Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
Spiperone Stimulates Regeneration in Pulmonary Endothelium Damaged by Cigarette Smoke and Lipopolysaccharide
Authors Skurikhin E, Pershina O ![]() , Zhukova M
, Zhukova M ![]() , Widera D
, Widera D ![]() , Pan E
, Pan E ![]() , Pakhomova A, Krupin V, Ermakova N
, Pakhomova A, Krupin V, Ermakova N ![]() , Skurikhina V, Sandrikina L, Morozov S, Kubatiev A, Dygai A
, Skurikhina V, Sandrikina L, Morozov S, Kubatiev A, Dygai A
Received 16 September 2021
Accepted for publication 6 December 2021
Published 30 December 2021 Volume 2021:16 Pages 3575—3591
DOI https://doi.org/10.2147/COPD.S336410
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Evgenii Skurikhin,1 Olga Pershina,1 Mariia Zhukova,1 Darius Widera,2 Edgar Pan,1 Angelina Pakhomova,1 Vyacheslav Krupin,1 Natalia Ermakova,1 Victoria Skurikhina,3 Lubov Sandrikina,1 Sergey Morozov,4 Aslan Kubatiev,4 Alexander Dygai1,4
1Laboratory of Regenerative Pharmacology, Goldberg ED Research Institute of Pharmacology and Regenerative Medicine, Tomsk National Research Medical Centre of the Russian Academy of Sciences, Tomsk, Russia; 2Stem Cell Biology and Regenerative Medicine Group, School of Pharmacy, University of Reading, Whiteknights Campus, Reading, RG6 6AP, UK; 3Siberian State Medical University, Tomsk, Russia; 4Institute of General Pathology and Pathophysiology, Moscow, Russia
Correspondence: Evgenii Skurikhin
Laboratory of Regenerative Pharmacology, Goldberg ED Research Institute of Pharmacology and Regenerative Medicine, Tomsk National Research Medical Centre of the Russian Academy of Sciences, 634028, 3, Lenin Street, Tomsk, Russia
Tel/Fax +7-3822-418-375
Email [email protected]
Background: Endothelial dysfunction and destruction of the pulmonary microcirculation are important pathogenic factors in chronic obstructive pulmonary disease (COPD). In COPD, bronchial obstruction is associated with endothelial dysfunction. Thus, new pharmacological treatment options aimed at restoring the pulmonary endothelium represent a clinical need in COPD therapy. Notch1 has been shown to protect cells against apoptosis, inflammation, and oxidative stress caused by cigarette smoke extract (CSE). Therefore, drug which effect on Notch1 may be a potential therapeutic target for COPD in the future.
Methods: In this study, we assessed the potential of spiperone to mediate regeneration of pulmonary endothelium in model of pulmonary emphysema induced by a CSE and lipopolysaccharide (LPS) in female C57BL/6 mice.
Results: Spiperone increased the number of capillaries as well as the expression of the CD31 in the alveolar tissue compared to the controls. Moreover, application of spiperone prevented alveolar wall destruction (DI), and reduced the area of emphysema. Lastly, we demonstrated that spiperone positively influenced mobilization and migration of endothelial progenitor cells (EPC, CD45−CD34+CD31+), CD309+-endothelial cells, and angiogenesis precursors (CD45−CD117+CD309+) into the lung. Spiperone administration significantly reduced the number Notch1 positive CD309+-endothelial cells and Notch1+ EPCs.
Conclusion: Overall, our results suggest that spiperone mediates endothelial regeneration in an animal model of COPD. Thus, it could represent a novel therapeutic approach for treatment of emphysema associated with COPD.
Keywords: pulmonary emphysema, endothelial progenitor cells, VEGF2 (CD309), spiperone, Notch1, endothelial regeneration
Introduction
COPD is a common and incurable disease chronic inflammatory lung disease that causes obstructed airflow from the lungs. According to WHO, COPD will become the third biggest global killer by 2030.1 Although air pollution and chemical fumes can be causative for developing COPD, smoking, and exposure to cigarette smoke are the most important risk factors.2,3
COPD is characterized by persistent respiratory symptoms caused by pathophysiological changes including chronic inflammation and narrowing of peripheral airways to emphysema. Inflammation is the main initiating process in COPD. Furthermore, persistent pulmonary inflammatory response is a prominent feature of COPD.4 In addition to inducing local inflammation, irritants within the tobacco smoke attract proinflammatory cells to the lungs and induce the release of superoxide anions and other molecules that actively destroy the extracellular matrix (eg, neutrophil elastase, proteinase – 3, cathepsin G, matrix metalloproteinases).5 Another important pathogenic process in COPD is the destruction of alveolar structures.6–9 Simultaneous with apoptosis of the alveolar epithelium, systemic endothelial dysfunction develops10 involving endothelial cell apoptosis,11 and reduction of alveolar vascularization.12 The emerging endothelial dysfunction is associated with bronchial obstruction.13 The reduction of the microvascular flow is more pronounced in patients with severe COPD compared to patients suffering from mild COPD.14 It was demonstrated that the number of endothelial progenitor cells (EPC) is decreased in patients with COPD and correlates with severity of airway obstruction, hypoxemia, and peak oxygen consumption.15 Some studies also suggest a possible link between a decrease in circulating EPCs and systemic inflammation.15,16 According to the vascular hypothesis of cigarette smoke-induced emphysema, endothelial cell death initiates the disappearance of alveolar cells in emphysema.7 It is well known that LPS directly increases the permeability of lung endothelial cells17 and enhances the severity of lung injury caused by cigarette smoke.18 The negative effects of cigarette smoke on pulmonary endothelial cells are well known. There is evidence supporting the injurious roles of endothelial cell injury in the development of emphysema, pulmonary hypertension, and acute respiratory distress syndrome (ARDS). However, the role of EPC in COPD is uncleaned.
Pathogenically, COPD is the result of abnormal lung injury repair caused by dysregulation of various signaling pathways (such as Wnt and Notch) involved in the regeneration of lung tissue. This eventually results in aberrant pulmonary inflammation, tissue remodeling, and functional impairment of the alveolar parenchyma (i.e. airway inflammation, emphysema, and remodeling in COPD).19,20
To date, there are no curative treatment options for COPD. Current therapeutical options aimed at symptom management are behavioral interventions (smoking cessation) as well as prescribing short and long-acting bronchodilators (beta2 muscarinic antagonists), inhaled corticosteroids, or a combination of these options.21 However, long-term management of COPD requires to interfere with the progression of endothelial dysfunction and to reduce the large-scale apoptosis of endothelial cells. Moreover, compounds capable of regenerating an already damaged endothelium are an unmet clinical need in management of COPD. As the regeneration of terminally differentiated endothelium is challenging, targeting EPCs may represent a promising therapeutic option.22
It is known Notch signaling is involved in cell fate determination affecting the proliferation, differentiation, stem cell maintenance, and apoptosis of diverse cell types and the development of numerous organ systems.20 Notch1/4 has been reported to play a defensive role in apoptosis of endothelial cell, inhibiting Notch1/4 leading to abundant apoptotic cell death,23,24 whereas overexpression of Notch1/4 protected cells from apoptosis.24,25 It has been demonstrated that Notch1 and Notch4 expression decreased both in vivo in COPD and in vitro in cigarette smoke-treated endothelial cells.20 Moreover, Notch1 signaling is a key regulator of angiogenesis,26 as well as apoptosis, inflammation, and oxidative stress in human pulmonary microvascular endothelial cells after exposure to CSE.27 Based on these studies, it is compelling to speculate that Notch1 could qualify as one such molecular mediator, and modulation of Notch1 signaling in the endothelium may be beneficial for patients with COPD. According to data, targeting the Notch1 signaling pathway presents as an effective therapeutic strategy for COPD treatment. However, the role of Notch1 signaling in cigarette-smoke-induced COPD is still unclear. The regulatory mechanism of airway endothelial cell injury-caused by cigarette smoke remains unclear too. At the same time, there is an urgent need to identify new therapeutic drugs for the prevention and treatment of cigarette-related emphysema.
There is a rich literature on both agonist and antagonists of dopamine receptors, but most studies focused on modulation of dopamine function in the central nervous system in the context of schizophrenia and other brain disorders. Dopamine receptor D2 (D2R) is found in human pulmonary and lobar arteries,28 and on the surface of EPCs29 and alveolar type II epithelial cells.30 In 2015, it has been demonstrated that stimulation of D2R increases apoptosis of CD31+ endothelial cells.31 Briefly, this study revealed that stimulation of D2R with dopamine resulted in a decrease of the numbers of circulating EPC in mice with S180 sarcoma. Furthermore, it has been shown that dopamine can inhibit VEGF-dependent mobilization of EPC from the bone marrow and reduce the migration of CD45−CD309+ cells in vitro.29 Notably, D2R antagonist ethiclopid eliminated the negative effects of dopamine on the EPC migration and mobilization.29 It was shown an enhanced activation of serotonergic signaling in pulmonary fibrosis, and the serotonin (5-HT)2 receptors have been implicated to have important roles in observed profibrotic actions.32 Thus, dopamine receptor antagonists might have the potential to stimulate angiogenesis and therefore represent a novel approach to tackle COPD.
Spiperone is a butyrophenone antipsychotic agent with dopamine and serotonin (5-HT) receptor antagonist properties.33–35 It is also a high-affinity ligand of sigma receptors.36 It has been demonstrated that spiperone is a calcium regulator and that it specifically blocks canonical Wnt signaling prior to the activation of β-catenin by enhancing intracellular calcium levels.35
In this study, we assessed the potential regenerative effects of spiperone in CSE and LPS-induced experimental pulmonary emphysema in female C57BL/6 mice. Additionally, effects of spiperone on endothelial progenitor cells were assessed in vitro and in vivo.
Materials and Methods
Animals
Experiments were performed on 80 female C57BL/6 mice obtained from the nursery of the Surgical Bio-modelling Department of the Goldberg ED Research Institute of Pharmacology and Regenerative Medicine (veterinary certificate available), housed under specific pathogen-free conditions with food and water ad libitum. All animal experiments were carried out in accordance with the European Convention on the protection of vertebrates used in experiments for other scientific purposes and was approved by the laboratory animal control Committee of the Goldberg ED Research Institute of Pharmacology and Regenerative Medicine, Tomsk NRMC (IACUC Protocol No.131092017). At the beginning of the experimental period, the age of all animals was 10 weeks. All experiments were carried out in the autumn-winter period. Material sampling was carried out in the morning.
