Back to Journals » The Application of Clinical Genetics » Volume 14
Spinal Muscular Atrophy: Mutations, Testing, and Clinical Relevance
Authors Keinath MC, Prior DE, Prior TW
Received 8 December 2020
Accepted for publication 15 January 2021
Published 25 January 2021 Volume 2021:14 Pages 11—25
DOI https://doi.org/10.2147/TACG.S239603
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Melissa C Keinath,1 Devin E Prior,2 Thomas W Prior1
1Pathology, University Hospitals Center for Human Genetics, Cleveland, OH, USA; 2Neurology, Mount Auburn Hospital, Cambridge, MA, USA
Correspondence: Thomas W Prior Email [email protected]
Abstract: Spinal muscular atrophy (SMA) is a heritable neuromuscular disorder that causes degeneration of the alpha motor neurons from anterior horn cells in the spinal cord, which causes severe progressive hypotonia and muscular weakness. With a carrier frequency of 1 in 40– 50 and an estimated incidence of 1 in 10,000 live births, SMA is the second most common autosomal recessive disorder. Affected individuals with SMA have a homozygous loss of function of the survival motor neuron gene SMN1 on 5q13 but keep the modifying SMN2 gene. The most common mutation causing SMA is a homozygous deletion of the SMN1 exon 7, which can be readily detected and used as a sensitive diagnostic test. Because SMN2 produces a reduced number of full-length transcripts, the number of SMN2 copies can modify the clinical phenotype and as such, becomes an essential predictive factor. Population-based SMA carrier screening identifies carrier couples that may pass on this genetic disorder to their offspring and allows the carriers to make informed reproductive choices or prepare for immediate treatment for an affected child. Three treatments have recently been approved by the Food and Drug Administration (FDA). Nusinersen increases the expression levels of the SMN protein using an antisense oligonucleotide to alter splicing of the SMN2 transcript. Onasemnogene abeparvovec is a gene therapy that utilizes an adeno-associated virus serotype 9 vector to increase low functional SMN protein levels. Risdiplam is a small molecule that alters SMN2 splicing in order to increase functional SMN protein. Newborn screening for SMA has been shown to be successful in allowing infants to be treated before the loss of motor neurons and has resulted in improved clinical outcomes. Several of the recommendations and guidelines in the review are based on studies performed in the United States.
Keywords: spinal muscular atrophy, carrier screening, newborn screening, SMA treatment
Clinical Manifestations
Spinal muscular atrophy, an autosomal recessive disorder, is the most common genetic cause of infant mortality, affecting 1 in 10,000 live births.1 The disorder causes progressive loss of the alpha motor neurons of the ventral spinal cord and motor nuclei of the lower brainstem resulting in hypotonia, muscle weakness and atrophy of variable severity depending on the underlying genotype.1 Weakness is predominantly proximal with greater involvement of the lower extremities and diffuse areflexia on examination. Bulbar and respiratory muscle weakness can occur particularly in more severe cases. Facial and ocular muscles are generally not involved.2
As shown in Table 1, the disorder has traditionally been classified into types 0–4 based on symptom severity and genotype, though with recent disease-modifying therapies, phenotypes have become more diverse and classifications have evolved to focus on functional status (non-sitters, sitters, walkers), or treatment response (decline, no change, improvement).3
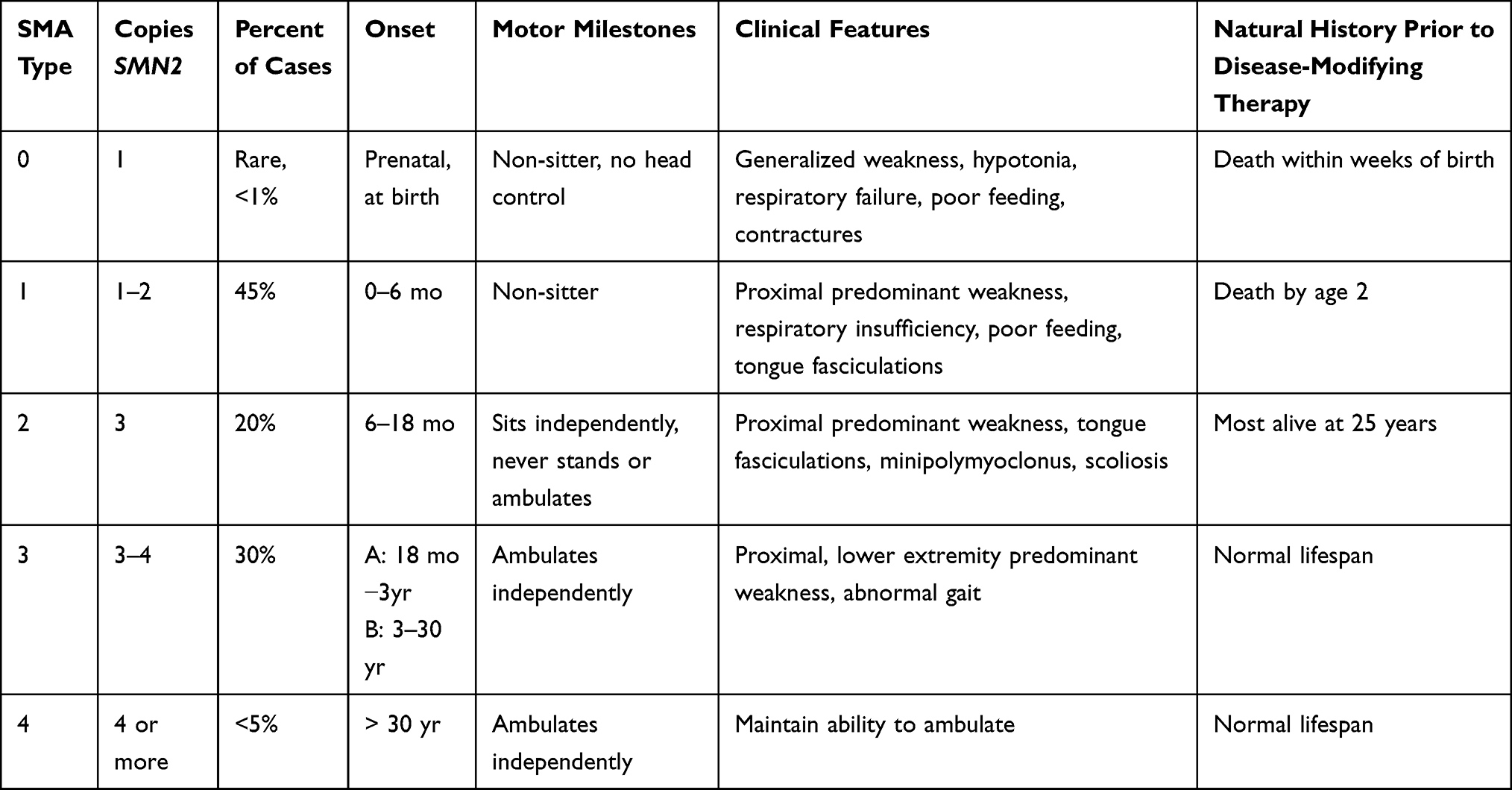 |
Table 1 Classification of Spinal Muscular Atrophy |
Under the traditional classification, type 0 SMA represents the most severe phenotype with one copy of SMN2. Patients present at birth with generalized weakness and hypotonia, respiratory distress, and poor feeding. Decreased intrauterine movements may be felt prenatally and lead to contractures. Prior to disease-modifying therapy, patients died within weeks of birth without achieving any motor milestones (non-sitters).2,4
Patients with type 1 SMA, the most common form and representing approximately 45% of cases, develop symptoms around 0–6 months of age, including flexion, proximal predominant limb weakness, respiratory insufficiency, and poor feeding. Given intercostal and chest wall muscle weakness, relative to preserved diaphragm strength, patients show a bell-shaped chest deformity with breathing and paradoxical breathing. Tongue fasciculations are present, but facial and ocular muscle strength remains intact. Cognitive function is normal to above average. Historically, patients did not achieve the ability to sit independently (non-sitters) with death often prior to age 2.2,5 Most of the type 1 patients have one to two copies of SMN2.
