Back to Journals » Clinical Ophthalmology » Volume 19
Sorsby Fundus Dystrophy in an Asian Pedigree: Pathogenic Timp3 P.Y191c Variant Impairs Its Binding with Mmp2/9 and Cellular Localization
Authors Li M, Peng H, Ding S, Wei Y, Zhang L, Zhou Z, Tang H, Shi P, Liang Y, Li G, Tao Y, Song Z ![]()
Received 27 July 2025
Accepted for publication 1 December 2025
Published 9 December 2025 Volume 2025:19 Pages 4551—4564
DOI https://doi.org/10.2147/OPTH.S556592
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yousef Fouad
Miao Li,1 Haiying Peng,1 Shenao Ding,1 Yuanmeng Wei,1 Leying Zhang,1 Zhongqiang Zhou,1 He Tang,1 Pingling Shi,1 Yingjuan Liang,1 Guanfeng Li,2 Ye Tao,1 Zongming Song1
1Henan Eye Institute, Henan Eye Hospital, People’s Hospital of Zhengzhou University, Henan, People’s Hospital, Zhengzhou, People’s Republic of China; 2Department of Ophthalmology, Henan Children’s Hospital, Zhengzhou, People’s Republic of China
Correspondence: Zhongqiang Zhou, Email [email protected] Zongming Song, Email [email protected]
Purpose: To characterize the clinical phenotype and elucidate the pathogenic mechanism of the novel TIMP3 p.Y191C variant in a multigenerational Asian pedigree with Sorsby Fundus Dystrophy (SFD).
Methods: Affected family members underwent comprehensive ophthalmic evaluations. Genetic analysis was performed via whole-exome and Sanger sequencing. An ARPE-19 cell models overexpressing wild-type or mutant TIMP3 were generated. Functional analysis including co-immunoprecipitation (Co-IP), MMP inhibition, and immunofluorescence were performed.
Results: A heterozygous TIMP3 p.Y191C variant was identified in seven affected members, co-segregating with bilateral choroidal neovascularization and disciform scarring. The tyrosine-191 residue is highly conserved, and structural/computational analyses predicted that the cysteine substitution introduces a smaller, hydrophobic residue and reduces protein stability. Functionally, the Y191C variant impaired TIMP3 binding to MMP2 and MMP9, reduced its inhibitory activity, and altered MMP2 localization following LPS stimulation. Consistent with this loss of function, the mutant TIMP3 significantly inhibited cell viability and promoted apoptosis in ARPE-19 cells under inflammatory stress.
Conclusion: The novel TIMP3 p.Y191C variant causes SFD in an Asian pedigree. Its pathogenicity arises from distinct disruptions in MMP2/9 binding and inhibition, coupled with altered MMP2 localization, thereby providing a mechanistic basis for the disease.
Keywords: sorsby fundus dystrophy, pathogenic variant, TIMP3, MMP2, MMP9
Introduction
Sorsby fundus dystrophy (SFD) is a rare, autosomal dominant degenerative maculopathy characterized by full penetrance.1 Its primary histopathological hallmarks include drusen-like deposits, atrophy of the retinal pigment epithelium (RPE) and photoreceptors, choroidal neovascularization (CNV), and disciform scarring.2 SFD is caused by variants in the TIMP3 gene (chromosome 22q), encoding tissue inhibitor of metalloproteinases-3.3 However, the precise molecular mechanisms linking TIMP3 variants to disease remain incompletely defined.
TIMP3 is a critical regulator of extracellular matrix (ECM) homeostasis, primarily through inhibition of matrix metalloproteinases (MMPs). It is predominantly localized within Bruch’s membrane in SFD. Beyond MMP inhibition, TIMP3 modulates angiogenesis by competitively binding VEGF and inhibiting its interaction with VEGFR2,4 and by suppressing endothelial cell proliferation, migration, and tube formation.5,6 TIMP3 also contributes to inflammation resolution, fibrosis suppression,7,8 and acts as a potent inhibitor of tumor-associated angiogenesis and metastasis.9–12
To date, twenty-one pathogenic TIMP3 variants have been associated with SFD. Among these, with the exception of the stop variant p.E 162*13,14 and the c.439–2dupA alteration at the intron 4/exon 5 junction,15 the remaining twenty variants are missense variants resulting in amino acid substitutions (Figure S1).16–25 The majority of these variants cluster within the final exon of the TIMP3 gene, predominantly altering the cysteine residue count within the corresponding pathogenic variant proteins. A prevailing hypothesis suggests that SFD pathogenesis arises not from loss of MMP inhibition, but from excessive deposition of pathogenic variant TIMP3 protein aggregates within Bruch’s membrane.2 This aggregation is mediated by intermolecular disulfide bonds involving the unpaired cysteine residues introduced by most variants, leading to high molecular weight complexes that persist bound to the ECM.15,26–28 However, this model faces challenges, as some variants, such as p.S156C, retain MMP inhibitory capacity and show no evidence of dimerization,29 indicating potential heterogeneity in pathogenic mechanisms.
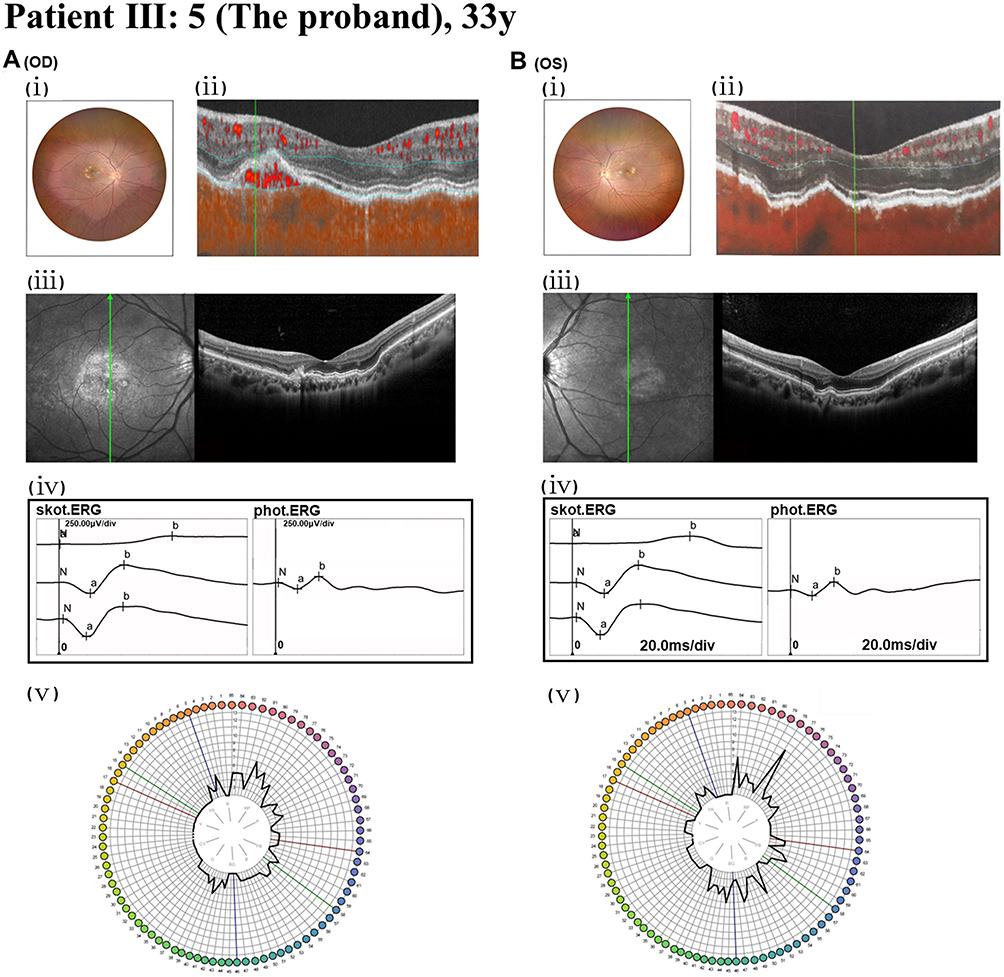 |
Figure 1 Fundus, angio-OCT, SD-OCT, FM-100, and ERG analyses of the proband (III:5). (A): The right eye (OD). (B) The left eye (OS). (A. i and B. i) Fundus imaging identified CNV and refractive error. (A. ii) Angio-OCT revealed abnormal blood flow signals in the outer retina. (B. ii) Angio-OCT revealed no significant changes observed. (A. iii) SD-OCT displayed macular atrophy of the retinal outer layers, structural disorganization, irregular RPE, and uneven hyperreflective clusters. The green line indicates the position and orientation of the corresponding cross-sectional OCT scan beam across the macula. (B. iii) Serrated alterations were observed in the outer retinal layer of the macula, with partially obscured and irregular light bands. (A. iv and B. iv) ERG examination indicated a moderate reduction in binocular cone and rod system activity. (N) Baseline, representing the resting potential prior to light stimulus. a-wave: The initial negative deflection, originating primarily from the hyperpolarization of photoreceptors (rods and/or cones). b-wave: The subsequent positive peak, reflecting the summed activity of ON-bipolar cells and Müller cells. (A. v and B. v) FM-100 testing demonstrated impaired color vision in both eyes. |
