Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Specifically targeted delivery of protein to phagocytic macrophages
Authors Yu M, Chen Z, Guo W, Wang J, Feng Y, Kong X, Hong Z ![]()
Received 16 October 2014
Accepted for publication 14 January 2015
Published 4 March 2015 Volume 2015:10(1) Pages 1743—1757
DOI https://doi.org/10.2147/IJN.S75950
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Carlos Rinaldi
Min Yu, Zeming Chen, Wenjun Guo, Jin Wang, Yupeng Feng, Xiuqi Kong, Zhangyong Hong
State Key Laboratory of Medicinal Chemical Biology, College of Life Sciences, Nankai University, Tianjin, People’s Republic of China
Abstract: Macrophages play important roles in the pathogenesis of various diseases, and are important potential therapeutic targets. Furthermore, macrophages are key antigen-presenting cells and important in vaccine design. In this study, we report on the novel formulation (bovine serum albumin [BSA]-loaded glucan particles [GMP-BSA]) based on β-glucan particles from cell walls of baker’s yeast for the targeted delivery of protein to macrophages. Using this formulation, chitosan, tripolyphosphate, and alginate were used to fabricate colloidal particles with the model protein BSA via electrostatic interactions, which were caged and incorporated BSA very tightly within the β-glucan particle shells. The prepared GMP-BSA exhibited good protein-release behavior and avoided protein leakage. The particles were also highly specific to phagocytic macrophages, such as Raw 264.7 cells, primary bone marrow-derived macrophages, and peritoneal exudate macrophages, whereas the particles were not taken up by nonphagocytic cells, including NIH3T3, AD293, HeLa, and Caco-2. We hypothesize that these tightly encapsulated protein-loaded glucan particles deliver various types of proteins to macrophages with notably high selectivity, and may have broad applications in targeted drug delivery or vaccine design against macrophages.
Keywords: macrophage, targeted drug delivery, vaccine, glucan particle, yeast-cell wall
Introduction
Macrophages are white blood cells of the innate immune system, and are widely distributed throughout the body. Macrophages perform various immunological functions in innate immunity, immune-cell activation, and other immunoreactions.1–3 Therefore, macrophages play important roles in the pathogenesis of various diseases, such as macrophage-activation syndrome,4 rheumatoid arthritis,5 atherosclerosis,6 inflammatory bowel disease,7 and cancer.8–11 Based on their role in mediating disease control and progression, macrophages represent important potential therapeutic targets. Studies have been performed to develop new drugs that target macrophages to combat these diseases.12–14
Furthermore, macrophages are important antigen-presenting cells, and have the potential to effectively induce specific CD8+ cytotoxic T-cell response to vaccines. They have a remarkable cross-presenting activity, and can present antigens to CD8+ T cells in a major histocompatibility complex I-dependent manner.15,16 The targeted delivery of protein and peptide antigens directly to macrophages may form an effective strategy for vaccine design against cancers and infectious diseases with high adjuvant activity.17–19
The targeted delivery of drug molecules specifically to macrophages to combat diseases remains a challenge. Intensive studies have been performed to fabricate new carrier systems for these purposes.20–22 These systems are primarily based on the pattern-recognition receptors on the cell surface of macrophages, such as the family of scavenger receptors, including the mannose receptor,23–28 and the Fc and complement receptors,29–32 which are used by macrophages for phagocytic clearance. In some cases, receptors for high-density lipoprotein,33 amyloid-β peptide,34 and hyaluronic acid35 were also successfully used for the targeting purpose. Developing targeted delivery systems may result in a reduction of the amount of therapeutic agent needed to obtain a clinical effect, and may effectively reduce drug-induced toxicity and other side effects. The encapsulation or incorporation of drug molecules into carrier systems may also provide protection against drug degradation or inactivation en route to the macrophages. However, the low cell selectivity and high heterogeneity of these artificial systems remain problems for practical applications. Moreover, complex processes are required to express pattern-recognition receptor ligands on the carrier surface.36
β-Glucan particles,37,38 particularly from the cell walls of baker’s yeast, are hollow and porous 2–4 μm microspheres, and are widely used as supplements in human nutrition and as adjuvants in vaccine design.39 They are the major yeast pathogen-associated molecular pattern, and are recognized by the pattern-recognition receptor dectin-1, a highly expressed lectin on the surface of phagocytic macrophages.40 β-Glucan particles are efficiently taken up by macrophages in vitro.41 The hollow, porous structure of the β-glucan particles also permits for high drug loading, which allows them to be used as a targeted delivery vehicle for macrophages.37,38,42 Recently, Aouadi et al demonstrated that β-glucan-encapsulated small interfering ribonucleic acid (siRNA) particles are efficient oral delivery vehicles for targeting macrophages, and that the particles potentially silence the Map4k4 gene and protect animals from lipopolysaccharide-induced lethality.43,44 However, this approach involved multiple components and a complex formulation process, and utilized cytotoxic polyethylenimine (PEI),45,46 which may decrease their reproducibility and cause adverse effects. Huang et al offered a simplified encapsulation process using the direct absorption of protein antigen inside the glucans, and these particles stimulated strong immunologic responses in vivo as an immune adjuvant.47,48 However, the direct absorption process may release the absorbed protein from the carrier too rapidly. New strategies for protein encapsulation inside glucan particles are therefore necessary for the efficient use of β-glucan particles as targeted protein-delivery systems for macrophages.
Here, we report a novel formulation based on β-glucan particles for the targeted delivery of protein to macrophages. We used the β-glucan particles from the baker’s yeast Saccharomyces cerevisiae to fabricate this carrier system. In this formulation, chitosan, tripolyphosphate (TPP), and alginate were used to fabricate colloidal particles with the model protein bovine serum albumin (BSA) via electrostatic interactions, which were caged and therefore incorporated BSA tightly within the β-glucan particle shells. This tight-encapsulation formulation greatly reduced the rate of protein release and decreased protein leakage. The in vitro and in vivo cellular uptake experiments were performed to determine the recognition properties of the prepared BSA-loaded glucan particles (GMP-BSA). Protein encapsulation did not affect the recognition property of the β-glucan itself. GMP-BSA are selectively taken up by macrophages, such as Raw 264.7 cells, primary bone marrow-derived macrophages (BMDMs) and peritoneal exudate macrophages (PEMs), whereas nonphagocytic cells, including NIH3T3, AD293, HeLa, and Caco-2, do not take up the particles. We anticipate that these tightly encapsulated protein-loaded glucan particles will deliver various types of proteins to macrophages with notably high selectivity, and may have broad applications for targeted drug delivery or vaccine design against macrophages.
Materials and methods
Materials
Low-guluronic-content, low-viscosity sodium alginate (100–200 mPa·s) and low-molecular-weight chitosan (average molecular weight 10–20 kDa) were purchased from Heowns (Tianjin, People’s Republic of China [PRC]). BSA was purchased from Solarbio (albumin bovine V; Beijing, PRC). A MicroBCA kit was purchased from CW Biotech (Tianjin, PRC). Modified Eagle’s Medium (MEM) was obtained from HyClone (Logan, UT, USA). Roswell Park Memorial Institute (RPMI) 1640 medium was purchased from Thermo Fisher Scientific (Waltham, MA, USA). A Barnstead Nanopure Water Purification System was purchased from Thermo Fisher Scientific and used to prepare ultrapure water. All other chemicals and reagents used were of analytical grade and obtained commercially unless stated otherwise.
Cells and animals
The murine macrophage cell line Raw 264.7, murine embryonic fibroblast cell line NIH3T3, and human cervical cancer cell line HeLa were purchased from Saierbio (Tianjin, PRC). The human embryonic kidney cell line AD293 was purchased from BioHermes(Wuxi, PRC), and the human colorectal adenocarcinoma cell line Caco-2 was purchased from Boster (Wuhan, PRC). Raw 264.7 cells were cultured in RPMI 1640 medium, Caco-2 cells were cultured in MEM, and HeLa, AD293, and NIH3T3 cells were cultured in Dulbecco’s MEM (DMEM). All cells were incubated in medium containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in humidified air supplemented with 5% CO2. Female C57BL/6J mice (6–8 weeks of age, 15–20 g weight) were purchased from the Academy of Military Science (Beijing, PRC) and maintained in a germ-free environment with free access to food and water. All animal procedures were conducted under a protocol approved by the Institutional Animal Care and Use Committee of Nankai University (Tianjin, PRC).
