Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 10
Species difference in paclitaxel disposition correlated with poor pharmacological efficacy translation from mice to humans
Authors Li YF ![]() , Zhang C, Zhou S, He M, Zhang H, Chen N, Li F, Luan X, Pai M
, Zhang C, Zhou S, He M, Zhang H, Chen N, Li F, Luan X, Pai M ![]() , Yuan H
, Yuan H ![]() , Sun D
, Sun D ![]() , Li Y
, Li Y
Received 27 August 2018
Accepted for publication 5 October 2018
Published 8 November 2018 Volume 2018:10 Pages 165—174
DOI https://doi.org/10.2147/CPAA.S185449
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Ying Fei Li,1 Chengyue Zhang,1 Simon Zhou,1 Miao He,2 Huixia Zhang,2 Nianhang Chen,1 Feng Li,2 Xin Luan,2 Manjunath Pai,3 Hebao Yuan,2 Duxin Sun,2 Yan Li1
1Translational Development and Clinical Pharmacology, Celgene Corporation, Summit, NJ 07901, USA; 2Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, MI 48109, USA; 3Department of Clinical Pharmacy, College of Pharmacy, University of Michigan, Ann Arbor, MI 48109, USA
Background: Paclitaxel (PTX) products currently approved by the Food and Drug Administration include Kolliphor EL-paclitaxel micelles (KoEL-paclitaxel, Taxol) and nanoparticle albumin-bound paclitaxel (nab-paclitaxel, Abraxane). Despite containing the same cytotoxic agent, different PTX formulations have distinct pharmacological responses and indications in patients with cancer. Several novel PTX delivery vehicles that have shown superior efficacy to Taxol in animal models failed to demonstrate efficacy in Phase II/III human clinical trials.
Materials and methods: A 10 mg/kg IV dose of KoEL-paclitaxel or nab-paclitaxel was administered to mice, and the pharmacokinetics (PK) profile of PTX in mice was then compared with the human PK profile from clinical studies. Population PK model and simulation was used to delineate the distribution and elimination characteristics in each species. In addition, tumor shrinkage was measured after weekly administration of both formulations in mouse xenograft model.
Results: Our pharmacokinetic modeling results suggested that elimination predominates over distribution in driving PTX disposition in mice, hence restricting the PTX tissue accumulation. Moreover, the rapid elimination of PTX in mice minimized the different formulation effects on PTX tissue distribution, which is believed to link to the superior efficacy of nab-paclitaxel over KoEL-paclitaxel seen in human. In contrast to mice, PTX distribution predominates over elimination in human, and the decline in plasma PTX concentration reflected the deeper tissue distribution by nab-paclitaxel.
Conclusion: This species difference in PTX distribution and elimination hinders a simple direct extrapolation from animals to humans. Therefore, species difference in drug distribution and elimination should be carefully assessed during translational drug development.
Keywords: paclitaxel, pharmacokinetics, tissue distribution, drug elimination, disposition, species difference
Introduction
Paclitaxel (PTX) is an effective chemotherapeutic agent with a unique mechanism of action and broad antitumor spectrum. However, development of an intravenous (IV) PTX formulation has been challenging due to its poor aqueous solubility.1,2 The first commercialized PTX product (Taxol®) is formulated in a vehicle composed of Kolliphor® EL (KoEL, formerly known as Cremophor® EL, polyoxyethylated castor oil) and dehydrated ethanol (1:1, v/v, US Pharmacopoeia). Although Taxol is still the most common taxane treatment in clinical utility, it is far from fully satisfactory due to its side effects, and premedication is often required to prevent these severe adverse effects. Specifically, hypersensitivity reactions and neurotoxicity are suspected to be caused by KoEL in the vehicle.3,4 In addition, the high amount of KoEL in the vehicle results in a large solution volume of the final product, which requires a rather long infusion duration. Therefore, considerable efforts have been put forth to develop novel PTX delivery systems that enable an easier administration, more favorable tolerability profiles, and greater antitumor efficacy.1,5
The purported solvent-associated side effects have motivated investigations on KoEL-free PTX formulations, such as polymer micelles, emulsions, nanoparticles, liposomes, and polymer conjugates. Surprisingly, various novel PTX delivery vehicles, which showed superior efficacy to Taxol in mouse xenograft models, yielded a motley of interesting results in clinical trials. Tocosol® PTX, the first vitamin E-based emulsion formulation used in clinical trials, did not meet the primary endpoint of noninferiority compared with KoEL-paclitaxel in Phase III clinical studies, in patients with metastatic breast cancer.6 A lower objective response rate was observed in Tocosol PTX compared with KoEL-paclitaxel (37% vs 45%; P=0.085). In addition, significantly greater neutropenia and febrile neutropenia were seen in Tocosol PTX. Genexol-PM is another KoEL-free polymeric micelle formulation, in which PTX is encapsulated in methoxypoly ethylene glycol-poly [D, L-lactide] diblock copolymer. Although methoxypoly ethylene glycol-poly [D, L-lactide] micelles have been shown to be biocompatible and nontoxic in a variety of in vitro and in vivo studies,7 patients receiving Genexol-PM regimens still experienced an unexpected high frequency of hypersensitivity reactions and a high rate of peripheral neuropathy and neutropenia.8 However, nanoparticle albumin-bound (nab) PTX is a solvent-free, human albumin–stabilized formulation of PTX approved by the Food and Drug Administration (FDA) for the treatment of metastatic pancreatic cancer, for which KoEL-paclitaxel is not indicated,9 nab-paclitaxel has a low incidence of hypersensitivity reactions, and the different safety profiles between the KoEL-free PTX formulations, including Genexol-PM and nab-paclitaxel, can be attributed to the different tissue distribution pattern, which is driven by the specific delivery vehicle interaction with tissues of distinct lipid and protein composition.
Although the different PTX formulations showed comparable plasma PTX exposures in clinical studies, they exhibited distinct efficacy and safety profiles, which posted challenges in evaluating the pharmacological responses during the development of novel PTX delivery vehicles. In addition, plasma concentrations are not likely to be the representative surrogates for pharmacological responses of different PTX formulations. This phenomenon was indeed observed in several studies of encapsulated drug delivery systems,10 in which equivalent drug changes in systemic circulation did not lead to corresponding changes in target tissues or organs, and consequently led to different efficacy and safety profiles for different delivery systems.11,12 Taking this into consideration, a semimechanistic pharmacokinetic (PK) model with explicit PTX-carrier tissue distribution and decomposition was developed to characterize the PK profiles of nab-paclitaxel and KoEL-paclitaxel.13 Compared to a traditional three-compartment model,14,15 this semimechanistic PK model provided additional insight on the critical factors driving PTX disposition. The rapid tissue distribution of the PTX carrier complex and its rapid decomposition in systemic circulation may result in similar plasma PTX exposure but distinct tissue disposition profiles in humans. The safety and efficacy profile of PTX is indeed a function of the integral drug carrier complex, including both the drug itself and its delivery system.