Here, we identified a heterozygous TIMP3 variant c.572A>G (p.Y191C) in a multigenerational Chinese pedigree with classic SFD. To elucidate the pathogenic mechanism driven by this variant, we established stable ARPE-19 cell models - a cell type central to SFD pathology - expressing wild-type or mutant TIMP3. This study thus aims to characterize the functional consequences of TIMP3 p.Y191C within a relevant RPE context and define its contribution to SFD pathogenesis.
Materials and Methods
Subjects and Clinical Assessment
The study adhered to the Declaration of Helsinki and received approval from the Ethics Committee of Henan Eye Hospital for the collection and use of clinical data, family histories, and blood samples for genetic analysis [IRB approval number: HNEECKY-2022-16]. Informed written consent was obtained from all participants.
Comprehensive family and medical histories were meticulously gathered from all accessible relatives. A series of ophthalmic evaluations was conducted, encompassing best corrected visual acuity (BCVA), refractive error, intraocular pressure (IOP) measurement, slit-lamp biomicroscopy, color vision testing, fundus photography, visual field analysis, and ophthalmoscopy. Additional advanced diagnostic procedures, including high-resolution fundus imaging (Zeiss), angiography optical coherence tomography (Angio-OCT, Zeiss, Heidelberg), spectral-domain optical coherence tomography (SD-OCT, Zeiss, Heidelberg) and full-field electroretinography (ERG, Diagnosys, LKC), Farnsworth-Munsell 100 Hue Test Results (FM-100, X-Rite), were performed on select family members for more detailed ophthalmological insights.
Whole Exome Sequencing
Genomic DNA was isolated from peripheral blood samples of all family members using the TIANGEN Blood DNA Kit (DP304, TIANGEN, China). High-throughput targeted sequencing was applied to examine the exon regions and flanking intronic junctions (50bp) of all recognized genes in the tested subjects. Whole-exome sequencing was employed to analyze genomic data, with alignment files generated through duplicate read removal and localized alignment to the hg19 (GRCh37) human reference genome. Variants were identified and annotated using rigorously validated bioinformatics pipelines, ensuring adherence to established analytical standards.
In Silico Molecular Genetic Analysis and Bioinformatics Analysis
The pathogenicity of the variants was evaluated using the following web applications: LRT (Likelihood Ratio Test), PolyPhen-2 (Polymorphism Phenotyping v2), variant Taster,30 SIFT (Sorting Intolerant from Tolerant),31 FATHMM (Functional Analysis Through Hidden Markov Models),31 and CADD (Combined Annotation Dependent Depletion).32 Multiple sequence alignments across species were performed with Clustal Omega.33 Protein tertiary structures were modeled through the Swiss-Model workspace (http://swissmodel.expasy.org),34 while domain analyses utilized the CDD online platform (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). The structural implications of variants were examined using HOPE (http://www.cmbi.umcn.nl/hope). Homology model superimpositions of TIMP3 were generated with PyMOL (https://pymol.org/2/). Protein stability changes associated with TIMP3 variants were predicted via MUpro (http://mupro.proteomics.ics.uci.edu/)35 and I-Mutant v2.0 (http://folding.biofold.org/i-mutant/i-mutant2.0).36 All listed resources are publicly accessible.
Cell Culture and Lentivirus Vector Mediated Overexpression
The human RPE cell line ARPE-19, sourced from the American Type Culture Collection (Rockville, MD, USA), was maintained in DMEM/Ham’s F-12 medium (1:1) supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C under a 5% CO2 atmosphere. Lentiviral vectors were engineered to facilitate the overexpression of TIMP3 in its wild-type (WT, pSLenti-SFH-EGFP-P2A-Puro-CMV-TIMP3-3xFLAG-WPRE) and mutant (MT, pSLenti-SFH-EGFP-P2A-Puro-CMV-TIMP3(Y191C)-3xFLAG-WPRE) forms, as well as a control empty vector (pSLenti-SFH-EGFP-P2A-Puro-CMV-MCS-3xFLAG-WPRE). Transfection was performed 24 hours prior using Lipofectamine 3000 (Thermo Fisher Scientific, Chicago, IL). Following proliferation, cells were selected with 5 μg/mL blasticidin for 3–4 weeks. Stable cell lines expressing the empty vector (GL120), wild-type TIMP3 (FLAG TIMP3-H21770), and mutant TIMP3 (FLAG TIMP3-H21827) proteins were isolated and cryopreserved.
LPS was applied to ARPE-19 cells to induce inflammation, and cultures were divided into eight groups: ARPE-19; ARPE-19 + LPS (2 μg/mL); ARPE-19 (GL120); ARPE-19 (GL120) + LPS (2 μg/mL); ARPE-19 (FLAG TIMP3-H21770); ARPE-19 (FLAG TIMP3-H21770) + LPS (2 μg/mL); ARPE-19 (FLAG TIMP3-H21827); and ARPE-19 (FLAG TIMP3-H21827) + LPS (2 μg/mL). After 24 hours, assessments were conducted to evaluate cell viability and apoptosis, analyze mRNA and protein expression, or perform immunofluorescence after fixation in 4% paraformaldehyde for 30 minutes.
Biochemistry Analysis and Immunofluorescence Staining
Total RNA was extracted from retinal tissue using Trizol (Invitrogen, Carlsbad, CA, USA), and RNA concentration was quantified with the NanoDrop 2000 (Thermo Scientific). Subsequently, mRNA (5 ng) was reverse-transcribed into cDNA using the Reverse Transcriptase Kit. The primers employed for cDNA amplification were listed in Table S1.
