Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
Spinal cord demyelination combined with hyperhomocysteinemia: a case report
Authors Hao M, Zhang Y, Hou S, Chen Y, Shi M, Zhao G, Deng Y
Received 11 July 2014
Accepted for publication 23 September 2014
Published 3 November 2014 Volume 2014:10 Pages 2057—2059
DOI https://doi.org/10.2147/NDT.S70963
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Meimei Hao, Yan Zhang, Shuangxing Hou, Yanling Chen, Ming Shi, Gang Zhao, Yanchun Deng
Department of Neurology, Xijing Hospital, Fourth Military Medical University, Xi’an, People’s Republic of China
Abstract: Hyperhomocysteinemia (HHcy) has been recognized as an independent risk factor for atherosclerotic vascular disease. Here we report a patient who suffered from spinal cord demyelination combined with HHcy. The patient was admitted to our hospital with a diagnosis of acute myelitis. However, hormone therapy was ineffective. Further investigations revealed that he had HHcy and a homozygous mutation of the gene encoding methylenetetrahydrofolate reductase (MTHFR) c.677C>T, which is a key enzyme involved in homocysteine metabolism. In view of these findings, we treated the patient with B vitamins and his symptoms gradually improved. Spinal magnetic resonance imaging performed 3 months after onset showed near recovery of the lesion. To our knowledge, similar reports are rare.
Keywords: demyelination, hyperhomocysteinemia, homocysteine, methylenetetrahydrofolate reductase, methylation
Introduction
Since McCully1 first reported the relationship between homocysteine and atherosclerosis, hyperhomocysteinemia (HHcy) has come to be recognized as an independent risk factor for atherosclerotic vascular disease.2 Further, some studies have demonstrated that HHcy might be involved in the pathophysiology of many neuropsychiatric disorders, including brain atrophy, epilepsy, and Alzheimer’s disease.3–5 Here we report a patient diagnosed with spinal cord demyelination who presented with HHcy and a homozygous mutation of the gene encoding methylenetetrahydrofolate reductase (MTHFR) c.677C>T. We suspect a relationship between HHcy and his spinal cord demyelination.
Case report
A 53-year-old man was admitted to our hospital with a 7-day history of numbness below thoracic level (T)10. His symptoms had started with lower limb weakness followed by slight difficulty with urination. He reported having had a cold before onset of these symptoms.
On admission his general examination was normal. Neurological examination showed weakness (4/5) in his lower limbs. Deep and superficial sensitivity were decreased below T10. Deep tendon reflexes were preserved in the lower limbs. Pathological reflexes were absent. Except for diabetes, his medical history and family history were unremarkable.
The results of a full blood count and serum biochemistry were normal. A cerebrospinal fluid examination showed a total protein concentration of 0.63 g/L and the cell count was normal. Cervical and thoracic spinal magnetic resonance imaging (MRI) demonstrated that the spinal cord was swollen at cervical level (C)7–T3, which was considered to represent inflammatory lesions (Figure 1A). Therefore, he was diagnosed with acute myelitis. We treated him with a 7-day course of intravenous prednisone 10 mg/day, and however, this was ineffective. To further distinguish it from the subacute combined degeneration of the cord, we tested his serum homocysteine, vitamin B12, and folate, which revealed normal serum vitamin B12 (268.00 pg/mL; normal range 211.00–911.00 pg/mL), elevated serum homocysteine (44.49 μmol/L; normal range 5–15 μmol/L), and decreased serum folate (2.70 ng/mL; normal range 3.1–17.5 ng/mL). Using a previously described method,6 we detected genetic polymorphisms rs1801133 (c.677C>T) and rs1801131 (c.1298A>C) on the MTHFR gene, which encodes the methylenetetrahydrofolate reductase, a key enzyme in homocysteine metabolism. The results showed that the patient carried the TT genotype of the MTHFR c.677C>T gene variant and no mutation was found in rs1801131 of the MTHFR gene (Figure 2).
