")
Back to Journals » International Journal of Nanomedicine » Volume 18
Sorafenib-Loaded PLGA Carriers for Enhanced Drug Delivery and Cellular Uptake in Liver Cancer Cells
Authors Caputo TM , Cusano AM , Principe S, Cicatiello P, Celetti G, Aliberti A, Micco A, Ruvo M, Tagliamonte M, Ragone C, Minopoli M, Carriero MV , Buonaguro L, Cusano A
Received 26 April 2023
Accepted for publication 29 June 2023
Published 26 July 2023 Volume 2023:18 Pages 4121—4142
DOI https://doi.org/10.2147/IJN.S415968
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yan Shen
Tania Mariastella Caputo,1,* Angela Maria Cusano,2,* Sofia Principe,1 Paola Cicatiello,1 Giorgia Celetti,1 Anna Aliberti,1 Alberto Micco,2 Menotti Ruvo,3 Maria Tagliamonte,4 Concetta Ragone,4 Michele Minopoli,5 Maria Vincenza Carriero,5 Luigi Buonaguro,4 Andrea Cusano1,2
1Optoelectronics Group, Department of Engineering, University of Sannio, Palazzo Dell’ Aquila Bosco Lucarelli, Benevento, Italy; 2CeRICTscrl Regional Center Information Communication Technology, Palazzo Ex Poste, Benevento, Italy; 3Institute of Biostructure and Bioimaging, National Research Council, Naples, Italy; 4Innovative Immunological Models Unit, Istituto Nazionale Tumori - IRCCS - “Fond G. Pascale”, Naples, Italy; 5Neoplastic Progression Unit, Istituto Nazionale Tumori IRCCS “Fondazione G. Pascale”, Naples, Italy
*These authors contributed equally to this work
Correspondence: Andrea Cusano; Anna Aliberti, Palazzo Dell’Aquila Bosco Lucarelli, Corso Garibaldi 107, Benevento, I-82100, Italy, Email [email protected]; [email protected]
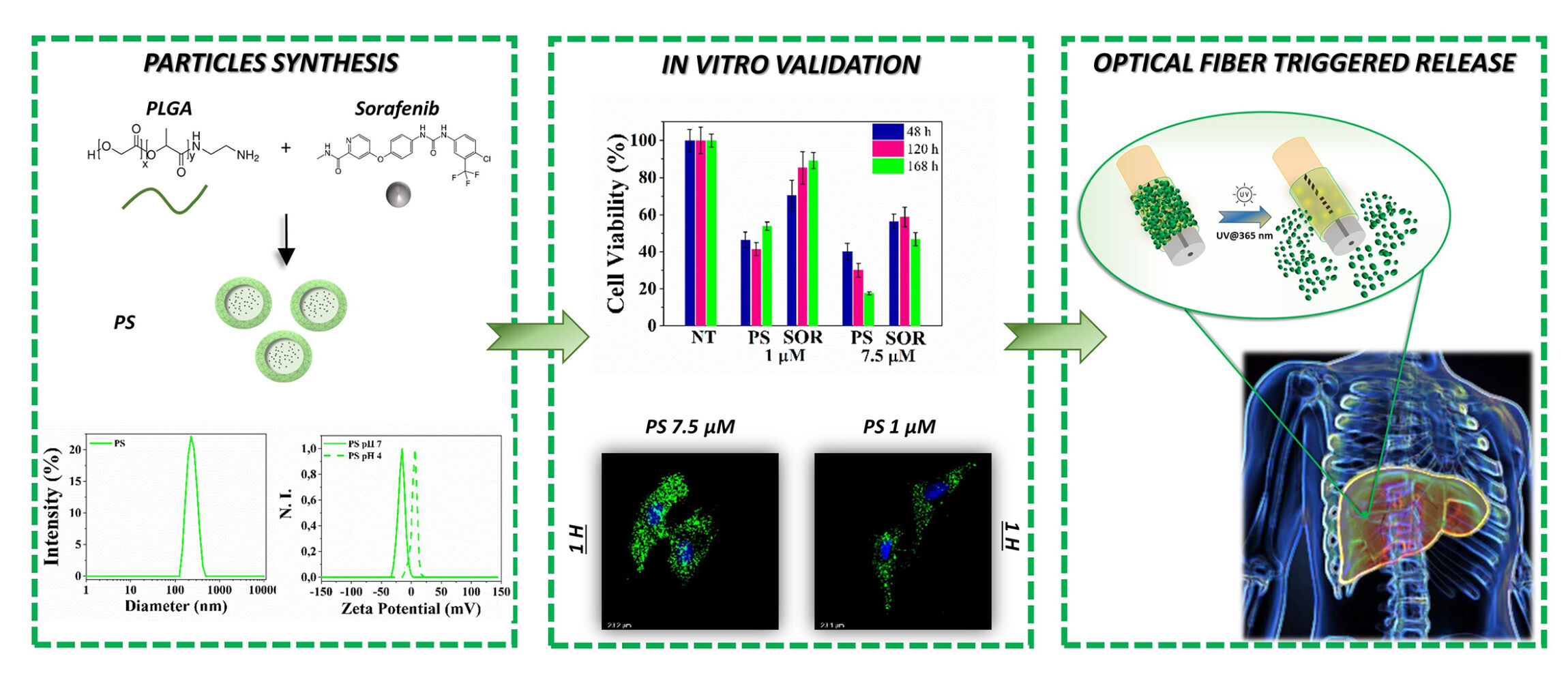
Introduction: Currently, conventional treatments of hepatocellular carcinoma (HCC) are not selective enough for tumor tissue and lead to multidrug resistance and drug toxicity. Although sorafenib (SOR) is the standard first-line systemic therapy approved for the clinical treatment of HCC, its poor aqueous solubility and rapid clearance result in low absorption efficiency and severely limit its use for local treatment.
Methods: Herein, we present the synthesis of biodegradable polymeric Poly (D, L-Lactide-co-glycolide) (PLGA) particles loaded with SOR (PS) by emulsion-solvent evaporation process. The particles are carefully characterized focusing on particle size, surface charge, morphology, drug loading content, encapsulation efficiency, in vitro stability, drug release behaviour and tested on HepG2 cells. Additionally, PLGA particles have been coupled on side emitting optical fibers (seOF) integrated in a microfluidic device for light-triggered local release.
Results: PS have a size of 248 nm, tunable surface charge and a uniform and spherical shape without aggregation. PS shows encapsulation efficiency of 89.7% and the highest drug loading (8.9%) between the SOR-loaded PLGA formulations. Treating HepG2 cells with PS containing SOR at 7.5 μM their viability is dampened to 40%, 30% and 17% after 48, 129 and 168 hours of incubation, respectively.
Conclusion: The high PS stability, their sustained release profile and the rapid cellular uptake corroborate the enhanced cytotoxicity effect on HepG2. With the prospect of developing biomedical tools to control the spatial and temporal release of drugs, we successfully demonstrated the potentiality of seOF for light-triggered local release of the carriers. Our prototypical system paves the way to new devices integrating microfluidics, optical fibers, and advanced carriers capable to deliver minimally invasive locoregional cancer treatments.
Keywords: poly(lactic-co-glycolic acid), sorafenib, emulsion solvent evaporation technique, hepatocellular carcinoma, optical fiber, microfluidics
Graphical Abstract:
Introduction
Liver cancer is one of the most common cancers and the fourth leading cause of cancer death worldwide in 2020. Hepatocellular carcinomas account for approximately 90% of primary liver cancers.1–3 Currently, conventional antitumor drugs lack selectivity for tumor tissue,4–6 and the main barriers to chemotherapy are multidrug resistance (MDR) and drug toxicity.7
In the past few decades, various drug delivery systems,8,9 such as lipid-based nanostructures (liposomes,10,11 lipid carriers12,13) polymer-based nanostructures (dendrimers,14,15 micelles,16,17 polymerosomes,18 nanogels19,20), inorganic nanostructured systems21,22 (gold,23,24 silver25,26 and iron nanoparticles27), and non-metallic nanoparticles (carbon28,29 and silica-based30,31 nanoparticles, quantum dots32) have been developed to: (i) improve the solubility and stability of insoluble drugs; (ii) prevent nonspecific distribution and degradation in vivo, and (iii) increase drug concentration in tumor lesions through the effect of enhanced permeation and retention (EPR).33,34 Most drug delivery systems thus reduce the therapeutic dose by passively delivering the drug into tumor tissue. Similarly, through a sustained and controlled drug delivery, nanoparticles provide more effective tumor cell killing than drug alone.