Preparation of β-glucan particles
β-Glucan particles were prepared from commercially available S. cerevisiae baker’s yeast (Angel Yeast, Chifeng, PRC) via a series of alkaline and acidic extraction steps as described previously.37 Briefly, 3.75 g baker’s yeast was resuspended in 50 mL water, and the pH was adjusted to 12.0–12.5 with 1.0 M NaOH. The suspension was heated to 60°C with stirring for 1 hour and then centrifuged at 2,000 g for 10 minutes to recover the insoluble material containing the cell walls. The material was then resuspended in 50 mL water, brought to pH 4–5 with HCl, and heated to 55°C with stirring for an additional 1 hour. The glucan particles were then collected after centrifugation at 8,000 rpm for 3 minutes, successive washes with water (three times), isopropanol (four times), and acetone (two times), and drying under a vacuum to yield 1.72 g slightly off-white powder.
Fluorescein and rhodamine B labeling of glucan particles and BSA
The glucan particles were labeled with fluorescein isothiocyanate (FITC) or rhodamine B isothiocyanate. Briefly, the glucan particles (500 mg) were incubated with FITC or rhodamine B isothiocyanate (5 mg, dissolved at 2.5 mg/mL in dimethyl sulfoxide [DMSO]) in 50 mL sodium carbonate buffer (0.1 M, pH 9.2) overnight at 37°C in the dark. Unreacted FITC or rhodamine B isothiocyanate was then quenched by incubation with Tris buffer (10 mL, 1.0 M, pH 8.3) for 30 minutes. The labeled glucan particles were extensively washed with sterile water until the color was removed, dehydrated with absolute ethanol and acetone, and then dried under vacuum in the dark at room temperature.
Protein BSA was similarly labeled with FITC or rhodamine B isothiocyanate. Briefly, BSA (10 mg) was dissolved in a mixture of 10 mL 5 mM ethylenediaminetetraacetic acid and 1.2 mL 0.1 M carbonate buffer (pH 9.2). FITC or rhodamine B isothiocyanate (1 mg, dissolved at 2.5 mg/mL in DMSO) was added to the buffered BSA solution and stirred at room temperature in the dark overnight. Tris buffer (2.0 mL, 1.0 M, pH 8.3) was then added, and the reaction mixture was stirred for an additional 15 minutes at room temperature to quench the free fluorescent labeling reagent. The product was purified by dialysis against water and lyophilized in the dark.
Preparation of GMP-BSA
GMP-BSA were prepared based on the ionic gelation of chitosan with TPP and alginate sodium inside the glucan particles. Briefly, 10 mg dry glucan particles were mixed with 20 μL BSA solution (50 mg/mL in phosphate-buffered saline [PBS]) at room temperature for 10 minutes, and then combined with 20 μL low-molecular-weight chitosan solution (0.4%, w/w, pH 5.0) at room temperature for 2 hours to allow the particles to swell and engulf the BSA and chitosan solution. Then, 1.0 mL TPP/alginate solution (1.0 mg/mL TPP, 0.4 mg/mL alginate sodium) was added in excess, and the formed particles were dispersed by ultrasonication and then stirred for 1 hour. The particles were centrifuged at 2,000 g for 5 minutes, washed with PBS three times, resuspended in 70% ethanol to sterilize the particles, and washed three times again with PBS. The washed particles were lyophilized under a vacuum to form the GMP-BSA.
Morphological characterization of the GMP-BSA
The shape and surface morphology of the GMP-BSA were observed using confocal microscopy and a JSM-7500F field-emission scanning electron microscope (SEM; JEOL, Tokyo, Japan). For the confocal laser-scanning microscopy (CLSM) observation, the particles were first fluorescently labeled with rhodamine B or FITC on the glucan particle shell or BSA as described earlier, and then particle morphology and protein distribution were observed by confocal microscopy (Nikon Eclipse TE300; MRC-1024; BioRad, Hercules, CA, USA). The particles were also characterized for size with a Malvern Zetasize Nano ZS90 (Malvern Instruments, Malvern, UK).
Determination of BSA-loading and encapsulation efficiency
The BSA-loading and encapsulation efficiency of the GMP-BSA were analyzed using a micro-bicinchoninic acid (micro-BCA) method to indirectly determine the free BSA in the supernatant. Briefly, the supernatants of the GMP-BSA formulations were collected after separation via centrifugation, and were then analyzed for BSA content with a micro-BCA protein-assay kit for 96-microwell plates according to the manufacturer’s instructions. Each supernatant (60 μL) was mixed with the same volume of micro-BCA test liquid in a 1.5 mL centrifuge tube. After incubation at 60°C for 1 hour, 100 μL of each mixture was transferred to a microplate, and the absorbance of the samples was measured at 570 nm using a microplate reader (EL808IU-PC; BioTek Instruments, Winooski, VT, USA). The concentration of BSA was calculated from a standard curve, prepared by measuring the intensity of known concentrations of free BSA. The blank glucan particles containing chitosan/TPP/alginate inside were prepared using the same method, but without the addition of BSA. This supernatant was collected and used to dissolve BSA to prepare the standard curve. Drug-loading (LE) and encapsulation efficiency (EE) were calculated as follows:
|
|

|
|
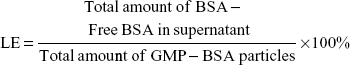
In vitro release of BSA from the GMP-BSA
The in vitro BSA-release profiles of the GMP-BSA were determined. Briefly, GMP-BSA (10 mg) were incubated with 1 mL of release medium (PBS, 0.02% sodium azide, pH 7.4) in an Eppendorf tube in a horizontal rotator at 150 rpm at 25°C. At predetermined intervals, the supernatant was collected after centrifugation at 8,000 rpm for 5 minutes, and was replaced with an equal volume of fresh release medium. The supernatant of each sample was analyzed for the amount of released BSA using the micro-BCA assay. The release profiles of the BSA were analyzed using analysis of variance with the Origin 8.0 software.
Protein-integrity verification using SDS-PAGE and CD spectroscopy
The primary structural integrity of the released BSA was examined by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE) compared to the native BSA and protein markers. The released BSA (15 μL) from the GMP-BSA with different storage times was used as sample. All gels were run using an EPS 300 electrophoresis unit from Tanon at a constant voltage mode of 180 V in Tris/glycine SDS buffer under nonreducing conditions, which preserved the aggregates linked by disulfide bonds. The gels were stained with Coomassie Brilliant Blue to detect the protein.
The secondary structure of the released BSA was measured using a circular dichroism (CD) spectroscope with a 2 mm quartz cylindrical cell compared to the native BSA. The samples were scanned over the wavelength range 200–260 nm.
Generation and culture of BMDMs
C57BL/6J mice (10 weeks old) were used for the preparation of primary BMDMs.49 Bone marrow cells were isolated from the femur and tibia bones of mice, and the tissues were filtered using nylon mesh. The cells from each mouse were seeded into two 10 cm tissue-culture dishes using BMDM medium containing 20% fetal bovine serum, 1% penicillin/streptomycin, 30% L929-conditioned medium, and DMEM. The medium was changed every 2 days, and after 5 days the cells were ready for the following experiments.
Generation and culture of PEMs
For the preparation of the PEMs,49 10-week-old C57BL/6J mice were injected intraperitoneally (IP) with 2 mL 4% (w/v) Brewer’s thioglycolate broth for 4 days, and then the peritoneal exudate cells from the peritoneal cavity were harvested using two sequential peritoneal lavages with serum-free PBS. The cells were washed and resuspended in RPMI 1640 medium containing 100 U/mL penicillin and 100 μg/mL streptomycin and 3% fetal bovine serum. Then, the cells were plated at a density of 6×105 per well in a 24-well plate in RPMI 1640 culture media supplemented with 10% fetal bovine serum. After 3 hours, nonadherent cells were washed away with PBS, and adherent macrophages were maintained on the culture plate for the following experiments.
Generation and culture of activated M1 and M2 macrophages
BMDMs isolated from 10-week-old C57BL/6J mice and cultured in 24-well plates (Corning) for 5 days were polarized over the next 36 hours by culturing with 30 ng/mL IFNγ (Peprotech, Rocky Hill, NJ, USA) and 10 ng/mL lipopolysaccharide (Sigma-Aldrich, St Louis, MO, USA) for M1 macrophages50 or 20 ng/mL IL4 (Peprotech) for M2 macrophages.51 The medium was changed, and the cells were ready for the following experiments.