Type 2 SMA, comprising 30% of cases, presents with weakness by 6–18 months and most often have three SMN2 copies. Patients achieve the ability to sit unsupported (sitters), often by 9 months, though may later lose this ability and are never able to stand or walk independently.2 Examination shows predominantly proximal weakness, more severe in the lower extremities, tongue atrophy and fasciculations, and sometimes fine distal tremor (minipolymyoclonus). Respiratory insufficiency and dysphagia are common, particularly in more severe phenotypes.2 Weak axial musculature may contribute to significant scoliosis which may worsen restrictive lung disease and respiratory insufficiency.3 Aggressive supportive treatments prior to the onset of disease-modifying therapy led to increased lifespan, with 68.5% of this historic cohort surviving to age 25.3,5
With onset ranging from 18 months to adulthood, type 3 SMA, representing 15% of cases, is defined by achieving the ability to stand or walk without support (walkers), though this ability may be lost with disease progression.2 Patients may present with symptoms of proximal weakness such as falls, abnormal gait, and difficulty climbing stairs. Unlike other phenotypes, type 3 patients have a normal life expectancy.2,6 Most patients do not develop significant respiratory muscle weakness.6 Walkers may be sub-grouped into type 3a with symptoms onset of 18 months-3 years, type 3b with onset 3–30 years, and type 4 with onset at 30 years or more.3 The milder type 3 and 4 patients usually present with 3 and 4 copies of SMN2.
Although SMA is defined as a disorder of the motor neurons, SMN protein is expressed in most tissues with different organs and tissues varying in their requirements for development and functioning. Research in animal models and patients has shown that SMA is a multisystem disease also affecting the skeletal muscle, heart, kidney, liver, pancreas, spleen, and immune system. Multiorgan features including congenital heart defects, cardiac rhythm abnormalities, sleep disturbances, impaired kidney function, and pancreatic defects, have previously been reported in SMA patients, particularly in the more severely affected type 0 or 1 subtypes. As patients live longer with new therapies, these multisystem comorbidities may become more common.7–11
Gene Structure
The majority of SMA cases are caused by mutations in the survival motor neuron 1 (SMN1) gene positioned at 5q13.12 SMN1 (also called SMNT, where T stands for telomere) spans 20-kb and lies in the telomeric portion of an inverted duplication of 500kb, a DNA architecture prone to rearrangements and deletions (Figure 1). The duplicated centromeric element, known as SMN2 (also called C-BCD541 and SMNC, where C stands for centromere) is highly homologous to SMN1 with more than 99% nucleotide identity. First thought to have 8 exons, both SMN1 and SMN2 contain 9 exons that encode the 294-amino acid protein, survival of motor neuron (SMN).12,13 The exons are numbered 1, 2a, 2b, 3, 4, 5, 6, 7 and 8. The stop codon for SMN occurs in exon 7, and exon 8 is left untranslated. SMN1 and SMN2 vary by 8 nucleotides, 5 of which are intronic and 3 of which occur in the last 3 exons. Of the differences, only a C-to-T transition in SMN2 exon 7 (specifically, c.840C>T) falls in a coding region and disrupts an exonic splice enhancer in exon 7. As a consequence of this change, most of the SMN2 transcripts lack exon 7, creating an incomplete and degraded SMN protein. An estimated 10% of the protein produced by each SMN2 copy is functional, making it a modifying gene (Figure 1). SMN is an RNA-binding protein that contributes to many cellular processes and pathways, most notably, playing a critical role in snRNP complex assembly in the cytoplasm.14
 |
Figure 1 Two genes are responsible for producing the survival motor neuron (SMN) protein, SMN1 and SMN2. SMN1 provides humans with the proper quantity of SMN protein necessary for a normal phenotype. SMN2 is an inverted duplicate of SMN1 lying closer to the centromere. A C>T transition in exon 7 of SMN2 causes the SMN2 gene to produce mostly (~90%) nonfunctional protein and a small amount (~10%) of the functional SMN protein. |
Pathogenic Variants
The majority of SMA cases are caused by an absence of the SMN1 gene. Ninety-five percent of SMA affected individuals have a homozygous deletion of SMN1 exon 7 or gene conversion from SMN1 to SMN2, and most of the remaining 5% are compound heterozygotes for an SMN1 exon 7 deletion and an SMN1 point mutation (normal in: Figure 2A, variants in: Figure 2B and C).5 Other intragenic mutations found in the compound heterozygous state with an SMN1 deletion include: missense, nonsense, splice site mutations, insertions, deletions and duplications (Figure 2C). Recurrent variants have been found in exons 3 and 6, making these two exons hot spots for small mutations and missense mutations, respectively (Figure 2D).15,16 Exon 6 codes for a domain in the protein which plays a role in protein oligomerization, and those patients with exon 6 missense mutations have decreased SMN protein self-oligomerization capacity.15 The exon 6 p.Tyr272Cys missense mutation is the most frequently reported mutation in the SMN1 gene. Public archives such as Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/) and Varsome (https://varsome.com/) report relationships between human genome variations and associated phenotypes along with reported clinical significance. Currently, Clinvar shows 49 pathogenic mutations reported in the SMN1 gene, 20 likely pathogenic, 62 benign, 7 likely benign, in addition to 40 variants of uncertain significance. The majority of pathogenic mutations reported to these databases are substitutions, deletions and duplications with several reports of small insertions.
 |
Figure 2 (A) A chromosome carrying a normal copy of SMN1 and SMN2. (B) The blank box indicates a deleted gene. A deletion can remove part or all of the SMN1 gene. (C) The curved arrow represents a conversion. With the C>T transition in SMN1, the SMN1 copy now closely resembles SMN2 and is considered SMN2-like. (D) Point mutations occurring in any of the SMN1 exons prior to the last exon can affect the SMN protein. |
Testing
Diagnostic Testing
With 95% of affected individuals having a homozygous absence of SMN1 exon 7, screening for the loss of exon 7 is the first tier in diagnostic testing. There are numerous DNA assays which allow for the detection of absence of SMN1 exon 7 and are based on the c.840C>T difference between SMN1 and SMN2. Multiplex ligation-dependent probe amplification (MLPA) is one of the most popular methods used as an initial deletion test in laboratories as it is convenient, highly sensitive, and capable of determining both SMN1 and SMN2 copy number.17
Prenatal testing for SMA for the SMN1 exon 7 deletion can be performed on DNA extracted from either chorionic villus sampling (CVS) specimens or amniotic fluids. Prenatal diagnosis of SMA often occurs when there is a previously identified homozygous deletion in the index case or a 25% risk of an affected fetus (when both parents are identified as carriers by family history or following carrier testing) or the presence of abnormal findings on fetal ultrasound, such as decreased fetal movements, contractures in-utero, or increased nuchal translucency. The presence of maternal cell contamination of the fetal specimen may result in a false-negative test result and therefore must be tested for and shown to be absent prior to reporting the prenatal test result.
The SMN1 exon 7 deletion test is currently being utilized as a reliable and accurate confirmatory test for the majority of patients suspected to have SMA. The test is highly sensitive, approximately 95%, and nearly 100% specific. Furthermore, results can be easily reported within 24 hours. The more invasive muscle biopsy test on patients presenting with an SMA-phenotype is no longer the first tier test and is unnecessary for the majority of cases. Alternative diagnoses should be considered when 2 normal copies of the SMN1 gene are detected in these individuals. In such cases, other motor neuron disorders that should be considered include congenital myopathies, muscular dystrophies, and metabolic disorders should be considered.