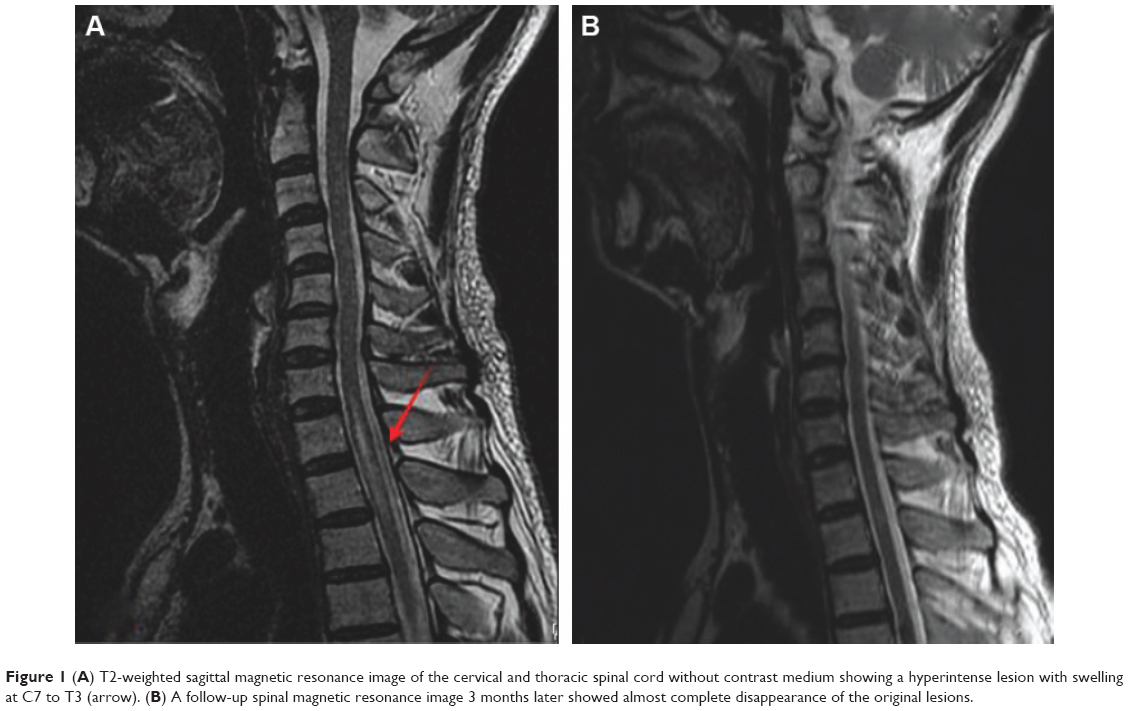 | Figure 1 (A) T2-weighted sagittal magnetic resonance image of the cervical and thoracic spinal cord without contrast medium showing a hyperintense lesion with swelling at C7 to T3 (arrow). (B) A follow-up spinal magnetic resonance image 3 months later showed almost complete disappearance of the original lesions. |
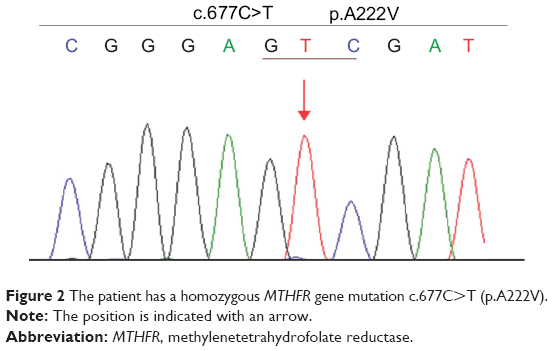 | Figure 2 The patient has a homozygous MTHFR gene mutation c.677C>T (p.A222V). |
In light of these findings, the patient was treated with intramuscular cobalamin injections (500 μg/day), vitamin B6 orally (30 mg/day), and folate orally (5 mg/day). Two weeks later, he showed moderate improvement of his sensory symptoms. Urination was normal. He was discharged on oral cobalamin 500 μg/day and oral vitamin B6 30 mg/day.
A subsequent spinal MRI performed 3 months after symptom onset showed near resolution of the original lesion (Figure 1B). Repeat serum homocysteine, vitamin B12, and folate analysis was normal (homocysteine 8.72 μmol/L, normal range 5–15 μmol/L; vitamin B12 544.10 pg/mL, normal range 211.00–911.00 pg/mL; folate 12.20 ng/mL, normal range 3.1–17.5 ng/mL). Treatment was then discontinued and the patient remains under follow-up.
Discussion
Here we describe a patient with spinal cord demyelination whose symptoms, initial laboratory findings, and imaging results suggested acute myelitis, and in whom prednisone was ineffective. He had neither anemia nor abnormal serum vitamin B12, and there were no other clinical or laboratory signs (red blood cell count, mean corpuscular volume, hematocrystallin, and albumin values were normal) of malabsorption. A spinal MRI then showed that the lesion was not confined to the dorsal and lateral spinal cord. Further, considering the acute onset and atypical clinical presentation, it seemed inappropriate to make a diagnosis of spinal cord demyelination, at least of a typical type. Further examination revealed that, except for the elevated plasma homocysteine and folate deficiency, the patient was homozygous for the gene encoding MTHFR c.677C>T. Based on these findings, we treated the patient with B vitamins, and his symptoms gradually improved.