Among the various polymer-based nanoparticles, those based on poly(lactic-co-glycolic acid) (PLGA) are the most safe and versatile for drug delivery applications. PLGA is a Food and Drug Administration (FDA)-approved biodegradable polymer widely used in cancer nanotechnology. It is also suitable for encapsulation of lipophilic drugs due to its hydrophobicity.35 In addition, the range of applications and the drug loading capacity of PLGA can be expanded by optimizing its structure.36
Since 2007, according to EASL HCC guidelines,37 SOR is recommended as the gold standard first-line systemic therapy for patients with advanced HCC or who fail to respond to loco regional therapies.38–40 SOR is the only systemic therapy proven to extend overall survival when used as a first-line treatment, showing a gradual median improvement over time (from 10.7 months in the SHARP study (2005–2006),38 to 12.3 months in the REFLECT study (2015)41 and over 13 months in the IMbrave-150 study (2018–2019)).42
SOR is a dual aryl multi-kinase inhibitor that inhibits tumor cell proliferation blocking the cell signaling pathway mediated by Raf/MEK/ERK.43 It also indirectly inhibits tumor cell growth interfering with the tyrosine kinase activity of vascular endothelial growth factor receptors, of serine-threonine kinases Raf-1, B-Raf44,45 and platelet-derived growth factor receptor β (PDGFR-β).46,47 Administration of SOR is the most effective single-drug therapy and along with levantinib, among the few systemic treatments48 capable of prolonging overall patient survival and reducing tumor progression in HCC patients. However, its severe side effects49 including hand-foot skin reaction, decreased heart blood flow, heart attack, intestinal perforation, alteration of thyroid hormone levels, loss of appetite, diarrhoea, rash, often lead to treatment discontinuation. Furthermore, SOR is poorly water soluble and is rapidly cleared and metabolized, resulting in low absorption efficiency in tumor tissue50 which severely limit its use for local treatments.51 The high toxicity and low bioavailability of SOR thus result in a narrow therapeutic window.
All these drawbacks may be overcome using nanocarriers like PLGA nanoparticles and targeted nanocomposites52,53 able to selectively deliver the drug to target tissues.54,55 Previous studies reported rather low SOR loading (1.4%) in PLGA nanoparticles using oil-in-water single emulsion solvent evaporation methods.56 Multiblock polymer nanoparticles composed of (poly(lactic acid)-poly(ethylene glycol)-poly(L-lysine)-diethylenetriamine-pentaacetic acid and the pH-sensitive material poly(L-histidine)-poly(ethylene glycol)-biotin were able to encapsulate 2.4% SOR.57 The loading efficiency was improved (5.3%) by the nanoprecipitation dialysis technique using a block copolymer of dextran and poly(D,L-lactide-co-glycolide).58
Drug combinations that provide synergistic effects have also been loaded in nanocarriers.59 As an example, polyvinyl alcohol (PVA)-doxorubicin nanocore particles covered with a thin shell of HSA-loaded SOR reached a drug loading of 2.4%. Also, lipid-polymer hybrid nanoparticles have been prepared for the co-delivery of doxorubicin and SOR to enhance efficacy of HCC therapy.60 However, despite the drug loading achieved in the above studies, cell viability (after in vitro treatment) still ranges from 75% to 50% (at 10 µM after 48 hours). Therefore, new biocompatible carriers with improved encapsulation and cytotoxic efficacy are continuously sought to improve drug bioavailability increasing their absorption in the target tissues and preventing quick degradation. To suppress the side effects of SOR as well as of other drugs,55,61,62 active tissue-specific targeting or controlled loco-regional delivery are the most attractive options that enable selective drug accumulation and minimise interaction with healthy cells. Controlled loco-regional delivery is an intriguing but challenging approach that requires stimuli responsive systems able to release drugs entrapped in suitable carriers following external triggers.
Against this background, a new SOR formulation based on amino-PLGAs and synthesized by an optimized emulsion-solvent evaporation process is here presented. A careful investigation of the morphological, structural, and physicochemical properties of the SOR-loaded PLGA particles (PS) has been carried out, focusing on particle size, surface charge, drug loading content, encapsulation efficiency, in vitro stability, and drug release behaviour. PSs exhibit the highest drug loading (9%) and improved cytotoxicity compared to the most significant SOR-loaded PLGA particles already found in the literature. With a view towards the realization of mini-invasive tools for loco regional delivery of SOR-loaded PLGA particles, a feasibility study has been conducted to demonstrate a light triggered side emitting fiber-optic (seOF) tool for loco regional drug delivery. Our proof of concept relies on a new light-triggered drug delivery system based on side-emitting OF (seOF) integrated with PLGA particles using a specific UV photocleavable linker. The light-triggered release of our PLGA particles has been also tested in a microfluidic device that can be easily integrated into different needle models. This study paves the way to the development of a novel light-triggered drug delivery system for on-demand and site-specific release of drugs.
Material and Methods
Materials
Poly lactic-co-glycolic acid (PLGA) (Amino terminated, 50:50 lactide: glycolide, MW 5.000 Da), Coumarin 6, Tetrahydrofuran (THF), Ethyl acetate (EtOAc), Ethanol (EtOH), Coumarin 6 (C6), phosphate buffer saline (PBS) tablets were purchased from Sigma Aldrich. Sorafenib (SOR) was obtained by Toronto research Chemicals. Water, acetonitrile, Trifluoroacetic acid (TFA) were LC-MS grade (ROMIL Ltd, UK). Deionized water (18.2 MΩ.cm) was obtained from a Milli-Q Plus system (Millipore) with a total organic content (TOC) < 3 ppb.
PLGA Particles Synthesis
PLGA particles were prepared using an oil-in-water (O/W) emulsion. Briefly, PLGA was dissolved in various organic solvents (EtOAc, THF, THF/EtOH) at a concentration of 10 mg/mL. The SOR was added to the oil phase (1 mg, 100 µg, 50 µg to obtain drug/polymer ratio of 0.1:1, 0.01:1, 0.005:1 (w/w) respectively). The oil phase (1 mL PLGA solution) was added dropwise to 10 mL of the water phase (2% (w/v) PVA solution). The emulsion was homogenized (ULTRA -TURRAX® IKA T 10, Germany) at maximum speed (30,000 rpm) for 60 seconds, followed by sonication (Branson Ultrasonics™ Sonifier™ SONIFIER SFX 150, USA) at 40% amplitude for 4 minutes on ice. The solution was then added dropwise to 10 mL of 0.5% (w/v) PVA and stirred overnight to allow the organic solvent to evaporate. Similarly, fluorescent PLGA particles were obtained by adding 0.5 mg of C6 to the oil phase, following the method described above. Finally, all the particles were recovered centrifuged (15,000 rpm, 30 min, 4°C, Heraeus Fresco 21, Thermo Fisher Scientific) and the pellet was washed three times with milliQ water.
Dynamic Light Scattering (DLS) Measurement and Particles Stability
The hydrodynamic diameter and surface potential of the particles were measured using a dynamic light scattering system (DLS, Malvern Zetasizer Nano ZS, 633 nm laser, 173° scattering angle) equipped with a temperature controller. Particles were diluted at a concentration of 50 µg/mL in 700 µL of MilliQ water, and analyses were performed at a fixed temperature of 25°C with an equilibration time of 120 seconds for a total of 5 runs.
Particle stability studies were performed by incubating 1 mg/mL SOR-loaded particles in PBS or DMEM at 4°C and 37°C. At specific times (0, 1, 2, 4, 7, 10, 14 days), the particle stock solutions were diluted and analysed as previously described. All experimental uncertainties represent the standard error of the mean of the replicate runs.
Morphological Analysis by Atomic Force Microscopy (AFM) and Transmission Electron Microscopy (TEM)
Morphological characterization of the PS particles by Atomic Force Microscopy (AFM) was performed directly on a glass slide using an Agilent 5420 AFM system (Agilent Technologies, Santa Clara, CA, USA). AFM images were obtained by scanning the particles in tapping mode to avoid their damage. Samples were prepared by drying a dilute drop of the PLGA dispersions on a 1 cm2 piece of glass slide at room temperature for 3 hours. Raw data collected by AFM were processed using Pico Image software (Keysight Technologies, Santa Rosa, CA, USA). In addition, the morphology of the particles was studied using transmission electron microscope (TEM). Samples were prepared by depositing 5 µL of the particle solution on special TEM copper grids with a carbon coated Formvar film. After evaporation of the solution, the grid was washed with deionized water, and analysis was performed using a TEM FEI Tecnai G2 Spirit TWIN.