Point Mutation Testing
Although the majority of molecular diagnoses for SMA cases will be through homozygous deletion screening, another 5% of cases will be caused by other subtle mutations in the SMN1 gene. As a consequence of the high deletion frequency, most of these cases will reveal a single SMN1 deletion on one allele and an intragenic type of mutation on the other allele. These patients are referred to as compound heterozygotes and have been shown to have a variety of different types of SMN1 mutations including missense mutations, nonsense mutations, splice site mutations, insertions and small deletions. Due to SMN1’s small size, it is a relatively straightforward procedure to Sanger sequence the gene and identify mutations in patients who are test negative for the homozygous deletion test. Furthermore, massive parallel DNA sequencing allows one to simultaneously sequence a number of genes involved in neuromuscular disorders along with the SMN1 gene.18 There are rare SMA affected patients with a single copy of SMN1 and an unidentified second mutation. These unidentified mutations may include a mutation in an intron, which could affect splicing or one within a regulatory region of the gene. In these cases, mRNA analysis may be helpful.
Verification of the occurrence of the intragenic mutations is located in the SMN1 gene, and not the SMN2 gene, should be performed. SMN1-specific long-range PCR amplification followed by either direct DNA sequencing of that long-range product or nested PCR sequencing is necessary when variants or mutations are identified. Direct sequencing of the SMN genes includes the following important limitations: 1) allele-specific sequencing requirements of all identified variants, 2) identification of variants of uncertain significance, 3) non-detection of mutations in patients with chromosomal rearrangements or mosaicism for the mutation and 4) non-detection of large deletions or insertions.19 All novel gene changes should be compared with variants with the human gene mutation database (HGMD) and Clinvar variant database. Additionally, the frequency of gene changes in the gnomAD database (https://gnomad.broadinstitute.org) should be considered during classification and the change should be analyzed by applicable predictive software for the effects of the change on the protein structure and function. If the patient is a compound heterozygote, the SMN1 deletion and the point mutation should be proven to be in the trans configuration, which may require parental testing (Figure 3). Sequence analysis of the SMN1 gene is of particular importance for patients with an SMA phenotype found to have 2 copies of the SMN1 gene, who originate from genetically isolated populations or are born to consanguineous parents. In families from genetically isolated populations, the point mutation may have a high frequency whereas in affected children born to consanguineous parents, the mutation would have only arisen once, and the affected received the same familial mutation from each parent. In either case, the affected individuals are homozygous for the mutation. Homozygous subtle mutations have been reported in some of these cases.20,21 Mutations should be classified following the American College of Medical Genetics and Genomics (ACMG) standards and guidelines.22
 |
Figure 3 (A) A wild type with 2 copies of SMN1 and SMN2 on each chromosome. (B) SMA carrier with only one copy of SMN1 on one chromosome and loss of SMN1 on the other. (C) A silent SMA carrier with a duplication of SMN1 on one chromosome and no SMN1 on the other chromosome. (D) SMA carrier with one normal copy of SMN1 on one chromosome and one copy that contains a point mutation on the other chromosome. |
Newborn Screening
The identification affected infants prior to the presentation of clinical symptoms has been accomplished by newborn screening (NBS) for a number of disorders and has allowed for newborns to be treated prior to irreversible changes that may take place. NBS has become one of the most successful public health initiatives in history and has improved the quality of life of many people with a variety of disorders. In recent years, the number of conditions included in many NBS panels has expanded. For a disorder to be included in an NBS program, they generally have to meet the following criteria: the condition is an important health problem, the disease can be detected in the early newborn period but is clinically silent, the test has appropriate sensitivity and specificity, and most importantly, there are clinical favorable outcomes associated with early therapeutic intervention.
In type 1 SMA infants, rapid loss of motor units has been shown to occur within the first three months of age resulting in severe denervation with loss of more than 95% of motor units within six months.23 For SMA type I patients, a very small window for beneficial therapeutic intervention exists. Therapies need to be administered within the newborn period for maximum benefit, before the loss of motor neurons, which can only be accomplished by identifying the infants through SMA newborn screening. In addition, identifying SMA in a newborn using NBS saves the family from the pain and cost of unnecessary testing in the future. Furthermore, the early diagnosis and subsequent genetic counseling can help identify other at-risk family members and prevent additional cases.
Treatment of SMA was previously limited to supportive care until December 2016 when the first disease-modifying therapy, nusinersen, was approved by the US Food and Drug Administration following a successful clinical trial. A single affected child was identified via the New York state SMA NBS pilot study and was enrolled in a presymptomatic nusinersen clinical trial.24 At three years of age, the child was meeting motor milestones appropriate for her age. Nusinersen was also the first drug that received approval for treatment of SMA by the European Medicine Agency (EMA). Gene therapy was subsequently approved by the FDA in May 2019. The most robust responses for both nusinersen treatment and gene therapy have been shown to occur when treatment is initiated pre-symptomatically.25 The EMA approved gene therapy for the treatment of patients with SMA having up to three copies of the SMN2 gene or the clinical presentation of SMA type 1.26
Since SMA does not have a biochemical marker, newborn patients undergo DNA testing for deletion of exon 7 in SMN1. The deletion was previously detected from DNA extracted from newborn blood spots using a liquid microbead array.27 A feasibility study involving 40,103 newborn blood spots and using the array approach identified four SMN1 homozygous deletions.28 Utilizing a high throughput newborn screen and qPCR, DNA from 165,525 blood spots were screened for the SMN1 deletion in a German Pilot Project and 22 cases of SMA were identified.29 All of the pre-symptomatic, nusinersen-treated children remained without motor symptoms, whereas the 2 untreated children with 2 SMN2 copies presented onset of disease before 3 months of age. NBS for SMA was implemented in New York State in 2018. Blood spots from 225,093 infants were tested in the first year, and 8 screened positive for the homozygous deletion. The asymptomatic infants with 2 or 3 SMN2 copies were treated with either nusinersen or gene therapy and were all asymptomatic at their last follow-up.30
An NBS program will also identify exon 7 deletion positive milder later-onset cases of SMA. Since these cases may not require early intervention as the more severe cases, the early diagnosis of the milder types of cases may not be as acceptable from both the medical community and families who may not desire to have this information. Positive SMA NBS results necessitate genetic counseling for the families; however, if the physicians and parents are aware that a child may develop SMA symptoms in the future, they can monitor for motor problems and prepare for treatment. The genotype/phenotype association between the SMN2 copy number and clinical severity is well supported in the literature (see SMN2 Testing Section) and will allow for selection of those SMA-affected individuals who require early therapeutic intervention.31 Lastly, it is worth reiterating that the clinical sensitivity of identifying a newborn with SMA is about 95%, as those compound heterozygotes with a single SMN1 exon 7 deletion and a point mutation will not be identified.