In vitro cellular uptake assays with confocal microscopy
The cellular uptake assays were performed both on the phagocytic and nonphagocytic cells. For assays with the phagocytic cells, Raw 264.7 cells, primary BMDMs, or PEMs were cultured at 1×106 cells/mL in 24-well plates. After 6 hours, BSA-loaded glucan particles were added to the culture medium at a variety of particle concentrations and incubated for 12 hours. Then, the cells were stained with iFluor™ 488 (IF488)-F4/80 primary antibody (clone BM8, lot AC709) to identify macrophages, and the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). For assays with the activated M1 and M2 macrophages, the GMP-BSA were added to the culture medium at 12 μg/mL and incubated for 6, 12, or 24 hours, with the nonactivated BMDM macrophages as the control. For assays with nonphagocytic cells, including NIH3T3, AD293, HeLa, and Caco-2 cells, the procedures were performed similarly to the phagocytic cells. After being washed with PBS, the cells were mounted and visualized on the confocal microscope (Nikon Eclipse TE300, MRC-1024) sequentially using the 350 nm (blue), 470–490 nm (green), and 515–560 nm (red) excitation filters. Each stain was performed on three individual groups, and the photographs in the figures are representative of each group.
Cytotoxicity assays
The cytotoxicity of the BSA-loaded glucan particles was determined by measuring the inhibition of cell growth using a tetrazolium dye (3-[4,5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide [MTT]) assay. Raw 264.7 cells harvested in a logarithmic growth phase were seeded on a 96-well plate at 5×103 cells/well. Next, the cells were incubated for 24 hours with various concentrations (3–400 μg/mL) of the BSA-loaded glucan particles. Then, 10 μL MTT solution (5.0 mg/mL) was added to each well. The plates were incubated with MTT for an additional 4 hours at 37°C. After the supernatant was removed, 100 μL DMSO was added to fully dissolve the crystals. Absorbance at 490 nm, which is related to the number of metabolically active cells, was measured using a microplate reader (EXL-800; BioTek Instruments).
In vivo GMP-BSA administration
Ten-week-old C57BL/6J male mice were IP injected with Brewer’s thioglycolate broth. On day 4, these mice were IP or intravenously (IV) injected with 30 mg/kg GMP-BSA with a rhodamine B label on BSA in 150 μL PBS, equal to 1.5 mg/kg BSA. After 3 hours, the peritoneal exudate cells and spleen, liver, and lungs were collected for macrophage separation, and the blood was collected for neutrophil separation.
Isolation of PEMs and tissue macrophages and CLSM observation
The PEMs were separated from the peritoneal exudate cells with centrifugation at 1,000 rpm and plated for 2 hours in DMEM plus 10% fetal bovine serum. The cells were washed with PBS to remove nonadherent cells, and the remaining macrophages were incubated with IF488-F4/80 primary antibody for 30 minutes. The cells were then fixed and visualized by microscopy. The spleen, liver, and lung were cut into small pieces, washed with PBS, and digested at 37°C for 30 minutes using 100 U/mL deoxyribonuclease I (RNase-free) and collagenase type 4. The digested tissues were filtered through 100 mm-pore nylon mesh, centrifuged, and the cells were plated in plastic dishes for 2–3 hours in DMEM plus 10% fetal bovine serum for attachment. After being washed with PBS to remove the nonadherent cells, the adherent cells were used as peritoneal macrophages for confocal microscopy observation.
Isolation of neutrophils and CLSM observation
Neutrophils were isolated from 750 μL of peripheral blood of the mice that were IV injected with GMP-BSA by centrifugation through an equal volume of density gradient media (neutrophil isolation kit, LZS1100; Haoyang, Tianjin, PRC) at 500 g for 30 minutes. The neutrophils were removed by aspiration, mixed with equal volumes of 0.45% NaCl, centrifuged at 500 g for 10 minutes, and washed twice with Hanks balanced salt solution. The cells were stained with DAPI and used for CLSM observation.
Results and discussion
Preparation of GMP-BSA particles
Baker’s yeast provides a hard shell containing a glucan layer as the skeleton and a mannan shell for targeting. After being processed with acid, alkali, isopropanol, and acetone, the nucleic acids, proteins, and organelles in the yeasts were washed away, and the hollow and porous glucan particles of the shells were retained. In previous studies, siRNA was entrapped inside the glucan particles because of the layer-by-layer electrostatic interaction between PEI and RNA. However, the cytotoxicity of PEI may cause serious side effects for in vivo applications. In this study, we used safer materials to encapsulate the proteins inside the hollow yeast glucan particles.
In the preliminary test, chitosan and heparin were used as the “fixing materials” to form particles with BSA caged inside the hollow glucan shells through anionic and cationic electrostatic interactions to form particles that sealed the holes of the glucan walls and prevented the protein from leaking out of the glucan-particle chamber. However, the protein-loaded particles easily aggregated, and the heparin used may also cause side effects, such as thrombocytopenia. Therefore, instead of heparin, TPP and alginate sodium were used together with chitosan as the cross-linkers to form the particles (Figure 1). The GMP-BSA were well dispersed, and the BSA was efficiently and tightly entrapped inside the particles. Chitosan and alginate are US Food and Drug Administration-approved additives for food, and TPP is a safe inorganic salt. The particles possessed safe characteristics for in vivo applications. The glucan particles were incubated with BSA mixed with chitosan for 2 hours, prior to the addition of the cross-linking agent TPP/alginate solution to form the chitosan complex. All samples were stirred at room temperature for up to 30 minutes and then dried under vacuum for 12 hours. Different concentrations of BSA and the fitting materials were tested (data not shown). Finally, the optimal parameters were used to prepare these inside cross-linked particles, as described in the Materials and methods section.
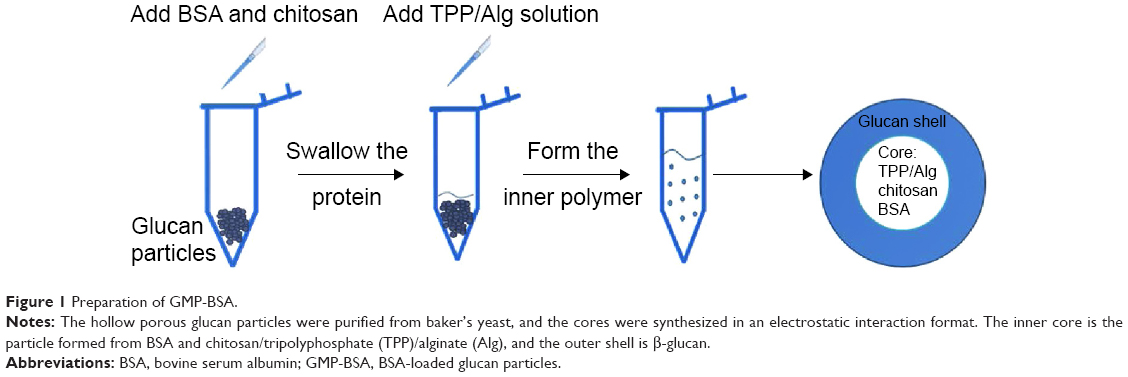 | Figure 1 Preparation of GMP-BSA. |
Morphology of GMP-BSA verified with CLSM and SEM
The surface and interior morphology of the GMP-BSA was visualized by CLSM and SEM. For the confocal experiments (Figure 2A), the glucan-particle shells and BSA were labeled with fluorescent dyes for visualization. We prepared two types of labeled GMP-BSA: in one, the glucan-particle shells were labeled with rhodamine B and the BSA was labeled with FITC (Figure 2A, left panel); in the other, the glucan-particle shells were labeled with FITC and the BSA was labeled with rhodamine B (Figure 2A, right panel). Both fluorescently labeled GMP-BSA showed strong FITC and rhodamine B fluorescence in the CLSM images, with a regular spherical shape and very smooth surfaces. This indicated that the BSA was encapsulated and uniformly distributed inside the glucan particles. Interestingly, the images from these two differently labeled particles showed some differences. As shown in Figure 2A, right panel, the spherical shape of the FITC fluorescence was larger than the rhodamine B fluorescence. This difference may have resulted from light-scattering phenomena. By SEM (Figure 2B), the majority of particles showed homogeneous morphological characteristics without any aggregation or adhesion. The GMP-BSA are distinguished by their characteristic ellipsoidal “wrinkled pea” morphology and irregular delineation. From the confocal and SEM data, the sizes of the GMP-BSA were estimated to be 2–4 μm. Laser-diffraction measurements were also used to determine the size, revealing a mean diameter of 3.2±0.2 μm (Figure 2C) and very homogeneous size and distribution (polydispersity index =0.023), which was consistent with the confocal and SEM data. It was also consistent with the size of the glucan particles originally from the yeast cell wall, which were 2–4 μm hollow porous microparticles.