Fourier Transform Infrared Spectroscopy (FTIR) Analysis
All PLGA particles (both naked and SOR-loaded) and the SOR were measured by using a Perkin-Elmer Spectrum 3 infrared spectrometer (Perkin-Elmer, Waltham, MA, USA) equipped with an attenuated total reflectance (UATR) accessory. Spectra were acquired by 10 scans with a resolution of 4 cm−1 in the range 650–4000 cm−1, and a series of three replicates of each formulation was analysed.
LC-MS Analysis of SOR
SOR quantification was performed by liquid chromatography-mass spectrometry (LC-MS) using a Thermo LTQ XL mass spectrometer with a Dionex™ UltiMate™ 3000 UHPLC system consisting of a Dionex™ UltiMate™ LPG-3400SD Standard Quaternary Pump, a Dionex™ UltiMate™ Standard Well Plate Autosampler (WPS-3000), and a column oven (TCC-3000) (Thermo Scientific, San Jose, CA, USA). Separation was performed on a reversed-phase C18 BioBasic™ column, 50×2.1 mm ID, 5 μm, equilibrated at 0.2 mL/min and in a column oven at 37°C. The drug was eluted with a gradient from 30% to 80% solvent B (0.1% v/v TFA in acetonitrile) over solvent A (0.1% v/v TFA in water) in 15 min. Under these conditions, the retention time of SOR was 11 min. The mass spectrometer was operated in positive ionization mode. The conditions were set as follows: Capillary temperature 275°C, sheath gas 25 (arbitrary units), auxiliary and sweep gas 10 (arbitrary units), spray voltage 3.5 kV, capillary voltage 7 V, and tube lens 65 V. For the full scan experiments, the mass range was set between m/z 200 and 600, and the most intense ion was selected to obtain the MS2 product ions. The MS2 scan was performed at normal scan rate in a range between 125 and 500 m/z, the isolation width was ± 3.5 m/z of the parent ion, and the activation times, activation Q, and normalized collision energies were set to 30 ms, 0.25, and 20%, respectively.
The calibration line, prepared in triplicate, was obtained by integrating the area of the MS2 peak at 425.19 m/z with respect to the concentration of the compound in a range between 31 ng/mL and 500 ng/mL.
Determination of the Encapsulation Efficiency and Drug Loading
Encapsulation efficiency (EE) and drug loading (DL) were quantified using LC-MS. Specifically, a given amount of particles was dissolved in DMSO (10 mg/mL) and the solution was centrifuged at 14,000 rcf (Fresco™ 21, Thermo Scientific™) for 20 minutes. The supernatant was then collected and 5 µL of the solution was injected for quantification. EE was calculated as the ratio between the amount of SOR contained in the particles and the theoretical drug content according to the following Equation (1):
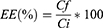
where Ci is the theoretical concentration of the added SOR and Cf is the concentration of the drug recovered in the supernatant after centrifugation of the particles.
DL was calculated according to the following Equation (2):
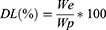
where We is the weight of the SOR encapsulated and Wp is the weight of particles used.
In vitro Drug Release Profile
In vitro drug release was evaluated by dialysis bag method. Briefly, 0.1 mg PLGA particles were mixed in 100 µL PBS and placed in a dialysis microfilter (3.5 K MWCO, Thermo Scientific™ Pierce™). The microfilter was immersed in 1 mL of PBS at 37°C and after a predetermined time interval (1.5, 2.5, 5.5, 7.5, 24, 72, 144, 216 hours) the solution was completely recovered and replaced with 1 mL of fresh solution. Finally, the particle solution was recovered from the microfilter, centrifuged to remove the supernatant, and the collected particles were dissolved in DMSO. All recovered aliquots were analysed by LC-MS.
In vitro Cytotoxic Effect by MTT Assay
The human HepG2 hepatocellular cell line (ATCC, HB-8065 -) was cultured in DMEM medium containing 10% heat-inactivated FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin (Invitrogen, Carlsbad, CA) at 37°C in an incubator humidified with 5% CO2. SOR was previously dissolved at a concentration of 100 mM in DMSO and then immediately before use in 150 µL of DMEM culture medium supplemented with 10% FBS for all dilution points.
HepG2 cells were plated out in 96-well plates at a density of 6000 cells/well in 100 μL of the appropriate complete medium and incubated overnight for adherence. At time 0, cells were treated with increasing doses of SOR (1 µM; 2.5 µM; 5 µM; 7.5 µM; 10 µM). Subsequently, the cells were incubated with each of the drug concentrations for 24 or 48 hours. After the specified time, the medium was removed and the viability of the cells was evaluated by MTT viability assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide. Specifically, a 10% MTT solution (12 mM) was added to the cells at 37°C for 4 hours. The solution was removed and 50 μL DMSO was added to the cells for 10 minutes. The plate was read at 595 nm using an ELISA reader.
The cytotoxic effect induced by PLGA particles was evaluated in HepG2 cell line with the drug encapsulated in. Specifically, 6×103 HepG2 cells were plated in 96 multiwells and incubated overnight. Cells were treated for 48 hours with an increasing concentration of naked particles (PN) corresponding to the particle amount required for loading with SOR of 500 nM, 1 µM, 2.5 µM, 5 µM, and 7.5 µM. Similarly, drug loaded particles (PS) and free drug (SOR) (1 µM and 7.5 µM) were incubated with HepG2 for 48, 120 and 168 hours. After incubation, the medium was removed and cell viability was assessed using the MTT viability assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) as previously described.
The percentage of viable cells was calculated using the equation below, with untreated cells considered as the control with 100% cell viability.
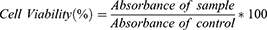
The results were expressed as mean values ± standard deviation of three independent measurements.
Statistical Analysis
Comparisons between individual data points were performed with the two-sided Student’s t-test and ANOVA, as appropriate. Normally distributed data were represented as mean ± S.E.M. Two-way ANOVA and Bonferroni post-hoc analysis were used to examine the significance of differences among groups. All P values were two tailed and considered significant if less than 0.05.
Cellular Uptake Analysis
HepG2 cells (2 x 104/sample) were seeded on glass coverslips and cultured for 24 hours in growth medium containing coumarin-6-loaded PLGA particles (PC6). Two PC6 concentrations corresponding to 1 μM and 7.5 μM SOR when encapsulated in the particles (PS) were used for this analysis. Internalization was assessed at different time points (1, 3, 6, 18, 24, and 48 hours). At each time point, slides were washed with PBS and fixed with 2.5% formaldehyde in PBS for 10 minutes at 4°C. Nuclear staining was performed with 4-6-diamidino-2-phenylindole dye (DAPI). Images were acquired using confocal laser scan microscopy (STELLARIS 8 -Leica Microsystems). A white light laser tunable between 440 and 790 nm was used for excitation and the samples were focused with a HC PL APO CS2 40x/1.10 water immersion objective. Emission signals were acquired using Power HyD detectors. The system was controlled using Leica Application Suite (LAS) X v4.3 software. The excitation wavelength and detection window for each fluorophore are given in parentheses: DAPI (405 nm; 425–499 nm); C6 (464 nm; 480–550 nm). All samples were acquired in triplicate.
For Z-stack analysis, cells were fixed with 2.5% formaldehyde to visualize the cytoskeleton, permeabilized with 0.1% Triton X-100 for 10 minutes at 4°C, and incubated with 0.1 μg/mL rhodamine-conjugated phalloidin (Sigma-Aldrich) for 40 minutes. In all cases, coverslips were mounted with 20% (w/v) Mowiol and visualized with the Axiovert 200 M inverted fluorescence microscope connected to a video camera or with the 510 confocal microscope META-LSM (Carl Zeiss).
Proof of Concept: “Towards Light Activated Drug Delivery Systems for Locoregional Drug Delivery”
This section described the main steps required to develop engineered OF to obtain light triggered drug delivery systems.
Fabrication and Characterization of the seOF Probe
To fabricate the seOF probes, a 3 cm trunk of a commercially available side-emitting fiber (Corning Fibrance 170) is spliced to a multimode fiber used as the launch fiber.
The side-emitting fiber has a core diameter of about 170 µm and a removable polymer cladding of 230 µm. The selected multimode fiber, on the other hand, has a core diameter of 200 µm and a cladding diameter of 220 µm (Thorlabs FC200UEA), which match the core of the side-emitting fiber. First, the OF were stripped from the coatings in the case of the launch fiber and from the cladding in the case of the side-emitting fiber, and then rinsed with isopropyl alcohol. In the second step, the multimode fiber and the side-emitting fiber were spliced (Figure 1a) using an arc fusion splicer (Fujikura FSM-100P+) capable of handling OF with cladding diameters larger than the usual 125 µm. In the final step, the side-emitting fiber was cleaved with an optical fiber cleaver (Fujikura CT −104) at a distance of 3 cm from the splicing point.
 |
Figure 1 Image of seOF splicing process (a) and schematics of the characterization setup (b). |
To obtain an optimized device and achieve the lowest possible power loss, the stripping, cutting, and splicing of the side-emitting fibers were optimized because the fibers are completely different from the standard single/multimode fiber in terms of both size and materials.