Carrier Testing
Quantitative SMN1 gene dosage analysis is used to identify carriers with one copy of SMN1 from non-carrier individuals with 2 or more copies of SMN1. Carrier testing has been valuable to families with a history of SMA who are at risk of having affected offspring. Learning the carrier status of a couple can allow informed family planning, lead them to test prenatally through non-invasive prenatal or in the setting of in vitro fertilization, preimplantation genetic testing. Genetic counseling is an important aspect of carrier testing, and individuals should receive counsel before the testing takes place. Unfortunately for most parents, their carrier status is discovered upon the birth of an affected child of an autosomal recessive disease. The goal of carrier screening is for all couples, even without a past history of the disease, to identify the risk of conceiving an affected child in advance of a pregnancy. The following criteria are used and considering a population-based carrier screening program: (1) the disorder is clinically severe, (2) there is a high frequency of carriers in the screened population, (3) availability of a reliable test with a high specificity and sensitivity, (4) availability of prenatal diagnosis and (5) and access to genetic counseling. SMA meets these criteria and is recommended by the American College of Medical Genetics and American College of Obstetricians and Gynecologists for inclusion in population-based genetic screening.32,33
The carrier test comes with some known limitations, including that approximately 2% of SMA cases are due to de novo rearrangement events rather than inheriting the mutation from a parent.34,35 Despite the genetic lethality of the disease, the carrier frequency is high owing at least in part to the high rate of de novo mutations in SMN1.34 It has also been shown that about 4% of the population possesses 3 SMN1 copies.35 As a result, carriers with two SMN1 copies on one chromosome and no SMN1 copies on the other chromosome is relatively common.36–39 This is referred to as the “2 + 0” genotype (Figure 3). A carrier with two SMN1 genes on one chromosome and an SMN1 deletion on the other chromosome cannot be distinguished from a noncarrier with one SMN1 copy on each chromosome 5. In most populations, approximately 3–4% of carriers have been shown to have the “2+0” genotype.40 However, in African Americans, the frequency of alleles with two or more copies of SMN1 has been shown to be 3–8 times more common when compared to other ethnic groups, which results in higher frequency of African American carriers with the “2+0” genotype.40 In certain populations, most notably in the Ashkenazi Jewish community, the presence of the g.27134T>G variant is associated with chromosomes carrying 2 SMN1 in cis.41 This variant is also informative in other specific populations, such as the Spanish population, where it is documented that 19.35% of cis carriers had the g.27134T>G variant.39 Consequently, the variant cannot be used to identify all cis carriers, as it is not always present. Family studies can also provide evidence toward identifying cis chromosomes. Finally, the dosage testing is a deletion-based test and does not identify carriers of other types of SMN1 intragenic mutations, which require sequencing. Ultimately, the detection of two SMN1 copies in an individual significantly reduces the risk of being a carrier, but a residual risk remains for those individuals found to have 2 SMN1 copies. Calculations using the Bayesian approach should be utilized for the accurate determination of residual risk.38
Genetic counseling is a fundamental component of any carrier screening program, and the concept of residual risk is not a new concept for genetic counselors, who regularly counsel couples regarding the cystic fibrosis carrier screening. It is important that individuals being tested acknowledge the limitations of the carrier test: 2 SMN1 genes in cis on the one chromosome 5, nondeletion types of mutations and the rare occurrence of de-novo mutations. The possibility of these false-negative results must be included in all carrier reports. As is true for all carrier screening programs, the testing must be confidential and voluntary. Preferably, carrier testing should be offered prior to conception when couples have the most reproductive options available.
SMN2 Testing
SMN2 copy number results provide probable information in predicting the likely clinical severity for an affected patient but should not be viewed as definitive (See Genotype Phenotype Association Section). As discussed in the newborn screening section, SMN2 copy number analysis is valuable for identifying those who require early treatment. The Biogen’s NURTURE clinical trial demonstrates the significant impact from early nusinersen treatment, with affected patients under six weeks of age with two or three copies of SMN2 had significantly better outcomes than when treatment was delayed beyond six weeks of age.25 Patients with two copies of SMN2 have a dramatically altered disease, with all patients surviving, sitting, 88% walking with assistance, 77% walking independently, and none requiring permanent ventilation assistance.25 Patients with three copies of SMN2 met motor milestones on schedule and did not show clinical SMA symptoms. In the SMA gene replacement therapy, 15 patients with two SMN2 copies were living event-free at 20 months of age, far better than the 8% rate of survival in a historical cohort.42
In 2018 Cure SMA enlisted a group of clinicians and expert scientists who were tasked with developing an algorithm for treatment of infants with positive SMA newborn screening results. A reiterative surveying modified Delphi technique was used.31 For those individuals with qualifying genotypes (two or three copies of SMN2), the decision to treat immediately was unanimously recommended based on the results of pre-symptomatic infants in the NURTURE trial.25 The working group recently updated their recommendation for infants diagnosed with SMA via NBS with four copies of SMN2. These infants should also receive immediate treatment, as even the loss of a small number of motor neurons is unacceptable when effective treatment is available.43
Genotype Phenotype Association
Since the loss of both copies of SMN1 exon 7 is found in the majority of patients, no phenotype-genotype correlation was initially observed in SMA. Several studies have now shown that the SMN2 copy number is the most important modifier of the SMA disease severity.3,35,44–47 At least one copy of SMN2 is retained in all patients and produces low levels of SMN protein but does not fully compensate for the loss of SMN1. Due to the presence of the splice mutation in exon 7, the SMN2 gene is only able to produce about 10% of full-length transcript. The copy number varies from zero to three copies in the normal population with approximately 10–15% of normals having no SMN2. The majority of patients with the severe type I form have one or two copies of SMN2, most patients with type II have three SMN2 copies, and milder patients with type III have three or four SMN2 copies. Three unrelated asymptomatic family members positive for the homozygous SMN1 deletion were reported to have five copies of SMN2.48 These cases support the modifying role of SMN2 copy number and reveal that the associated expression levels of five SMN2 copies may compensate for the loss of the SMN1 gene expression. This same inverse dosage relationship was shown in an SMA mouse model.49,50 Mice with two copies of human SMN2 but lacking the endogenous mouse Smn gene develop severe SMA and die within the first week of life; however, mice with multiple copies of SMN2 do not manifest the disease.
Exceptions to the inverse correlation between the SMN2 copy number and disease severity have occurred. Three SMN2 copies are the most common genotype in both type 2 and type 3 SMA, thus indicating that the copy number does not always accurately predict the phenotype in milder patients. The discordance between the SMN2 copy number and disease severity in some cases is consistent with the existence of disease modifiers that may contribute to the phenotypic outcome. Identification of SMA modifiers not only allows for a better understanding of the pathogenesis of the disorder but also may identify potential targets for therapeutic interventions.
The loss of the SMN1 gene can occur via deletion or by gene conversion to SMN2. The conversions in effect increase the SMN2 copy number and often result in milder phenotypes. However, exceptions exist and may be the result of incomplete deletions of the SMN2. As a result of the high sequence homology of the SMN loci, exact deletion breakpoints have been hard to identify. In the event SMN2 genes are truncated, they may not produce any full-length transcripts and thus, only nonfunctional SMN protein. Some of the conversion events are partial and result in different hybrid genes, which often consist of SMN2 exon 7 and SMN1 exon 8 but differ in the extent of the conversion.51 Partial conversions have been observed in approximately 5–7% of SMA patients. The smallest conversion, which was confined to SMN2 exon 7, with all other sequences remaining as the SMN1 version, was shown to increase exon 7 inclusion and produced milder phenotypes in five patients with 3 SMN2 copies. It was shown that the A-44G in intron 6 was the major positive modifier resulting in improved exon 7 splicing by reducing the RNA binding affinity of the splicing repressor HuR.52
It has generally been assumed that all SMN2 genes are equivalent; however, variants within the gene may also modify the phenotype. Modifiers within the SMN2 gene may affect splicing by either disrupting a slice silencer or creating a splice enhancer. The SMN2 exon 7 c.859G>C variant promotes exon 7 inclusion and thus, accounts for a milder phenotype than expected by the SMN2 copy number.53,54 Although the C to T at +6 in SMN2 exon 7 normally results in the production of SMN2 transcripts predominantly lacking exon 7 (by disrupting an exon splicing enhancer or creating an exon splicing silencer), the c.859G>C was shown to partly restore normal exon splicing and produce more full-length SMN2 transcript. The variant has never been reported in a patient with type 1 SMA but has been seen in patients with type 2 and 3 when the 2 SMN2 copies are heterozygous or homozygous for the c.859G>C variant, respectively.53,55 Importantly, these cases again support the therapeutic benefit of increasing the SMN2 gene expression in order to decrease the severity of the disorder (see in Treatment section).