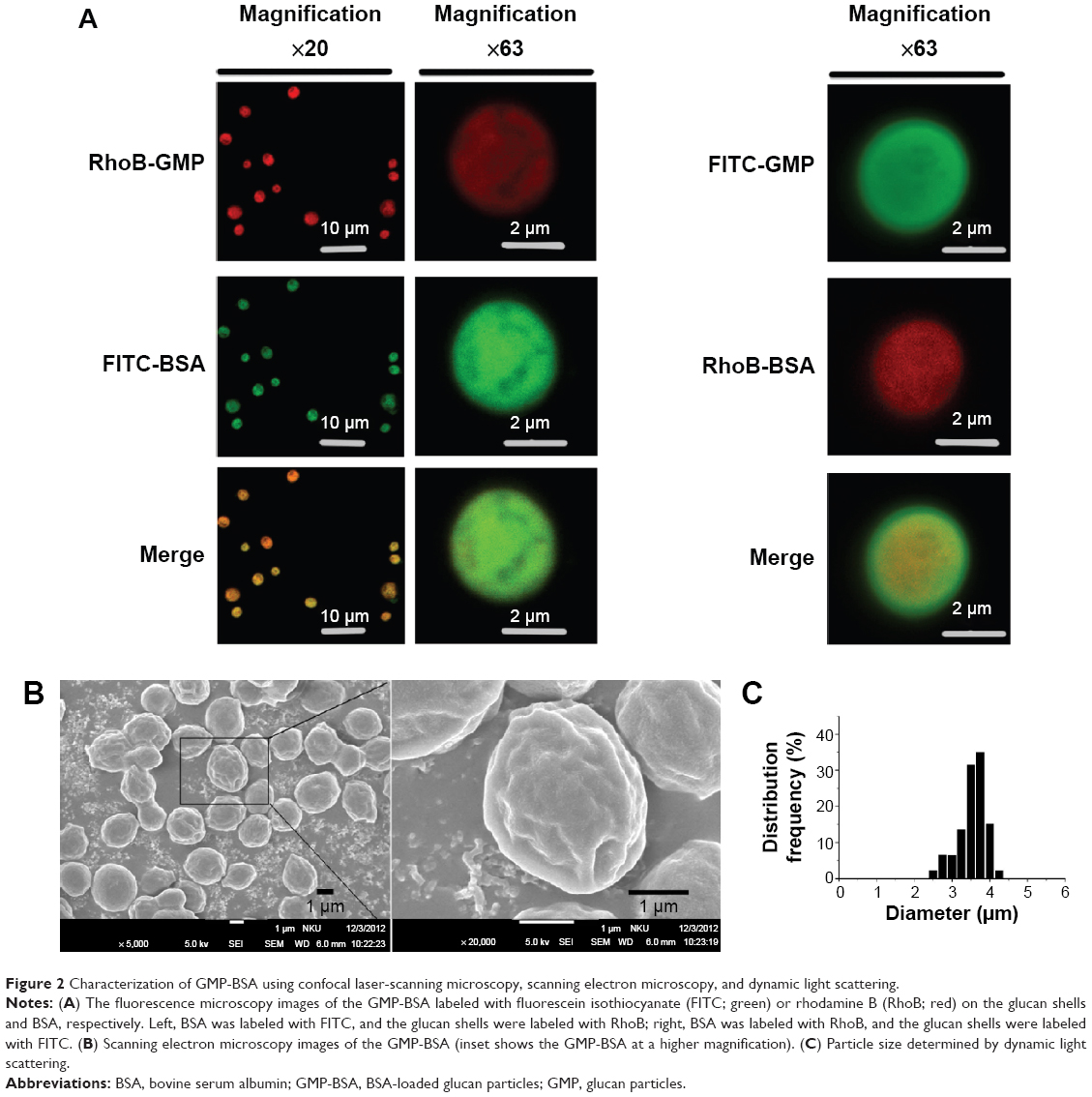 | Figure 2 Characterization of GMP-BSA using confocal laser-scanning microscopy, scanning electron microscopy, and dynamic light scattering. |
Loading and encapsulation efficiency of the GMP-BSA complex particles
We used a micro-BCA assay to determine the loading and encapsulation efficiency of BSA in the GMP-BSA. The data were obtained with three tests. The loading efficiency was 5.00%±0.25%, and the encapsulation efficiency was 49.67%±2.12%. Good loading and encapsulation efficiency were achieved in this formulation.
In vitro release behavior of loaded protein
Figure 3A shows the release profiles of BSA from the glucan particles in PBS (pH 7.4) in vitro. The GMP-BSA were stable in a liquid buffer environment. The release behavior was determined for 2 weeks, and the loaded BSA was released constantly and slowly from the particles during these 2 weeks with an adequate release rate. The overall release followed biphasic release kinetics, with a very low initial burst release in the first day, followed by a constant slow release over 14 days. The initial burst release was low, less than 20% in the first 12 hours. The slow release rate of BSA from the particles was dependent on the degradation of chitosan, alginate, or glucan polymer, because their degradation is slow. The release rate showed that it could be used as a long-term drug-delivery vehicle.
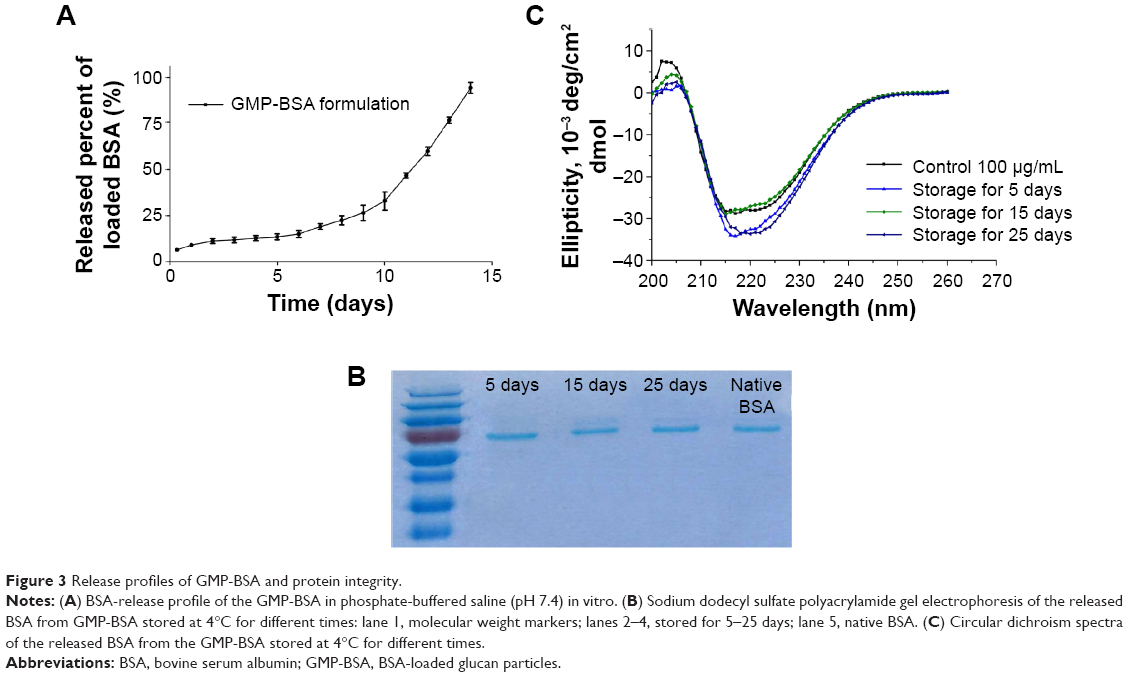 | Figure 3 Release profiles of GMP-BSA and protein integrity. |
Integrity of the released protein verified using SDS-PAGE and CD spectrum
Protein drugs may degrade because of irreversible protein degradation or aggregation due to deleterious factors, such as the encapsulating preparation process and the microenvironment within the polymeric matrix. Therefore, we investigated the integrity of the BSA released from the particles using SDS-PAGE and CD-spectrum analysis.
Figure 3B shows the SDS-PAGE results of the native BSA and BSA released from the GMP-BSA stored at 4°C for 5–25 days. The same band corresponding to a molecular weight of 66 kDa was observed in the released samples and the native BSA, without any lower- or higher-molecular-weight bands, which indicates that the primary structure of the BSA inside the GMP-BSA was retained.
The secondary structure of the native and released BSA was analyzed using far-ultraviolet CD (200–260 nm). As shown in Figure 3C, there were no significant changes in the CD spectrum of the BSA released from the GMP-BSA compared to the native BSA. Each spectrum displayed two minima at 208 and 222 nm, indicating that the native secondary structure of the released BSA was preserved.