While the distinct drug disposition of PTX in target tissues and organs explains the different safety and efficacy profiles across the various delivery systems in human trials, it is still puzzling to see the similar efficacy and/or safety profile with mouse xenograft model, regardless of the delivery systems. In our study, we aimed to examine aspects of PTX pharmacology in mice and humans to identify the cause for the discrepancy in efficacy between mouse xenograft models and human clinical trials. PK profiles were characterized following a 10 mg/kg IV dose of KoEL-paclitaxel and nab-paclitaxel in mice, while tumor shrinkage was measured after weekly administrations of both formulations in a mouse xenograft model of human pancreatic cancer. The PK of PTX in mice and humans was then analyzed and compared to delineate distribution and elimination characteristics. While rapid tissue distribution of a PTX–carrier complex decreases the plasma exposure of PTX in humans,14,15 elimination predominates over distribution in driving the decline in plasma exposure of PTX in mice. The much faster clearance (CL) of PTX in mice diminished the difference in PTX distribution, and ultimately the associated effect on tumor shrinkage by different delivery vehicles. The difference in efficacy between KoEL-paclitaxel and nab-paclitaxel indeed correlated with the different PTX dispositions in mice and humans.
Materials and methods
Chemicals and reagents
KoEL-paclitaxel (Taxol, Hospira Inc.) was obtained from University of Michigan Hospital, USA. nab-Paclitaxel was supplied by Celgene Corporation (Summit, NJ, USA). High-performance liquid chromatography–grade acetonitrile and pure water were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Ultrapure deionized water was supplied by a Milli-Q water system (Millipore, Bedford, MA, USA). Sodium chloride injections (0.9%) and heparin sodium injections (1,000 U/mL) were purchased from Hospira Inc. (Lake Forest, IL, USA).
Animal experiments
All animal experiments were performed in accordance with University of Michigan guidelines covering the humane care and use of animals in research. All animal procedures used in this study were approved by the University Committee on Use and Care Animals at the University of Michigan. CD-1 IGS mice (strain code: 022, 6–8 weeks old) were purchased from Charles River Laboratories (Wilmington, MA, USA). KoEL-paclitaxel and nab-paclitaxel were administrated to cohorts of mice via IV injection at 10 mg/kg. Serial blood samples were collected at 0 (predose), 0.08, 0.17, 0.25, 0.5, 0.75, 1, 2, 4, 7, 16, 24, 48, and 72 hours following IV administration. At each time point, three mice were euthanized, and blood samples were immediately collected via cardiac puncture using a 25 G needle and 1 mL syringe, which were pretreated with a heparin sodium solution. Plasma samples were separated from whole blood using a bench-top centrifuge at a speed of 14,500 rpm for 10 minutes and stored at −80°C until further analysis.
Male, nonobese, diabetic/severe combined immunodeficient (NOD/SCID) mice (6–8 weeks old) were obtained from Jackson Laboratory (Bar Harbor, ME, USA). Pancreatic cancer PANC-1 cells (1×106 cells/0.1 mL/mouse) suspended in a Matrigel/DMEM (50/50) mixture were injected subcutaneously into the mice. The mice were injected IV with saline control, KoEL-paclitaxel (10 mg/kg), and nab-paclitaxel (10 mg/kg) once weekly starting when average tumor sizes reached ~125 mm3 and continuing for 30 days. Tumor size (1/2×length×width×width, mm3) and body weight were measured every fifth day.
Stock solution, working solution, and quality control (QC)
PTX and docetaxel (internal standard [IS]) stock solutions were prepared at 9 mg/mL in acetonitrile and stored at −20°C. The stock solution of PTX was further diluted with acetonitrile to a series of concentrations from 2.44 to 5000 ng/mL as working solution. Quality controlled PTX working solutions at low, medium, and high concentrations were diluted from a separately prepared stock solution. Docetaxel stock solution was diluted with acetonitrile to the concentration of 1,000 ng/mL for sample preparation. QC samples were evenly distributed among samples of each batch.
Sample preparation
Aliquots of plasma samples (40 µL) from different time points were mixed with 40 µL acetonitrile and 120 µL IS solution in a 96-well plate. The plate was vortexed for 10 minutes and then centrifuged at 3,500 rpm for 10 minutes at 4°C to precipitate proteins. The supernatant was transferred, and 5 µL was injected into a liquid chromatography–tandem mass spectrometer (LC-MS/MS) for analysis.
Calibration curve
Calibration standard solutions were prepared by mixing 40 µL blank plasma with a 40 µL PTX working solution and 120 µL IS solution. Calibration curves were established using 12 nonzero calibration standards covering the lower limit of quantification (LLOQ) to 5,000 ng/mL for plasma. The LLOQ was 5 ng/mL for plasma. The calibration curves were generated by plotting the peak area ratios (PTX peak area/IS peak area) against the concentrations of PTX using a linear regression and 1/concentration2 (1/X2) weighting factor. The linearity of the relationship between peak area ratio and concentration was demonstrated by the correlation coefficients.
LC-MS/MS
Quantification of PTX was conducted on an LC-MS platform consisting of a Shimadzu High-performance liquid chromatography system and an AB-5500 QTrap (Sciex, Ontario, Canada) tandem mass spectrometer equipped with an electrospray ionization source. The sample injection volume was 5 µL, and chromatographic separation was performed on an Xbridge C18 column (3.5 µm, 50 mm × 2.1 mm internal diameter; Waters, Milford, MA, USA) at a flow rate of 400 µL/min. Mobile phase A consisted of 0.1% formic acid in water, and mobile phase B consisted of 0.1% formic acid in acetonitrile. Gradient elution started with 25% B for 0.5 minute, linearly increased to 65% at 2 minutes, to 95% B at 2.5 minute, kept at 95% B for 2 minutes, decreased to 25% B at 5 minutes, and kept at 25% for 2 minutes. The MS system was operated in positive-ion mode while a multiple reaction monitoring method was used for PTX (m/z 854.40 → 286.10) and IS (m/z 808.00 →226.00) detection. The gas temperature was 300°C with an ion-spray voltage of 5500 V, nitrogen and argon of 60 psi, declustering potential of 190.00 V for PTX and 173.00 V for IS, curtain gas of 30 psi, and collision energy of 21.00 V for PTX and 18.80 V for IS. LC-MS system control, data acquisition, peak integration, and quantitation were performed using the Analyst software package MDS SCIEX (version 1.6; Applied Biosystems, Toronto, Canada).