Reagents
Selective, competitive antagonist of dopamine D2-receptors spiperone was obtained from Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO, USA). In the animal experiments, spiperone was administered intraperitoneally once a day at a dose of 0.15 mg/kg/100 mL of saline solution from d1 to d45. The time points for the experiments and the concentration of spiperone were selected as described previously.37
Dopamine hydrochloride (Sigma-Aldrich, St. Louis, MO, USA) was used as dopamine receptor agonist in vitro at a concentration of 10−7 М. In order to block dopamine receptors in vitro, spiperone was used at 10−7 М. The concentration of spiperone was selected based on previous reports.38,39
Pulmonary Emphysema Model
Multiple studies showed that experimental COPD model can be established by a combination of exposure to cigarette smoke and intratracheal administration of LPS.4,40–42 Briefly, the administration of CSE and LPS accelerates the airway inflammatory response in mice similar to clinical manifestation of COPD in patients.4,41,43 Pulmonary emphysema was induced by a course of intratracheal CSE administration. CSE was isolated from L&M Red Label cigarettes at 2 cigarettes per mL (resin: 10 mg/cig, nicotine: 0.8 mg/cig, CO: 10 mg/cig) as described earlier.44,45
Inflammation has been recognized as the most important mechanism both at the beginning of COPD and the progress of COPD.46 Bacterial endotoxins, including LPS, are potential inducers of inflammation.47 Lipopolysaccharide (Escherichia coli O111: B4, Sigma-Aldrich, St. Louis, MO, USA) was used as an adjuvant. General anesthesia (pentobarbital) was used at the time point of the CSE and LPS administration.
CSE was administrated in a volume of 100 μL/mouse. LPS was intratracheal administered at a dose 3 μg/mouse in 50 μL of phosphate buffer (Figure 1).
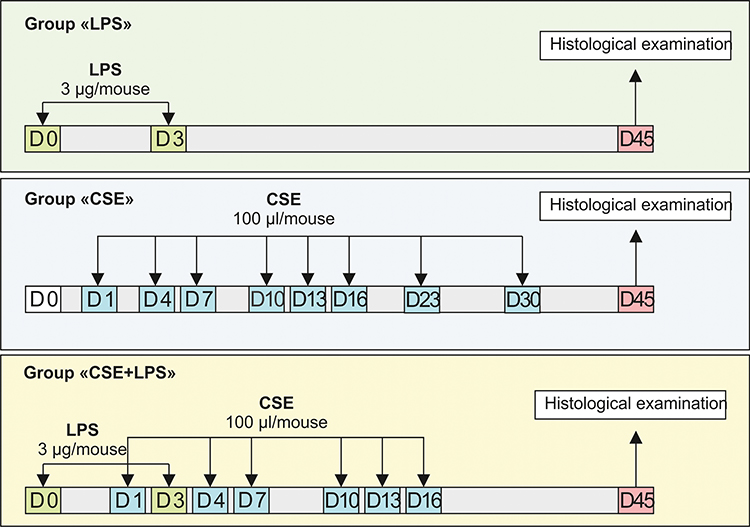 |
Figure 1 Graphical scheme of the protocol for cigarette smoke extract (CSE) or/and lipopolysaccharide (LPS) inducing pulmonary emphysema. |
Experimental Groups
Initially, we studied the possibility of modeling pulmonary emphysema by administering LPS or CSE. In this 1 series of experiment, mice were divided into 4 groups: mice of the control group (group 1 – control, n = 10), mice treated LPS (group 2 – LPS, n = 10), mice treated CSE (group 3 – CSE, n = 10), mice treated LPS and CSE (group 4 – CSE+LPS, n = 10) (Figure 1). All mice were removed from the experiment on the d45 by an overdose of CO2.
In the next 2 series of experiments on a model of LPS/CSE-induced pulmonary emphysema, we investigated the effects of spiperone in vivo. For the in vivo experiments, mice were divided into 3 groups: group 1 – control group (control, n = 10), group 2 – mice with emphysema caused by CSE and LPS (CSE/LPS, n = 20), and group 3 – mice with emphysema caused by CSE and LPS treated with spiperone (CSE/LPS/S, n = 10) (Figure 2). Mice of group 2 were injected with equivalent volume of saline solution. The spiperone treatment was once a day from d1 to d45. All mice were removed from the experiment on the d45 by an overdose of CO2.
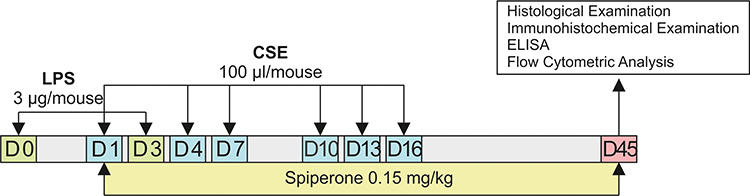 |
Figure 2 Graphical scheme of the protocol for studying the spiperone effects in vivo. |
Direct effects of dopamine and spiperone on endothelial cells were investigated in cultured cells isolated from mice with emphysema caused by CSE and LPS (Figure 3). Cell separation was performed to enrich + endothelial cells. For the in vitro experiments involving CD31+ cells 5 groups were assessed: group 1 – control group (CD31+cells before culture), group 2 – CD31+ cells after d5 culture, group 3 – CD31+ cells after culture with spiperone (10−7 M), group 4 – CD31+ cells after culture with dopamine (10−7 M), and group 5 – CD31+ cells after culture with spiperone (10−7 M) and dopamine (10−7 M).
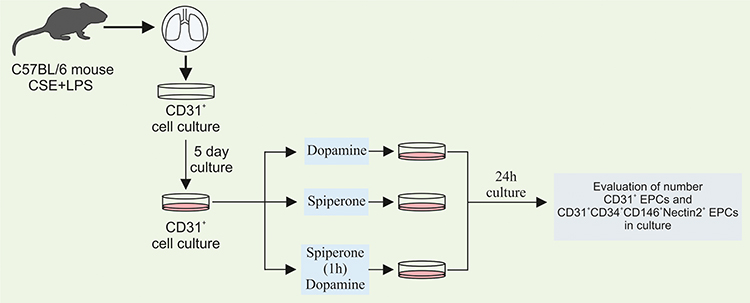 |
Figure 3 Graphical scheme of the protocol for CD31+ lung endothelial cells cultivation with dopamine and spiperone. |
Histological Examination of Lung Tissue
Histological examination of left lung lobes was carried in three areas of lung tissue (upper, middle, and lower) out on d45 as described previously.45,48 Sections of the lung were stained with hematoxylin and eosin. Lung structure, presence of edema, infiltration by pro-inflammatory cells as well as venous hyperemia, and vascular and bronchial wall thickening were assessed. Moreover, the degree of alveolar septa destruction49–53 and pulmonary parenchyma destruction degree (DI)53,54 were determined. Additionally, we calculated the number of capillaries on micro preparations from each experimental animal as described.53,55
Immunohistochemistry
Immunohistochemical analysis of the lung tissue was performed on d45. Lungs were manually inflated with paraformaldehyde (4%) until they reached maximal volume, then left lung was placed in paraformaldehyde (4%) solution and embedded in paraffin blocks for immunohistochemical staining. Manual lung inflation was performed according to the method described earlier.56,61 To detect specific cell markers, the following primary antibodies were used: rabbit polyclonal anti CD16 (ab203883, 1/100, species reactivity: Mouse, Rat, Human, Abcam, USA) and rabbit polyclonal anti CD31 (ab28364, 1/50, species reactivity: Mouse, Human, Pig, Abcam, USA). The sections were contrasted with hematoxylin. Visualization system was used in accordance with the manufacturer’s instructions for antibody detection (Spring bioscience, USA). The immunostaining procedure followed has been described previously.45 An Axio Lab.A1 microscope (Carl Zeiss, Germany) with an AxioCam ERc5s camera (Carl Zeiss, Germany) was used to obtain micrographs. Image analysis and calculation of cells expressing detectable antigens were performed with FIJI.
Enzyme-Linked Immunosorbent Assay
Serum concentration of α1-antitrypsin was determined by ELISA using appropriate kits for mice laboratory diagnosis in accordance with manufacturer’s instructions (Cusabio Biotech CO., LTD, China). The sensitivity for determining α1-antitrypsin is 0.195 μg/mL.
Concentrations of MMP9, MMP12, and α1-antitrypsin in lung homogenates of right lung lobes were determined by ELISA according to manufacturer’s instructions (Cusabio Biotech CO., Ltd., Wuhan, China). Sensitivities were >0.552 ng/mL for MMP9 and >1.95 pg/mL for MMP12.
Flow Cytometry
Mononuclear cells from blood and lungs were isolated as described earlier37,45,48 and the surface marker expression on mononuclear cells derived from blood and lungs was analyzed using flow cytometry. Surface Mouse Fc receptors were blocked by incubating the cells with unconjugated anti-CD16/CD32 antibodies for 10 minutes (eBioscience, San Diego, CA, Clone: 93, Cat# 14-0161-85, 1/50 dilution) in 50 μL of 0.1% saponin (Sigma-Aldrich, St. Louis, MO, Cat# S4521) and 1% bovine serum albumin (BSA, Sigma-Aldrich, St. Louis, MO, Cat# A3059-100G) in phosphate buffered saline (PBS) per tube. Then, cell suspensions were stained with fluorophore-conjugated monoclonal antibodies: CD45 PerCP (QC Testing: Mouse, Clone: 30-F11, Cat# 557235, 1/100 dilution) CD31 APC (QC Testing: Mouse, Clone: MEC 13.3, Cat# 551262, 1/50 dilution), CD34 FITC (QC Testing: Mouse, Clone: RAM34, Cat# 560238, 1/50 dilution), CD309 (Flk-1) APC (QC Testing: Mouse, Clone: Avas 12alpha1, Cat# 560070, 1/50 dilution), CD117 PeCy7 (QC Testing: Mouse, Clone: 2B8, Cat# 558163, 1/50 dilution), CD146 PerCP-Cy5.5 (QC Testing: Mouse, Clone: ME-9F1, Cat# 562231, 1/50 dilution), and Notch1 PE (QC Testing: Mouse, Clone: mN1A, Cat# 552768, 1/50 dilution) (all Becton Dickinson, San Jose, CA, USA). All antibodies were titrated to determine their optimal staining concentration and appropriate isotype controls were used. Labeled cells were washed thoroughly with 500 μL of FACSFlow (Becton Dickinson, Franklin Lakes, NJ, Cat# 342003).