Western blot (WB), Co-immunoprecipitation (Co-IP) and immunofluorescence (IF) staining were carried out following established protocols. Primary antibodies included TIMP3 (Proteintech, 10858-1-AP, WB: 1:2000, IP: 1:1000, IF: 1:400), MMP9 (Proteintech, 10375-2-AP, WB: 1:600, IP:1:500, IF: 1:100), MMP2 (Proteintech, 66366-1-Ig, WB: 1:1000, IP:1:500, IF: 1:200), and GAPDH (Abcam, ab8245, 1:5000). Secondary antibodies were species-matched HRP-conjugated for WB, light chain-specific for Co-IP (to exclude heavy chain interference), and fluorophore-conjugated for IF.
Cells were labeled with distinct fluorescent dyes, and fluorescence intensity was quantified across 10 randomly selected fields per well using the PerkinElmer Operetta CLS High Content Screening platform integrated with Harmony 4.5 software (PerkinElmer, Waltham, MA, USA). Cell viability was assessed via the cell counting kit‐8 (CCK‐8) assay, while apoptosis was analyzed using the annexin V-fluorescein isothiocyanate/propidium iodide detection kit (BD Biosciences, San Diego, CA) according to the manufacturer’s guidelines.
Cell Viability and Apoptosis Assay
Cell viability was assessed using the CCK‐8 assay. ARPE‐19 cells (1×105 per well) were seeded in 96‐well plates and cultured for 24h before treatment as described. Spent medium (100 μL) was replaced with an equal volume of fresh medium containing 10% CCK-8 (WST‐8, Dojindo Laboratories, Tokyo, Japan), and cells were incubated at 37°C for 1 to 4 hours. Absorbance at 450 nm was measured using a microplate reader, and viability was calculated as: Cell viability (%) = (A450sample - A450blank) / (A450control - A450blank) × 100.
To evaluate apoptosis and necrosis, the annexin V-FITC/PI apoptosis detection kit (BD Biosciences, San Diego, CA) was employed according to the manufacturer’s protocol. Cell suspensions containing >10,000 cells were prepared in 500 μL of binding buffer and incubated with annexin V-FITC and PI for 15 minutes. Flow cytometric analysis was performed using a FACSCalibur system (BD Biosciences). All experiments were conducted in triplicate.
Statistical Analysis
Statistical analysis was conducted using GraphPad Prism 8.0. Differences between groups were assessed through Student’s t-test or one-way analysis of variance (ANOVA), with a significance threshold set at P<0.05. Quantitative results were expressed as mean ± standard deviation (SD), ensuring clarity and consistency in data presentation.
Results
Clinical Manifestations
A Chinese SFD family comprising 23 members, including 7 affected individuals, was analyzed. The proband (III:5), a 26-year-old female, presented with impaired central vision and high myopia. Her BCVA was 1.0 in both eyes. Fundus photography revealed CNV in the right eye (Figure 1. A.i), corroborated by AngioOCT showing abnormal blood flow signals in the outer retina (Figure 1. A.ii). The left eye appeared unremarkable on these modalities (Figure 1. B.i, ii). SD-OCT of the right eye demonstrated macular outer retinal atrophy, structural disorganization, and irregularities in the RPE layer, partially accompanied by uneven hyperreflective foci (Figure 1. A. iii). In contrast, the left eye exhibited serrated RPE/outer retinal changes and blurred/irregular hyperreflective bands (Figure 1. B. iii). ERG indicated moderate bilateral reduction in cone and rod responses (Figure 1. A. iv and Figure 1. B. iv), and the FM-100 test revealed abnormal color vision in both eyes (Figure 1. A.v and Figure 1. B. v).
Among other family members, Patient II:3 exhibited RPE degeneration near the posterior pole in both eyes and pronounced choroidal vasculopathy, as shown by fundus photography (Figure 2. A. a.i and A. b. i). SD-OCT revealed disorganized retinal architecture with evident RPE abnormalities (Figure 2. A. a.ii and A. b. ii). Patient II:6 displayed bone spicule-like retinal pigmentation in the macular region and optic nerve papilla on fundus photography (Figure 2. B. a.i and B. b. i), with SD-OCT showing retinal thinning and marked disarray of layers (Figure 2. B. a. ii and B. b. ii). Patient II:11 demonstrated extensive macular pigmentation, scarring, and retinal atrophy on fundus imaging (Figure 2. C. a.i and C. b. i). SD-OCT revealed macular retinoschisis, structural disruptions, and irregular RPE (Figure 2. C. a. ii and C. b. ii). Detailed clinical features and TIMP3 variant data for family members were provided in Table 1, with additional information on patients II:7, II:10, and III:3 available in Supplementary Results (Figure S2).