Mouse PK model development
The plasma/blood and tissue concentration-time data were compiled and visualized using S-Plus (TIBCO, Seattle, WA, USA). Population PK analysis of plasma PTX concentration vs time data from mice was conducted using the nonlinear mixed-effect modeling program NONMEM (version 7.2; ICON Development Solution, Ellicott City, MD, USA), with first-order conditional estimation using the interaction option throughout data analysis. PTX concentration data were natural log transformed. The model building aid Perl-Speaks-NONMEM (PsN, version 3.5.3; Kajsa Harling and Andrew Hooker) postprocessing software was used for graphic processing. Mouse blood PTX concentration-time data were best described by a two-compartment structure PK model with volume of distribution in the central compartment (V1), volume of distribution in the peripheral compartment (V2), intercompartmental clearance (Q) between the central compartment and the peripheral compartment, and blood clearance (CL). Interindividual variability was modeled using an exponential error model, whereas the residue variability was described with a log error structure. Model selection was based on the minimum value of objective function, as calculated by NONMEM, the reliability of variable estimates, and the fit of the model to the data as approached by various graphical plots.
PK simulations of PTX formulations in mouse and in human
A simulation approach was used to compare the PTX disposition properties in mice and humans. The human PK models to describe nab-paclitaxel and KoEL-paclitaxel concentration-time profiles were previously established,14,15 which were three-compartment models with saturable elimination from the central compartment, saturable transport to the first peripheral compartment, and linear distribution to the second peripheral compartment. The following PKs variables were estimated: volumes of the central compartment (V1), first peripheral compartment (V2) and second peripheral compartment (V3), maximal elimination rate (VMEL), Michaelis–Menten constant (KMEL), maximal transport rate to the first peripheral compartment (VMTR), total plasma concentration of PTX at half VMTR (KMTR), and intercompartmental clearance between the central and second peripheral compartment (Q). Interindividual variability was modeled using a proportional model for both nab-paclitaxel and KoEL-paclitaxel, and residual variability was described using a log error structure. Final models were selected based on value of objective function, stability of parameter estimates and how well the model was able to describe the observed data through visual predictive check (VPC). The drug concentration-time profile in plasma (the central compartment) and peripheral tissues/organs (the peripheral compartments) for a “typical” patient was simulated using the published typical model parameters14,15 at the approved maximum dose, which is 175 mg/m2 over a 3-hour infusion for KoEL-paclitaxel and 260 mg/m2 over a 0.5-hour infusion for nab-paclitaxel. Mouse PK parameters were obtained from the mouse population PK model. The amount of drug in each peripheral compartment was calculated by multiplying the simulated drug concentration in the peripheral compartment with the corresponding volume of distribution. For mice, the total amount of drug in the peripheral compartment