These results suggest that the GMP-BSA provides a mild environment for the BSA, at least for the protein-encapsulation process and during 25 days’ storage. Because proteins may be degraded and are easily inactivated, this mild ionotropic gelation method is suitable for the preparation of protein-loaded particles.
In vitro cellular uptake of GMP-BSA by Raw 264.7 cells
Previous studies showed that β-glucans from yeast-cell walls possess high specificity and selectivity for macrophages. To show that the protein encapsulation inside the glucan particles does not change the uptake behavior of β-glucans, in vitro and in vivo cellular uptake tests were performed.
We first investigated the uptake behavior of the β-glucan-based GMP-BSA with macrophage Raw 264.7 cells. These cells are commonly used for in vitro modeling of macrophages and pharmaceutical intervention. For visualization, FITC (green) was used to label the BSA, and rhodamine B (red) was used to label the glucan shells. These particles were incubated with Raw 264.7 cells at different concentrations of 6, 12, and 24 μg/mL for 12 hours, and the uptake of these particles was then visualized with confocal microscopy. Two replicates were performed for each sample, and three different fields for each replicate were imaged. The exact localization of the particles within the cell, or whether the particles were internalized or extracellularly bound to the cell surface, was clearly distinguished using CLSM. As shown in Figure 4, the GMP-BSA, identified as green and red fluorescent spots, were efficiently internalized by Raw 264.7 cells and primarily localized to the apical and perinuclear regions of the cells. The cellular uptake of the particles was dose-dependent. After 12 hours of incubation at 12 or 24 μg/mL, confocal microscopy showed that approximately 90% of the macrophages internalized at least one particle, whereas most cells had internalized multiple particles.
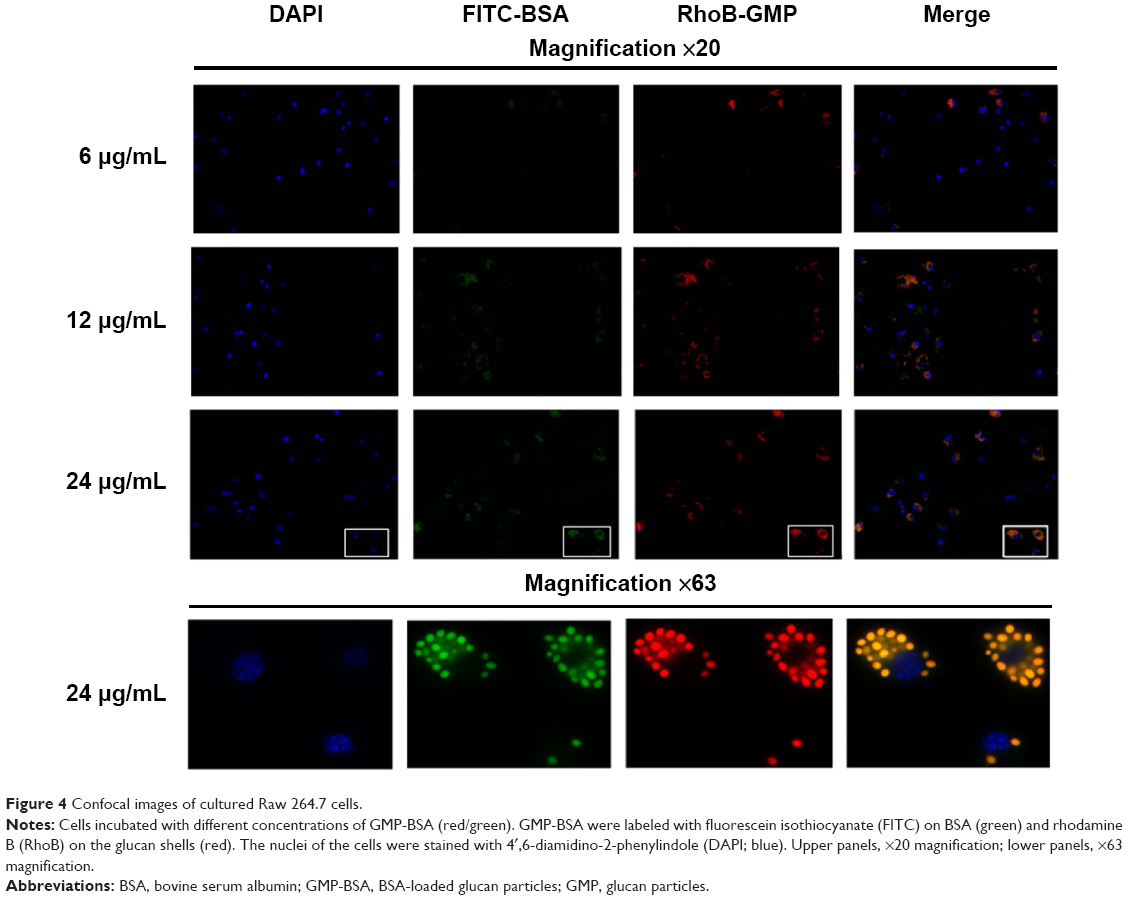 | Figure 4 Confocal images of cultured Raw 264.7 cells. |
Cellular uptake of GMP-BSA with primary BMDMs and PEMs in vitro
We also investigated the uptake behavior of GMP-BSA with primary BMDMs and PEMs, which are two types of commonly used primary macrophages. The GMP-BSA were incubated with these primary macrophages at a variety of particle concentrations (6/12/24 μg/mL), and then the cellular uptake of these particles was visualized with a fluorescence microscope.
Primary BMDMs took up each concentration of GMP-BSA (Figure 5A). When incubated at a low particle concentration (6 μg/mL), the fluorescence of both the GMP shell (rhodamine B-labeled) and BSA core (FITC-labeled) was observed inside the cells, and 54% of the BMDMs took up the GMP-BSA. At 12 μg/mL, approximately 84% of the cells took up the particles, and 74% of those cells absorbed two or more particles. At 24 μg/mL, 92% of the BMDMs took up the particles, and over 99% had two or more particles in one cell.
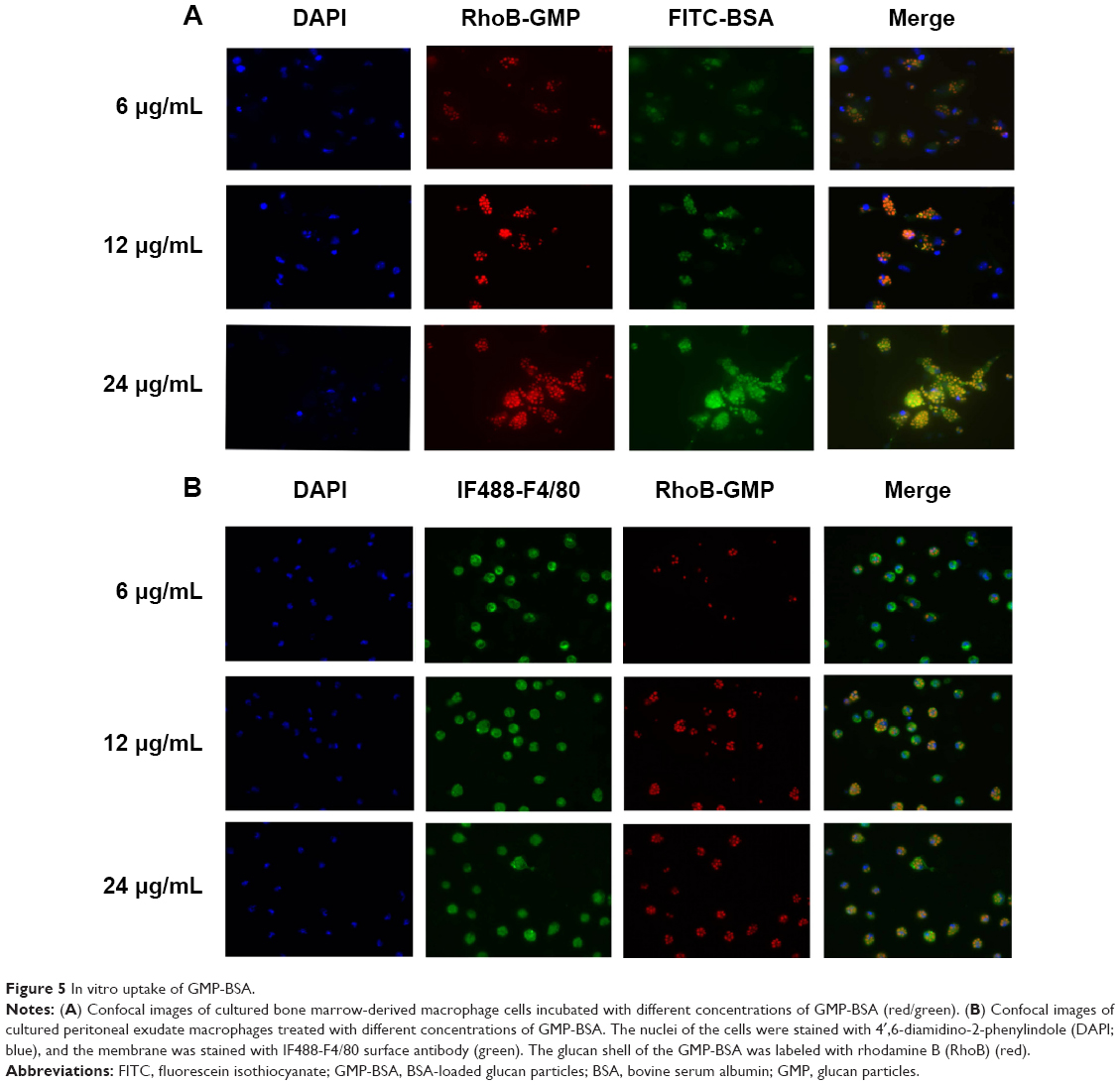 | Figure 5 In vitro uptake of GMP-BSA. |
The GMP-BSA were also incubated with primary PEMs in vitro at 6, 12, or 24 μg/mL for 12 hours to visualize cellular uptake behavior (Figure 5B). The confocal images showed that approximately 80% of the PEMs took up the GMP particles and the majority of cells took up more than one particle, even at a low particle concentration (6 μg/mL). When the concentration of particles was increased, PEMs took up more particles. Compared to the BMDM macrophages, PEMs had higher phagocytosis activity for the GMP-BSA.