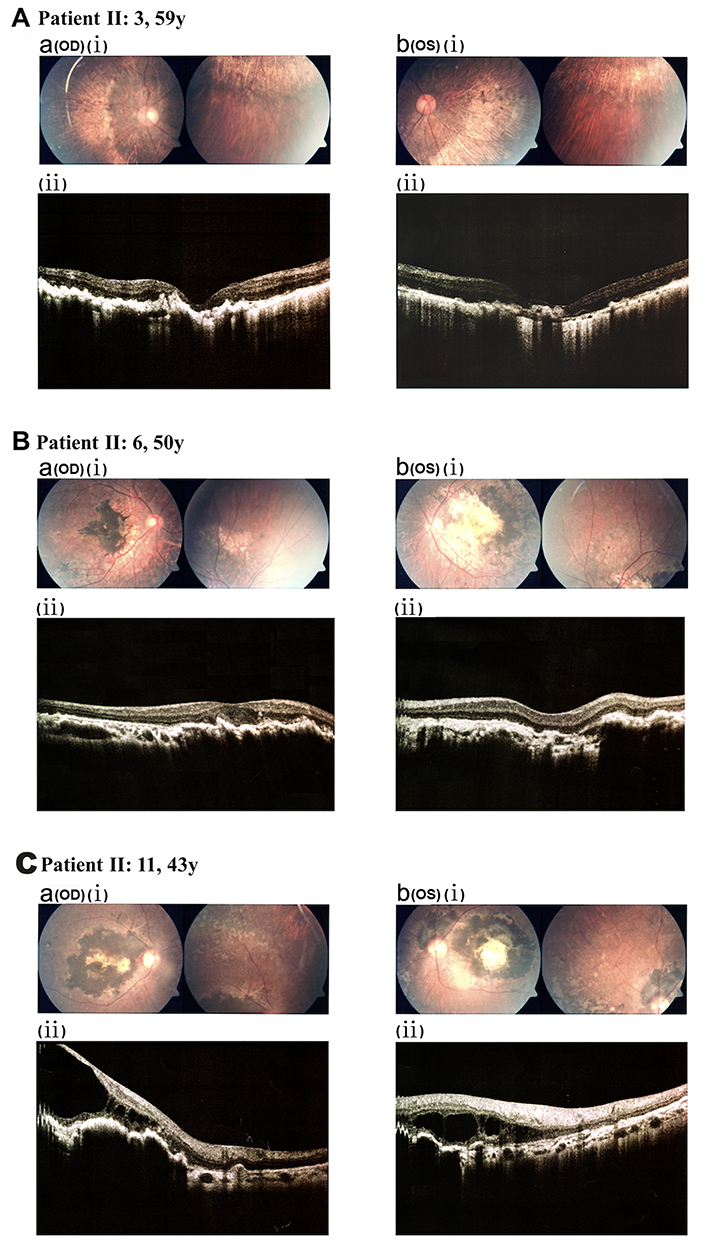 |
Figure 2 Fundus and SD-OCT characteristics of other family members (partial representative data). (a) The right eye (OD). (b) The left eye (OS). (A) Patient II:3. (A. a. i and A. b.i) Fundus photography showed RPE degeneration near the posterior pole bilaterally and pronounced choroidal vasculopathy. (A. a.ii and A. b.ii) SD-OCT revealed disrupted retinal architecture and RPE irregularities. (B) Patient II:6. (B. a.i and B. b.i) Fundus photography exhibited bone spicule-like pigmentation in the macular region and around the optic nerve head. (B. a.ii and B. b.ii) SD-OCT showed thinning of the retinal layers with pronounced structural disarray. (C) Patient II:11. (C. a.i and C. b.i) Fundus photography identified extensive macular pigmentation, scarring, and atrophy. (C. a.ii and C. b.ii) SD-OCT revealed macular retinoschisis, structural disorganization, and irregularities in the RPE. |
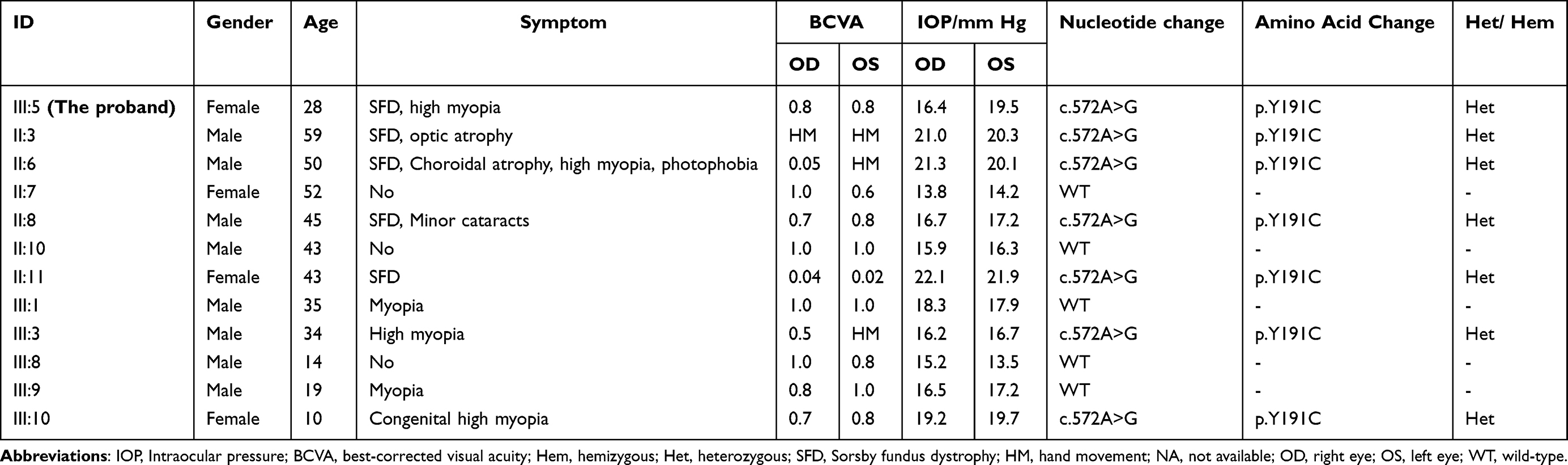 |
Table 1 Clinical Features and TIMP3 Variant Information Among the Family Members |
Identification of the Variants in TIMP3
The pedigree of the family was presented in Figure 3A. Targeted NGS identified a novel heterozygous TIMP3 variant (c.572A>G, p.Y191C) in the proband. Sanger sequencing confirmed this variant and confirmed its co-segregation with the SFD phenotype across affected family members (Figure 3B). Multiple sequence alignment revealed high evolutionary conservation of Tyr191 (Figure 3C). Structural prediction using HOPE software indicated that the p.Y191C substitution introduces a smaller, more hydrophobic residue, altering local protein structure (Figure 3D, E). The variant has not been previously cataloged in the human gene variant database (HGMD). Allele frequencies for c.572A>G (p.Y191C) were reported at approximately 0.00009, 0.00008, and 0.0001 in the ExAC, GnomAD, and 1000 Genome Aggregation Database, respectively, predominantly in East Asian populations. Predictive analyses using SIFT, Polyphen-2, LRT, Mutation Taster, FATHMM, and CADD classified the Y191C variant as “Deleterious” (Table 2).
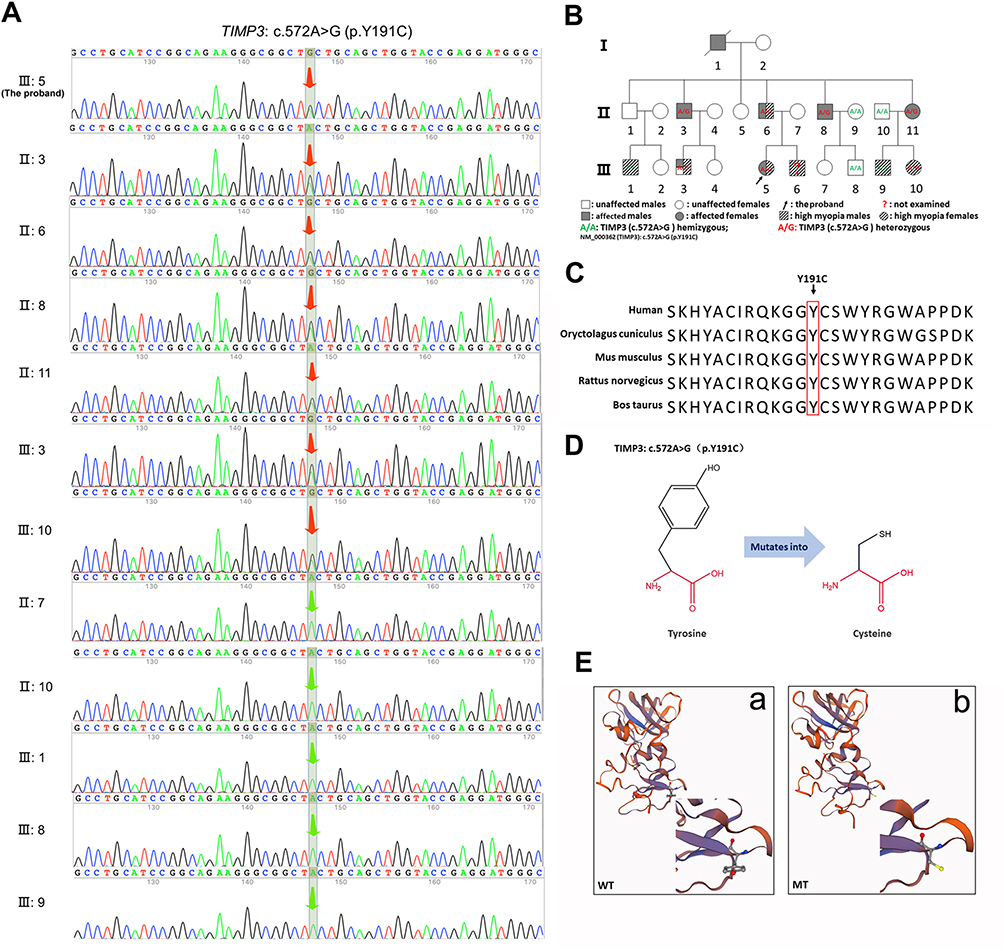 |
Figure 3 Whole exome sequencing. (A) Sanger sequencing chromatograms of twelve family members identified the heterozygous c.572A>G (p.Y191C) TIMP3 variant in the proband (III:5), his father (II:6), uncle (II:3, II:8), aunt (II:11), and cousins (III:3, III:10). (B) Pedigree analysis illustrating the inheritance of the c.572A>G (p.Y191C) variant. (C) Multiple sequence alignments of TIMP3 Tyr191 across species revealed that the variant occurred in highly conserved residues. (D) Schematic structures of the original and mutant amino acids, highlighting the backbone in red and the side chain in black. (E) Comparative 3D structures of the wild-type (a) and mutant (b) proteins. |
 |
Table 2 Pathogenicity Prediction and Population Distribution Frequencies |
Y191C Variant Disrupts TIMP3 Function
Computational analysis using MUpro (ΔΔG = −0.100) and I-Mutant v2.0 (ΔΔG = −0.500) predicted that the p.Y191C substitution destabilizes the TIMP3 protein (ΔΔG < 0 indicates decreased stability). To experimentally assess the variant, we established ARPE-19 cell lines stably expressing empty vector (GL120, control), wild-type TIMP3 (FLAG TIMP3-H21770, WT), and mutant TIMP3 (FLAG TIMP3-H21827, MT) vectors (Figure S3A). RT-PCR analysis confirmed successful transfection, showing significantly elevated TIMP3 expression in the WT group (P= 0.0074, 1.62 ± 0.40-fold) and the MT group (P= 0.0112, 1.23 ± 0.52-fold) compared to control group, with WT expression being significantly higher than MT (P = 0.00112) (Figure S3B). Western blot analysis corroborated these findings, showing increased TIMP3 protein levels in the WT (P= 0.0006, 2.24 ± 0.09-fold) and MT groups (P= 0.0001, 2.08 ± 0.12-fold) relative to the control group. Furthermore, protein levels in the WT group were higher than those in the MT group (P= 0.0231) (Figure S3C and D). Collectively, Together, these results suggest that the Y191C variant compromises TIMP3 stability and expression, pointing to a loss-of-function mechanism underlying its pathogenicity.