is presented, while for humans, the total amount of drug in the two peripheral compartments is presented. The amount of drug eliminated was estimated by subtracting the amount of drug in the central and peripheral compartments from the dose administered, and the percentage eliminated was calculated by dividing the amount of drug eliminated by the dose administered. In addition to estimating the amount of drug in various compartments, the dynamic distribution and elimination rates were calculated by multiplying the plasma concentrations by Q and CL, respectively.
Results
Accuracy and precision of analytical method assay
The LC-MS/MS calibration curves were generated with the blank of each tissue/organ and validated. The intraday and interday accuracies were within 85%–115% for QC and within 80%–120% for LLOQ (data not shown). The precisions of the intra- and interday accuracies were also within the acceptable limit. The LLOQ for most tissues was set at 25 ng/g; the peak area was >5-fold that of the blank, except for spleen (50 ng/g) and brain (10 ng/g). The calibration curve had a mean correlation coefficient R2>0.9897 across different organs.
PTX plasma concentration-time profile in mouse xenograft model
Following an IV administration of 10 mg/kg KoEL-paclitaxel and nab-paclitaxel, the concentration-time profiles of PTX in plasma were determined and are shown in Figure 1. In plasma, the highest concentration was observed at the first sampling time point (5 minutes), when KoEL-paclitaxel resulted in a sixfold higher concentration level (about 18,000 ng/mL) than nab-paclitaxel. In both treatment arms, the concentration started to decrease precipitously afterward and was below the LLOQ (5 ng/mL) after 7 hours for KoEL-paclitaxel and nab-paclitaxel.
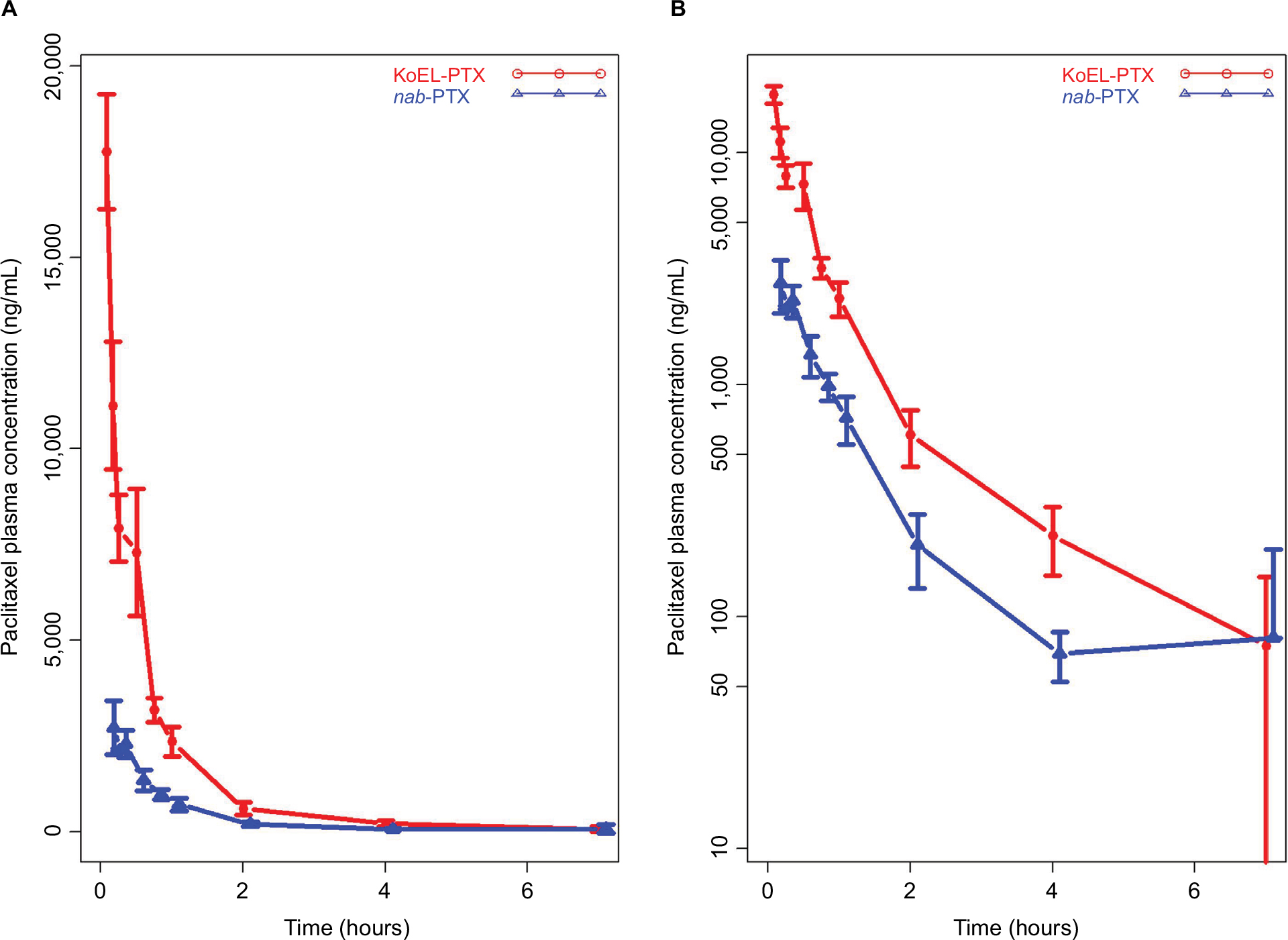 | Figure 1 Total paclitaxel plasma concentration–time profiles on a linear scale (A) and a semilog scale (B) for Kolliphor EL-paclitaxel micelles (KoEL-PTX) and nanoparticle albumin-bound paclitaxel (nab-PTX) following IV administration of 10 mg/kg in mice. |
Efficacy of nab-paclitaxel and KoEL-paclitaxel in mouse xenograft model
Tumor size was used to evaluate the efficacy of both the KoEL-paclitaxel and nab-paclitaxel formulations. As shown in Figure 2, KoEL-paclitaxel shrank the tumor to 28.8% of the control tumor size, whereas nab-paclitaxel shrank the tumor to 33.6% of the control size. Interestingly, no statistically significant difference in efficacy was observed between the KoEL-paclitaxel and nab-paclitaxel treatment groups.
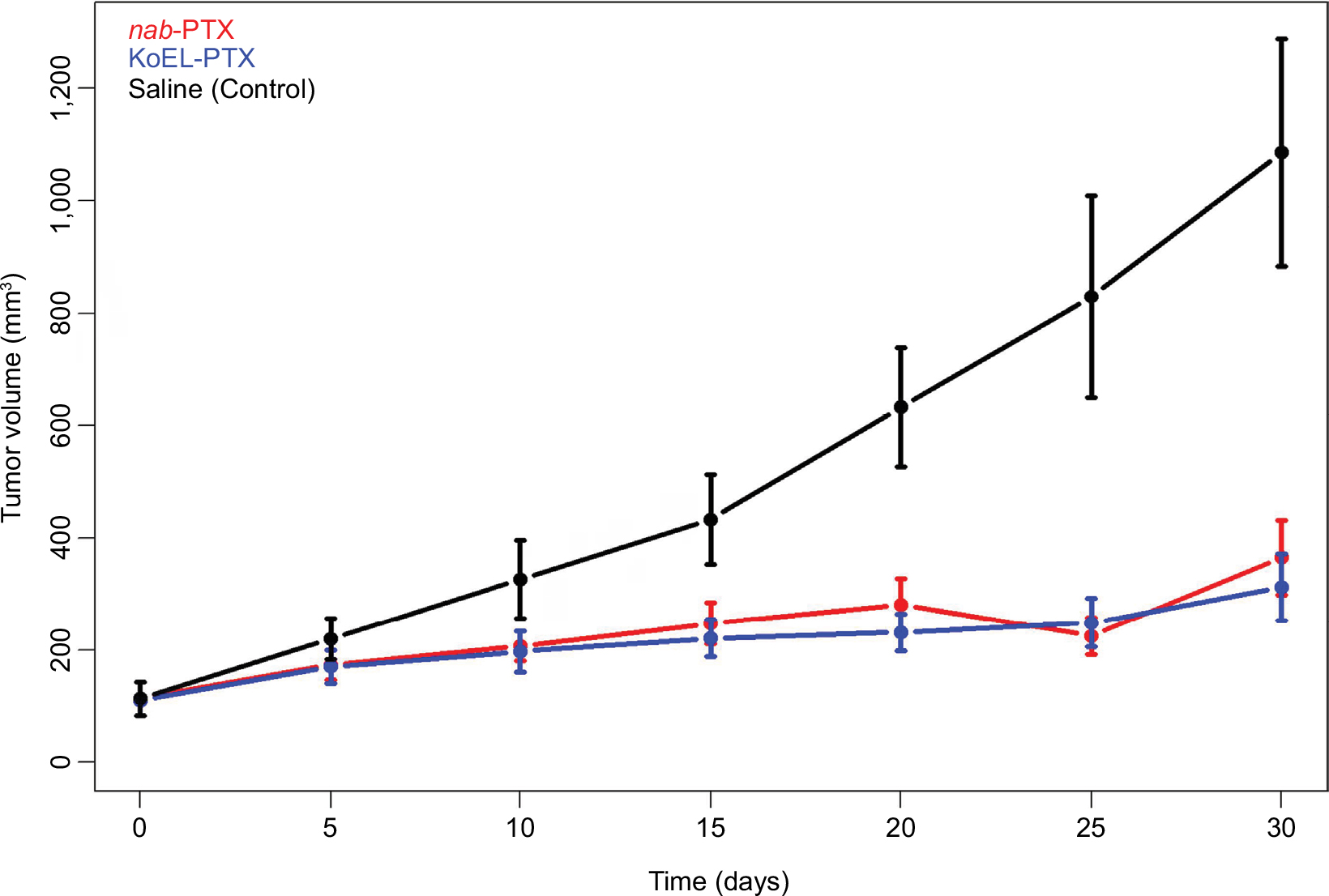 | Figure 2 Tumor size vs time profiles by formulation in mouse xenograft model. |
Mouse PK model estimates
Final PK parameters estimated from two different formulations in mouse are presented in Table 1. The PK parameters for KoEL-paclitaxel were different from those of nab-paclitaxel. The rates of PTX distribution (Q) were higher when administered as albumin-bound nanoparticle, compared with KoEL-paclitaxel. In addition, V1 and V2 were larger when administered as nab-paclitaxel, suggesting that nab-paclitaxel has more extensive tissue distributions than KoEL-paclitaxel. However, the much higher elimination (CL) than distribution (Q) for both formulations in mice diminished such difference in tissue distributions between the two formulations seen in human.