These results show that primary macrophages, including BMDMs and PEMs, efficiently take up GMP-BSA, and that the particle fabrication had little influence on the cellular uptake behavior of β-glucans by macrophages.
Cellular uptake of GMP-BSA with activated M1 and M2 macrophages in vitro
Macrophages change their cellular morphology and secretory pattern as a result of appropriate stimulation. Activated macrophages are routinely classified into two different phenotypes: classically activated M1 macrophages and alternatively activated M2 macrophages. Both phenotypes are important components of the innate and adaptive immune systems. M1 macrophages comprise immune effector cells with an acute inflammatory phenotype and are highly aggressive against bacteria, whereas M2 macrophages tend to resolve inflammation and facilitate wound healing. In order to test further the interaction of GMP-BSA with macrophages, both M1 and M2 macrophages were obtained through activation of murine BMDM cells according to published procedures.50,51 Then, they were treated with rhodamine B-labeled GMP-BSA with different incubation times. As shown in Figure 6, a very interesting result was obtained. Although all the macrophages, including BMDM, M1, and M2, took up the GMP-BSA very efficiently, most of the particles inside the M2 cells were degraded, with rhodamine B fluorescence full of the whole cells. Some of the particles were also degraded in the M1 macrophages, while nonactivated BMDM cells did not show such capability. This result gave some clues that the proteins or peptides encapsulated inside the particles could be released efficiently once taken up by macrophages.
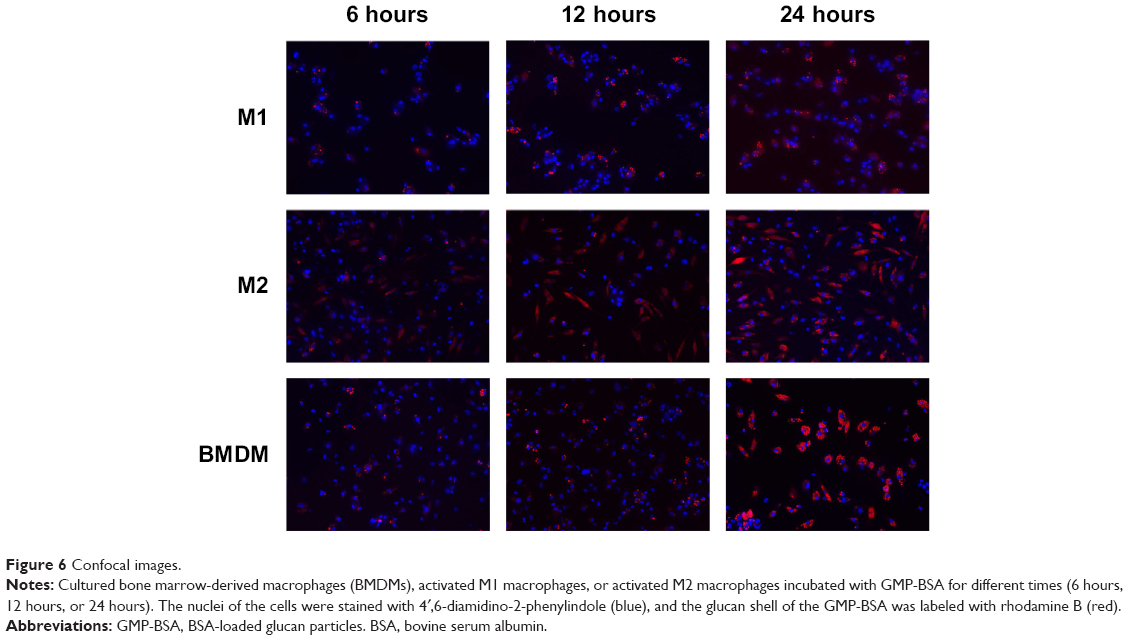 | Figure 6 Confocal images. |
Nonphagocytic cells (NIH3T3, AD293, HeLa, and Caco-2) do not take up GMP-BSA particles
To verify that our GMP-BSA have high specificity for macrophages, these particles were also incubated with nonphagocytic cells, including NIH3T3, AD293, HeLa, and Caco-2 cells, at 24 μg/mL for 12 hours. Their uptake behaviors were examined using confocal microscopy. The fluorescence microscopy images showed that these cells did not take up any particles (Figure 7). This 100% selectivity makes these particles useful for targeted drug delivery to macrophages.
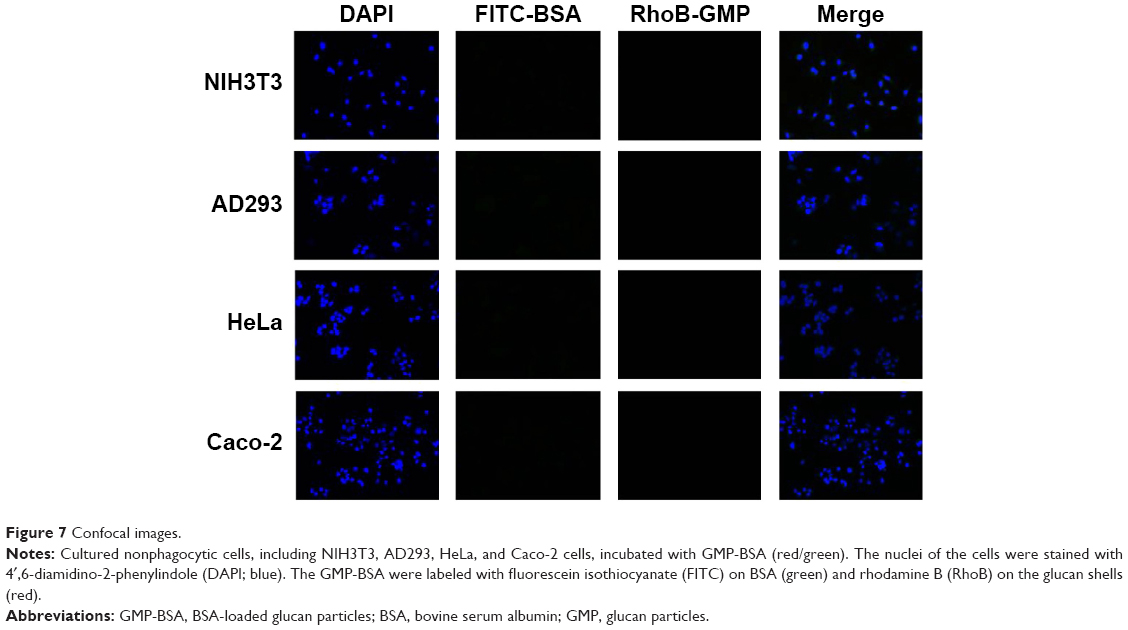 | Figure 7 Confocal images. |
In vitro cytotoxicity of the GMP-BSA
To verify that these NPs are not cytotoxic upon uptake by macrophages, we performed a standard MTT viability assay against Raw 264.7 cells and the nonphagocytic cells. As shown in Figure 8, Raw 264.7 cells were nearly 100% viable compared to the untreated cells after 24 hours’ exposure to the GMP-BSA or blank glucan particles up to 0.4 mg/mL, suggesting good biocompatibility with macrophages. Similarly, we demonstrated that these particles were not cytotoxic to nonphagocytic cells, including NIH3T3, AD293, HeLa, and Caco-2 cells (data not shown). This is explained by the observation that these particles were not taken up by nonphagocytic cells.