CCK-8 assays indicated no significant difference in cell viability among the empty vector control, WT, and MT groups under basal conditions (P>0.05). Following LPS treatment, a marked reduction in cell viability was observed in the MT group compared with the empty vector control (P=0.0096), while the WT group showed no significant difference relative to the control (P=0.7128, Figure 4A, n=3/group). Flow cytometry analysis revealed a significant increase in apoptosis in the normal control group upon LPS exposure. Moreover, the MT group exhibited a substantially higher apoptosis rate compared to the empty vector control group post-LPS treatment (30%, P=0.0005; Figure 4B, C). These findings demonstrate that p.Y191C TIMP3 diminishes ARPE-19 cell resilience to stress, reducing viability and promoting apoptosis.
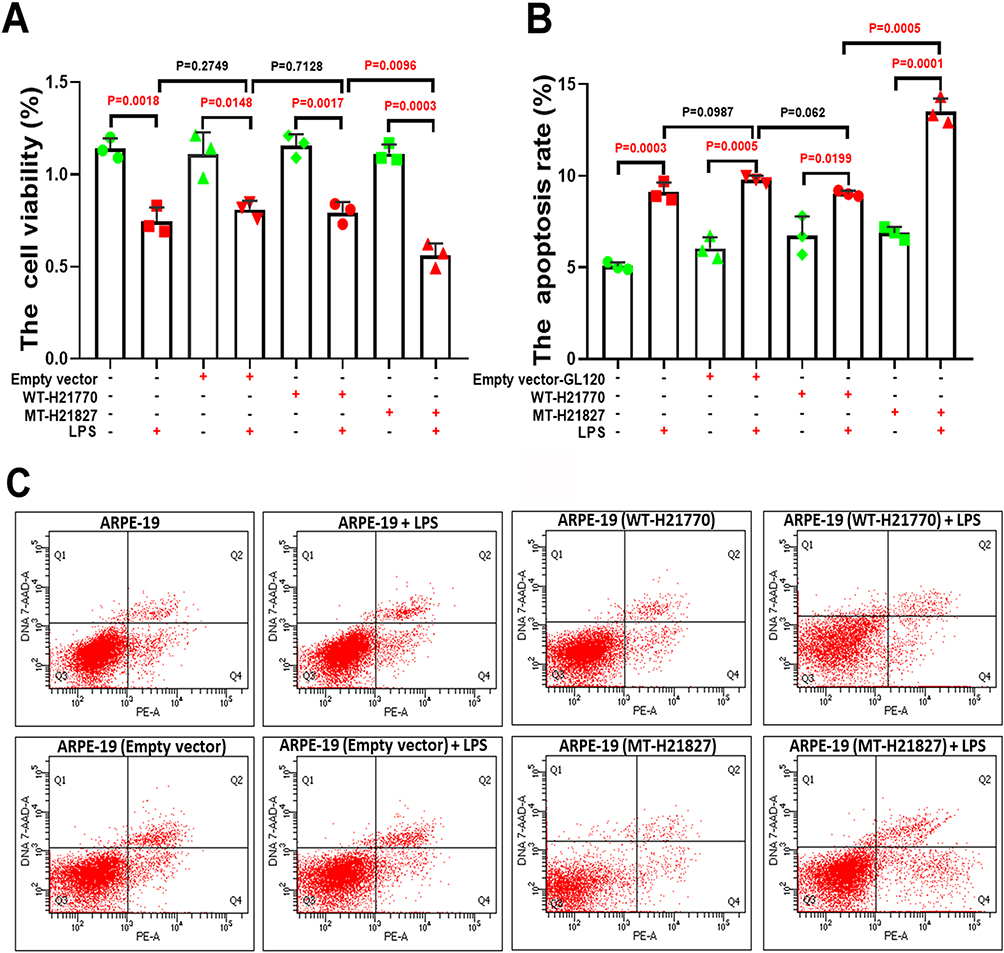 |
Figure 4 The TIMP3 variant Y191C mitigated the cell viability and promoted the apoptosis of ARPE-19. (A) CCK-8 assay demonstrated a significant reduction in cell viability in the MT group compared to the empty vector control group 24 hours post-LPS treatment (P=0.0096), while no significant difference was observed in the WT group (P=0.7128). (B and C) Flow cytometry analysis indicated a significant increase in apoptosis rates in the MT group compared to the empty vector control group following LPS treatment (P=0.0005). In contrast, the WT group exhibited a 10% reduction in apoptosis rates compared to the empty vector control group. FIGURE 4 The TIMP3 variant Y191C mitigated the cell viability and promoted the apoptosis of ARPE-19. (A) CCK-8 assay demonstrated a significant reduction in cell viability in the MT group compared to the empty vector control group 24 hours post-LPS treatment (P=0.0096), while no significant difference was observed in the WT group (P=0.7128). (B and C) Flow cytometry analysis indicated a significant increase in apoptosis rates in the MT group compared to the empty vector control group following LPS treatment (P=0.0005). In contrast, the WT group exhibited a 10% reduction in apoptosis rates compared to the empty vector control group. n=3/group. “n” denotes biological replicates, defined as independently assayed aliquots derived from the same lentiviral infection and differentiation batch. |
Y191C Variant Impairs Protein Interactions and Alters MMP2 Subcellular Localization
To elucidate the molecular interactions between TIMP3 and its target metalloproteinases, we performed Co-IP assays using an anti-FLAG magnetic beads on lysates from ARPE-19 cells expressing empty vector, WT or MT TIMP3. IP analysis (Figure 5A) confirmed efficient precipitation of FLAG-tagged proteins in both WT and MT groups, indicating a reliable experimental system. Further analysis revealed that the Y191C mutation not only promoted the formation of FLAG-TIMP3 complexes but also altered its binding selectivity: it significantly enhanced the interaction with the 45~55 kDa active fragment of MMP9, while concurrently weakening the association with MMP2. Input lysate analysis (Figure 5A) revealed a substantial increase in TIMP3 levels in WT cells and a moderate elevation in MT cells compared with the empty vector group, thus confirming the successful transfection and partial destabilization of the mutant protein. MMP9 immunoblotting detected both the 92 kDa glycosylated pro-form and the 45~55 kDa active form in all groups. Notably, the 45~55 kDa fragment was specifically enriched in the Co-IP samples (Figure 5B), with a markedly stronger binding intensity in the MT group than that in the WT group. These findings suggest that the abnormally strong capture of active MMP9 by mutant TIMP3 may compromise its broad-spectrum inhibitory capacity, rendering it unable to effectively regulate other metalloproteinases such as MMP2, thereby leading to an overall imbalance in the protease inhibition network. Furthermore, the increased level of the 45~55 kDa active MMP9 in MT cells may reflect an activated proteolytic environment, which could be exacerbated by the abnormal enrichment of TIMP3. Given the crucial role of MMP9 in degrading Bruch’s membrane and promoting choroidal neovascularization, this enhanced interaction is likely to disrupt the homeostasis of the ocular tissue. In contrast, the co-precipitation of MT TIMP3 with MMP2 was significantly reduced compared with the WT. Input analysis further verified the reduced MMP2 expression in WT cells but elevated levels in MT cells, implying that a compensatory regulatory mechanism which may be activated in response to the dysfunctional state of TIMP3.