The fabricated seOF were optically characterized to determine the power density on the probe side surface and the scattering efficiency. Measurements were performed using an integrated sphere (Thorlabs S140C) in conjunction with an optical power meter (Thorlabs PM100D).
Based on:
- the output power of the launch fiber, measured before splicing the side-emitting fiber and labelled Pin in Figure 1b;
- the powers measured at the output of the probe after the fabrication process, indicated by Ptip and Ptot in Figure 1b, indicating the powers emanating from the tip only and the total power emanating from the entire probe, respectively;
the power scattered at the probe Pscatt was calculated as the difference Ptot - Ptip, and the scattering efficiency was then evaluated as the ratio between Pscatt and Pin.
SeOF Functionalization
The functionalization strategy used for anchoring the carriers (PC6) to the seOF surface involves four main steps: i) hydroxylation, ii) silanization, iii) photocleavable linker anchoring, and iv) covalent binding of the carriers. For the hydroxylation step, the seOF was first incubated in the piranha solution (H2O2:H2SO4 30:70) for 30 minutes at 25°C, then immersed in 12 M HCl for 1 hr at 25°C and 0.1 M NaOH for 1 hr at 50°C. Finally, the hydroxylated fiber was washed with pure ethanol. Silanization was performed by incubating the seOF in 3-azidopropyltriethoxysilane (AZTES) 67 mM, previously dissolved in pure ethanol, overnight at 4°C. The photocleavable linker was anchored by strain-promoted azide-alkyne cycloaddition (SPAAC) reaction between the azide molecules on the seOF and the dibenzocyclooctyne (DBCO) of the photocleavable DBCO-PC-NHS ester reagent (PC DBCO-NHS Ester I, Click chemistry tools, Cat# 1160). The latter was concentrated 0.5 mM in DMSO and incubated with the seOF overnight at 25°C. Finally, in the fourth step, the seOF was immersed in the amino PC6 solution (2.5% w/v in borate buffer 50 mM pH 8) through a dip coater (KSV NIMA KN4001, Biolin Scientific Oy, Espoo, Finland – speed 1 mm/min, speed 0.5 mm/min) for 30 minutes. The seOF was carefully rinsed three times with milliQ water and analysed using CLSM and an optical microscope (Olympus BXFM upright).
Particle Release Mediated by Photocleavage in Batch and in Microfluidic Chip
Particle release was tested by both batch and microfluidic methods. For batch testing, the PC6-functionalized seOF was connected to a fibre-coupled high-power light source (Mightex FCS −0365-201) emitting UV radiation with a wavelength of 365 nm and a maximum radiant flux of 200 mW inside a fiber with 1 mm core diameter and 0.22 NA. Then, the seOF was directly immersed in a quartz cuvette (10 mm low-volume quartz - 400 µL) and the laser source was activated (at maximum power) to allow photocleavage of the linker and release of PC6. At specific time points, the laser was turned off, and the solution was collected and replaced with fresh medium. All aliquots were analysed using a fluorometer (Fluoromax-Plus -Horiba Scientific). Fluorescence emission spectra were recorded in the spectral range from 475 to 600 nm with an integration time of 0.1 s and a step size of 1 nm at an excitation wavelength of 464 nm. All spectra were recorded with a slit width of 5 nm bandpass in both the excitation and emission.
The 3D-printed microfluidic device (SanChip S.r.l, Italy) consists of two inlets and one outlet. The main channel is used to integrate the seOF into the chip and the side channel for the buffer solution. The main channel is 4 cm long, 3 mm in diameter, and has an inlet with a plug that has a single (or multiple) hole (250 µm diameter) to allow central positioning of the fiber in the chip. The buffer solution flowing through the side channel is used to ensure hydration of the polymer carriers present on the surface of the seOF probe and to expel them through the outlet once the seOF is activated. In short, the seOF functionalized with the carriers remains hydrated in the central channel for a certain time, and once the photocleavage is activated the solution containing the carriers is collected at the outlet and analysed, as described for the batch procedure.
The outlet is equipped with an adapter for a needle (or catheter) to deliver the vectors to the site of interest. The entire device is 6.5 cm long, 3 cm wide and 1 cm high.
Results
Synthesis, Characterization and Drug Release Profiles of PLGA Particles
PLGA particles were synthesized by oil-in-water (O/W) emulsion method. The PLGA used for particle synthesis is characterized by low molecular weight (MW 5000 Da) with a 50:50 ratio of lactic and glycolic acid. Additionally, the copolymer is terminated with a primary amine for further functionalization. In order to obtain monodisperse particles with high encapsulation efficiency, the synthetic parameters were carefully selected, especially the best solvents, the presence of co-solvents, the concentration of surfactant, the ratio between the oil/water phases and between polymer/drug concentration.
As shown in Figure 2a, two organic solvents were used as oil phase: PSs prepared using EtOAc have a diameter of 300 ± 9 nm, while the particles prepared with THF have a size of 267 ± 13 nm. The addition of ETOH as co-solvent further reduces the particle diameter to 248 ± 6 nm. The ratio between the drug and the polymer influences the amount of SOR loaded in the particles, as shown in Figure 2b. The more SOR is contained in the oil phase, the more drug is introduced into the core of the particles, resulting in encapsulation efficiencies (EE) of 89.7 ± 13.5%, 70.8 ± 4.8%, 48.4 ± 1%, and drug loading (DL) of 8.9 ± 1.3%, 0.7 ± 0.05%, 0.2 ± 0.01% for the formulations prepared with SOR:PLGA ratio of 0.1:1, 0.01:1, and 0.005:1, respectively. Once ultimate the emulsion formulation, all particles were analysed by Dynamic Light Scattering (DLS) to check their size, homogeneity, surface charge and stability (Figure 3 and Table 1).
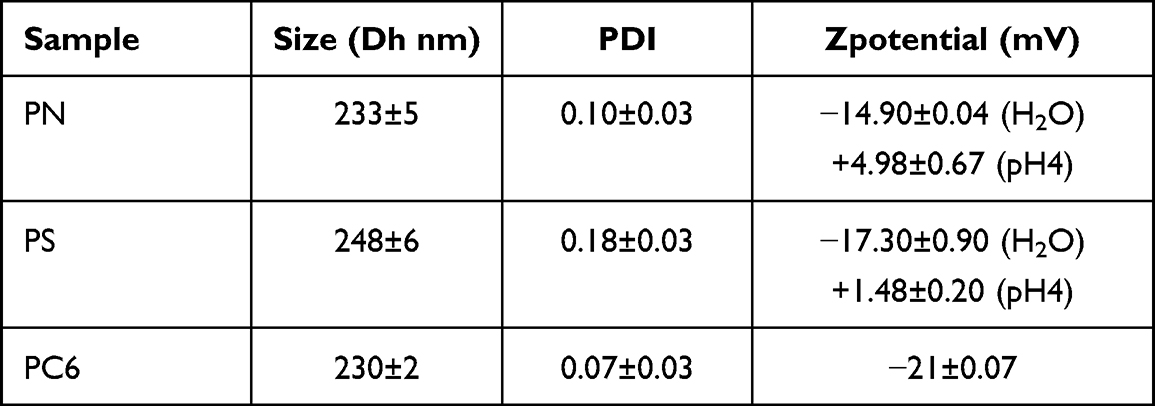 |
Table 1 DLS Measurements of PLGA Particles |
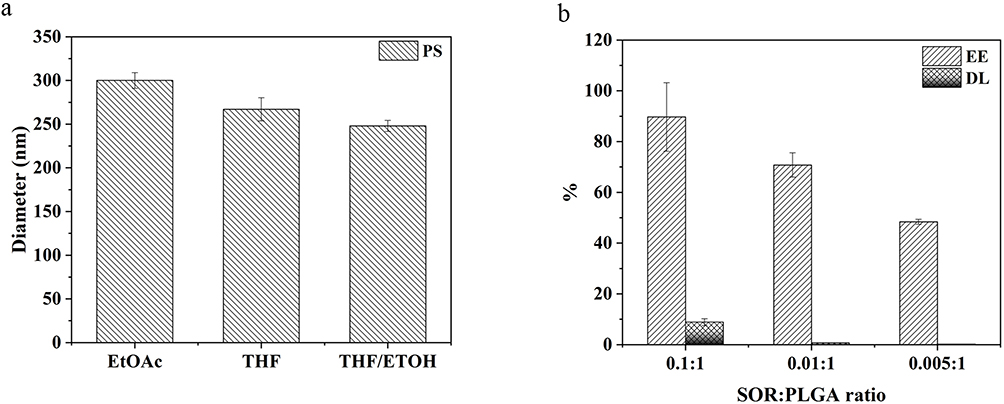 |
Figure 2 (a) Solvent effect on the particle size; (b) encapsulation efficiency (EE) and drug loading (DL) measured for different SOR: PLGA ratio. |
 |
Figure 3 PLGA particles analysis by DLS: (a) PN, (b) PS, (c) PC6 Particle diameter measurements; (d) PN, (e) PS surface charge quantification and (f) PS stability in DMEM and PBS at different temperatures and over time. |
In particular, particles not loaded with the drug (PN-naked or simply PN) have a diameter of 233 ± 5 nm and a polydispersion index (PDI) of 0.1. The surface charge of PN particles in milliQ water is −14.90 ± 0.04 mV, while in an acidic medium (pH 4) the protonation of the primary amines causes an increase in the surface charge to + 4.98 ± 0.67 mV (Δ = + 19.88). Compared to PN, PS particles are characterized by a larger size (248 ± 6 nm) and a lower surface charge (−17.30 ± 0.90) as further indication of drug encapsulation. The surface charge of PS changes from −17.30 ± 0.90 mV in milliQ water to +1.48 ± 0.2 mV in acidic medium. Finally, particles loaded with the dye coumarin 6 (PC6), synthesized with the aim of studying the particle’s kinetics of internalization in cells, have a diameter of 230 ± 2 nm, a polydispersion index (PDI) of 0.07, and a surface charge of −21 ± 0.07 mV.