For the intracellular staining of the Notch1, a Fixation/Permeabilization Solution Kit (Becton Dickinson, San Jose, CA, USA, Cat# 554714) was used in accordance with the manufacturer’s instructions.
For the in vitro experiments, the antibodies mentioned above were supplemented with an unconjugated Nectin2 (CD112) antibody (Clone; 829038, MAB3869, 1/100 dilution, R&D Systems, Minneapolis, MN). After incubating for 30 minutes at RT, cells were washed with 500 μL of 0.1% saponin and 1% BSA buffer, centrifuged at 500g for 5 minutes, and aspirated. 50 μL of PE conjugated donkey anti-rabbit (DαR) secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, 1/100 dilution) was added and cells were incubated for 30 minutes. A final wash and centrifugation were performed, and cells were resuspended in 500 μL of FACSFlow (Becton Dickinson, Franklin Lakes, NJ, Cat# 342003). Appropriate isotype controls were used.
All samples were run on a Becton Dickinson FACSCanto II flow cytometer. FACS analysis was performed as described above.45,53
Dissociation of Lung Tissue and Magnetic Separation of CD31+ Cells
The effects of dopamine and spiperone on CD31+ lung endothelial cells isolated from mice in group 2 were investigated on d45. Lung endothelial cells were isolated using a standard protocol.45,53,57,58 Briefly, lungs were isolated from the surrounding tissue, rinsed with PBS, mechanically dispersed, and minced with scissors. 2.5 mL digestion buffer containing 100 mg/mL Dispase II (Roche Diagnostics, Indianapolis, IN), 10 mg/mL Collagenase A (Roche Diagnostics, Indianapolis, IN), 1500 kU/mL DNase I (Sigma-Aldrich, St. Louis, MO), and 0.025M CaCl2 was used per lung on an orbital shaker at 37°C for 1 hour. The suspension was gently mixed using a pipette and further incubated for 30 minutes before being filtered through a 40μm filter followed by a centrifugation at 300g for 5 minutes. The cell suspension isolated from the lung was subjected to magnetic sorting to enrich CD31+ cells.
Magnetic sorting was performed using EasySep™ Mouse Biotin Positive Selection Kit (Catalog #18556, StemCell Technologies, Canada) and Biotin Rat Anti-Mouse CD31 antibodies (QC Testing: Mouse, Clone: MEC 13.3, Cat# 553371, 1/50 dilution, Becton Dickinson, San Jose, CA, USA). The fraction of CD31+ cells was isolated using EasySep™ Magnet (StemCell Technologies, Canada).
Assessment of the EasySep ™ cell separation efficiency was carried out using flow cytometry as described above.
Cultivation of CD31+ Cells
The fraction of CD31+ cells (106 cells/1 mL of medium) obtained after magnetic sorting was cultured on gelatin-coated plastic tablets for T-25 cell cultures in the medium consisted of 50% DMEM (Sigma-Aldrich, St. Louis, MO), 10% fetal bovine serum (FBS, Sigma-Aldrich, USA), 2 mM L-glutamine (Sigma-Aldrich, St. Louis, MO), 100 U/mL penicillin and 100 μg/mL streptomycin (Sigma-Aldrich, St. Louis, MO). The next day, we have replaced medium on medium with M199 (Sigma-Aldrich, St. Louis, MO) with 15% FBS (Sigma-Aldrich, St. Louis, MO), 1 mM l-glutamine (Sigma-Aldrich, St. Louis, MO), 100 μg/mL heparin (Sigma-Aldrich, St. Louis, MO) and endothelial growth factor 50 μg/mL (Sigma-Aldrich, St. Louis, MO). Cells were cultured for 5 days in standard gas (3.5% CO2) and temperature conditions (37°C). The medium was changed every 1–2 days. Evaluation of the effectiveness of the cultivation carried out to increase the mass of CD31+ cells was performed using flow cytometry.
Cultivation of CD31+ Lung Endothelial Cells with Dopamine and Spiperone
After 5 days of cultivation, CD31+ cells were detached by trypsinization followed by reseeding on T-25 gelatin-coated plastic plates at 3×105 cells/1 mL. Prior to further cultivation, cells we pre-incubated with dopamine (10−7 M) or spiperone (10−7 M) for 1 hour. The effects of pretreatment with dopamine and spiperone (10−7 M) were assessed separately. Following the pre-incubation, all CD31+ cells were cultivated for 24h at 3.5% СО2 and 37°C.
At the end of the cultivation period, the effects of dopamine, spiperone, and dopamine against background of spiperone administration on CD31+ cells and CD31+CD34+CD146+ Nectin2+ cells isolated from group of mice exposed CSE and LPS were assessed using flow cytometry (Figure 3).
Statistical Analysis
Statistical analysis was performed using the SPSS software package (version 15.0, SPSS Inc., Chicago, IL, USA). Data were analyzed and presented as means ± standard error of the mean. Statistical significance was evaluated by Mann–Whitney test (for nonparametric data). A p-value of less than 0.05 (by two-tailed testing) was considered statistically significant.
Results
Histomorphological Changes in Lung Tissues of Mice with Pulmonary Emphysema Induced by CSE or/and LPS
Initially, we studied the possibility of modeling pulmonary emphysema by administering LPS or CSE (Figure 1). The mice in the control group were active and had stable respiration. The mice in groups 2 (treated with LPS), 3 (treated with CSE) and 4 (treated with LPS and CSE) usually stayed still with gathered fur. Their respiration was not stable. In order to assess the morphology of the lung tissue, sections were prepared on d45 followed by H&E staining. In the first series of experiments, LPS administration caused hyperemia and plethora of blood vessels in the microvasculature of the lungs, lymph-macrophage infiltration into the lung parenchyma of group 2 mice compared to group 1 mice. In addition, inflammatory cells were detected perivascular and peribronchial (d45) (Figure 4B). We observed a decrease in the number of capillaries in all parts of the lungs from mice treated with LPS (P < 0.05) compared to the control group. Additionally, we evaluated the linear interception (Lm) and destructive index (DI). LPS administration did not affect Lm, but caused destruction of the alveoli.
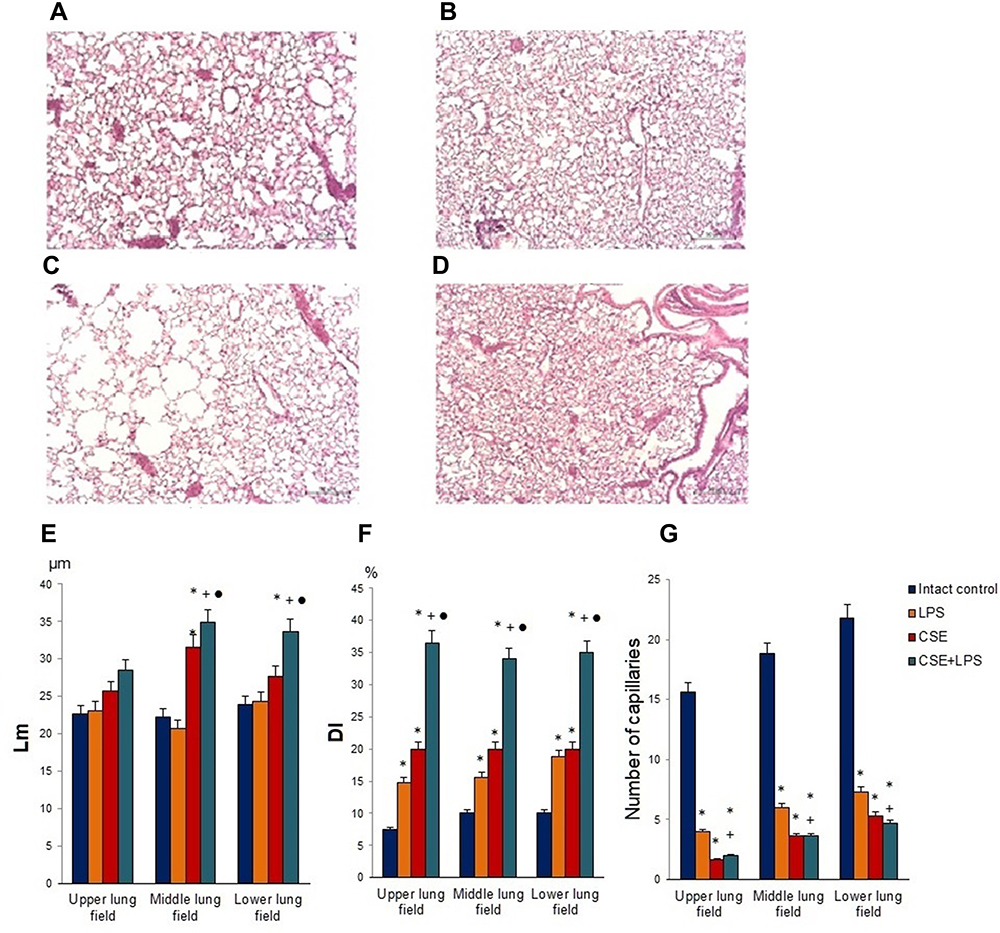 |
Figure 4 Micrographs of lung sections (middle pulmonary field) obtained from female C57BL/6 (A) mice of intact control; (B) mice treated with LPS; (C) mice treated with CSE; (D) mice treated with LPS and CSE (d) on d45. Tissues were stained with hematoxylin-eosin. × 100. Scale bar 50 μm. Morphometry of the lungs of females C57BL/6 mice on d45: (E) Mean linear intercept (Lm); (F) destructive index (DI); (G) the number of capillaries in 5 consecutive fields of lungs sections (upper, middle and lower pulmonary field). Groups: intact control – group from intact mice (Intact control), mice treated with LPS (LPS), mice treated CSE (CSE), mice treated with LPS and CSE (LPS + CSE). * – for comparison with the intact control group by Mann–Whitney test; + – for comparison with the LPS control group by Mann–Whitney test; ● – for comparison with the CSE group by Mann–Whitney test. |
CSE administration caused much less inflammation in the lungs of group 3 mice compared to group 1 mice (d45) (Figure 4C). Meanwhile, in group 3, greater destruction of the microvasculature was observed (P < 0.05), compared to group 2. On the other hand, CSE caused more significant alveolar destruction compared to LPS, as indicated by the Lm and DI values (P < 0.05) and diffuse emphysema (Figure 4E and F).