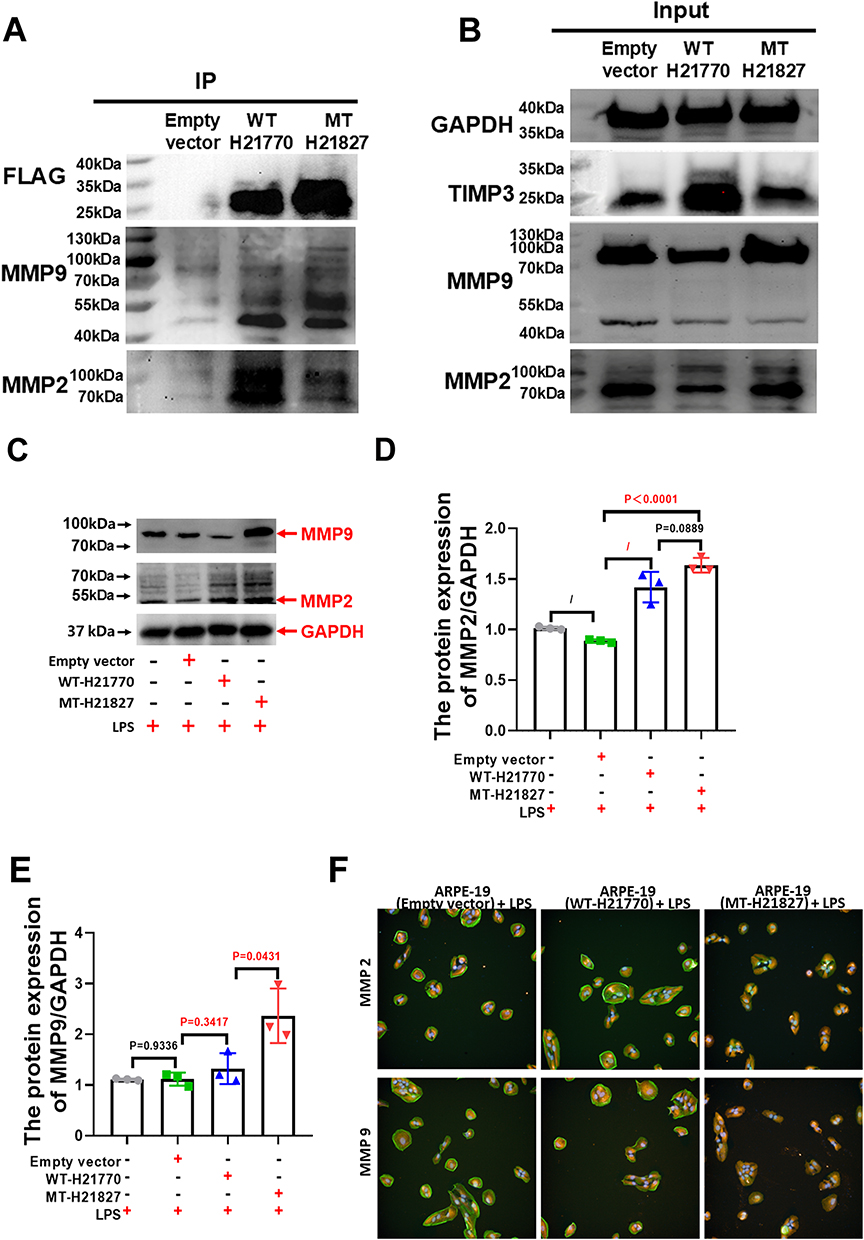 |
Figure 5 The Y191C variant inhibited the interaction between TIMP3 and MMP proteins. (A and B) The Y191C mutation increases FLAG-TIMP3 complex formation, selectively enhancing interaction with MMP9 proteolytic fragments (45~55 kDa) while reducing MMP2 binding. (C–E) Following LPS administration, MMP2 expression levels in the empty vector control group remained comparable to those in the normal control group. However, in the MT group, MMP2 expression was significantly elevated compared to the empty vector group (P < 0.0001, 1.84 ± 0.21-fold) and showed a modest increase compared to the WT control group (P= 0.0889, 1.15 ± 0.13-fold). Additionally, MMP9 expression in the MT group was markedly higher than in the empty vector group after LPS treatment (P < 0.0172, 2.12 ± 0.41-fold). (F) IF staining revealed nuclear localization of MMP2 in ARPE-19 cells of the WT group, whereas in the MT group, MMP2 expression extended into the cytoplasm of ARPE-19 cells. n=3/group. “n” refers to the number of independent biological replicates. |
As illustrated in Figure 5C–E, MMP2 protein levels in the empty vector group remained comparable to those in the normal control group following LPS administration. In contrast, the MT group exhibited a significant elevation in MMP2 levels compared to both the empty vector group (P<0.0001, 1.84±0.21-fold) and the WT control group (P=0.0889, 1.15±0.13-fold) under the same conditions. Similarly, MMP9 protein levels in the MT group were markedly higher than in the empty vector control group after LPS exposure (P<0.0172, 2.12±0.41-fold) and significantly exceeded those in the WT control group (P=0.0431, 1.79±0.23-fold). These observations indicate that the TIMP3 Y191C variant impairs the TIMP3-MMP9 interaction, destabilizing TIMP3 and compromising its inhibitory regulation of MMP9 activity. Conversely, the TIMP3-MMP2 interaction appears to be less affected. Immunofluorescence staining further revealed that MMP2 localized primarily within the nucleus of ARPE-19 cells in the empty vector and WT groups, whereas in the MT group, MMP2 was predominantly localized in the cytoplasm (Figure 5F), suggesting that the Y191C variant profoundly alters the intracellular distribution of MMP2. Collectively, our findings establish that the TIMP3 Y191C variant: (i) destabilizes TIMP3-MMP9 binding interactions, (ii) compromises metalloproteinase regulatory capacity, and (iii) drives pathological MMP2 mislocalization - mechanisms underlying its disease pathogenesis.