The particle stability was studied in different media (DMEM and PBS) and at different temperatures (4°C and 37°C). As shown in Figure 3f, the PS particles remain stable in both buffers and at both temperatures for 6 days at least in terms of overall size. Then, those incubated in DMEM (4°C/37°C) and in PBS (4°C) gradually decrease in size, undergoing a diameter reduction of 5.26%, 6.88% and 4.05%, respectively, after 14 days. Conversely, particles solubilized in PBS at 37°C show a slight increase in their size with a maximum diameter of 259 ± 6 nm after 14 days of analysis.
The PS particles were imaged using Atomic Force Microscopy (AFM) (Figure 4a). The strong interaction between PLGA and glass allowed the AFM analysis on dried nanoparticles, as already reported in previous studies.63 The morphological studies show uniform and spherical shaped discrete particles without aggregation and smooth in surface morphology. The AFM height profiles (Figure 4b) reveal the spherical shape as well as the low polydispersity. A comparison of AFM imaging technique to DLS measurements shows that the particle diameters obtained from AFM (≈ 420 nm) are slightly different compared to those measured by DLS (≈ 250 nm). This effect can be mainly attributed to the fact that the AFM measurements were performed against air at room temperature and in dried state where different effects like interactions with the substrate, the stiffness of the nanoparticles can occur; instead, DLS measurements were performed in buffered solutions and at constant temperatures.64
 |
Figure 4 (a) AFM analyses of PLGA particles deposited on a gold surface. (b) Height profiles of the selected PLGA particles (white dot lines in the topographic images) obtained from AFM measurements in tapping mode. (c) Transmission Electron Microscope images of PLGA particles (scale bar 200 nm and 500 nm). |
Moreover, Transmission Electron Microscope (TEM) images (Figure 4c) show PLGA particles spherical and homogeneous in their morphology without superficial fractures or superficial anomalies. In this case, the particle diameter is consistent with the one previously evaluated by DLS.
The successful synthesis of the particles and encapsulation of the drug were also confirmed by Fourier Transform Infrared Spectroscopy (FTIR) measurements. In Figure 5a are reported the infrared spectra of PN and PS particles and of the pure SOR. As predictable in an FTIR spectrum, there are many peaks in the range between 1600 and 600 cm−1 due to the stretching and bending vibrations of C-C and C-O bonds. The FTIR spectrum of the free SOR (Figure 5 - green line) shows two characteristic bands at 3335 and 3299 cm−1 that are due to the N-H stretching (Figure 5b). In addition, there is a band at 3079 cm−1 related to the C-H stretching and one at 1706 cm−1 accounting for the amide C=O group. Several additional bands between 1643, 1553, 1505, 1481 and 1330 cm−1 are visible and are related to C=C, N-H, C-N stretching, respectively (Figure 5c). All these bands are also found in the spectrum of PS particles (see Figure 5c - red line). The black line (PN) and the red line (PS) have a common large peak at about 1750 cm−1 corresponding to the stretching vibrations of the ester C=O bond, which is very abundant in the PLGA structure.
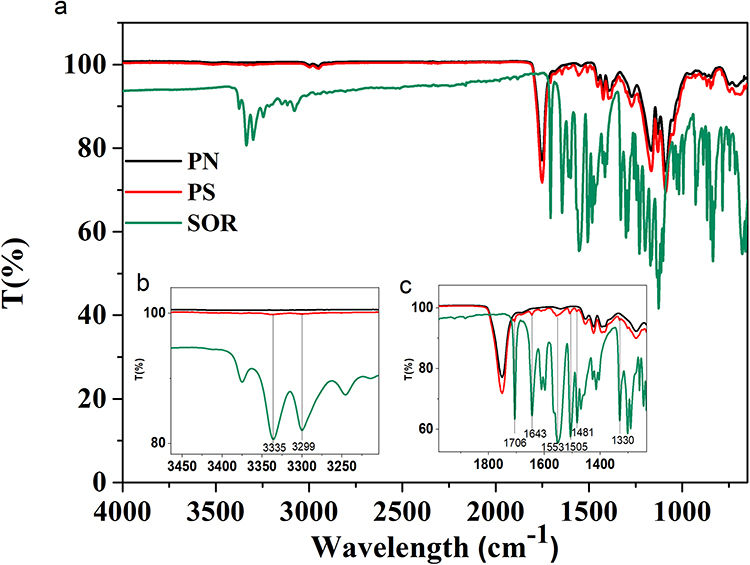 |
Figure 5 FTIR-ATR analysis of PLGA particles and pure SOR (a). The insets show a spectrum zoom between 3500–3200 (b) cm−1 and 2000–1230 cm−1 (c). |
An appropriate LC-MS/MS method has been set up to quantify the amount of SOR released from PS by monitoring the drug fragment at 425.19 m/z. The in vitro release profile is shown in Figure 6. The main graph shows that 100 µg of particles PS (SOR:PLGA 0.1:1) dissolved in 1 mL of medium can release about 90 ng/mL (193.4 nM) in the first 1.5 hours and 272 ng/mL (585.2 nM) after 24 hours. In the following days, the drug release is slow and characterized by a constant release of 25 ng/mL drug per day up to 72 hours. After 9 days, the total amount of drug released is 342 ng/mL (735.8 nM, Figure 6) corresponding to about 3.8% of the total encapsulated SOR (8.9 µg). In the inset of Figure 6 the cumulative time-dependent release of SOR normalized to the drug loading is also reported and compared to other two PS preparations differing in the drug/polymer ratio (SOR:PLGA 0.01:1 and 0.005:1). These data show that, although the absolute amount of SOR cumulatively released is similar for the three preparations (around 0.3 µg/mL; see also Table S1), the values normalized to the drug loading vary from 3.8% for SOR:PLGA 0.1:1, to about 40% for SOR:PLGA 0.01:1 and to 100% for SOR:PLGA 0.005:1. The overall data show that, using SOR:PLGA 0.1:1, only a very small amount of drug is lost through passive diffusion, while approximately 96% is retained in the particles after 220 hours and can therefore function as a stable reserve to be released by alternative and more controlled methods, such as the one proposed in this paper (see below). On the basis of these considerations, the formulation PS (SOR:PLGA 0.1:1) (hereafter abbreviated with only PS) was selected for further studies on the SOR-sensitive HCC cell line.