On the d45 after the CSE and LPS administration in the lungs of mice from group 4, we found hyperemia and plethora of the vessels of the microvasculature as well as thinning of the alveolar capillaries and their desolation (Figure 4). In the parenchyma and the lumen of the alveoli, macrophages and lymphocytes were found; the walls of the alveoli were thickened due to inflammatory infiltration. Diffuse pulmonary emphysema developed (Figure 4). In addition, there were ruptures of the alveolar septa and atelectasis of a part of the lungs and the number of capillaries decreased compared to group 1. On lung preparations, we observed the expansion of the alveoli. Additionally, we evaluated the increase in airspace (Lm) and the destruction of the alveolar walls (DI). In all parts of the lung in group 2 mice, there was a significant (p < 0.05) increase in Lm and DI compared to group 1 mice (Figure 4E and F). We compared the parameters of the lungs of mice in groups 2, 3 and 4 (Figure 4). It turned out that in group 4, the infiltration of the lungs with inflammatory cells exceeded that in group 3, while the area of emphysema, destruction of the capillary network of the lungs and alveoli were more significant than in groups 2 and 3. Thus, the effects of spiperone should be studied in a model of LPS and CSE induced pulmonary emphysema.
Spiperone Has Positive Effects on Histomorphological Changes in Lung Tissues of Mice with LPS/CSE-Induced Pulmonary Emphysema
In the next 2 series of experiments on a model of LPS/CSE-induced pulmonary emphysema, we investigated the effects of spiperone in vivo. Compared with the normal alveolar architecture (intact control group), the staining of lung tissue sections from group of mice treated with LPS and CSE (Figure 5B) showed histologically advanced emphysema in 2nd series of experiment. The lungs from the experimental group (LPS/CSE) revealed signs of hyperemia, microvasculature vascular congestion, thinning and desolation of alveolar capillaries, and a decreased number of capillaries (Figures 5 and 6). In contrast to the lungs of group 1, we found macrophages and lymphocytes in alveoli parenchyma and lumen as well as thickened alveoli walls in mice of group 2. Further analysis revealed the development of diffuse pulmonary emphysema (Figure 5), ruptures of alveolar septa, and atelectasis of the lungs. In addition, alveoli expansion has been detected as well as an increase in alveolar wall destruction (DI). Notably, a considerable increase (p < 0.05) of DI has been observed in all lung compartments of animals in group 2 compared to the control animals (Figure 5D–I). Application of spiperone decreased formation of alveolar septa ruptures in the middle and lower sections of the lungs in mice of group 3 (LPS/CSE/S) compared to group 2 (Figure 5D–I). Moreover, the administration of spiperone prevented the development of emphysema and increased the number of capillaries in group 3 compared to group 2 (Figure 6C).
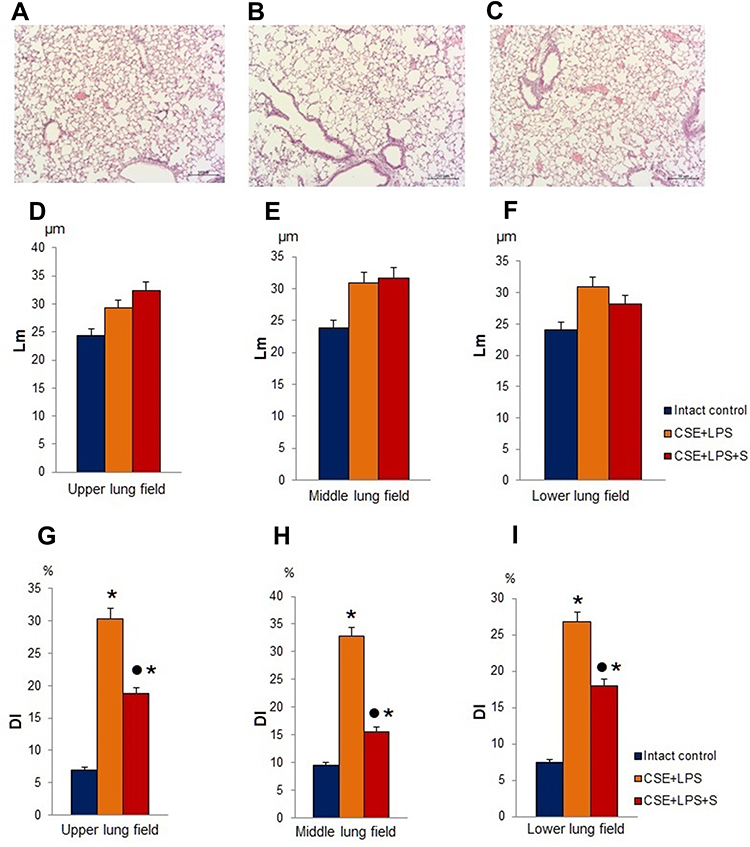 |
Figure 5 Photomicrographs of lungs sections (lower pulmonary field) obtained from female C57BL/6 mice on d45 (A–C). Tissues were stained with hematoxylin-eosin. Scale bar 10 μm. The arrows indicate the rupture of the alveoli. (A) Section from control group; (B) section from mice with CSE/LPS-induced emphysema; (C) section from mice with emphysema treated with Spiperone between d1-d45. At least 10 photomicrographs of the lung tissue at × 100 magnification were taken for each experimental animal from all experimental groups. (D–I) Quantification of pulmonary emphysema and morphometry of the lungs of females C57BL/6 mice on d45: (D–F) Lm and (G–I) DI values of mice; (D and G) Upper; (E and H) Middle; (F and I) Lower pulmonary field. Results are expressed as means ± SEM. n =10 animals/group; * - for comparison with the Intact control group by Mann–Whitney test; ● - for comparison with the CSE+LPS group by Mann–Whitney test. |
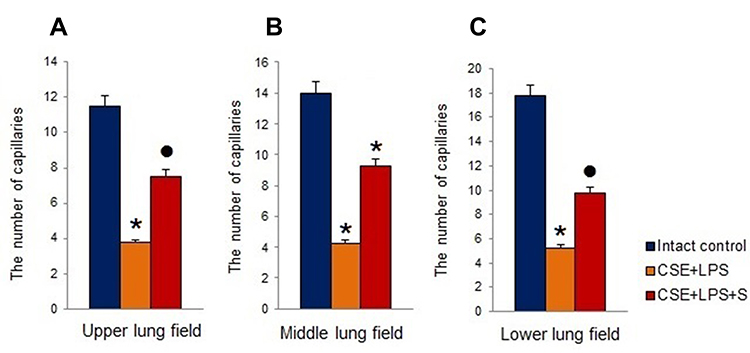 |
Figure 6 The number of capillaries in 5 consecutive fields of lungs sections (upper, middle and lower pulmonary field) obtained from female C57BL/6 mice on d45. (A) Upper lung field; (B) middle lung field; (C) lower lung field. Groups: intact control – control group from intact mice, CSE+LPS – mice with CSE/LPS-induced emphysema, CSE+LPS+S – mice with emphysema treated with Spiperone. Results are presented as the mean±SEM. *- for comparison with the Intact control group by Mann–Whitney test; ● - for comparison with the CSE+LPS group by Mann–Whitney test. |
Immunohistochemical Examination of the Lung Tissue
Immunohistochemical analysis of tissue slices prepared on d45 revealed an increase of CD16 expression in group 2 compared to group 1 (Figure 7A–D). In contrast, a decrease in CD31 expression was observed (Figure 7E–H).
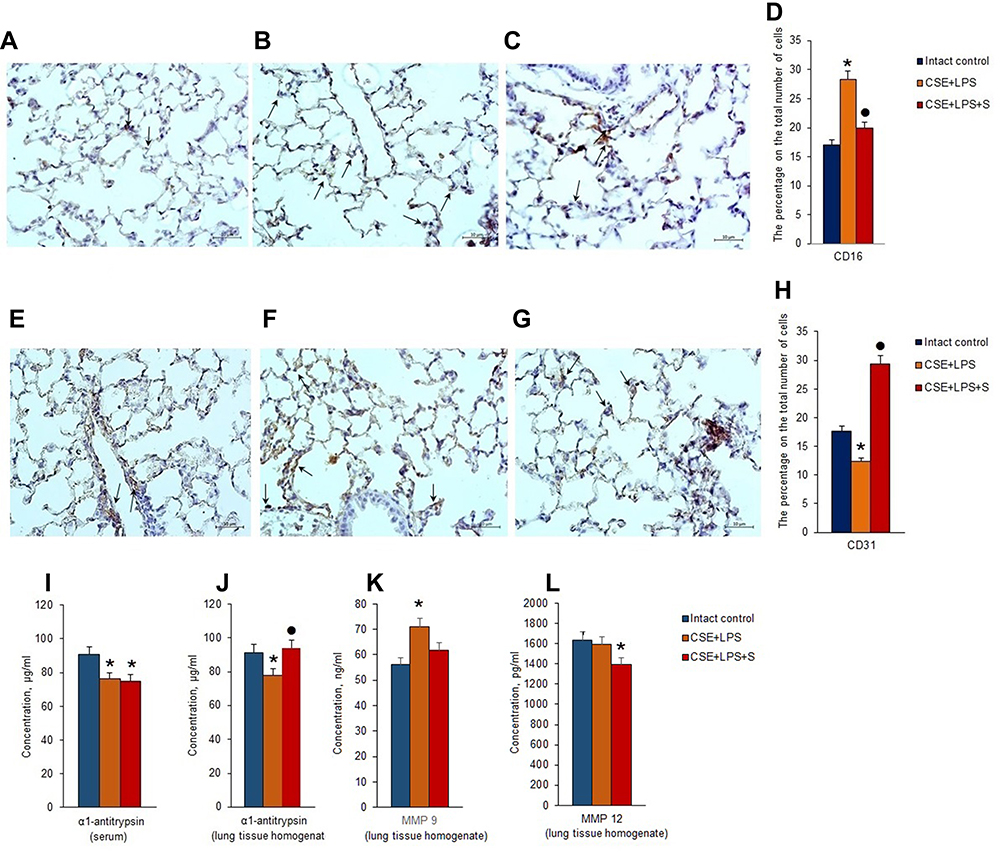 |
Figure 7 Immunohistochemical expression of specific cellular markers: (A–C) CD16 and (E–G) CD31 in the lungs (lower pulmonary field) from female C57BL/6 mice on d45. All sections were counterstained with hematoxylin. Scale bar 10 μm. The arrows indicate positive cells. (A and E) section from control group; (B and F) section from mice with CSE/LPS-induced emphysema; (C and G) Section from mice with emphysema treated with Spiperone between d1-d45; (D) immunohistochemical staining for CD16 in the lungs from female C57BL/6 mice on d45; (H) immunohistochemical staining for CD31 in the lungs from female C57BL/6 mice on d45. The α1-antitrypsin concentration in the serum (I) and the levels of α1-antitrypsin (J), MMP 9 (K), and MMP 12 (L) in lung homogenate of female C57BL/6 mice with lung damage caused by intratracheal administration of CSE and LPS on d45. Groups: intact control – control group from intact mice, CSE+LPS – mice with CSE/LPS –induced emphysema, CSE+LPS+S – mice with emphysema treated with Spiperone. Results are presented as the mean±SEM. * - for comparison with the Intact control group by Mann–Whitney test (p < 0.05); • - for comparison with the CSE+LPS group by Mann–Whitney test p < 0.05. |
Administration of spiperone significantly increased the number of CD31+ cells in alveoli of group 3 mice compared to group 2. Moreover, CD16 expression was significantly reduced (Figure 7D).