Discussion
The present study identifies a novel TIMP3 variant (c.572A>G, p.Y191C) in a Chinese family with Sorsby fundus dystrophy (SFD), expanding the genotypic and phenotypic spectrum of this rare autosomal dominant disorder. Clinically, affected individuals exhibited features reminiscent of both early and advanced AMD, including choroidal neovascularization (CNV), outer retinal atrophy, and RPE abnormalities. The proband (III:5) demonstrated asymmetric involvement, with CNV in the right eye and serrated RPE changes in the left, suggesting variable expressivity. ERG and color vision deficits further corroborated functional impairment. Notably, the Y191C variant was absent in East Asian population databases (ExAC-EAS), and computational analyses unanimously classified it as pathogenic, supported by its high evolutionary conservation and predicted structural destabilization.
The Y191C variant disrupts TIMP3 function through multiple mechanisms: (1) Impaired MMP Regulation: Co-IP assays revealed weakened TIMP3-MMP2 binding but enhanced interaction with a ~55kDa MMP9 proteolytic fragment, suggesting aberrant complex formation that depletes functional inhibitors. This aligns with prior reports that TIMP3 variants compromise MMP inhibitory capacity, leading to ECM dysregulation.37 (2) Altered Subcellular Localization: Immunofluorescence demonstrated cytoplasmic mislocalization of MMP2 in mutant TIMP3 cells, contrasting with nuclear retention in wild-type controls. Given the established role of nuclear MMP2 in cleaving DNA damage sensors like Ku70 to facilitate apoptosis under genotoxic stress,38,39 its absence from the nucleus likely undermines the RPE’s ability to manage genomic damage. This novel mechanism may thereby critically exacerbate RPE vulnerability to environmental insults and accelerate SFD progression. (3) Dominant-Negative Effects: Despite comparable protein levels, mutant TIMP3 exhibited reduced cell viability and increased apoptosis under LPS-induced stress, indicating a toxic gain-of-function mechanism independent of expression levels.40
The Y191C variant drives SFD pathogenesis through a feedforward loop of MMP dysregulation and inflammatory activation. Loss of MMP9 inhibition delays its clearance, permitting synergistic interaction with MMP2 to promote CNV.41 Concurrently, MMP9-mediated NF-κB activation exacerbates neuroinflammation and oxidative stress, culminating in retinal thinning and RPE degeneration.42 Our findings reconcile prior paradoxes in TIMP3-associated pathology by demonstrating that conformational hijacking of mutant TIMP3-rather than mere haploinsufficiency-underlies disease. This mechanistic framework is further supported by the recapitulation of AMD-like phenotypes in TIMP3 S179C mice, where MMP2/9 hyperactivity increased bFGF/VEGF secretion and laser-induced CNV.37
The clinical failure of broad-spectrum MMP inhibitors (eg, doxycycline) in TIMP3 disorders underscores the need for precision therapeutics.6 Our data suggest two targeted strategies: (1) Small-molecule disruptors of mutant TIMP3/MMP9 fragment complexes to restore free inhibitor pools, and (2) CRM1 inhibitors to correct MMP2 nuclear-cytoplasmic trafficking. Future studies should delineate SFD-specific inflammatory cascades to optimize intervention timing, particularly in early-stage disease where RPE resilience may still be salvageable.
In conclusion, this study identifies the TIMP3 p.Y191C variant in an Asian SFD pedigree and elucidates novel pathogenic mechanisms, which include the selective impairment of MMP9 inhibition and the severe disruption of MMP2 interaction. Furthermore, we demonstrate a pathological cytoplasmic mis localization of MMP2. Collectively, these defects synergistically disrupt ECM homeostasis, promote inflammation and angiogenesis, and compromise RPE cell resilience, culminating in the characteristic features of SFD. By elucidating the novel pathogenic mechanism of the TIMP3 p.Y191C variant, our work lays the necessary molecular groundwork for the future development of potential targeted therapies.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Ethics Statement
The research protocol involving human participants received approval from the Ethics Committee of Henan Eye Hospital (Approval No. HNEECA-2022-16). Written informed consent was obtained from all participants or their legal guardians/next of kin, including specific consent for the publication of potentially identifiable images or data featured in this article.
Acknowledgments
We would like to thank all the patients and their families for their invaluable participation in this study.
Funding
This research was funded by grants from the National Natural Science Foundation of China (82071008), the Zhong yuan Technology Leading Talent Project (224200510013), the Major Science and Technology Projects of Henan Province (221100310200), the Natural Science Foundation of Henan Province (252300421269), and the important project of science and technology of the Henan province (LHGJ20240640).
Disclosure
The authors declare no conflicts of interest, financial or personal, that could have influenced the outcomes or interpretations presented in this work.
References
1. Sorsby A, Mason ME, Gardener N. A fundus dystrophy with unusual features. Br J Ophthalmol. 1949;33(2):67–97. doi:10.1136/bjo.33.2.67
2. Lin RJ, Blumenkranz MS, Binkley J, et al. A novel His158Arg mutation in TIMP3 causes a late-onset form of Sorsby fundus dystrophy. Am J Ophthalmol. 2006;142(5):839–848. doi:10.1016/j.ajo.2006.06.003
3. Jacobson SG, Cideciyan AV, Regunath G, et al. Night blindness in Sorsby’s fundus dystrophy reversed by vitamin A. Nat Genet. 1995;11(1):27–32. doi:10.1038/ng0995-27
4. Handsley MM, Edwards DR. Metalloproteinases and their inhibitors in tumor angiogenesis. Int, J, Cancer. 2005;115(6):849–860. doi:10.1002/ijc.20945
5. Clark IM, Swingler TE, Sampieri CL, et al. The regulation of matrix metalloproteinases and their inhibitors. Int J Biochem Cell Biol. 2008;40(6–7):1362–1378. doi:10.1016/j.biocel.2007.12.006
6. Qi JH, Ebrahem Q, Ali M, et al. Tissue inhibitor of metalloproteinases-3 peptides inhibit angiogenesis and choroidal neovascularization in mice. PLoS One. 2013;8(3):e55667. doi:10.1371/journal.pone.0055667
7. Fiorentino L, Cavalera M, Menini S, et al. Loss of TIMP3 underlies diabetic nephropathy via FoxO1/STAT1 interplay. EMBO Mol Med. 2013;5(3):441–455. doi:10.1002/emmm.201201475
8. Gill SE, Huizar I, Bench EM, et al. Tissue inhibitor of metalloproteinases 3 regulates resolution of inflammation following acute lung injury. Am J Pathol. 2010;176(1):64–73. doi:10.2353/ajpath.2010.090158