The clinical severity observed in approximately 5% of patients with SMN1 intragenic mutations is not only determined by the SMN2 copy number but also by the type and location of the mutation. The observed phenotype in patients carrying specific point mutations has not always correlated with the SMN2 copy number. Mendonca et al recently identified 16 patients with the exon 3 nonsense mutation c.460C>T (p.Gln154*).16 All of the patients presented with a milder phenotype (SMA types 3 and 4), including 2 patients with 1 SMN2 copy and 10 patients with 2 SMN2 copies. The missense mutation p.Ala2Val has been found in several type 3 patients in the presence of 1 or 2 copies of SMN2 while type 1 patients with p.Trp92Ser were shown to have 3 SMN2 copies.56
Other genes outside the SMN1/SMN2 loci may modify the disease severity. There are very rare families reported in the literature in which the disease severity is not consistent between affected siblings with identical SMN2 copy number.35,57–62 These patients are referred to as “discordant siblings,” and they have been shown to occur with all types of SMA. Thus, there are type 1 SMA cases where the sibling has type 2 SMA, type 2 cases where the sibling has SMA type 3b, and type 3a cases where the sibling is phenotypically normal. Some of the variability among discordant sibs may be explained by differences in splicing factors, allowing some SMN2 copies to express more full-length transcripts.63 It has also been reported that in some families with clinically unaffected females with SMN1 deletions, plastin 3 (PLS3, T-plastin or T-fimbrin; MIM 300,131) expression was higher than in their affected siblings.64 PLS3 plays a key role in axonogenesis and may act as another gene modifier. Overexpression of PLS3 rescues the axonal growth defects observed in the zebrafish with reduced SMN levels. However, in a severe SMA mouse model, the transgenic expression of PLS3 did not restore motor function or lifespan.65 Another disease modifier identified in at least five asymptomatic individuals with an SMN1 deletion and 4 copies of SMN2 is Neurocalcin delta (NCALD), a negative regulator of endocytosis.66 These gene modifiers are significant for understanding the pathogenesis of SMA and may be important targets for future therapeutic efforts.
Treatment
Understanding the underlying genetic mechanism of SMA has led to the development of two targeted therapies that increase functional SMN protein production, the first by altering SMN2 RNA to produce a full length SMN protein and the second through direct delivery of the SMN1 gene via a viral vector.67 Table 2 contains treatment types, mechanism of action, route of administration, related clinical trials and status for each drug used in the treatment of SMA for both SMN dependent and independent pathways.
 |
Table 2 SMA Treatments |
SMN1 Gene Therapy
Onasemnogene abeparvovec uses an adeno-associated virus capsid to deliver a copy of the SMN1 gene with a cytomegalovirus enhancer and chicken beta-actin promoter to motor neurons, muscle, and other peripheral tissues where SMN1 is expressed through a single intravenous injection. It was approved for treatment of SMA by the FDA in May 2019.42 START, a Phase I trial treated 15 infants with SMA type 1 with varying doses of gene therapy and compared outcomes to a historical control group to evaluate safety, adverse events, time until death or permanent ventilation, motor milestones and motor function. All treated patients by the age of 20 months were alive without requiring permanent mechanical ventilation with significant improvement in motor milestones in the high dose cohort.42 Longer term follow-up has shown sustained survival and improved motor function of treated patients, with better outcomes and baseline function in patients treated earlier.68–70 Liver function tests were found to be elevated after drug administration, likely due to an immune response to the viral vector. Daily steroid (1mg/kg) administration for one month and monitoring of liver function tests prevented further liver toxicity. During clinical trials, transient decreases in platelet counts, some meeting the criteria for thrombocytopenia, were also reported.71 No other adverse effects of the gene therapy have been reported.42,68–70 STRONG, a Phase I clinical trial, investigated intrathecal gene therapy administration to 28 SMA type 2 patients aged 6 months to 60 years. Review of data 6–12 months after therapy showed no treated patients had lost motor milestones and some had gained milestones.70,72 The trial was recently placed on a partial hold by the FDA recently given safety concerns from an animal model.73 SPR1NT is an ongoing Phase III trial evaluating safety and efficacy of gene therapy administration to pre-symptomatic SMA patients with 2–3 copies of SMN2. Preliminary data have shown that all treated patients are alive without requiring ventilation and are achieving motor milestones.74
SMN2 Modulators
Nusinersen is an antisense oligonucleotide that modifies that splicing of SMN2 pre-mRNA by binding to a specific region to block splicing and promote inclusion of exon 7, thus resulting in production of a full length SMN protein. Nusinersen became the first drug approved for treatment of SMA by the US FDA in December 2016 and the European Medicines Agency in June 2017 after multiple trials supporting its safety and efficacy.67 As this antisense oligonucleotide does not cross the blood brain barrier, administration is by intrathecal injection.67
A Phase I study (NCT01494701 and NCT01780246), which included 28 patients with SMA types 2 and 3, ages 2–14 years, showed safety and tolerability with no serious adverse events found following single intrathecal injections of nusinersen (1–9 mg doses). SMN protein was found to be increased in CSF and patients receiving the higher doses showed improved motor function.75 A Phase II trial (NCT01839656) with escalating doses of nusinersen administered to 20 SMA type 1 patients, ages 3 weeks to 7 months similarly showed safety, tolerability and efficacy, with improved motor function in patients receiving higher doses and significantly increased overall survival with decreased need for mechanical ventilation.76 Following these trials, two large, multicenter, randomized controlled, Phase III studies, ENDEAR in SMA type 1 patients (including 122 patients 7 months or younger) and CHERISH in SMA type 2 patients (126 patients, 2–9 years old) showed significant improvement in motor milestones and survival in the treated group, with more benefit seen in infants with shorter disease duration in the ENDEAR trial.76,77 NURTURE, an ongoing Phase II trial is investigating the optimal timing of nusinersen treatment in 25 pre-symptomatic SMA patients with 2 or 3 copies of SMN2. In an interim report, all patients were alive without need for ventilation and able to sit without support. Most had achieved independent ambulation.25 SHINE, an ongoing Phase III trial, is currently assessing the long-term effects of nusinersen treatment including patients previously enrolled in the abovementioned ENDEAR trial, and has found the treatment to be safe, well tolerated, and most effective when administered early.78
Nusinersen is delivered as a loading dose of four 12 mg intrathecal injections over 2 months, followed by maintenance doses every 4 months.3 Complications can result from the lumbar puncture including rare reports of hydrocephalus.67 The lumbar puncture may also be challenging in patient with scoliosis or spinal fusion, requiring use of radiographic-image-based guidance and intrathecal catheters.67 The lack of systemic delivery is also a concern as the SMN protein may have important functions in the peripheral tissues.11