As an important enzyme in folate and homocysteine metabolism, MTHFR c.677C>T polymorphisms have been associated with HHcy.7 Many studies have demonstrated that high levels of homocysteine can increase the risk of vascular disease,2,8 but reports of spinal cord demyelination and HHcy are rare. Our case suggests that people with mutation of the MTHFR gene combined with HHcy and folate deficiency might be highly susceptible to demyelination. Some likely mechanisms for the pathophysiological damage are as follows.
First, MTHFR mutation-induced disturbances in methylation process can result in a lower methylation rate of myelin basic protein, which plays an important role in the tight compaction of myelin sheath through association with acidic lipids on the cytosolic side of the membrane bilayer, and the decreased methylation of it could cause conformational change and splitting of the myelin sheet.9 Jaiser and Winston10 have proposed that methylation of myelin protein is limited by disorders of the methylation cycle, which also contribute to both copper deficiency myelopathy and spinal cord demyelination. Second, HHcy is proatherogenic and prothrombotic, so increases the risk of ischemic disease, and might indirectly promote demyelination. Further potential mechanisms for demyelinating neuropathy may relate to HHcy, which can promote neuronal hypersensitivity to excitotoxicity and apoptosis.5,11 Successful treatment with vitamin B supports such speculation. The mutation was found by chance in our patient and we still do not know how it occurred. We suspect that the causes of this mutation are complex. In addition to heredity, internal and external environmental factors, such as infection and radiation during growth and development, may also contribute to this mutation. However, the possibility that the MTHFR c.677C>T genotype, especially in the presence of folate deficiency, could promote HHcy and affect the spinal cord cannot be ignored.12,13
In summary, it is important to pay attention to plasma homocysteine, vitamin B12, and folate levels in the patient with spinal cord demyelination or other myeleterosis. If these are abnormal, treatment with B vitamins should be started promptly, particularly in those who also have MTHFR gene mutation.
Acknowledgment
We are grateful to the patient for his cooperation in preparing this case report.
Disclosure
The authors report no conflicts of interest in this work.
References
McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol. 1969;56(1):111–128. | ||
Boushey CJ, Beresford SA, Omenn GS, et al. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. JAMA. 1995;274(13):1049–1057. | ||
Sachdev P. Homocysteine and neuropsychiatric disorders. Rev Bras Psiquiatr. 2004;26(1):50–56. | ||
Baldelli E, Leo G, Andreoli N, et al. Homocysteine potentiates seizures and cell loss induced by pilocarpine treatment. Neuromolecular Med. 2010;12(3):248–259. | ||
Sachdev PS. Homocysteine and brain atrophy. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(7):1152–1161. | ||
Caccamo D, Condello S, Gorgone G, et al. Screening for C677T and A1298C MTHFR polymorphisms in patients with epilepsy and risk of hyperhomocysteinemia. Neuromolecular Med. 2004;6(2–3):117–126. | ||
Bailey LB, Gregory JR. Polymorphisms of methylenetetrahydrofolate reductase and other enzymes: metabolic significance, risks and impact on folate requirement. J Nutr. 1999;129(5):919–922. | ||
Temple ME, Luzier AB, Kazierad DJ. Homocysteine as a risk factor for atherosclerosis. Ann Pharmacother. 2000;34(1):57–65. | ||
Frattini D, Fusco C, Ucchino V, et al. Early onset methylmalonic aciduria and homocystinuria cblC type with demyelinating neuropathy. Pediatr Neurol. 2010;43(2):135–138. | ||
Jaiser SR, Winston GP. Copper deficiency myelopathy. J Neurol. 2010;257(6):869–881. | ||
Kruman II, Culmsee C, Chan SL, et al. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J Neurosci. 2000;20(18):6920–6926. | ||
Bhargava S, Tyagi SC. Nutriepigenetic regulation by folate-homocysteine-methionine axis: a review. Mol Cell Biochem. 2014;387(1–2):55–61. | ||
Lever EG, Elwes RD, Williams A, et al. Subacute combined degeneration of the cord due to folate deficiency: response to methyl folate treatment. J Neurol Neurosurg Psychiatry. 1986;49(10):1203–1207. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.