ELISA of Serum α1-Antitrypsin and α1-Antitrypsin, MMP9, and MMP12 in Lung Homogenate
Alpha1-antitrypsin is believed to be secreted by alveolocytes, expressed on leukocytes and plasma cells, and found in the blood. It is believed that MMP9 is secreted by alveolar macrophages and granulocytes, while MMP12 is secreted by alveolar macrophages. ELISA revealed that the experimental emphysema reduced the concentrations of α1-antitrypsin in the serum and lung homogenate in the group with pulmonary emphysema compared to the control group (Figure 7I and J). Modeling of LPS/CSE-related emphysema caused a significant increase in the levels of MMP9 in the lungs of group 2 mice compared to intact controls (d45). However, the level of MMP12 was not changed (Figure 7K and L). Spiperone administration resulted in a higher a level of lung homogenate α1-antitrypsin in group compared to group with pulmonary emphysema (Figure 7J). Treatment with Spiperone caused a significant decrease of the levels MMP9 and MMP12 in the lungs of mice after LPS/CSE administration compared to untreated mice in group 2 (Figure 7K and L).
Cytometric Examination of Endothelial Progenitor Cells, VEGF2+ Endothelial Cells, and Angiogenesis Precursors in Lung and Blood
Spiperone Positive Effects on Endothelial Cells Populations in the Lung of Mice with LPS/CSE-Induced Pulmonary Emphysema
To assess potential changes in the numbers of endothelial cells, endothelial precursor cells and angiogenesis precursors, flow cytometric analysis was applied. In the lungs of mice in group with pulmonary emphysema, a decrease in the numbers of EPCs (CD45−CD34+CD31+) and VEGF2+ endothelial cells (CD45−CD309+) was observed compared with group 1 (Figure 8A, C and E). Moreover, the number of angiogenesis precursors (CD45−CD117+CD309+) was reduced by 30% (p < 0.05). Spiperone significantly increased number of all these immature endothelial cells in the lungs of mice compared to group with pulmonary emphysema (Figure 8A, C and E).
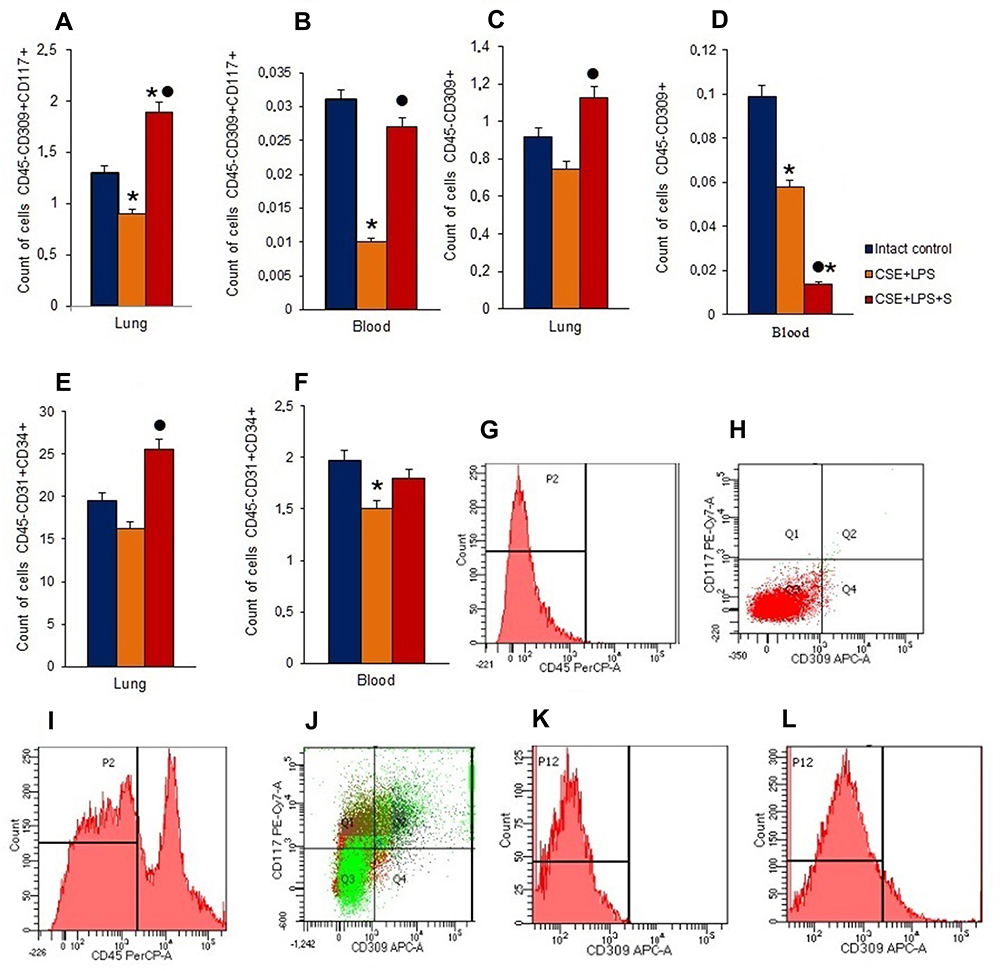 |
Figure 8 Characterization of endothelial progenitor cells, VEGF2+ endothelial cells and angiogenesis precursors in lungs and blood of female C57BL/6 mice on d45. Cells were analyzed by flow cytometry using antibodies against CD45, CD31, CD34, CD117, and CD309. Dot plots are representative for three independent experiments with the mean from three independent experiments. (A) The number of angiogenesis precursors (CD45−СD117+CD309+) in lungs of female C57BL/6 mice; (B) the number of angiogenesis precursors (CD45−СD117+CD309+) in blood of female C57BL/6 mice; (C) the number of VEGF2+ endothelial cells (CD45−СD309+) in lungs of female C57BL/6 mice; (D) the number of VEGF2+ endothelial cells (CD45-СD309+) in blood of female C57BL/6 mice; (E) the number of endothelial progenitor cells (CD45−СD34+CD31+) in lungs of female C57BL/6 mice; (F) the number of endothelial progenitor cells (CD45−СD34+CD31+) in blood of female C57BL/6 mice. Groups: intact control – control group from intact mice, CSE+LPS – mice with CSE/LPS –induced emphysema, CSE+LPS+S – mice with emphysema treated with Spiperone. Results are presented as the mean±SEM. * - for comparison with the Intact control group by Mann–Whitney test (p <0.05); ● - for comparison with the CSE+LPS group by Mann–Whitney test (p < 0.05). (G) Histogram of isotype control for IgG2b (PerCP); (H) Dot plot of isotype control for IgG2b (PE-Cy7), and IgG2b (APC); (I) Histogram of CD45 (PerCP) expression; (J) Phenotype establishment and qualitative analysis of CD117 (PE-Cy7) and CD309 (APC) expression; (K) Isotype control for IgG2b (APC); (L) Histogram of CD309 APC expression. |
Spiperone Positive Effects on Endothelial Cells Populations in the Blood of Mice with LPS/CSE-Induced Pulmonary Emphysema
In blood of animals with pulmonary emphysema (group 2), we observed a significant decrease in the number of angiogenesis precursors, EPCs, and VEGF2+ endothelial cells compared with group 1 (Figure 8B, D and F). Spiperone increased the number of circulating angiogenesis precursors in the blood of group 3 mice by 170% (p < 0.05) compared with group 2, while VEGF2+ endothelial cell number decreased by 76% (p < 0.05) (Figure 8B, D and F).
Cytometric Study of Notch1+ Endothelial Cells in Lungs
Pulmonary emphysema induced by CSE and LPS in group 2 mice was accompanied by a significant decrease in the number of angiogenesis precursors and EPCs expressing Notch1 (Figure 9A–C). Administration of spiperone (group 3) significantly reduced the number Notch1 positive VEGF2+ endothelial cells by 65% (p < 0.05) and Notch1+ EPCs by 59% (p < 0.05) compared to group 2. In contrast, no significant changes were observed in the numbers of Notch1+ angiogenesis precursors (Figure 9).