Small molecules therapies that modify SMN2 mRNA splicing to produce increased full length SMN protein have also been developed and in contrast to nusinersen, have benefits of systemic distribution and oral bioavailability. Risdiplam, one of these small molecules taken as a liquid formulation daily, became the third approved disease-modifying treatment for SMA by the US FDA on August 7, 2020 given results of ongoing clinical trials showing safety and efficacy.79 FIREFISH, an ongoing trial for SMA type 1 patients (set to finish 2023), showed improved motor function, attainment of motor milestones, and lack of dysphagia and ventilatory requirement in treated patients.80,81 SUNFISH studying SMA types 2 and 3 (ongoing trial finishing in 2023) also showed improvement of motor function in treated patients, particularly in younger patients.82–84 JEWELFISH, a Phase II open label trial (finishing in December 2024), is currently investigating the safety and tolerability of risdiplam in SMA patients ages 6 months–60 years who have previously been treated with nusinersen or onasemnogene abeparvovec.85 RAINBOWFISH (finishing in October 2025) is assessing risdiplam safety and efficacy in presymptomatic newborns and infants up to 6 weeks old.86
RG7800 small molecule therapy was previously investigated in Phase I trials (MOONFISH), but studies were ended after eye toxicity was found.3 Phase I–II clinical trials of branaplam were also terminated after evidence of blood vessel, kidney, spinal cord, and peripheral nerve toxicity was found in animal studies.87,88
Celecoxib, a cyclooxygenase 2 inhibitor, was shown to increase SMN protein in SMA cells and animal models. A Phase II trial is currently recruiting SMA 2 and 3 patients, in hopes that it may serve as an adjunctive SMA treatment.89
SMN Independent and Combination Pathways
Other therapies under investigation have targeted pathways independent of the SMN1 and 2 genes and focused on enhancing survival and functioning of motor neurons and muscle through alternative mechanisms.3
Olesoxime is a cholesterol-like molecule that preserves mitochondrial functioning and inhibits release of apoptotic factors that lead to motor neuron death. By restoring mitochondrial homeostasis, olesoxime is thought to maintain motor neuron integrity, reduce denervation of muscle tissue, and reduce reactive astroglia and microglia activation.90 An initial Phase II trial in patients with SMA II and III did not reach the primary outcome of improved motor function and a follow up Phase II trial showed a decline in motor function after 18 months resulting in cessation of drug development.91–93 Reldesemtiv is a skeletal muscle troponin activator. It slows calcium release from the troponin complex and sensitizes the sarcomere response to calcium in order to increase the skeletal muscle force response to nerve stimulation. A Phase II trial in patients with SMA II–IV showed no safety or tolerability issues. A Phase III trial is planned.94 SK-015 is a monoclonal antibody that inhibits latent myostatin to promote muscle cell growth and division. It is currently being investigated in SMA2 and 3 patients in the TOPAZ trial.95,96 Pyridostigmine, an acetylcholinesterase drug used for treating myasthenia gravis, is theorized to improve muscle strength and fatigue in SMA. It is currently being trialed in patients with types 2–4 SMA.89
With the development and approval of multiple SMA disease-modifying therapies, the use of combination therapy is being investigated. As adequate functional SMN protein is required for optimal function of many dividing cells, it is unclear if a single dose of onasemnogene abeparvovec or maintenance nusinersen will be sufficient.3 Given the drugs' different mechanisms of actions, combination use of onasemnogene abeparvovec and nusinersen could maximize therapeutic effect with little risk of drug interaction and toxicity.89 Small case series and reports showed patients treated with both onasemnogene abeparvovec and nusinersen tolerated the combination, though efficacy and long-term effect remain unclear.97,98 As mentioned previously, JEWELFISH, a Phase II open label trial, is currently investigating the safety and tolerability of risdiplam in SMA patients’ ages 6 months–60 years who have previously been treated with nusinersen or onasemnogene abeparvovec.85 Combined use of nusinersen and SMN-independent myostatin inhibitor therapy has shown a positive effect in animal models that may translate to another clinical approach.89
Conclusion
The most robust response to SMA treatments has clearly been shown to occur in treating presymptomatic patients. Thus, early detection through newborn screening is paramount to ensuring efficient treatment access prior to manifesting symptoms of the disease. Prenatal cases identified as a result of carrier screening will also allow for early treatment. Biomarkers and outcome measures to assess disease response to therapy are needed, particularly in determining use of additional or combination treatments. Future research is needed to understand the long-term effects of the therapies alone or in combination.67
Acknowledgments
The authors would like to thank Demitrios Dedousis for his edits.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Groen EJN, Talbot K, Gillingwater TH. Advances in therapy for spinal muscular atrophy: promises and challenges. Nat Rev Neurol. 2018;14(4):214–224. doi:10.1038/nrneurol.2018.4
2. Arnold WD, Kassar D, Kissel JT. Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle Nerve. 2015;51(2):157–167. doi:10.1002/mus.24497
3. Wirth B, Karakaya M, Kye MJ, Mendoza-Ferreira N. Twenty-five years of spinal muscular atrophy research: from phenotype to genotype to therapy, and what comes next. Annu Rev Genomics Hum Genet. 2020;21(1):231–261. doi:10.1146/annurev-genom-102319-103602
4. Dubowitz V. Very severe spinal muscular atrophy (SMA type 0): an expanding clinical phenotype. Eur J Paediatr Neurol. 1999;3(2):49–51. doi:10.1016/S1090-3798(99)80012-9
5. Kolb SJ, Kissel JT. Spinal muscular atrophy. Neurol Clin. 2015;33(4):831–846.
6. Zerres K, Rudnik-Schoneborn S. Natural history in proximal spinal muscular atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol. 1995;52(5):518–523. doi:10.1001/archneur.1995.00540290108025
7. Pera MC, Romeo DM, Graziano A, et al. Sleep disorders in spinal muscular atrophy. Sleep Med. 2017;30:160–163. doi:10.1016/j.sleep.2016.11.012
8. Rudnik-Schoneborn S, Heller R, Berg C, et al. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J Med Genet. 2008;45(10):635–638. doi:10.1136/jmg.2008.057950
9. Takahashi N, Shimada T, Ishibashi Y, et al. Cardiac involvement in Kugelberg-Welander disease: a case report and review. Am J Med Sci. 2006;332(6):354–356. doi:10.1097/00000441-200612000-00009
10. Palladino A, Passamano L, Taglia A, et al. Cardiac involvement in patients with spinal muscular atrophies. Acta Myol. 2011;30(3):175–178.
11. Yeo CJJ, Darras BT. Overturning the paradigm of spinal muscular atrophy as just a motor neuron disease. Pediatr Neurol. 2020;109:12–19. doi:10.1016/j.pediatrneurol.2020.01.003
12. Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–165. doi:10.1016/0092-8674(95)90460-3
13. Burglen L, Lefebvre S, Clermont O, et al. Structure and organization of the human survival motor neurone (SMN) gene. Genomics. 1996;32(3):479–482. doi:10.1006/geno.1996.0147
14. Kolb SJ, Battle DJ, Dreyfuss G. Molecular functions of the SMN complex. J Child Neurol. 2007;22(8):990–994. doi:10.1177/0883073807305666
15. Hahnen E, Schonling J, Rudnik-Schoneborn S, Raschke H, Zerres K, Wirth B. Missense mutations in exon 6 of the survival motor neuron gene in patients with spinal muscular atrophy (SMA). Hum Mol Genet. 1997;6(5):821–825. doi:10.1093/hmg/6.5.821
