585
10. Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. JNCI J Natl Cancer Inst. 2005;97(5):339–346. doi:10.1093/jnci/dji055
11. Schmid S, Li JJN, Leighl N. Mechanisms of osimertinib resistance and emerging treatment options. Lung Cancer. 2020;147:123–129. doi:10.1016/j.lungcan.2020.07.014
12. Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol. 2012;13(1):e23–e31. doi:10.1016/S1470-2045(11)70129-2
13. Bauml JM, Viteri S, Minchom A, et al: FP07.12 underdiagnosis of EGFR exon 20 insertion mutation variants: Estimates from NGS-based real-world datasets. J Thorac Oncol. 2021;16:S208–S209
14. Robichaux JP, Elamin YY, Tan Z, et al. Mechanisms and clinical activity of an and HER2 exon 20–selective kinase inhibitor in non–small cell lung cancer. Nat Med. 2018;24(5):638–646. doi:10.1038/s41591-018-0007-9
15. Naidoo J, Sima CS, Rodriguez K, et al. Epidermal growth factor receptor exon 20 insertions in advanced lung adenocarcinomas: clinical outcomes and response to erlotinib: EGFR Exon 20 Insertions. Cancer. 2015;121(18):3212–3220. doi:10.1002/cncr.29493
16. Kim TM, Ock C-Y, Kim M, et al. Phase II study of osimertinib in NSCLC patients with EGFR exon 20 insertion mutation: a multicenter trial of the Korean Cancer Study Group (LU17-19). Ann Oncol. 2019;30:v628. doi:10.1093/annonc/mdz260.051
17. van Veggel B, Madeira R, Santos JFV, et al. Osimertinib treatment for patients with EGFR exon 20 mutation positive non-small cell lung cancer. Lung Cancer. 2020;141:9–13. doi:10.1016/j.lungcan.2019.12.013
18. Reiss JW, Gandara DR, Frampton GM, et al. J Thorac Oncol. 2018; 13(10): 1560–1568. doi:10.1016/j.jtho.2018.06.019
19. Vyse S, Huang PH. Targeting EGFR exon 20 insertion mutations in non-small cell lung cancer. Signal Transduct Target Ther. 2019;4(1):5. doi:10.1038/s41392-019-0038-9
20. Oxnard GR, Lo PC, Nishino M, et al. Natural history and molecular characteristics of lung cancers harboring EGFR Exon 20 insertions. J Thorac Oncol. 2013;8(2):179–184. doi:10.1097/JTO.0b013e3182779d18
21. Girard N, Bazhenova L, Minchom A, et al. Comparative clinical outcomes for patients with NSCLC harboring EGFR Exon 20 insertion mutations and common EGFR mutations. J Thorac Oncol. 2021;16(3):S145–S146. doi:10.1016/j.jtho.2021.01.228
22. Vijayaraghavan S, Lipfert L, Chevalier K, et al. Amivantamab (JNJ-61186372), an Fc Enhanced EGFR/cMet bispecific antibody, induces receptor downmodulation and antitumor activity by monocyte/macrophage trogocytosis. Mol Cancer Ther. 2020;19(10):2044–2056. doi:10.1158/1535-7163.MCT-20-0071
23. Yun J, Lee S-H, Kim S-Y, et al. Antitumor activity of amivantamab (JNJ-61186372), an EGFR-cMet bispecific antibody, in diverse models of EGFR Exon 20 insertion-driven NSCLC. Cancer Discov. 2020:CD-20-0116. doi:10.1158/2159-8290.CD-20-0116
24. Grugan KD, Dorn K, Jarantow SW, et al. Fc-mediated activity of EGFR x c-Met bispecific antibody JNJ-61186372 enhanced killing of lung cancer cells. mAbs. 2017;9(1):114–126. doi:10.1080/19420862.2016.1249079
25. Park K, Haura EB, Leighl NB, et al. Amivantamab in EGFR Exon 20 insertion–mutated non–small-cell lung cancer progressing on platinum chemotherapy: initial results from the CHRYSALIS Phase I Study. J Clin Oncol. 2021:JCO.21.00662. doi:10.1200/JCO.21.00662
26. Moores SL, Chiu ML, Bushey BS, et al. A novel bispecific antibody targeting EGFR and cMet is effective against EGFR inhibitor–resistant lung tumors. Cancer Res. 2016;76(13):3942–3953. doi:10.1158/0008-5472.CAN-15-2833
27. Neijssen J, Cardoso RMF, Chevalier KM, et al. Discovery of amivantamab (JNJ-61186372), a bispecific antibody targeting EGFR and MET. J Biol Chem. 2021;296:100641. doi:10.1016/j.jbc.2021.100641
28. Sabari JK, Shu CA, Park K, et al. Amivantamab in post-platinum EGFR exon 20 insertion mutant non–small cell lung cancer. WorldConf Lung Cancer. 2021;16:s108–s109.
29. Le X, Goldman J, Clarke J, et al. Abstract CT081: poziotinib activity and durability of responses in previously treated EGFR exon 20 NSCLC patients - a Phase 2 study. Tumor Biol. 2020:CT081–CT081. doi: 10.1158/1538-7445.AM2020-CT081
30. Zhou C. Mobocertinib in NSCLC with EGFR Exon 20 insertions: results from EXCLAIM and platinum-pretreated patient populations; 19.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.