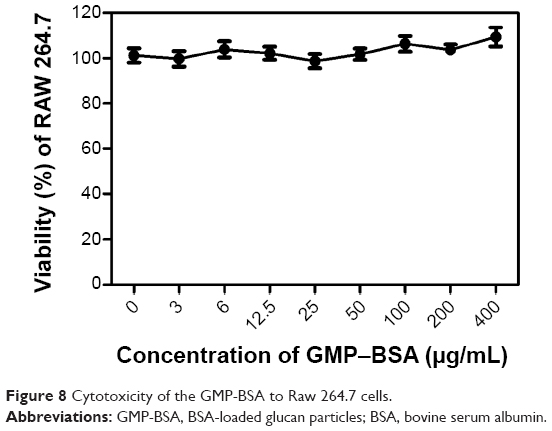 | Figure 8 Cytotoxicity of the GMP-BSA to Raw 264.7 cells. |
GMP shells containing a BSA core induce cell uptake in macrophages in mice through IP or IV injection, but not in neutrophils
To determine whether these particles also showed efficient uptake behavior in vivo, 10-week-old C57BL/6J female mice were IP or IV injected with GMP-BSA labeled with rhodamine B. Then, the macrophages in the organs and peritoneal exudate cells were isolated for confocal observation.
In the IP injected mice, the particles showed good uptake (Figure 9A). A large number of particles were found in the PEMs, and several were found in the macrophages in the liver, lung, and spleen, although fewer compared to the PEMs. In the IV injected mice, the PEMs and the macrophages in the organs, including the liver, lung, and spleen, also showed high particle uptake (Figure 9B). However, compared to the PEMs from the IP injected mice, the PEMs from the IV injected mice took up fewer particles. In the IP injection, the particles were first taken up by PEMs and then transferred to other organs through the blood circulation, which may have resulted in more particles in the PEMs compared to the IV injection. In the IV injected mice, the spleen showed slightly higher uptake than the lung and liver. The high uptake efficiency indicates that these particles can be used for in vivo applications.
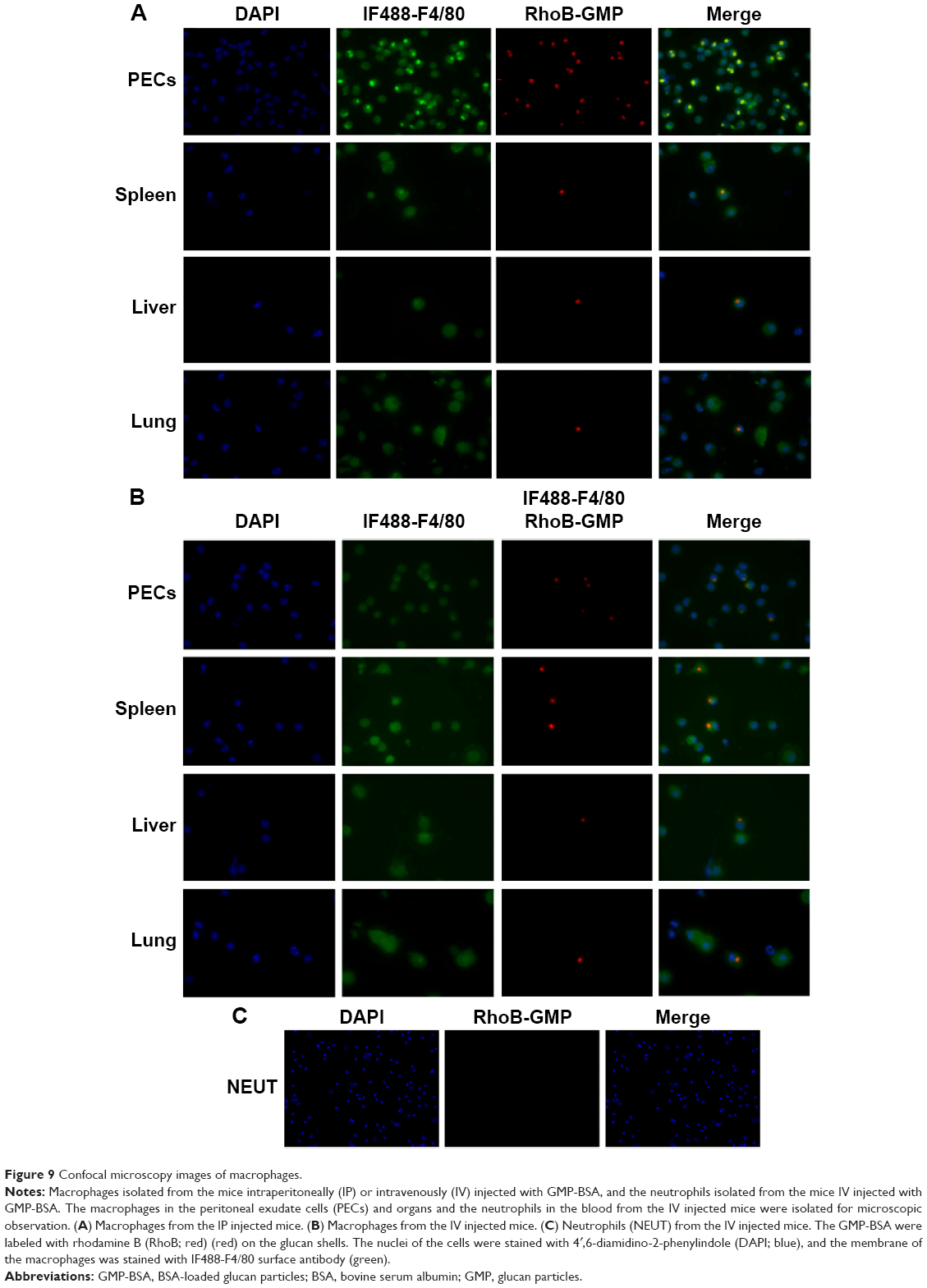 | Figure 9 Confocal microscopy images of macrophages. |
Neutrophils are also phagocytes, capable of ingesting microorganisms or particles. They account for approximately 50%–70% of all white blood cells, and are much more numerous than macrophages. In order to compare the selectivity of the GMP-BSA to neutrophils and to macrophages, the neutrophils in the peripheral blood of the mice that were IV injected with GMP-BSA were isolated for CLSM observation. As shown in Figure 9C, the neutrophils did not take up the particles. It seems that the GMP-BSA have quite good selectivity for macrophages.
Conclusion
In this study, we developed a novel macrophage-targeted protein-delivery system based on β-glucan polymers. The model protein BSA was tightly encapsulated inside the glucan particles. This system delivers proteins efficiently and specifically to macrophages without affecting nonphagocytic cells, such as NIH3T3, AD293, HeLa, and Caco-2. Due to the high stability and good biocompatibility of this formulation, it may have broad applications for targeted protein drug delivery or vaccine design against macrophages.
Acknowledgments
This work was supported by the Research Fund for the Doctoral Program of Higher Education of China (20110031110019) and the Natural Science Foundation of Tianjin (13JCZDJC28300 and 11JCYBJC25600).
Disclosure
The authors report no conflicts of interest in this work.