 |
Figure 6 SOR cumulative release in PBS at 37°C measured for the PS formulation with highest drug loading (SOR: PLGA 0.1:1). In the inset are reported the cumulative release expressed as percentage for all the formulations studied in this work. |
In vitro Cytotoxic Effect of Free SOR on HCC Cell Line
The HCC cell line HepG2 was used to investigate the sensitivity of HCC cells to SOR. Cells were treated with a gradient of SOR concentrations at different time points and then cell growth inhibition was determined using the MTT assay. SOR is capable of inducing cell death in a dose- and time-dependent manner (Figure 7a). In particular, the EC50 value is reached at 7.5 µM after 48 hours (Figure 7b). The EC50 value observed at 48 hours is fully consistent with a previous study conducted by Wei et al.65 Considering the cytotoxic concentration range analysed for the free SOR, the lower value tested (1 µM) and the one corresponding to the EC50 value (7.5 µM) were used to further evaluate the particle cellular uptake and cytotoxicity.
 |
Figure 7 SOR EC50 determination. (a) Free SOR cytotoxic effect at different times (24h and 48h) with different drug concentrations, (b) “Dose – response curve” of the cells treated with different concentrations of SOR after 48h. 100% represents the viability of untreated cells. |
PLGA Particles Uptake in Human HCC Cell Line by Confocal Laser Scan Microscopy (CLSM)
In order to demonstrate the ability of PLGA particles to enter cells and thus carry the drug within them, the intracellular distribution of fluorescent PLGA particles (PC6) was assessed by CLSM in HepG2 cells. Fluorescence microscopy analysis revealed that PC6s show time- and concentration-dependent internalization, as indicated by the appearance of a green fluorescent signal that progressively pervades the entire cytoplasm. Significant internalization of PC6 at a particle concentration corresponding to PS 7.5 μM (Figure 8a) is already observed after only one hour of incubation.
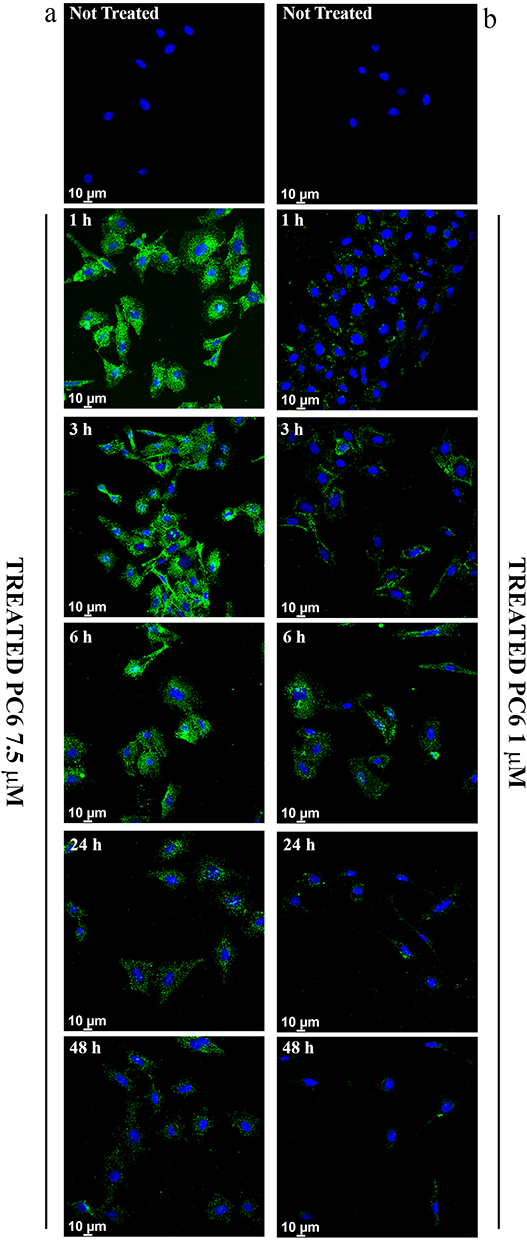 |
Figure 8 Cellular Uptake analysis of PC6 by HepG2 at 7.5 µM (a) and 1 µM (b) particles concentration for different time points (1h to 48h). |
Massive internalization is observed during the first 6 hours at a concentration of 7.5 μM. The time-dependent internalization of PC6 by HepG2 exposed to 1 μM (Figure 8b) and 7.5 μM PC6 for 1 hour and 6 hours was further confirmed by z-stack analysis of confocal images acquired at 0.1 μm intervals through the cell (Figure 9 and Supporting Information SI 1. Figure S1). Internalization of 7.5 μM PC6 occurs after only 1 hour (Figure 9b). After 6 hours, the cells exhibit diffused intra-cytoplasmic fluorescence, as shown in the 3D projections (Figure 9d). In contrast, the images collected after 1 hour of incubation with 1 μM PC6 show a slower internalization of the particles, which takes about 6 hours to invade the cytoplasm (Figure 8b) as also observed in the 3D projections (Figure 9a–c).
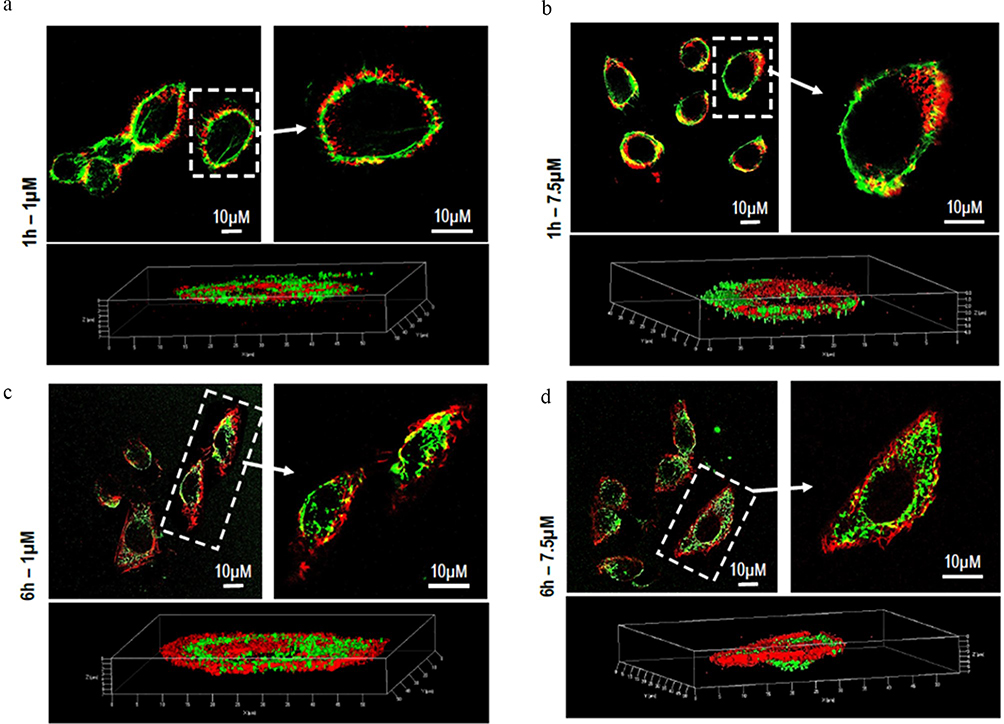 |
Figure 9 Z-series images of PC6 Particle internalization by HepG2 at 1µM (a–c) and 7.5 µM (b–d) after 1h and 6h of treatment. |
In vitro Cytotoxic Effect of PLGA Particles on HCC Cell Line
To investigate the safety of PNs and the capability of PSs to enhance SOR toxicity, the cytotoxic effect of PLGA particles on the HepG2 cell line was evaluated. For this purpose, cells were treated for 48, 120 and 168 hours with particles at concentrations corresponding to 1 µM and 7.5 µM of free SOR.
Although PNs have a minimal effect on cell viability at the highest concentration tested (88%) (Figure 10a), PS particles induce a significant increase in cytotoxic effect at both concentrations tested. Compared to cells treated with free SOR, those exposed to the PS formulation at 1 µM undergo a more extensive reduction of cell viability after 48 hours (46% vs 70% of cell viability). After 168 hours of treatment, PS at 1 µM, cell viability was reduced to 53.7% compared to 89% attained with free SOR (Figure 10b). Moreover, PSs at 7.5 µM cause cell death overtime and reach 17.4% of cell viability after 168 hours of treatment, while free SOR at the same concentration reduces viability at only 50%. Altogether, the data suggest the strengthened ability of the particle to efficiently protect the SOR inside its hydrophobic core and stably deliver the drug into cells (Figure 10b).