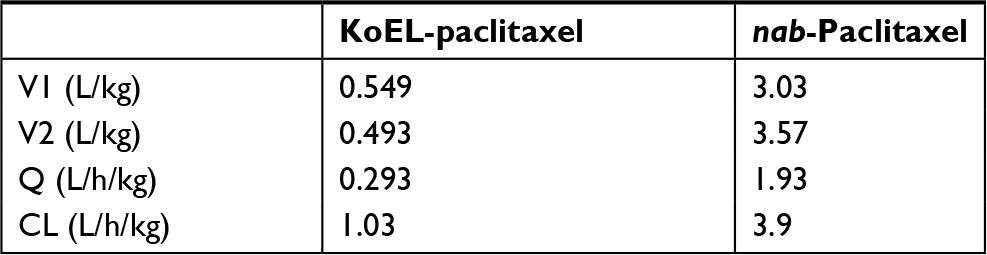 | Table 1 Mouse population pharmacokinetic parameters of KoEL-paclitaxel and nab-paclitaxel Abbreviations: CL, blood clearance; Q, inter-compartmental clearance between the central compartment and the peripheral compartment; V1, volume of distribution of the central compartment; V2, volume of distribution of peripheral compartment. |
PK simulations of PTX distribution and elimination in humans and mice
A three-compartment PK model was previously reported to describe the plasma concentration vs time data of KoEL-paclitaxel or nab-paclitaxel in patients with solid tumors, and the final PK parameters estimated from two different formulations in human were presented in Table 2.14,15 Simulations were performed based on the estimated parameters to produce dynamic rates of concomitant distribution (defined as the product of Q and concentration) and eliminations (defined as the product of CL and concentration), as shown in Figure 3. In humans, the saturable elimination rate is far below the saturable “transporter-driven” distribution rate in a clinical relevant concentration range for both formulations. In contrast, the rate of PTX elimination is much higher than the distribution in mice for both formulations in this study (Table 1). To further compare the PTX disposition properties in mice and human, simulation was conducted to understand the plasma time-concentration, tissue distribution, and drug elimination profiles in each species (Figure 4). While KoEl-PTX and nab-PTX show comparable plasma PTX concentration profiles in both mice and humans (Figure 4A, D), an opposite trend is observed in the peripheral tissues. Specifically, nab-paclitaxel exhibits a drastically higher peripheral tissue exposure compared with KoEL-paclitaxel in humans, whereas minimal difference between the two formulations is observed in mice (Figure 4B vs E). Such distinct drug disposition pattern in tissue distribution may be attributed to the fact that mice exhibit a much rapid elimination compared with human, as demonstrated by the fact that more than 80% parental PTX is eliminated from mice within 4 hours post dose for both formulations, while it takes much longer time for parental PTX to eliminate from human (Figure 4C vs F). The drug elimination in mice was too rapid to allow for sufficient tissue distribution to take place, and hence, comparable tissue accumulation is observed between the two formulations. Such negligible difference between the tissue distribution in mice is indeed consistent with the lack of statistical significant difference in efficacy between KoEL-paclitaxel and nab-paclitaxel from the mouse xenograft model (Figure 2).
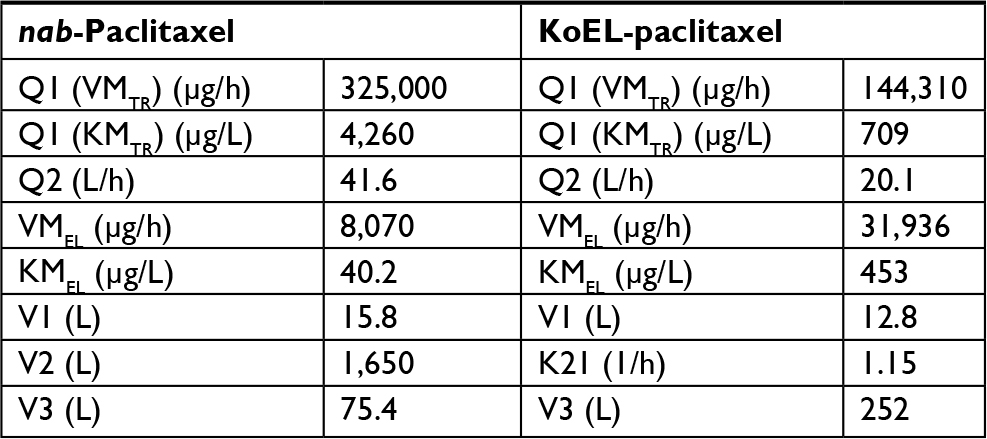 | Table 2 Human population pharmacokinetics parameters of nab-paclitaxel and KoEL-paclitaxel Notes: K21, the rate constant from the second peripheral compartment to the central compartment; KMEL, PTX concentration in the central compartment at 50% of VMEL; KMTR, PTX concentration in the central compartment at 50% of VMTR; Q1, intercompartmental clearance between the central compartment and the first peripheral compartment; Q2, intercompartmental clearance between the central compartment and the second peripheral compartment; V1, volume of distribution for the central peripheral compartment; V2, volume of distribution for the first peripheral compartment; V3, volume of distribution for the second peripheral compartment; VMEL, maximum elimination rate from the central compartment; VMTR, maximum intercompartmental distribution rate between the central compartment and the first peripheral compartment. |