References
Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327(5966):656–661. | ||
Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604. | ||
Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. | ||
Grom AA, Mellins ED. Macrophage activation syndrome: advances towards understanding pathogenesis. Curr Opin Rheumatol. 2010;22(5):561–566. | ||
Lebre MC, Tak PP. Macrophage subsets in immune-mediated inflammatory disease: lessons from rheumatoid arthritis, spondyloarthritis, osteoarthritis, Behçet’s disease and gout. Open Arthritis J. 2010;3:18–23. | ||
Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145(3):341–355. | ||
Zhang J, Tang C, Yin C. Galactosylated trimethyl chitosanecysteine nanoparticles loaded with Map4k4 siRNA for targeting activated macrophages. Biomaterials. 2013;34(14):3667–3677. | ||
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. | ||
Lin EY, Li JF, Gnatovskiy L, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006;66(23):11238–11246. | ||
Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196(3):254–265. | ||
Galdiero MR, Garlanda C, Jaillon S, Marone G, Mantovani A. Tumor associated macrophages and neutrophils in tumor progression. J Cell Physiol. 2012;228(7):1404–1412. | ||
Hristodorov D, Mladenov R, Huhn M, Barth S, Thepen T. Macrophage-targeted therapy: CD64-based immunotoxins for treatment of chronic inflammatory diseases. Toxins. 2012;4(9):676–694. | ||
Prakash SP, Modarai B, Humphries J, et al. The monocyte/macrophage as a therapeutic target in atherosclerosis. Curr Opin Pharmacol. 2009;9(2):109–118. | ||
Kohichi C, Inagawa H, Hino M, et al. Utilization of macrophages in anticancer therapy: the macrophage network theory. Anticancer Res. 2004;24(5C):3311–3320. | ||
Asano K, Nabeyama A, Miyake Y, et al. CD169-positive macrophages dominate antitumor immunity by cross-presenting dead cell-associated antigens. Immunity. 2011;34(1):85–95. | ||
Schliehe C, Redaelli C, Engelhardt S, et al. CD8– dendritic cells and macrophages cross-present poly(d,l-lactate-co-glycolate) acid microsphere-encapsulated antigen in vivo. J Immunol. 2011;187(5):2112–2121. | ||
Muraoka D, Harada N, Hayashi T, et al. Nanogel-based immunologically stealth vaccine targets macrophages in the medulla of lymph node and induces potent antitumor immunity. ACS Nano. 2014;8(9):9209–9218. | ||
Irvine DJ, Swartz MA, Szeto GJ. Engineering synthetic vaccines using cues from natural immunity. Nat Mater. 2013;12(11):978–990. | ||
D’Argenio DA, Wilson CB. A decade of vaccines: integrating immunology and vaccinology for rational vaccine design. Immunity. 2010;33(4):437–440. | ||
Lameijer MA, Tang J, Nahrendorf M, Beelen RH, Mulder WJ. Monocytes and macrophages as nanomedicinal targets for improved diagnosis and treatment of disease. Expert Rev Mol Diagn. 2013;13(6):567–580. | ||
Moghimi SM, Parhamifar L, Ahmadvand D, et al. Particulate systems for targeting of macrophages: basic and therapeutic concepts. J Innate Immun. 2012;4(5–6):509–528. | ||
Chellata F, Merhi Y, Moreau A, Yahia L. Therapeutic potential of nanoparticulate systems for macrophage targeting. Biomaterials. 2005;26(35):7260–7275. | ||
Asthana GS, Asthana A, Kohli DV, Vyas SP. Mannosylated chitosan nanoparticles for delivery of antisense oligonucleotides for macrophage targeting. Biomed Res Int. 2014;2014:526391. | ||
Gao J, Chen P, Singh Y, et al. Novel monodisperse PEGtide dendrons: design, fabrication, and evaluation of mannose receptor-mediated macrophage targeting. Bioconjug Chem. 2013;24(8):1332-1344. | ||
Yu SS, Lau CM, Barham WJ, et al. Macrophage-specific RNA interference targeting via “click”, mannosylated polymeric micelles. Mol Pharm. 2013;10(3):975-987. | ||
Zhu S, Niu M, O’Mary H, Cui Z. Targeting of tumor-associated macrophages made possible by PEG-sheddable, mannose-modified nanoparticles. Mol Pharm. 2013;10(9):3525-3530. | ||
Pruthi J, Mehra NK, Jain NK. Macrophages targeting of amphotericin B through mannosylated multiwalled carbon nanotubes. J Drug Target. 2012;20(7):593–604. | ||
Tiwari S, Chaturvedi AP, Tripathi YB, Mishra B. Macrophage-specific targeting of isoniazid through mannosylated gelatin microspheres. AAPS Pharm Sci Tech. 2011;12(3):900–908. | ||
Wood TR, Chow RY, Hanes CM, et al. PKC pseudosubstrate and catalytic activity are necessary for membrane delivery during IgG-mediated phagocytosis. J Leukoc Biol. 2013;94(1):109–122. | ||
Zhao C, Fana T, Yang Y, et al. Preparation, macrophages targeting delivery and anti-inflammatory study of penta-peptide grafted nanostructured lipid carriers. Int J Pharm. 2013;450(1–2):11–20. | ||
Jain S, Amiji M. Tuftsin-modified alginate nanoparticles as a noncondensing macrophage-targeted DNA delivery system. Biomacromolecules. 2012;13(4):1074-1085. | ||
Betageri GV, Black CD, Szebeni J, Wahl LM, Weinstein JN. Fc-receptor-mediated targeting of antibody-bearing liposomes containing dideoxycytidine triphosphate to human monocyte/macrophages. J Pharm Pharmacol. 1993;45(1):48–53. | ||
Gu X, Zhang W, Liu J, et al. Preparation and characterization of a lovastatin-loaded protein-free nanostructured lipid carrier resembling high-density lipoprotein and evaluation of its targeting to foam cells. AAPS Pharm Sci Tech. 2011;12(4):1200–1208. | ||
Richman M, Perelman A, Gertler A, Rahimipour S. Effective targeting of Aβ to macrophages by sonochemically prepared surface-modified protein microspheres. Biomacromolecules. 2013;14(1):110–116. | ||
Kamat M, El-Boubbou K, Zhu DC, et al. Hyaluronic acid immobilized magnetic nanoparticles for active targeting and imaging of macrophages. Bioconjug Chem. 2010;21(11):2128–2135. | ||
Etzerodt A, Maniecki MB, Graversen JH, Møller HJ, Torchilin VP, Moestrup SK. Efficient intracellular drug-targeting of macrophages using stealth liposomes directed to the hemoglobin scavenger receptor CD163. J Control Release. 2012;160(1):72–80. | ||
Soto ER, Ostroff GR. Characterization of multilayered nanoparticles encapsulated in yeast cell wall particles for DNA delivery. Bioconjug Chem. 2008;19(4):840–848. | ||
Hong F, Yan J, Baran JT, et al. Mechanism by which orally administered beta-1,3-glucans enhance the tumoricidal activity of antitumor monoclonal antibodies in murine tumor models. J Immunol. 2004;173(2):797–806. | ||
Huang H, Ostroff GR, Lee CK, Wang JP, Specht CA, Levitz SM. Distinct patterns of dendritic cell cytokine release stimulated by fungal beta-glucans and Toll-like receptor agonists. Infect Immun. 2009;77(5):1774–1781. | ||
Herre J, Gordon S, Brown GD. Dectin-1 and its role in the recognition of beta-glucans by macrophages. Mol Immunol. 2004;40(12):869–876. | ||
Figueiredo S, Moreira J, Geraldes CG, Rizzitelli S, Aimeb S, Terreno E. Yeast cell wall particles: a promising class of nature-inspired microcarriers for multimodal imaging. Chem Commun. 2011;47(38):10635–10637. | ||
Soto ER, Kim YS, Lee J, Kornfeld H, Ostroff GR. Glucan particle encapsulated rifampicin for targeted delivery to macrophages. Polymers. 2010;2(4):681–689. | ||
Aouadi M, Tesz GJ, Nicoloro SM, et al. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature. 2009;458(7242):1180–1185. | ||
Tesz GJ, Aouadi M, Prot M, et al. Glucan particles for selective delivery of siRNA to phagocytic cells in mice. Biochem J. 2011;436(2):351–362. | ||
Reibetanz U, Claus C, Typlt E, Hofmann J, Donath E. Defoliation and plasmid delivery with layer-by-layer coated colloids. Macromol Biosci. 2006;6(2):153–160. | ||
Thomas M, Lu JJ, Ge Q, Zhang C, Chen J, Klibanov AM. Full deacylation of polyethylenimine dramatically boosts its gene delivery efficiency and specificity to mouse lung. Proc Natl Acad Sci U S A. 2005;102(16):5679–5684. | ||
Huang H, Ostroff GR, Lee CK, et al. Relative contributions of dectin-1 and complement to immune responses to particulate β-glucans. J Immunol. 2012;189(1):312–317. | ||
Smet RD, Demoor T, Verschuere S, et al. β-Glucan microparticles are good candidates for mucosal antigen delivery in oral vaccination. J Control Release. 2013;172(3):671–678. | ||
Schleicher U, Bogdan C. Generation, culture and flow-cytometric characterization of primary mouse macrophages. Methods Mol Biol. 2009;531:203–224. | ||
Vats D, Mukundan L, Odegaard JI, et al. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 2006;4(1):13–24. | ||
Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, et al. Macrophage-specific PPAR controls alternative activation and improves insulin resistance. Nature. 2007;447(7148):1116–1120. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.