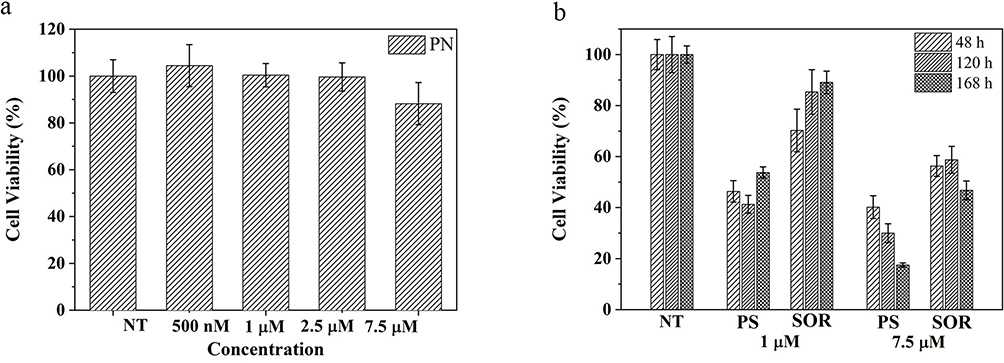 |
Figure 10 In vitro cytotoxicity studies: (a) Viability of HepG2 cells cultured with: (a) empty PN without SOR after 48 hours, and (b) various concentrations of PS and free SOR after 48, 120 and 168 hours. NT correspond to not treated cells. |
Proof of Concept: “Towards Light Activated Drug Delivery Systems for Loco-Regional Drug Delivery”
Light is a promising release stimulus because of its tunable features (wavelength and power density). However, the lack of reliable procedures to use light as triggering mechanism for drug release limits its use for loco regional drug delivery platforms in clinically relevant scenarios.
Among light waveguides, optical fibers (OF) represent an attractive option to guide light and trigger drug release. They benefit from their size, biocompatibility, and the capability to be easily integrated into needles, catheters, and nanoendoscopes.66 Moreover, OFs can be easily integrated into microfluidic devices, providing a solid basis for the development of mini-invasive smart platforms for light-activated loco-regional drug delivery.67,68
Although OF tip was used as an intrinsically light coupled platform for light mediated drug delivery, the reduced payload poses severe limitation for its practical use in clinically relevant scenarios.69,70 A larger payload can be achieved by immobilizing carriers on the cylindrical surface of optical fibers (larger surface area compared to the optical fiber tip), but special light coupling mechanisms are required to efficiently spread light from the fiber core to the cladding and activate the carrier release. Different coupling methods were developed, including core offset, Fiber Bragg Gratings (FBG) with tapers, Long Period Gratings (LPG), tilted FBGs, etc.71–79
Here, we propose, for the first time, the use of seOFs as a coupling method to efficiently scatter light towards the outer surface of the optical fiber and activate PLGA particles through a suitable photocleavable linker, as schematically shown in Figure 11.
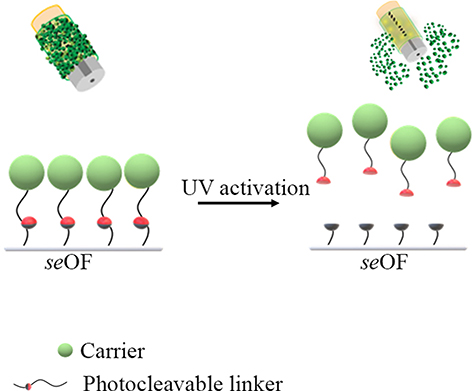 |
Figure 11 Scheme of the light activated drug delivery systems for loco-regional drug delivery. |
The seOFs are characterized by the presence of scatterers inside the core, which promote the dispersion of the transmitted radiation from the core through the lateral surface enabling the light-mediated release of our particles. Due to the intrinsic capability of seOF to scatter guided light through the fiber towards the surroundings, we designed the overall fiber architecture to achieve the required optical power density at the fiber surface to activate the cleavage of a spacer arm containing a photocleavable moiety. In this way, carriers linked by amine-reactive crosslinker chemistry can be efficiently photoreleased using low-intensity UV light (at 365 nm, 1–5 mW/cm2 from product datasheet).
To achieve this goal, a careful design was required to take into account all the parameters involved, followed by a careful validation and optimization phase.
Fabrication and Characterization of the seOF Probe
The seOF fabricated and used for the tests were characterized in terms of scattering efficiencies (Figure 1b). The samples used for the batch tests show an average efficiency (and power density) of 32 ± 1.8% (6 ± 1.5 mW/cm2) for the light-activated controls and 29.8 ± 6.3% (4.5 ±1.2 mW/cm2) for the non-light-activated controls. These values are highly reproducible for all prepared seOFs. Similarly, the optical fibers used for the microfluidic experiment reported in Figure 1f achieve average efficiencies and power densities of 29.5 ± 4.7% (5.5 ± 1.8 mW/cm2) and 31.2 ± 3.4% (4.7 ± 0.8 mW/cm2) for the light-activated and non-activated fibers, respectively.
These efficiencies were obtained using the standard splicing technique (Figure 1a) described in the Methods section. In Figure 12c, a spotlight can be seen near the splicing point. This spotlight can be attributed to the scattering losses that occur at the interface between the multimode optical fiber used to launch UV light and the side-emitting fiber. Further improvements can be achieved by optimizing the light coupling between the multimode fiber and the side-emitting one. Indeed, these optical fibers have different core diameters (see Methods section for details) and require advanced splicing procedures to taper the diameter of the multimode fiber from 220 µm to 170 µm, which is the diameter of the side-emitting fiber responsible for the light-activated release of carriers.
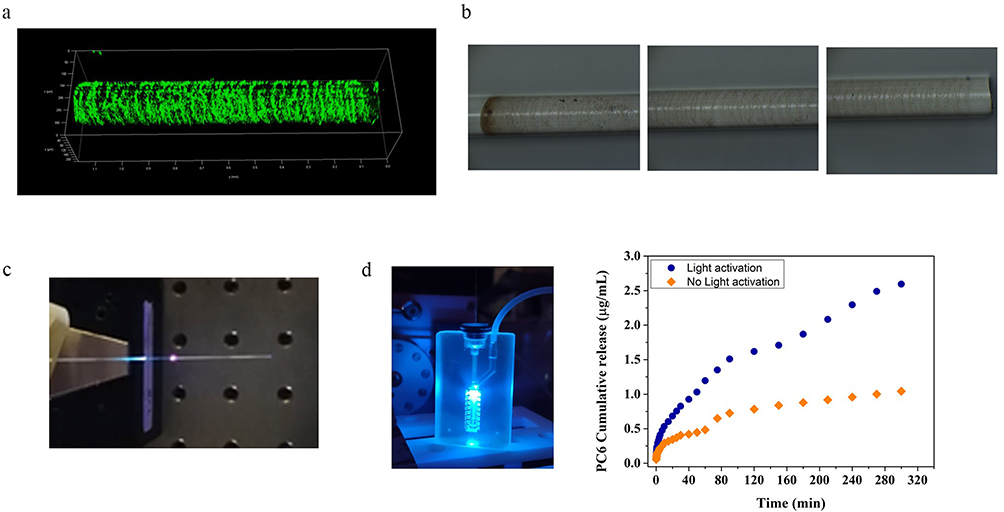 |
Figure 12 Development of particle loaded seOF-microfluidic integrated systems for light triggered locoregional drug delivery: (a) CLSM and (b) optical microscope image of seOF after PC6 immobilization; (c) image of the assembled seOF; (d) particle loaded seOF microfluidic device integrated with seOF and correspondent particle release profile. |
Using this method, a taper with a length of 7 mm was realized, resulting in an increase of the scattering efficiency to about 47% with a power density at the surface of the fiber of about 9 mW/cm2.
SeOF Functionalization with Particles
The protocol required for fiber functionalization resulted from a careful study, first performed on planar surface (see Supporting SI 2 and Figure S2) and then successfully transferred to the seOF surface. To monitor the first two steps of functionalization (hydroxylation and silanization), the wettability of the glass surface was evaluated using the water contact angle (WCA). The analyses indicate that the optimised protocol increases the hydrophilicity after the hydroxylation step (WCA of 17°) and the hydrophobicity after silanization (WCA 70°) compared to the other tested conditions (Figure S3). Silanization is further confirmed both by ELISA assay (Figure S4) and the distinctive N3 band at 2100 cm−1 detected by FTIR measurements (Figure S5). The PC DBCO-NHS ester reagent was selected to act as a linker between the fiber and the particles, as envisaged in the third step. It contains a dibenzocyclooctyne (DBCO) moiety that reacts with the azide (N3) group of the 3-Azidopropyltriethoxysilane (AZTES) by a strain-promoted azide-alkyne cycloaddition (SPAAC) reaction, and a reactive amine group connected through a spacer arm containing a photocleavable moiety. The FTIR spectra (Figure S5) collected after the SPAAC reaction show, as expected, a decrease in the intensity of the characteristic N3 band, due to the formation of the stable triazoles. The last step consists in the integration of the fluorescent amino-PLGA particles – PC6. After 30 minutes of incubation, successful integration of PLGA carriers was confirmed both on the planar surface by AFM and fluorescence analysis (Figure S6), and on seOF by CLSM and optical microscopy images (Figure 12a–b).