9. Spurbeck WW, Ng CY, Strom TS, et al. Enforced expression of tissue inhibitor of matrix metalloproteinase-3 affects functional capillary morphogenesis and inhibits tumor growth in a murine tumor model. Blood. 2002;100(9):3361–3368. doi:10.1182/blood.V100.9.3361
10. Das AM, Seynhaeve AL, Rens JA, et al. Differential TIMP3 expression affects tumor progression and angiogenesis in melanomas through regulation of directionally persistent endothelial cell migration. Angiogenesis. 2014;17(1):163–177. doi:10.1007/s10456-013-9385-2
11. Das AM, Bolkestein M, van der Klok T, et al. Tissue inhibitor of metalloproteinase-3 (TIMP3) expression decreases during melanoma progression and inhibits melanoma cell migration. Eur J Cancer. 2016;66:34–46. doi:10.1016/j.ejca.2016.06.020
12. Das AM, Koljenović S, Oude Ophuis CM, et al. Association of TIMP3 expression with vessel density, macrophage infiltration and prognosis in human malignant melanoma. Eur J Cancer. 2016;53:135–143. doi:10.1016/j.ejca.2015.09.014
13. Langton KP, McKie N, Smith BM, et al. Sorsby’s fundus dystrophy mutations impair turnover of TIMP-3 by retinal pigment epithelial cells. Hum Mol Genet. 2005;14(23):3579–3586. doi:10.1093/hmg/ddi385
14. Arris CE, Bevitt DJ, Mohamed J, et al. Expression of mutant and wild-type TIMP3 in primary gingival fibroblasts from Sorsby’s fundus dystrophy patients. Biochim Biophys Acta. 2003;1638(1):20–28. doi:10.1016/s0925-4439(03)00036-x
15. Tabata Y, Isashiki Y, Kamimura K, et al. A novel splice site mutation in the tissue inhibitor of the metalloproteinases-3 gene in Sorsby’s fundus dystrophy with unusual clinical features. Hum Genet. 1998;103(2):179–182. doi:10.1007/pl00008707
16. Guan B, Huryn LA, Hughes AB, et al. Early-onset timp3-related retinopathy associated with impaired signal peptide. JAMA Ophthalmol. 2022;140(7):730–733. doi:10.1001/jamaophthalmol.2022.1822
17. DeBenedictis MJ, Gindzin Y, Glaab E, et al. A novel TIMP3 mutation associated with a retinitis pigmentosa-like phenotype. Ophthalmic Genet. 2020;41(5):480–484. doi:10.1080/13816810.2020.1795889
18. Vergaro A, Pankievic M, Jedlickova J, et al. Disease-causing timp3 variants and deep phenotyping of two czech families with sorsby fundus dystrophy associated with novel p.(tyr152cys) mutation. Int J Mol Sci. 2024;25(7):3744. doi:10.3390/ijms25073744
19. Gliem M, Müller PL, Mangold E, et al. Sorsby fundus dystrophy: novel mutations, novel phenotypic characteristics, and treatment outcomes. Invest Ophthalmol Vis Sci. 2015;56(4):2664–2676. doi:10.1167/iovs.14-15733
20. Felbor U, Stöhr H, Amann T, et al. A novel Ser156Cys mutation in the tissue inhibitor of metalloproteinases-3 (TIMP3) in Sorsby’s fundus dystrophy with unusual clinical features. Hum Mol Genet. 1995;4(12):2415–2416. doi:10.1093/hmg/4.12.2415
21. Felbor U, Suvanto EA, Forsius HR, et al. Autosomal recessive Sorsby fundus dystrophy revisited: molecular evidence for dominant inheritance. Am J Hum Genet. 1997;60(1):57–62.
22. Weber BH, Vogt G, Pruett RC, et al. Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby’s fundus dystrophy. Nat Genet. 1994;8(4):352–356. doi:10.1038/ng1294-352
23. Barbazetto IA, Hayashi M, Klais CM, et al. A novel TIMP3 mutation associated with Sorsby fundus dystrophy. Arch Ophthalmol. 2005;123(4):542–543. doi:10.1001/archopht.123.4.542
24. Jacobson SG, Cideciyan AV, Bennett J, et al. Novel mutation in the TIMP3 gene causes Sorsby fundus dystrophy. Arch Ophthalmol. 2002;120(3):376–379. doi:10.1001/archopht.120.3.376
25. Rogers MF, Shihab HA, Mort M, et al. FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Bioinformatics. 2018;34(3):511–513. doi:10.1093/bioinformatics/btx536
26. Schoenberger SD, Agarwal A. A novel mutation at the N-terminal domain of the TIMP3 gene in Sorsby fundus dystrophy. Retina. 2013;33(2):429–435. doi:10.1097/IAE.0b013e318263d3b4
27. Saihan Z, Li Z, Rice J, et al. Clinical and biochemical effects of the E139K missense mutation in the TIMP3 gene, associated with Sorsby fundus dystrophy. Mol Vis. 2009;15:1218–1230.
28. Riera M, Navarro R, Ruiz-Nogales S, et al. Whole exome sequencing using Ion Proton system enables reliable genetic diagnosis of inherited retinal dystrophies. Sci Rep. 2017;7:42078. doi:10.1038/srep42078
29. Sivaprasad S, Webster AR, Egan CA, et al. Clinical course and treatment outcomes of Sorsby fundus dystrophy. Am J Ophthalmol. 2008;146(2):228–234. doi:10.1016/j.ajo.2008.03.024
30. Schwarz JM, Rödelsperger C, Schuelke M, et al. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7(8):575–576. doi:10.1038/nmeth0810-575
31. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi:10.1038/nprot.2009.86
32. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi:10.1038/ng.2892
33. Sievers F, Higgins DG. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018;27(1):135–145. doi:10.1002/pro.3290
34. Biasini M, Bienert S, Waterhouse A, et al. Swiss-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014.Vol. 42;W252–W258. doi10.1093/nar/gku340
35. Cheng J, Randall A, Baldi P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins. 2006;62(4):1125–1132. doi:10.1002/prot.20810
36. Capriotti E, Fariselli P, Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005;33:W306–W310. doi:10.1093/nar/gki375
37. Qi JH, Bell B, Singh R, et al. Sorsby fundus dystrophy mutation in tissue inhibitor of metalloproteinase 3 (TIMP3) promotes choroidal neovascularization via a fibroblast growth factor-dependent mechanism. Sci Rep. 2019;9(1):17429. doi:10.1038/s41598-019-53433-6
38. Cauwe B, Opdenakker G. Intracellular substrate cleavage: a novel dimension in the biochemistry, biology and pathology of matrix metalloproteinases. Crit Rev Biochem Mol Biol. 2010;45(5):351–423. doi:10.3109/10409238.2010.501783
39. Baghirova S, Hughes BG, Poirier M, et al. Nuclear matrix metalloproteinase-2 in the cardiomyocyte and the ischemic-reperfused heart. J Mol Cell Cardiol. 2016;94:153–161. doi:10.1016/j.yjmcc.2016.04.004
40. Yeow KM, Kishnani NS, Hutton M, et al. Sorsby’s fundus dystrophy tissue inhibitor of metalloproteinases-3 (TIMP-3) mutants have unimpaired matrix metalloproteinase inhibitory activities, but affect cell adhesion to the extracellular matrix. Matrix Biol. 2002;21(1):75–88. doi:10.1016/s0945-053x(01)00180-9
41. Pouw AE, Greiner MA, Coussa RG, et al. Cell-matrix Interactions in the eye: from cornea to choroid. Cells. 2021;10(3):687. doi:10.3390/cells10030687
42. Panda SP, Soni U. A review of dementia, focusing on the distinct roles of viral protein Corona and MMP9 in dementia: potential pharmacotherapeutic priorities. Ageing Res Rev. 2022;75:101560. doi:10.1016/j.arr.2022.101560
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.