16. Mendonca RH, Matsui C
17. Huang CH, Chang YY, Chen CH, et al. Copy number analysis of survival motor neuron genes by multiplex ligation-dependent probe amplification. Genet Med. 2007;9(4):241–248. doi:10.1097/GIM.0b013e31803d35bc
18. Tan CA, Westbrook MJ, Truty R, et al. Incorporating spinal muscular atrophy analysis by next-generation sequencing into a comprehensive multigene panel for neuromuscular disorders. Genet Test Mol Biomarkers. 2020;24(10):616–624. doi:10.1089/gtmb.2019.0282
19. Cheng S, Fockler C, Barnes WM, Higuchi R. Effective amplification of long targets from cloned inserts and human genomic DNA. Proc Natl Acad Sci U S A. 1994;91(12):5695–5699. doi:10.1073/pnas.91.12.5695
20. Bussaglia E, Clermont O, Tizzano E, et al. A frame-shift deletion in the survival motor neuron gene in Spanish spinal muscular atrophy patients. Nat Genet. 1995;11(3):335–337. doi:10.1038/ng1195-335
21. Cusco I, Lopez E, Soler-Botija C, Jesus Barcelo M, Baiget M, Tizzano EF. A genetic and phenotypic analysis in Spanish spinal muscular atrophy patients with c.399_402del AGAG, the most frequently found subtle mutation in the SMN1 gene. Hum Mutat. 2003;22(2):136–143. doi:10.1002/humu.10245
22. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
23. Swoboda KJ, Prior TW, Scott CB, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol. 2005;57(5):704–712. doi:10.1002/ana.20473
24. Kraszewski JN, Kay DM, Stevens CF, et al. Pilot study of population-based newborn screening for spinal muscular atrophy in New York state. Genet Med. 2018;20(6):608–613. doi:10.1038/gim.2017.152
25. De Vivo DC, Bertini E, Swoboda KJ, et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: interim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord. 2019;29(11):842–856. doi:10.1016/j.nmd.2019.09.007
26. Kirschner J, Butoianu N, Goemans N, et al. European ad-hoc consensus statement on gene replacement therapy for spinal muscular atrophy. Eur J Paediatr Neurol. 2020;28:38–43. doi:10.1016/j.ejpn.2020.07.001
27. Pyatt RE, Mihal DC, Prior TW. Assessment of liquid microbead arrays for the screening of newborns for spinal muscular atrophy. Clin Chem. 2007;53(11):1879–1885. doi:10.1373/clinchem.2007.092312
28. Prior TW, Snyder PJ, Rink BD, et al. Newborn and carrier screening for spinal muscular atrophy. Am J Med Genet A. 2010;152A(7):1608–1616. doi:10.1002/ajmg.a.33474
29. Vill K, Kolbel H, Schwartz O, et al. One year of newborn screening for SMA - results of a german pilot project. J Neuromuscul Dis. 2019;6(4):503–515. doi:10.3233/JND-190428
30. Kay DM, Stevens CF, Parker A, et al. Implementation of population-based newborn screening reveals low incidence of spinal muscular atrophy. Genet Med. 2020;22(8):1296–1302. doi:10.1038/s41436-020-0824-3
31. Glascock J, Sampson J, Haidet-Phillips A, et al. Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J Neuromuscul Dis. 2018;5(2):145–158. doi:10.3233/JND-180304
32. Prior TW, Professional P, Guidelines C. Carrier screening for spinal muscular atrophy. Genet Med. 2008;10(11):840–842. doi:10.1097/GIM.0b013e318188d069
33. Committee on Genetics. committee opinion no. 691: carrier screening for genetic conditions. Obstet Gynecol. 2017;129(3):e41–e55. doi:10.1097/AOG.0000000000001952
34. Wirth B, Schmidt T, Hahnen E, et al. De novo rearrangements found in 2% of index patients with spinal muscular atrophy: mutational mechanisms, parental origin, mutation rate, and implications for genetic counseling. Am J Hum Genet. 1997;61(5):1102–1111. doi:10.1086/301608
35. McAndrew PE, Parsons DW, Simard LR, et al. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet. 1997;60(6):1411–1422. doi:10.1086/515465
36. Wirth B, Herz M, Wetter A, et al. Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am J Hum Genet. 1999;64(5):1340–1356. doi:10.1086/302369
37. Mailman MD, Hemingway T, Darsey RL, et al. Hybrids monosomal for human chromosome 5 reveal the presence of a spinal muscular atrophy (SMA) carrier with two SMN1 copies on one chromosome. Hum Genet. 2001;108(2):109–115. doi:10.1007/s004390000446
38. Ogino S, Wilson RB. Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum Genet. 2002;111(6):477–500. doi:10.1007/s00439-002-0828-x
39. Alias L, Bernal S, Calucho M, et al. Utility of two SMN1 variants to improve spinal muscular atrophy carrier diagnosis and genetic counselling. Eur J Hum Genet. 2018;26(10):1554–1557. doi:10.1038/s41431-018-0193-4
40. Hendrickson BC, Donohoe C, Akmaev VR, et al. Differences in SMN1 allele frequencies among ethnic groups within North America. J Med Genet. 2009;46(9):641–644. doi:10.1136/jmg.2009.066969
41. Luo M, Liu L, Peter I, et al. An Ashkenazi Jewish SMN1 haplotype specific to duplication alleles improves pan-ethnic carrier screening for spinal muscular atrophy. Genet Med. 2014;16(2):149–156. doi:10.1038/gim.2013.84
42. Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713–1722. doi:10.1056/NEJMoa1706198
43. Glascock J, Sampson J, Connolly AM, et al. Revised recommendations for the treatment of infants diagnosed with spinal muscular atrophy via newborn screening who have 4 copies of SMN2. J Neuromuscul Dis. 2020;7(2):97–100. doi:10.3233/JND-190468
44. Mailman MD, Heinz JW, Papp AC, et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med. 2002;4(1):20–26. doi:10.1097/00125817-200201000-00004
45. Crawford TO, Paushkin SV, Kobayashi DT, et al. Evaluation of SMN protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PLoS One. 2012;7(4):e33572. doi:10.1371/journal.pone.0033572
46. Wadman RI, Stam M, Gijzen M, et al. Association of motor milestones, SMN2 copy and outcome in spinal muscular atrophy types 0–4. J Neurol Neurosurg Psychiatry. 2017;88(4):365–367. doi:10.1136/jnnp-2016-314292
47. Calucho M, Bernal S, Alias L, et al. Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord. 2018;28(3):208–215. doi:10.1016/j.nmd.2018.01.003
48. Prior TW, Swoboda KJ, Scott HD, Hejmanowski AQ. Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am J Med Genet A. 2004;130A(3):307–310. doi:10.1002/ajmg.a.30251
49. Hsieh-Li HM, Chang JG, Jong YJ, et al. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24(1):66–70. doi:10.1038/71709
50. Monani UR, Coovert DD, Burghes AH. Animal models of spinal muscular atrophy. Hum Mol Genet. 2000;9(16):2451–2457. doi:10.1093/hmg/9.16.2451
51. Ruhno C, McGovern VL, Avenarius MR, et al. Complete sequencing of the SMN2 gene in SMA patients detects SMN gene deletion junctions and variants in SMN2 that modify the SMA phenotype. Hum Genet. 2019;138(3):241–256. doi:10.1007/s00439-019-01983-0
52. Wu X, Wang SH, Sun J, Krainer AR, Hua Y, Prior TW. A-44G transition in SMN2 intron 6 protects patients with spinal muscular atrophy. Hum Mol Genet. 2017;26(14):2768–2780. doi:10.1093/hmg/ddx166
53. Prior TW, Krainer AR, Hua Y, et al. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet. 2009;85(3):408–413. doi:10.1016/j.ajhg.2009.08.002
54. Vezain M, Saugier-Veber P, Goina E, et al. A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum Mutat. 2010;31(1):E1110–E1125. doi:10.1002/humu.21173
55. Bernal S, Alias L, Barcelo MJ, et al. The c.859G>C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. J Med Genet. 2010;47(9):640–642. doi:10.1136/jmg.2010.079004
56. Yamamoto T, Sato H, Lai PS, et al. Intragenic mutations in SMN1 may contribute more significantly to clinical severity than SMN2 copy numbers in some spinal muscular atrophy (SMA) patients. Brain Dev. 2014;36(10):914–920. doi:10.1016/j.braindev.2013.11.009
57. Cobben JM, van der Steege G, Grootscholten P, de Visser M, Scheffer H, Buys CH. Deletions of the survival motor neuron gene in unaffected siblings of patients with spinal muscular atrophy. Am J Hum Genet. 1995;57(4):805–808.