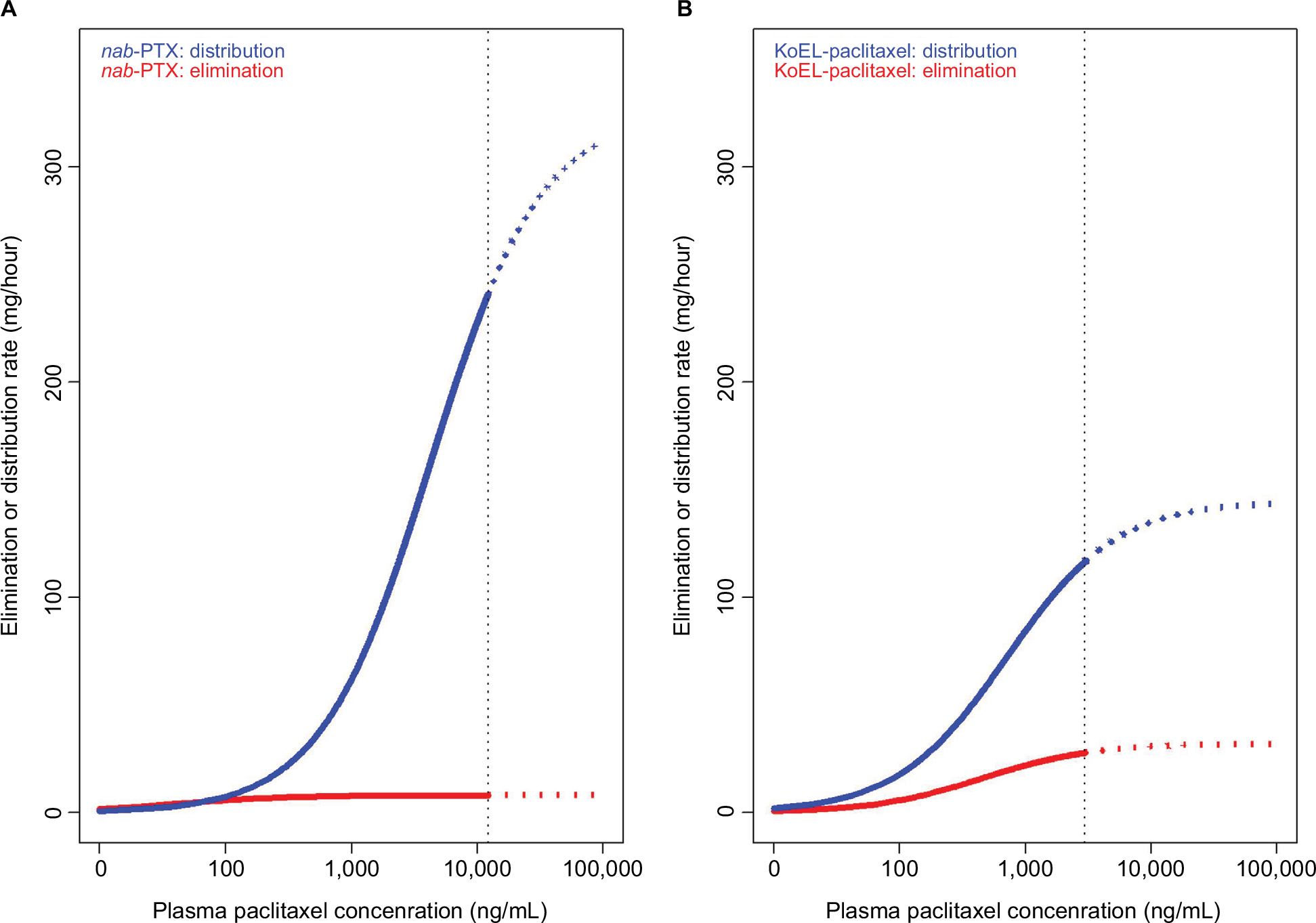 | Figure 3 Distribution rate vs elimination rate of nanoparticle albumin-bound paclitaxel (nab-PTX) (A) and Kolliphor EL-paclitaxel micelles (KoEL-PTX) (B) in clinically relevant concentration ranges in human. The vertical dash line represents maximal plasma paclitaxel concentrations predicted from previous population pharmacokinetic analysis of each formulation. |
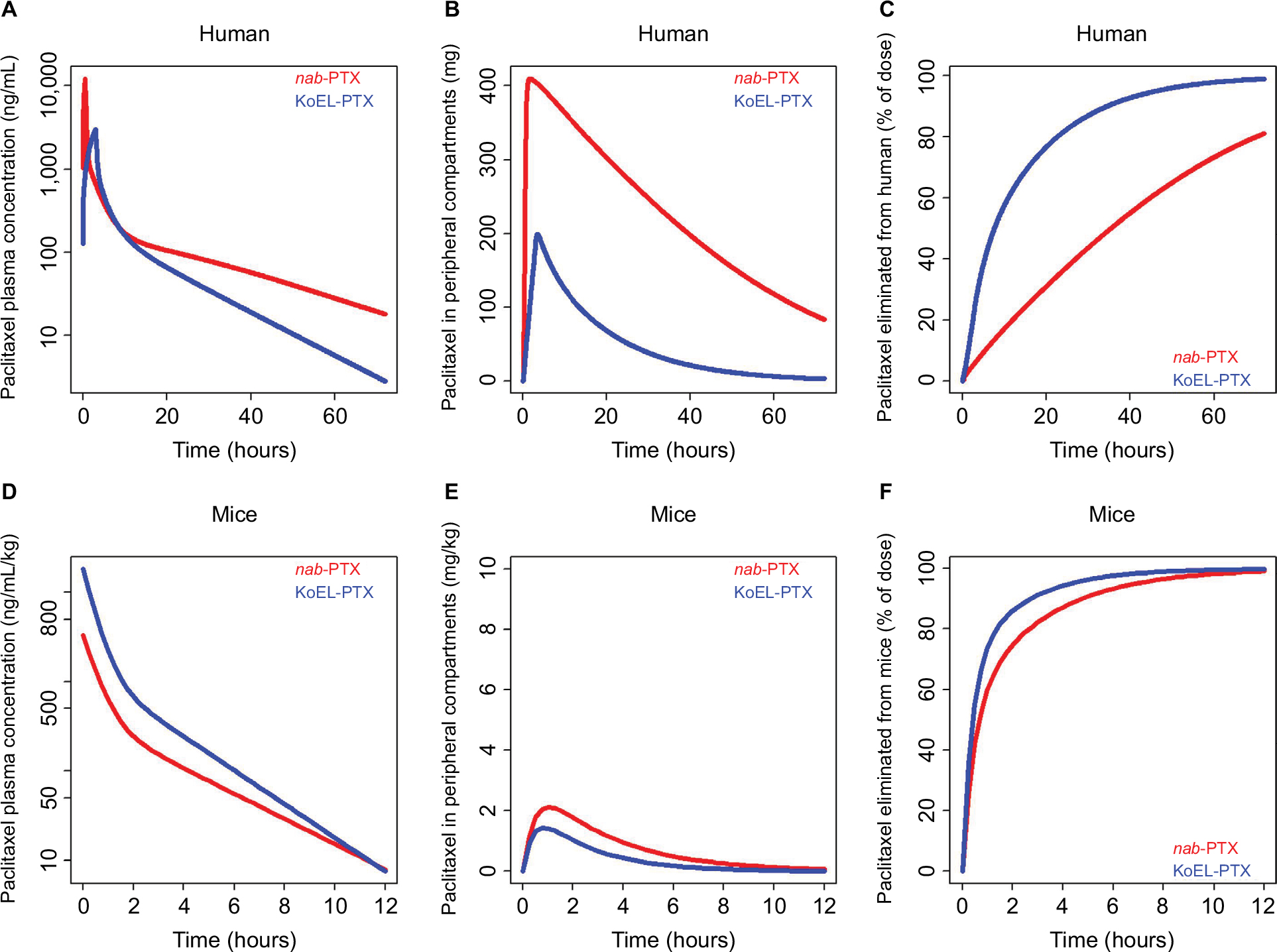 | Figure 4 Model-predicted paclitaxel disposition in plasma and peripheral tissues and accumulative drug elimination in humans and mice. Predicted drug exposure vs time profiles in plasma (A and D). Predicted drug amount vs time profiles in tissues (B and E). Predicted percentage of drug eliminated from the body vs time profiles (C and F). |
Discussion
PTX is a chemotherapeutic agent that has played a critical role in the treatment of breast, ovary, and non-small-cell lung cancer.16–18 More recently, the nab formulation has made important progress in PTX-based treatment of metastatic pancreatic cancer.19 Several novel PTX delivery vehicles were developed initially attempting to optimize clinical utility while overcoming the limitations of the conventional KoEL formulation.20 Although these formulations exhibited comparable efficacy profiles in mouse xenograft models, mixed results were observed in human clinical trials.6–9 Specifically, when examining the efficacy of KoEL-paclitaxel and nab-paclitaxel in mouse xenograft model of pancreatic cancer cells, the tumor size measurement during treatment period in the nab-paclitaxel arm did not show statistically significant difference from the KoEL-paclitaxel arm (Figure 2). However, in human clinical trials, the nab-paclitaxel arm demonstrated significantly higher response rates compared with KoEL-paclitaxel (33% vs 19%, respectively; P=0.001) and significantly longer time to tumor progression (23.0 vs 16.9 weeks, respectively; hazard ratio =0.75; P=0.006) in patients with metastatic breast cancer.21 In addition, nab-paclitaxel demonstrated its superiority to standard of care chemotherapy in patients with metastatic pancreatic cancer.19
Furthermore, although Tocosol PTX showed an improved antitumor activity and tolerability when compared with KoEL-paclitaxel preclinically,22 inferior objective response and higher toxicity were observed with Tocosol PTX in a pivotal 2-arm Phase III clinical trial.23 Hence, when evaluating the different delivery systems for PTX, results from animal studies were not predictive of the efficacy and safety profiles seen in human. Such discrepancy in efficacy between mice and human has motivated us to study the underlying mechanism that drives the species difference.