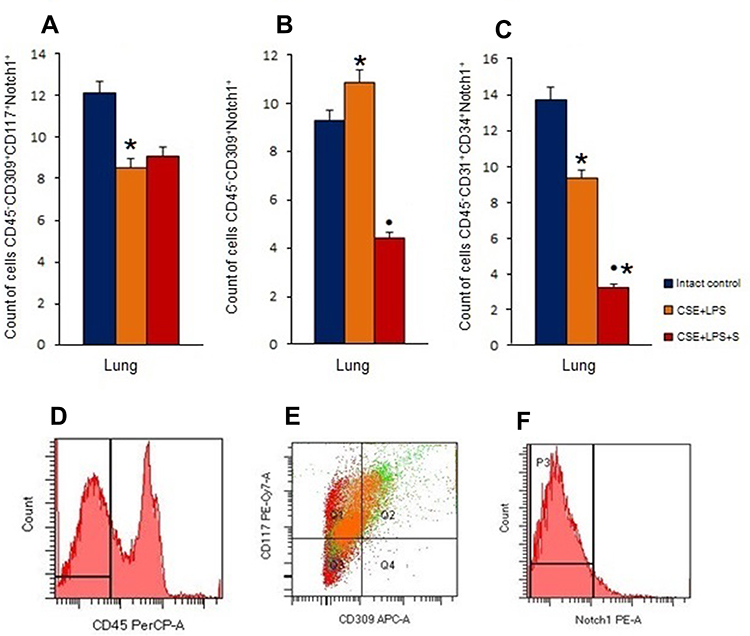 |
Figure 9 Characterization of Notch1+ endothelial cells in lungs female C57BL/6 mice on d45. Cells were analyzed by flow cytometry using antibodies against CD45, CD31, CD34, CD117, CD309 and Notch1. Dot plots are representative for three independent experiments with the mean from three independent experiments. (A) The number of angiogenesis precursors (CD45−СD117+CD309+Notch1+) in lungs of female C57BL/6 mice; (B) the number of VEGF2+ endothelial cells (CD45−СD309+Notch1+) in lungs of female C57BL/6 mice; (C) the number of endothelial progenitor cells (CD45−СD34+CD31+Notch1+) in lungs of female C57BL/6 mice. Groups: intact control – control group from intact mice, CSE+LPS – mice with CSE/LPS-induced emphysema, CSE+LPS+S – mice with emphysema treated with Spiperone. Results are presented as the mean±SEM. * - for comparison with the Intact control group by Mann–Whitney test (p < 0.05); ● - for comparison with the CSE+LPS group by Mann–Whitney test (p < 0.05). (D) Histogram of CD45 (PerCP) expression; (E) Phenotype establishment and qualitative analysis of CD117 (PE-Cy7), and CD309 (APC) expression; (F) Histogram of Notch1 PE expression. |
Spiperone and Dopamine Effects on Pulmonary CD31+ Cells Culture in vitro
We assessed the effects of dopamine and spiperone on the population of CD31+ cells isolated from mice with emphysema by CSE and LPS. Additionally, we evaluated the number of CD31+CD34+CD146+Nectin2+ cells before and after cultivation. Cultivation for 5 days increased the proportion of CD31+CD34+CD146+Nectin2+ cells in CD31+ endothelial cell culture of group 2 by more than 100% (p < 0.05) compared to group 1 (Figure 10E). Notably, spiperone (group 3) had no effects on the numbers of CD31+CD34+CD146+Nectin2+ cells in vitro (Figure 10E). In contrast, dopamine application resulted in a 6-fold increase of the CD31+CD34+CD146+Nectin2+ cell population in group 4 (p < 0.05) compared to group 2.
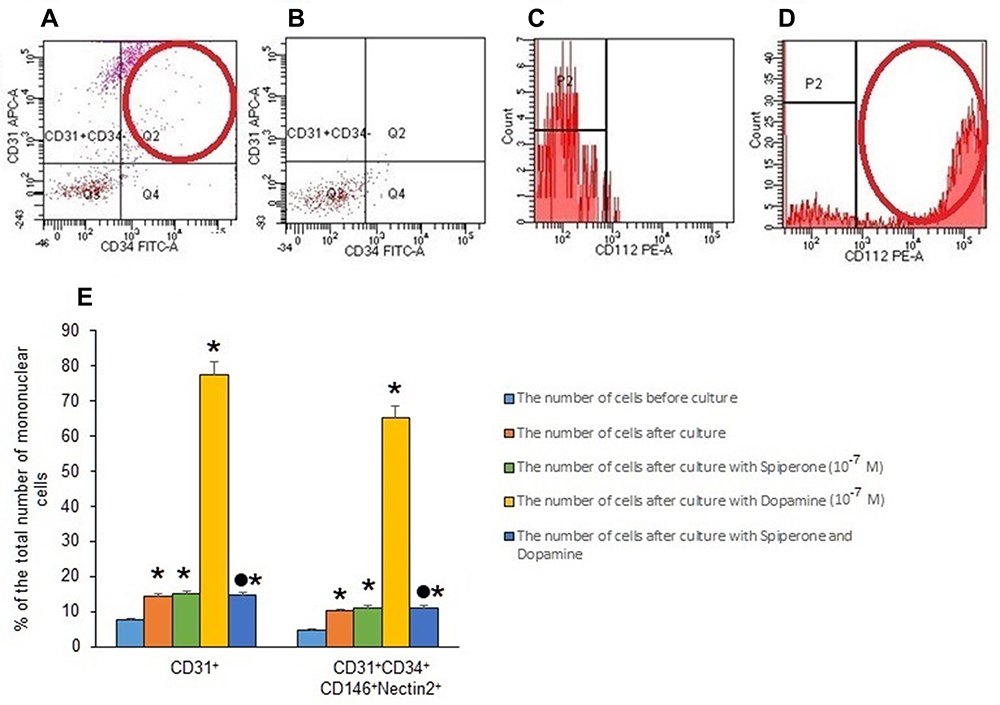 |
Figure 10 Characterization of Nectin2+endothelial cells in lungs from female C57BL/6 mice on d45. Cells were analyzed by flow cytometry using antibodies against CD45, CD31, CD34, CD146, and CD112 (Nectin2). (A) Phenotype establishment and qualitative analysis of CD31 (APC) and CD34 (FITC) expression; (B) Dot plot of isotype control for IgG2b (APC) and IgG2a (FITC); (C) Histogram of isotype control for IgG2b (PE); (D) Histogram of CD112 (Nectin 2) (PE) expression; (E) the effect of dopamine and spiperone on the content of mature endothelial cells and their precursors (% of the total number of mononuclear cells) in a culture of mononuclear cells isolated from lungs of female C57BL/6 mice with emphysema by CSE and LPS on d45 (M±m). Results of 3 independent experimental series are presented. Groups: group 1 - control group (CD31+cells before culture), group 2 – CD31+ cells after d5 culture, group 3 – CD31+ cells after culture with spiperone (10−7 M), group 4 – CD31+ cells after culture with dopamine (10−7 M), and group 5 – CD31+ cells after culture with spiperone (10−7 M) and dopamine (10−7 M). * - p < 0.05 significance of difference compared with group 1. ● - p < 0.05 significance of difference compared with group 2. |
Pretreatment of CD31+ cells with spiperone reversed the dopamine-induced increase of the numbers of CD31+CD34+CD146+Nectin2+ cells in group 5 compared to group 4 (Figure 10E). We assessed the effects of dopamine and spiperone effect on CD31+ cells isolated from mice with CSE/LPS-induced emphysema.
Discussion
Development and progression of COPD are associated with many factors including genetic disposition, gender, inhalation or exposure to toxic substances, bacterial and viral infections, and last but not least the socio-economic status.59 Chronic cigarette smoke stimulation causes lung inflammation and emphysema. Airway remodeling and emphysema are the main characteristics of COPD, and exposure to cigarette smoke is the most important risk factor for airway restriction and emphysema.60 It is known that COPD is accompanied by systemic endothelial dysfunction10 which worsens during the disease and, apparently, is an important factor in its progression. Thus, achieving a restoration of the endothelium may lead to alveolar tissue regeneration representing an attractive target in COPD. Moreover, growing data provide evidence that endothelial repair is impaired in COPD and suggest that this pathological process is associated with EPC.45,61 Taken together, we hypothesized that EPCs play an important role in the pathogenesis of COPD, and the possible protective role of increasing EPCs in a smoking-induced COPD model can provide more insight into the treatment of COPD.
In this study, intratracheal administration of CSE and LPS caused inflammatory infiltration into the lungs, enlargement of alveolar air spaces, destruction of the lung parenchyma, confirming the efficiency and efficacy of intratracheal CSE injection for establishing a smoking-related rodent emphysema model (Figures 4 and 5). Interestingly, CSE and LPS reduced the numbers of angiogenesis precursors (CD45−CD117+CD309+), VEGF2+ endothelial cells, and EPCs (CD45−CD34+CD31+) in blood and lungs (Figure 8). Notably, this correlated with the decreased number of capillaries in the alveolar tissue. Thus, the partial, CSE/LPS-induced destruction of the microvasculature could inhibit cell migration through the bloodstream and trans-endothelial transition of the cells following the migration.
It is likely that inflammation exerted additional inhibitory effects on these processes. Combined, this could lead to a disturbance of endothelial regeneration as indicated by a decrease of CD31 expression in the lungs.
We observed a mortality of 20% in mice with CSE/LPS-induced emphysema and an absence of mortality after spiperone treatment. We did not observe the mortality of mice with CSE/LPS-induced pulmonary emphysema treated with spiperone. In addition, the treatment reduced the levels of inflammation, prevented the destruction of interalveolar septa and reduced the area of emphysema (Figure 5). In addition, we observed an increase in the number of capillaries in the lungs, CD31 expression in the alveolar tissue (Figure 7) as well as an increase in the concentration of lung α1-antitrypsin in group 3 mice compared with group of mice with pulmonary emphysema (Figure 7I). It was shown that cigarette smoking to induce the functional inactivation of α1-antitrypsin, a neutrophil elastase inhibitor, which leaves smokers vulnerable to lung tissue destruction.62,63
Based on these data, we concluded that spiperone stimulated regeneration of alveolar tissue after CSE and LPS administration. This vascular response to spiperone improved stem cell migration and their trans-endothelial transition. This was confirmed by an increase of the numbers of EPСs, VEGF2+ endothelial cells, and angiogenesis precursors in the lungs of mice in group 3 mice compared to untreated mice in group with pulmonary emphysema (Figure 8A, C and E). We also observed an increase of the numbers of angiogenesis precursors in the blood of mice treated with spiperone. Here, we do not exclude potential positive effects of spiperone on mobilization of cells from the bone marrow (Figure 8B, D and F).
Notch signaling plays a significant role in the development and homeostasis of lung tissue.64,65 Briefly, activation of Notch signaling pathway inhibits cell differentiation and maturation and its expression is downregulated during cell differentiation.20,66–68 In this context, it is noteworthy that the levels of Notch1 expression in mature endothelial cells decrease in COPD patients.20 However, little is known about the impact of emphysema on immature Notch1+ endothelial cells.