58. Hahnen E, Forkert R, Marke C, et al. Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: evidence of homozygous deletions of the SMN gene in unaffected individuals. Hum Mol Genet. 1995;4(10):1927–1933. doi:10.1093/hmg/4.10.1927
59. Jedrzejowska M, Borkowska J, Zimowski J, et al. Unaffected patients with a homozygous absence of the SMN1 gene. Eur J Hum Genet. 2008;16(8):930–934. doi:10.1038/ejhg.2008.41
60. Bernal S, Also-Rallo E, Martinez-Hernandez R, et al. Plastin 3 expression in discordant spinal muscular atrophy (SMA) siblings. Neuromuscul Disord. 2011;21(6):413–419. doi:10.1016/j.nmd.2011.03.009
61. DiDonato CJ, Ingraham SE, Mendell JR, et al. Deletion and conversion in spinal muscular atrophy patients: is there a relationship to severity? Ann Neurol. 1997;41(2):230–237. doi:10.1002/ana.410410214
62. Pane M, Lapenta L, Abiusi E, et al. Longitudinal assessments in discordant twins with SMA. Neuromuscul Disord. 2017;27(10):890–893. doi:10.1016/j.nmd.2017.06.559
63. Hofmann Y, Lorson CL, Stamm S, Androphy EJ, Wirth B. Htra2-beta 1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2). Proc Natl Acad Sci U S A. 2000;97(17):9618–9623. doi:10.1073/pnas.160181697
64. Oprea GE, Krober S, McWhorter ML, et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008;320(5875):524–527. doi:10.1126/science.1155085
65. McGovern VL, Massoni-Laporte A, Wang X, et al. Plastin 3 expression does not modify spinal muscular atrophy severity in the 7 SMA mouse. PLoS One. 2015;10(7):e0132364. doi:10.1371/journal.pone.0132364
66. Riessland M, Kaczmarek A, Schneider S, et al. Neurocalcin delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. Am J Hum Genet. 2017;100(2):297–315. doi:10.1016/j.ajhg.2017.01.005
67. Ramdas S, Servais L. New treatments in spinal muscular atrophy: an overview of currently available data. Expert Opin Pharmacother. 2020;21(3):307–315. doi:10.1080/14656566.2019.1704732
68. Al-Zaidy S, Pickard AS, Kotha K, et al. Health outcomes in spinal muscular atrophy type 1 following AVXS-101 gene replacement therapy. Pediatr Pulmonol. 2019;54(2):179–185. doi:10.1002/ppul.24203
69. Lowes LP, Alfano LN, Arnold WD, et al. Impact of age and motor function in a phase 1/2A study of infants with SMA type 1 receiving single-dose gene replacement therapy. Pediatr Neurol. 2019;98:39–45. doi:10.1016/j.pediatrneurol.2019.05.005
70. Mendell JR, Lehman KJ, McColly M, et al. AVXS-101 Gene-Replacement Therapy (GRT) in Spinal Muscular Atrophy Type 1 (SMA1): long-term follow-up from the phase 1 clinical trial (S25.006). Neurology. 2019;92(15 Supplement):
71. Darras BT, Farrar MA, Mercuri E, et al. An integrated safety analysis of infants and children with symptomatic Spinal Muscular Atrophy (SMA) treated with nusinersen in seven clinical trials. CNS Drugs. 2019;33(9):919–932. doi:10.1007/s40263-019-00656-w
72. Finkel RS, Day JW, Darras BT, et al. Phase 1 study of intrathecal administration of AVXS-101 Gene-Replacement Therapy (GRT) for Spinal Muscular Atrophy type 2 (SMA2) (STRONG) (P1.6–059). Neurology. 2019;92(15Supplement):
73. Novartis Announces AVXS-101 Intrathecal Study Update [press release]. 2019.
74. Strauss KA, Farrar MA, Swoboda KJ, et al. Onasemnogene abeparvovec-xioi gene-replacement therapy in presymptomatic spinal muscular atrophy: SPR1NT Study update (2384). Neurology. 2020;94(15 Supplement):2384.
75. Chiriboga CA, Swoboda KJ, Darras BT, et al. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology. 2016;86(10):890–897. doi:10.1212/WNL.0000000000002445
76. Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723–1732. doi:10.1056/NEJMoa1702752
77. Mercuri E, Darras BT, Chiriboga CA, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018;378(7):625–635. doi:10.1056/NEJMoa1710504
78. Hache M, Swoboda KJ, Sethna N, et al. Intrathecal injections in children with spinal muscular atrophy: nusinersen clinical trial experience. J Child Neurol. 2016;31(7):899–906. doi:10.1177/0883073815627882
79. FDA Approves Oral Treatment for Spinal Muscular Atrophy [press release]. 2020.
80. Seabrook T, Baranello G, Servais L, et al. FIREFISH part 1: early clinical results following an increase of SMN protein in infants with type 1 spinal muscular atrophy (SMA) treated with risdiplam (RG7916).
81. Baranello G, Servais L, Day J, et al. l. P.353FIREFISH part 1: 16-month safety and exploratory outcomes of risdiplam (RG7916) treatment in infants with type 1 spinal muscular atrophy. Neuromuscul Disord. 2019;29:S184. doi:10.1016/j.nmd.2019.06.515
82. Mercuri E, Baranello G, Kirschner J, et al. Update from SUNFISH part 1: safety, tolerability and PK/PD from the dose-finding study, including exploratory efficacy data in patients with type 2 or 3 Spinal Muscular Atrophy (SMA) treated with risdiplam (RG7916) (S25.007). Neurology. 2019;92(15 Supplement):
83. Mercuri E, Baranello G, Kirschner J, et al. O.41Sunfish part 1: 18-month safety and exploratory outcomes of risdiplam (RG7916) treatment in patients with type 2 or 3 spinal muscular atrophy. Neuromuscul Disord. 2019;29:S208. doi:10.1016/j.nmd.2019.06.595
84. Mercuri E, Kirschner J, Baranello G, et al. Clinical studies of RG7916 in patients with spinal muscular atrophy: SUNFISH part 1 study update. Neuromuscul Disord. 2017;27:S209.
85. Chiriboga CA, Mercuri E, Fischer D, et al. P.363JEWELFISH: safety and pharmacodynamic data in patients with spinal muscular atrophy (SMA) receiving treatment with risdiplam (RG7916) that have previously been treated with nusinersen. Neuromuscul Disord. 2019;29:S187. doi:10.1016/j.nmd.2019.06.525
86. Bertini E, Day J, Muhaizea M, et al. P.362RAINBOWFISH: a study of risdiplam (RG7916) in newborns with pre-symptomatic spinal muscular atrophy (SMA). Neuromuscul Disord. 2019;29:S187. doi:10.1016/j.nmd.2019.06.524
87. Jevtic S, Carr D, Dobrzycka-Ambrozevicz A, et al. Branaplam in type 1 spinal muscular atrophy: second part of a Phase I/II study.
88. Parente V, Corti S. Advances in spinal muscular atrophy therapeutics. Ther Adv Neurol Disord. 2018;11:1756285618754501. doi:10.1177/1756285618754501
89. Chen TH. New and developing therapies in spinal muscular atrophy: from genotype to phenotype to treatment and where do we stand? Int J Mol Sci. 2020;21:9.
90. Sunyach C, Michaud M, Arnoux T, et al. Olesoxime delays muscle denervation, astrogliosis, microglial activation and motoneuron death in an ALS mouse model. Neuropharmacology. 2012;62(7):2346–2352. doi:10.1016/j.neuropharm.2012.02.013
91. Bordet T, Buisson B, Michaud M, et al. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J Pharmacol Exp Ther. 2007;322(2):709–720. doi:10.1124/jpet.107.123000
92. Bertini E, Dessaud E, Mercuri E, et al. Safety and efficacy of olesoxime in patients with type 2 or non-ambulatory type 3 spinal muscular atrophy: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017;16(7):513–522. doi:10.1016/S1474-4422(17)30085-6
93. Roche Stops Work on Olesoxime After Disappointing Long-Term Results in Phase 2 Trial [press release]. SMA News Today; 2018.
94. Rudnicki S, Andrews J, Malik F, Wolff A, Day J. CY 5021 a Phase 2, Double-Blind, Randomized, Placebo-Controlled, Multiple-Dose Study of Reldesemtiv 2 Ascending-Dose Cohorts of Patients with Spinal Muscular Atrophy (SMA). The Cure SMA; 2018.
95. Long KK, O’Shea KM, Khairallah RJ, et al. Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum Mol Genet. 2019;28(7):1076–1089. doi:10.1093/hmg/ddy382
96. Chyung Y. Interim results from a phase 1 study of SRK-015, a fully human monoclonal antibody that inhibits myostatin activation.
97. Lee BH, Collins E, Lewis L, et al. Combination therapy with nusinersen and AVXS-101 in SMA type 1. Neurology. 2019;93(14):640–641. doi:10.1212/WNL.0000000000008207
98. Harada Y, Rao VK, Arya K, et al. Combination molecular therapies for type 1 spinal muscular atrophy. Muscle Nerve. 2020;62(4):550–554. doi:10.1002/mus.27034
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.