In our study, after administering a 10 mg/kg IV dose of either KoEL-paclitaxel or nab-paclitaxel to mice, we observed a rapid decline in plasma concentration for both KoEL-paclitaxel and nab-paclitaxel (Figure 1). A 2-compartmental model was used to characterize the PKs profiles of PTX. As shown in Table 1, nab-paclitaxel exhibited a more rapid tissue distribution (drug distribution/elimination [Q/CL] ratio of ~0.5) compared to KoEL-paclitaxel (Q/CL ratio of 0.3). This is in accordance with a previous finding that nab-paclitaxel had a more efficient transport across endothelial cells and greater penetration in xenograft tumors.24
While both PTX formulations showed different tissue distributions in mice, the model suggested that the rapid decline in PTX concentrations was a result of rapid elimination (CL) rather than distribution (Q) (Table 1). In fact, the model estimates of drug elimination (CL) and distribution rates (Q) in mice showed an opposite pattern from the human parameters,14 in which PTX elimination rate (CL) is far below the distribution rate (Q) in clinical relevant concentration range for both KoEL-paclitaxel and nab-paclitaxel (Table 2). The faster distribution of PTX than elimination in human (Figure 3) is augmented by the ability of nab-paclitaxel to exploit the physiological transporter properties of albumin.24
These findings are indeed consistent with the different intrinsic capacities of drug metabolism/excretion between mice and humans. In vitro and in vivo absorption, distribution, metabolism, and excretion (ADME) studies have demonstrated that majority of PTX undergoes hepatic metabolism and biliary excretion, while renal excretion of parent drug is minimal (<10%).25 More intensive biliary excretion has been found in rodents compared with human patients receiving PTX.26 As shown in the study by Monsarrat et al,26 PTX and its metabolites were excreted into bile at a much faster rate in rats than in human. At 12-hour post-dosing, more than 40% PTX and its metabolites were excreted into bile in rat, whereas only 7% was excreted in human. There was 25% parental PTX in rat bile compared with 2% in human bile, implying that PTX was metabolized and eliminated faster in rat than in human. Furthermore, this study also found that there was interspecies difference in the type and the chemical structure of the hydroxylated metabolites between rats and human. These interspecies differences in the sites of hydroxylation suggested that different CYP450 enzymes may be involved in PTX metabolism in rats and humans. Taken together, we concluded that there was a flipped distribution vs elimination kinetics between mice and humans.
Although both the mouse and human PK model predicted nab-paclitaxel to have a more rapid tissue distribution than KoEL-paclitaxel, the rapid elimination of PTX in mice marginalized such difference. Hence, distinct tissue distribution profiles between KoEL-paclitaxel and nab-paclitaxel are only observed in human, but not in mice (Figure 4B, E). Specifically, in human, majority of the circulating drug decline is governed by distribution rather than elimination (Figure 4A–C). While the plasma AUCs are comparable for both formulations, nab-paclitaxel produces drastically higher drug accumulation in the peripheral compartment. In mice, however, drug elimination is the main factor that drives the initial decline plasma PTX concentration (Figure 4D–F). Despite that the PK parameter estimates suggested that nab-paclitaxel could penetrate tissues more efficiently in mice, the elimination force competes with drug distribution, resulting in minimal difference in tissue accumulation (Figure 4E) between the two formulations. This result, combined with our previous finding that drug disposition in tissue, rather than plasma, is a better surrogate for efficacy,13 explains the reason that the two formulations showed different efficacy in human but comparable efficacy in mice.
Animal models, such as mouse models, have been used extensively in preclinical setting to predict dose, efficacy, and safety in humans. Although in many occasions we observe a certain degree of translation from preclinical animal model to clinical outcomes, in our specific case with complex drug delivery systems, the mouse model failed to differentiate the efficacy between KoEL-paclitaxel and nab-paclitaxel seen in human trials. On the basis of our experimental and modeling analyses, this phenomenon can be attributed to the fact that mice exhibited a much rapid elimination rate compared with humans, as seen in Figure 4C, F, as well as in the study by Monsarrat et al.26 Consequently, the impact of formulation on tissue distribution would only be reflected in humans, in which the elimination rate is much lower than the distribution rate (Figure 3), thus allowing time for tissue penetration. As a result of the different tissue distribution patterns by the formulation effect, different efficacy profiles are observed in humans with KoEL-paclitaxel compared with nab-paclitaxel.
While noninvasive drug concentration measurement in human tissue would be ideal to understand the different clinical outcomes from different PTX formulations, it is still of great challenge with the existing technology. Since there is a limitation with using preclinical model to distinguish the effects of different PTX formulations in human, simulation could serve as an alternative approach to provide kinetic insight in understanding how the drug disposition links to its efficacy and safety.