As shown in Figure 9A–C, CSE/LPS -induced pulmonary emphysema was accompanied by a decrease in the numbers of EPCs and angiogenesis precursors expressing Notch1. This could be explained by an induction of differentiation of these unspecialised cells.
A multitude of studies suggested a crosstalk between Notch and VEGF signaling.69–71 Moreover, a dopamine-induced direct interaction between VEGFR2 and D2R disrupts VEGF-mediated angiogenesis.72 Thus, the regenerative effect of spiperone may be mediated by dopamine-induced direct interaction between Notch and VEGF. In our study, we performed flow cytometric assessment of the numbers of pulmonary VEGF2+ endothelial cells co-expressing Notch1. According to our data, spiperone reduced the number of VEGF2+/Notch1+ endothelial cells more than two-fold in the lungs of mice within group 3 compared to group 2 (Figure 9). This could be explained by a potential dissociation of the D2 receptor and VEGFR2 by blocking dopamine receptors on cell membrane. Thus, spiperone presumably eliminates dopamine inhibitory effect on angiogenesis.
Helms and colleagues reported that D2Rs are expressed by EPCs.30 The presence of these receptors suggests a direct regulatory effect of dopamine on lung EPCs in emphysema and COPD. Our in vitro results showed that dopamine exerted a stimulating effect on EPCs with CD31+CD34+CD146+Nectin2+ phenotype in endothelial cell culture obtained from emphysema dilated lungs (Figure 10E). Notably, Nectin2 is a marker of actively proliferating cells. Pretreatment of endothelial cells with spiperone reduced dopamine-induced proliferation of EPCs. In addition, in the in vivo experiments, spiperone reduced the amount of Notch1+ EPCs in the group 3 mice lungs compared with group 2 mice (Figure 9). From our point of view, a blockade of D2Rs inhibits dopamine-mediated proliferation of EPCs expressing CD31 and CD34. Moreover, their differentiation could be promoted by neglecting the effects of dopamine on the Notch.
Taken together, we demonstrated multiple positive effects of spiperone in a model of COPD. We showed that spiperone induces its positive vascular effects by facilitating mobilization and migration of EPCs (CD45−CD34+CD31+), VEGF2+ (CD309+) endothelial cells and angiogenesis precursors (CD45−CD117+CD309+) to injured lungs. Endothelial regeneration caused by spiperone administration can be explained by the fact that a blockade of D2Rs eliminates negative effects of dopamine on Notch signaling-mediated differentiation of EPCs and on VEGF-mediated angiogenesis. Future research will assess the details of these complex cross-couplings and could lead to novel strategies in COPD therapy based on dopamine receptor inhibition.
Moreover, spiperone can affect canonical Wnt signaling.35 The Wnt inhibitory function of spiperone is not associated with its dopamine- and serotonin-receptor antagonist properties. Instead, spiperone increases intracellular calcium levels.35 This could be another mechanism of spiperone action at the COPD. Previously, it was shown that spiperone can reverse key changes in gene expression associated with regulation of hydrolase activity pathway in epithelial cells and monocytes subjected to cigarette smoke stimulation.73 Therefore, further studies are required to understand the mechanisms underlying the action of spiperone in pulmonary injury repair and pathogenesis of COPD.
Despite the positive effects of spiperone, we understand that our COPD model (LPS/CSE-induced pulmonary emphysema) does not fully reproduce the clinical picture of the disease. It must be noted that the processes involved in the pathogenesis of COPD that we mentioned earlier are complicatedly connected, interacted, and networked in the development of COPD, and the cause-effect relationships are still unknown. In addition, when evaluating the therapeutic implications of spiperone administration, we were able to show only a protective and not a curative effect of spiperone treatment because of the experimental murine model with CSE/LPS-induced COPD. Nevertheless, the therapeutic strategies aimed at protecting the lung from COPD may prove useful for slowing the presently unstoppable progression of COPD.
Conclusion
Our research has identified that treatment of spiperone could represent a novel therapeutic approach for the treatment of patients with COPD.
Disclosure
The authors declare no conflict of interest. This research was funded by grants from the MINISTRY OF SCIENCE AND HIGHER EDUCATION RUSSIAN FEDERATION, grant number 075-15-2020-773.
References
1. World Health Organization. Global health estimates 2016: deaths by cause, age, sex, by country and by region, 2000–2016; 2018. Available from: www.who.int/ru/news-room/fact-sheets/detail/the-top-10-causes-of-death.
2. Rycroft CE, Heyes A, Lanza L, et al. Epidemiology of chronic obstructive pulmonary disease: a literature review. Int J Chron Obstruct Pulmon Dis. 2012;7:457–494. doi:10.2147/COPD.S32330
3. De Cunto G, Cavarra E, Bartalesi B, et al. Innate immunity and cell surface receptors in the pathogenesis of COPD: insights from mouse smoking models. Int J Chron Obstruct Pulmon Dis. 2020;15:1143–1154. doi:10.2147/COPD.S246219
4. Lee JW, Ryu HW, Lee SU, et al. Pistacia weinmannifolia ameliorates cigarette smoke and lipopolysaccharide-induced pulmonary inflammation by inhibiting interleukin-8 production and NF-κB activation. Int J Mol Med. 2019;44:949–959. doi:10.3892/ijmm.2019.4247
5. Bagdonas E, Raudoniute J, Bruzauskaite I, et al. Novel aspects of pathogenesis and regeneration mechanisms in COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:995–1013. doi:10.2147/COPD.S82518
6. Segura-Valdez L, Pardo A, Gaxiola M, et al. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest. 2000;117:684–694. doi:10.1378/chest.117.3.684
7. Kasahara Y, Tuder RM, Cool CD, et al. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med. 2001;163:737–744. doi:10.1164/ajrccm.163.3.2002117.5
8. Yokohori N, Aoshiba K, Nagai A; Respiratory Failure Research Group in Japan. Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest. 2004;125:626–632. doi:10.1378/chest.125.2.626
9. Imai K, Mercer BA, Schulman LL, et al. Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur Respir J. 2005;25:250–258. doi:10.1183/09031936.05.00023704
10. Green CE, Turner AM. The role of the endothelium in asthma and chronic obstructive pulmonary disease (COPD). Respir Res. 2017;18:20. doi:10.1186/s12931-017-0505-1
11. Peinado VI, Ramírez J, Roca J, et al. Identification of vascular progenitor cells in pulmonary arteries of patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2006;34:257–263. doi:10.1165/rcmb.2005-0255OC
12. Wedzicha JA, Calverley PM, Rabe KF. Roflumilast: a review of its use in the treatment of COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:81–90. doi:10.2147/COPD.S89849
13. Moro L, Pedone C, Scarlata S, et al. Endothelial dysfunction in chronic obstructive pulmonary disease. Angiology. 2008;59:357–364. doi:10.1177/0003319707306141
14. Estepar RS, Kinney GL, Black-Shinn JL, et al. Computed tomographic measures of pulmonary vascular morphology in smokers and their clinical implications. Am J Respir Crit Care Med. 2013;188:231–239. doi:10.1164/rccm.201301-0162OC
15. Palange P, Testa U, Huertas A, et al. Circulating haemopoietic and endothelial progenitor cells are decreased in COPD. Eur Respir J. 2006;27:529–541. doi:10.1183/09031936.06.00120604
16. Kim EK, Lee JH, Jeong HC, et al. Impaired colony-forming capacity of circulating endothelial progenitor cells in patients with emphysema. Tohoku J Exp Med. 2012;227:321–331. doi:10.1620/tjem.227.321
17. Sakhatskyy P, Wang Z, Borgas D, et al. Double-hit mouse model of cigarette smoke priming for acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2017;312:56–L67. doi:10.1152/ajplung.00436.2016
18. Gotts JE, Abbott J, Fang X, et al. Cigarette smoke exposure worsens endotoxin-induced lung injury and pulmonary edema in mice. Nicotine Tob Res. 2017;19:1033–1039. doi:10.1093/ntr/ntx062
19. Li S, Hu X, Wang Z, et al. Different profiles of notch signaling in cigarette smoke-induced pulmonary emphysema and bleomycin-induced pulmonary fibrosis. Inflamm Res. 2015;64:363–371. doi:10.1007/s00011-015-0816-y
20. Zong D, Li J, Cai S, et al. Notch1 regulates endothelial apoptosis via the ERK pathway in chronic obstructive pulmonary disease. Am J Physiol Cell Physiol. 2018;315:C330–C340. doi:10.1152/ajpcell.00182.2017
21. Rabe KF, Hurd S, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176:532–555. doi:10.1164/rccm.200703-456SO
22. Kamei N, Atesok K, Ochi M. The use of endothelial progenitor cells for the regeneration of musculoskeletal and neural tissues. Stem Cells Int. 2017;2017:1960804. doi:10.1155/2017/1960804
23. Quillard T, Coupel S, Coulon F, et al. Impaired Notch4 activity elicits endothelial cell activation and apoptosis: implication for transplant arteriosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:2258–2265. doi:10.1161/ATVBAHA.108.174995
24. Dabral S, Tian X, Kojonazarov B, et al. Notch1 signalling regulates endothelial proliferation and apoptosis in pulmonary arterial hypertension. Eur Respir J. 2016;48:1137–1149. doi:10.1183/13993003.00773-2015
25. MacKenzie F, Duriez P, Wong F, et al. Notch4 inhibits endothelial apoptosis via RBP-Jkappa-dependent and -independent pathways. J Biol Chem. 2004;279:11657–11663. doi:10.1074/jbc.M312102200