In summary, we characterized the PK of two IV-delivered PTX formulations in mice in conjunction with their in vivo performance to inhibit tumor growth in mouse xenograft models. The results from the mouse PK and xenograft experiments were compared with respective PK and efficacy data in human trials. While KoEL-paclitaxel had a plasma PTX exposure in mice that was four times higher than nab-paclitaxel, the two formulations had a nearly comparable tissue exposure and tumor shrinkage in a mouse xenograft model. In addition, nab-paclitaxel showed a more efficient tissue drug delivery in both mice and humans based on population PK model. However, the dispositional differences in PTX distribution and elimination produced diametrically opposite tissue PTX levels in these two species, and consequently opposite efficacy and adverse effects. Hence, species difference in drug disposition should be carefully evaluated before a meaningful efficacy and/or safety model is selected during translational drug development.
Acknowledgments
Editorial assistance was provided by MediTech Media, Ltd, through funding by Celgene Corporation. The authors are fully responsible for all content and editorial decisions for this manuscript. The authors acknowledge the financial support for this study from Celgene Corporation.
Disclosure
Ying Fei Li, Chengyue Zhang, Simon Zhou, Nianhang Cheng, and Yan Li are employees of and hold equity ownership in Celgene Corporation. The authors report no other conflicts of interest in this work.
References
Hennenfent KL, Govindan R. Novel formulations of taxanes: a review. Old wine in a new bottle? Ann Oncol. 2006;17(5):735–749. | ||
Sparreboom A, Wolff AC, Verweij J, et al. Disposition of docosahexaenoic acid-paclitaxel, a novel taxane, in blood: in vitro and clinical pharmacokinetic studies. Clin Cancer Res. 2003;9(1):151–159. | ||
Fransson MN, Gréen H, Litton JE, Friberg LE. Influence of Cremophor EL and genetic polymorphisms on the pharmacokinetics of paclitaxel and its metabolites using a mechanism-based model. Drug Metab Dispos. 2011;39(2):247–255. | ||
Sparreboom A, van Zuylen L, Brouwer E, et al. Cremophor EL-mediated alteration of paclitaxel distribution in human blood: clinical pharmacokinetic implications. Cancer Res. 1999;59(7):1454–1457. | ||
Bernabeu E, Cagel M, Lagomarsino E, Moretton M, Chiappetta DA. Paclitaxel: what has been done and the challenges remain ahead. Int J Pharm. 2017;526(1–2):474–495. | ||
A Multicenter Phase 3, Randomized Comparison of the Safety and Efficacy of Weekly TOCOSOL(R) Paclitaxel vs. Weekly Paclitaxel Injection in the Treatment of Metastatic Breast Cancer; 2018. Available from: https://ichgcp.net/clinical-trials-registry/NCT00251095. Accessed August 26, 2018. | ||
Liggins RT, Burt HM. Polyether-polyester diblock copolymers for the preparation of paclitaxel loaded polymeric micelle formulations. Adv Drug Deliv Rev. 2002;54(2):191–202. | ||
Lee KS, Chung HC, Im SA, et al. Multicenter phase II trial of Genexol-PM, a Cremophor-free, polymeric micelle formulation of paclitaxel, in patients with metastatic breast cancer. Breast Cancer Res Treat. 2008;108(2):241–250. | ||
Saif MW. U.S. Food and Drug Administration approves paclitaxel protein-bound particles (Abraxane(R)) in combination with gemcitabine as first-line treatment of patients with metastatic pancreatic cancer. JOP. 2013;14(6):686–688. | ||
Pathak SM, Ruby PK, Aggarwal D. In vitro and in vivo equivalence testing of nanoparticulate intravenous formulations. Drug Res (Stuttg). 2014;64(4):169–176. | ||
Müller M, dela Peña A, Derendorf H. Issues in pharmacokinetics and pharmacodynamics of anti-infective agents: distribution in tissue. Antimicrob Agents Chemother. 2004;48(5):1441–1453. | ||
Yokoi K, Chan D, Kojic M, et al. Liposomal doxorubicin extravasation controlled by phenotype-specific transport properties of tumor microenvironment and vascular barrier. J Control Release. 2015;217:293–299. | ||
Li Y, Chen N, Palmisano M, Zhou S. Pharmacologic sensitivity of paclitaxel to its delivery vehicles drives distinct clinical outcomes of paclitaxel formulations. Mol Pharm. 2015;12(4):1308–1317. | ||
Chen N, Li Y, Ye Y, Palmisano M, Chopra R, Zhou S. Pharmacokinetics and pharmacodynamics of nab-paclitaxel in patients with solid tumors: disposition kinetics and pharmacology distinct from solvent-based paclitaxel. J Clin Pharmacol. 2014;54(10):1097–1107. | ||
Joerger M, Huitema AD, van den Bongard DH, Schellens JH, Beijnen JH. Quantitative effect of gender, age, liver function, and body size on the population pharmacokinetics of Paclitaxel in patients with solid tumors. Clin Cancer Res. 2006;12(7 Pt 1):2150–2157. | ||
McGuire WP, Rowinsky EK, Rosenshein NB, et al. Taxol: a unique antineoplastic agent with significant activity in advanced ovarian epithelial neoplasms. Ann Intern Med. 1989;111(4):273–279. | ||
Nowak AK, Wilcken NR, Stockler MR, Hamilton A, Ghersi D. Systematic review of taxane-containing versus non-taxane-containing regimens for adjuvant and neoadjuvant treatment of early breast cancer. Lancet Oncol. 2004;5(6):372–380. | ||
Ramalingam S, Belani CP. Paclitaxel for non-small cell lung cancer. Expert Opin Pharmacother. 2004;5(8):1771–1780. | ||
Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–1703. | ||
Singla AK, Garg A, Aggarwal D. Paclitaxel and its formulations. Int J Pharm. 2002;235(1-2):179–192. | ||
Gradishar WJ, Tjulandin S, Davidson N, et al. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23(31):7794–7803. | ||
Spigel S, Jones S, Greco F, et al. S-8184 vitamin E paclitaxel emulsion: preclinical and phase I data [abstract]. In Proceedings of the American Society of Clinical Oncology. 2002, May 18–21; Orlando, FL; Abstract number 406. | ||
Constantinides PP, Han J, Davis SS. Advances in the use of tocols as drug delivery vehicles. Pharm Res. 2006;23(2):243–255. | ||
Chen N, Brachmann C, Liu X, et al. Albumin-bound nanoparticle (nab) paclitaxel exhibits enhanced paclitaxel tissue distribution and tumor penetration. Cancer Chemother Pharmacol. 2015;76(4):699–712. | ||
Sonnichsen DS, Relling MV. Clinical pharmacokinetics of paclitaxel. Clin Pharmacokinet. 1994;27(4):256–269. | ||
Monsarrat B, Alvinerie P, Wright M, et al. Hepatic metabolism and biliary excretion of Taxol in rats and humans. J Natl Cancer Inst Monogr. 1993;(15):39–46. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.