26. Wu J, Wang X, Yao Z, et al. Allyl isothiocyanate may reverse the expression of MRP1 in COPD rats via the Notch1 signaling pathway. Arch Pharm Res. 2019;42(11):1000–1011. doi:10.1007/s12272-019-01183-4
27. Luo J, Li L, Hu D, et al. LINC00612/miR-31-5p/Notch1 axis regulates apoptosis, inflammation, and oxidative stress in human pulmonary microvascular endothelial cells induced by cigarette smoke extract. Int J Chron Obstruct Pulmon Dis. 2020;15:2049–2060. doi:10.2147/COPD.S255696
28. Ricci A, Mignini F, Tomassoni D, et al. Dopamine receptor subtypes in the human pulmonary arterial tree. Auton Autacoid Pharmacol. 2006;26:61–369. doi:10.1111/j.1474-8673.2006.00376.x
29. Chakroborty D, Chowdhury UR, Sarkar C, et al. Dopamine regulates endothelial progenitor cell mobilization from mouse bone marrow in tumor vascularization. J Clin Invest. 2008;118:1380–1389. doi:10.1172/JCI33125
30. Helms MN, Chen XJ, Ramosevac S, et al. Dopamine regulation of amiloride-sensitive sodium channels in lung cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L710–L722. doi:10.1152/ajplung.00486.2004
31. Hoeppner LH, Wang Y, Sharma A, et al. Dopamine D2 receptor agonists inhibit lung cancer progression by reducing angiogenesis and tumor infiltrating myeloid derived suppressor cells. Mol Oncol. 2015;9:270–281. doi:10.1016/j.molonc.2014.08.008
32. Löfdahl A, Tornling G, Wigén J, et al. Pathological insight into 5-HT2B receptor activation in fibrosing interstitial lung diseases. Int J Mol Sci. 2020;22:225. doi:10.3390/ijms22010225
33. Leysen JE, Gommeren W, Laduron PM. Spiperone: a ligand of choice for neuroleptic receptors. 1. Kinetics and characteristics of in vitro binding. Biochem Pharmacol. 1978;27:307–316. doi:10.1016/0006-2952(78)90233-2
34. Gundlach AL, Largent BL, Snyder SH. 125I-Spiperone: a novel ligand for D2 dopamine receptors. Life Sci. 1984;35:1981–1988. doi:10.1016/0024-3205(84)90479-x
35. Lu D, Carson DA. Spiperone enhances intracellular calcium level and inhibits the Wnt signaling pathway. BMC Pharmacol. 2009;9:13. doi:10.1186/1471-2210-9-13
36. Snyder SH, Largent BL. Receptor mechanisms in antipsychotic drug action: focus on sigma receptors. J Neuropsychiatry Clin Neurosci. 1989;1:7–15. doi:10.1176/jnp.1.1.7
37. Skurikhin EG, Pershina OV, Reztsova AM, et al. Modulation of bleomycin-induced lung fibrosis by pegylated hyaluronidase and dopamine receptor antagonist in mice. PLoS One. 2015;10:e0125065. doi:10.1371/journal.pone.0125065
38. Liu Y, Wolfe SA. Haloperidol and spiperone potentiate murine splenic B cell proliferation. Immunopharmacology. 1996;34:147–159. doi:10.1016/0162-3109(96)00120-8
39. Liang L, MacDonald K, Schwiebert EM, et al. Spiperone, identified through compound screening, activates calcium-dependent chloride secretion in the airway. Am J Physiol Cell Physiol. 2009;296:C131–C141. doi:10.1152/ajpcell.00346.2008
40. Hardaker EL, Freeman MS, Dale N, et al. Exposing rodents to a combination of tobacco smoke and lipopolysaccharide results in an exaggerated inflammatory response in the lung. Br J Pharmacol. 2010;160:1985–1996. doi:10.1111/j.1476-5381.2010.00857
41. Lee JW, Ryu HW, Lee SU, et al. Protective effect of polyacetylene from Dendropanax morbifera Leveille leaves on pulmonary inflammation induced by cigarette smoke and lipopolysaccharide. J Funct Foods. 2017;32:358–366. doi:10.1016/j.jff.2017.03.007
42. Zhou Y, Tan X, Kuang W, et al. Erythromycin ameliorates cigarette-smoke-induced emphysema and inflammation in rats. Transl Res. 2012;159:464–472. doi:10.1016/j.trsl.2011.09.007
43. Moazed F, Burnham EL, Vandivier RW, et al. Cigarette smokers have exaggerated alveolar barrier disruption in response to lipopolysaccharide inhalation. Thorax. 2016;71:1130–1136. doi:10.1136/thoraxjnl-2015-207886
44. He ZH, Chen P, Chen Y, et al. Comparison between cigarette smoke-induced emphysema and cigarette smoke extract-induced emphysema. Tob Induc Dis. 2015;13:6. doi:10.1186/s12971-015-0033-z
45. Skurikhin EG, Pershina OV, Pakhomova AV, et al. Endothelial progenitor cells as pathogenetic and diagnostic factors, and potential targets for GLP-1 in combination with metabolic syndrome and chronic obstructive pulmonary disease. Int J Mol Sci. 2019;20:1105. doi:10.3390/ijms20051105
46. Liang GB, He ZH. Animal models of emphysema. Chin Med J (Engl). 2019;132:2465–2475. doi:10.1097/CM9.0000000000000469
47. Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–891. doi:10.1038/nature01326
48. Skurikhin EG, Krupin VA, Pershina OV, et al. Endothelial progenitor cells and Notch-1 signaling as markers of alveolar endothelium regeneration in pulmonary emphysema. Bull Exp Biol Med. 2018;166:201–206. doi:10.1007/s10517-018-4314-4
49. Thurlbeck WM. Measurement of pulmonary emphysema. Am Rev Respir Dis. 1967;95:752–764. doi:10.1164/arrd.1967.95.5.752
50. Bracke KR, D’hulst AI, Maes T, et al. Cigarette smoke-induced pulmonary inflammation and emphysema are attenuated in CCR6-deficient mice. J Immunol. 2006;177:4350–4359. doi:10.4049/jimmunol.177.7.4350
51. Muñoz-Barrutia A, Ceresa M, Artaechevarria X, et al. Quantification of lung damage in an elastase-induced mouse model of emphysema. Int J Biomed Imaging. 2012;2012:734734. doi:10.1155/2012/734734
52. Xiong J, Tian J, Zhou L, et al. Interleukin-17A deficiency attenuated emphysema and bone loss in mice exposed to cigarette smoke. Int J Chron Obstruct Pulmon Dis. 2020;15:301–310. doi:10.2147/COPD.S235384
53. Skurikhin E, Nebolsin V, Widera D, et al. Antifibrotic and regenerative effects of treamid in pulmonary fibrosis. Int J Mol Sci. 2020;21:8380. doi:10.3390/ijms21218380
54. Saetta M, Shiner RJ, Angus GE, et al. Destructive index: a measurement of lung parenchymal destruction in smokers. Am Rev Respir Dis. 1985;131:764–769. doi:10.1164/arrd.1985.131.5.764
55. Tilton RG, Miller EJ, Kilo C, et al. Pericyte form and distribution in rat retinal and uveal capillaries. Invest Ophthalmol Vis Sci. 1985;26:68–73.
56. Watson JK, Sanders P, Dunmore R, et al. Distal lung epithelial progenitor cell function declines with age. Sci Rep. 2020;10:10490. doi:10.1038/s41598-020-66966-y
57. Lim YC, Garcia-Cardena G, Allport JR, et al. Heterogeneity of endothelial cells from different organ sites in T-cell subset recruitment. Am J Pathol. 2003;162:1591–1601. doi:10.1016/S0002-9440(10)64293-9
58. Fehrenbach ML, Cao G, Williams JT, et al. Isolation of murine lung endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2008;296:L1096–L1103. doi:10.1152/ajplung.90613.2008
59. Kessler R, Partridge MR, Miravitlles M, et al. Symptom variability in patients with severe COPD: a pan-European cross-sectional study. Eur Respir J. 2011;37:264–272. doi:10.1183/09031936.00051110
60. Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summary. Am J Respir Crit Care Med. 2017;195:557–582. doi:10.1164/rccm.201701-0218PP
61. Doyle MF, Tracy RP, Parikh MA, et al. Endothelial progenitor cells in chronic obstructive pulmonary disease and emphysema. PLoS One. 2017;12:e0173446. doi:10.1371/journal.pone.0173446
62. Gadek J, Fells G, Crystal R. Cigarette smoking induces functional antiprotease deficiency in the lower respiratory tract of humans. Science. 1979;206:1315–1316. doi:10.1126/science.316188
63. Kapellos TS, Bassler K, Aschenbrenner AC, et al. Dysregulated functions of lung macrophage populations in COPD. J Immunol Res. 2018;2018:2349045. doi:10.1155/2018/2349045
64. Radtke F, Fasnacht N, Macdonald HR. Notch signaling in the immune system. Immunity. 2010;32:14–27. doi:10.1016/j.immuni.2010.01.004
65. Xu K, Moghal N, Egan SE. Notch signaling in lung development and disease. Adv Exp Med Biol. 2012;727:89–98. doi:10.1007/978-1-4614-0899-4_7
66. Walker L, Carlson A, Tan-pertel HT, et al. The Notch receptor and its ligands are selectively expressed during hematopoietic development in the mouse. Stem Cells. 2001;19:543–552. doi:10.1634/stemcells.19-6-543
67. Sainson RC, Aoto J, Nakatsu MN, et al. Cell-autonomous notch signaling regulates endothelial cell branching and proliferation during vascular tubulogenesis. FASEB J. 2005;19:1027–1079. doi:10.1096/fj.04-3172fje
68. Zanotti S, Canalis E. Mechanisms in endocrinology: notch signaling in skeletal health and disease. Eur J Endocrinol. 2013;168(6):R95–R103. doi:10.1530/EJE-13-0115
69. Blanco R, Gerhardt H. VEGF and Notch in tip and stalk cell selection. Cold Spring Harb Perspect Med. 2013;3:a006569. doi:10.1101/cshperspect.a006569
70. Fish JE, Cantu Gutierrez M, Dang LT, et al. Dynamic regulation of VEGF-inducible genes by an ERK/ERG/p300 transcriptional network. Development. 2017;144:2428–2444. doi:10.1242/dev.146050
71. Kühn C, Checa S. Computational modeling to quantify the contributions of VEGFR1, VEGFR2, and lateral inhibition in sprouting angiogenesis. Front Physiol. 2019;10:288. doi:10.3389/fphys.2019.00288
72. Sinha S, Vohra PK, Bhattacharya R, et al. Dopamine regulates phosphorylation of VEGF receptor 2 by engaging src-homology-2-domain-containing protein tyrosine phosphatase 2. J Cell Sci. 2009;122:3385–3392. doi:10.1242/jcs.053124
73. Zhua D, Maa N, Chen L, et al. Verification of the role of spiperone in the treatment of COPD through bioinformatics analysis. Int Immunopharmacol. 2021;101(A):108308. doi:10.1016/j.intimp.2021.108308
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.