The particle release was assessed by quantifying the fluorescence emission of PC6 after each run of activation. Assays with the PC6 functionalized seOFs prove that light activation results in the release of 2.67 ± 0.38 µg/mL of particles compared to the 0.9 ± 0.12 µg/mL observed with the not activated control in batch (Figure S7). The results observed with the not activated sample can be explained considering two aspects: i) a small amount of dye can be released from the bound particles during the incubation due to polymer hydrolysis in aqueous phosphate buffer, and ii) some particles that are not covalently bound can be detached from the seOF surface.
The seOF was then integrated into a microfluidic device (Figure S8) designed to precisely position the seOF in its main channel (Figure 12d). The seOF was inserted in the chip through a first inlet, appropriately modified to ensure tightness of the fiber and avoid fluid leakage, while the buffer solution was perfused through a second inlet at a flow rate of 50 µL/min using a peristaltic pump (403U/VM2 10 RPM - Watson-Marlow). The outlet of the device features an integrated adapter to which the needle can be hooked. This integrated seOF- microfluidic system allowed to control the light mediated carrier release and their delivery to the site of interest thanks to the modified outlet. Thanks to its optimized design, the device was able to accommodate multiple seOFs (up to 5) in its main channel, allowing for customizable carrier release.
Remarkably, the results of the release experiments performed in microfluidics are in full agreement with those of the batch experiments. Light activation leads to higher particle release (2.6 ± 0.27 µg/mL) compared to the amount obtained without activation (1.04 ± 0.1 µg/mL) (Figure 12d).
Discussion
In this work, polymeric biodegradable PLGA-based particles with improved drug loading and rapid cell internalisation were synthesised and tested in vitro to enhance the therapeutic effect of SOR on HepG2 cells of hepatocellular carcinoma. Among the various available techniques, PLGA particles were prepared by emulsion solvent evaporation. The choice of the technique is critical as it impacts on particle stability, shape, size, and encapsulation efficiency.80,81 DLS measurements proved that the presence of the co-solvent plays an important role in the particle size and polydispersion index, leading to smaller and monodisperse particles compared to those obtained with the other solvents tested (Figure 2a). Among the possible co-solvents, EtOH was chosen because of its high solubilizing capacity for SOR, as evidenced by its wide application in the preparation of different types of SOR-encapsulated particle formulations. The ratio between the amount of drug and polymer was also optimized to obtain particles with improved encapsulation efficiency (EE) and drug loading (DL). The formulation here presented indeed shows 9% of DL, which to the best of our knowledge, is the highest ever observed for SOR-loaded PLGA particles (typically below 5%.)56–60 (See also SI 5 Table S2).
Particle size, EE, DL, degradation rate, and surface charge all depend on the physico-chemical characteristics of the polymer. Commonly, PLGA particles are anionic, but for our formulation, a polymer with low molecular weight and amino-terminated groups was opportunely selected to obtain tunable and smart biodegradable particles. At physiological pH, these particles are indeed negatively charged and stable over 14 days (Figure 3f), whereas under acidic conditions – a condition close to that found in the tumour microenvironment – the amino groups are readily protonated and lead to an overall positive surface charge (Figure 3d–e). It should be remarked that particle surface charge has a major impact on cellular interaction and uptake, since cancer cells are usually negatively charged, therefore positively charged surfaces are desirable to maximise particle–cell interactions. To impart these features, several authors have strived in recent years to conjugate the particles with cationic molecules82 (ie, polymers, biomaterials, proteins, small molecules), whereas in our formulation, the positive charges are integrated in the particle structure thus not requiring any additional component or modification step to favourably match the acidic tumour site. Interestingly, the amino groups in our PLGA particles, in a not-so-long-term vision, can be easily used as anchor sites for specific ligands for enabling active targeting.
For pharmaceutical purposes, characterization of particles is essential to fully understand their properties. Particle size and shape can influence the efficacy of the therapeutic agent in terms of release profile, cellular uptake, and tissue penetration.83 The deep morphological analysis carried out in this study proves that our PLGA particles exhibit a highly homogeneous spherical shape with a narrow size distribution, characteristics that, as shown in a recent review on the role of particle shape in drug delivery, are well suited to the purpose of hepatic drug delivery.84 Indeed, it is well known that spherical particles in the range of 50 to 250 nm accumulate in the liver by a mechanism called “Enhanced Permeability and Retention” (EPR). Also, the particle spherical shape promotes escape from the Reticuloendothelial System (RES) and Kupffer cells, which are responsible for rapid elimination of particles from the bloodstream and phagocytosis, respectively.84 We find that in our formulation, the release rate (Figure 6) is strongly influenced by the drug/polymer ratio. Particles with higher drug encapsulation level exhibit slower release rates with a lower percentage of drug released in buffer. As a matter of fact, the PS having SOR:PLGA 0.01:1 and PS with SOR:PLGA 0.005:1 show a sharp release kinetics in the first 9 hours, with a final percentage of drug released of 40% and 100%, respectively. In contrast, for the optimal formulation with PS having SOR:PLGA 0.1:1, after the initial boost observed during the first 24 hours, a small and sustained drug release is observed over 220 hours in buffer, with a total drug release percentage of 3.4%. This value is remarkably lower compared to the release percentages of all PLGA-based preparations listed in Table S2, which are never below 20%. It is worth to note that the reported DexbLG nanoparticles58 follow a similar trend of release as drug loading increases. This phenomenon could be due to the hydrophobic interaction in the core of the nanoparticles, which is especially favoured at higher drug contents.85,86 Under these conditions, the hydrophobic drug likely crystallizes in the core of the nanoparticles and may remain stably entrapped.85 Accordingly, our PS formulation prevents the release of the drug into the medium and preserves the SOR in the hydrophobic core for a longer time interval. This hypothesis is supported by the cellular uptake analysis carried out by CLSM on HepG2 cells treated with particles loaded with coumarin 6 (PC6). In our experiments, the kinetics of internalization is not only time-dependent, but also exhibits a concentration-dependency, as evidenced by the lower signal recorded with PC6 1 µM at 1 hour compared with that at 7.5 µM.
Due to the fast cellular internalization, PS particles may continuously release the SOR inside the cell, avoiding the extracellular drug degradation and unwanted interactions, thus increasing drug efficacy at a given dose. This favourable mechanism may explain the higher cytotoxicity observed with PS on HepG2 cells compared to those described in other works where SOR-loaded PLGA particles used at 10 µM for 48 hours reduce cell viability in a range comprised between 75% and 50% (see SI 5 Table S3).56–58
Other authors investigated co-delivery strategies to enhance the cytotoxic effect of SOR-Doxorubicin (Dox) core-shell particles.59,60 They measured a cell viability of 80% with 5 µM SOR in the absence of Dox and achieved 40% in the presence of Dox. Interestingly, compared with free SOR, our PS shows significant antiproliferating activity at both 1 and 7.5 µM for 48 to 168 hours, demonstrating the enhancement of drug stability by encapsulation. After 48 hours, PSs at 1 µM and 7.5 µM reduce cell viability at 46% and 40%, respectively. Moreover, treatment at 7.5 µM causes a continuous toxic effect for 168 hours (viability 17%), while viability at the lowest concentration reaches an almost constant value of 50% (Figure 10b). Overall, these results strongly demonstrate that our amino PLGA-based carriers are suitable for the transport and release of SOR and have an enhanced toxic effect on HepG2 cells.
Starting from the high potential of PS carriers, a step ahead toward the development of a new system for light triggered drug delivery based on seOFs was demonstrated. The goal of the study was to demonstrate the possibility of controlling the spatial and temporal release of carriers by a miniaturized light-driven device. Particle release from the seOFs is controlled by modifying the fiber surface through the insertion of a heterobifunctional UV-cleavable linker, which is responsible for anchoring the particles to the seOF surface. Upon activation, the light guided by the seOF is scattered through the cladding toward the outer surface of the seOF, enabling the cleavage of the linker and releasing the particles. This system is integrated into a microfluidic device to control the transport of the particles to the site of interest. The results show that the integrated seOF microfluidic system achieves the same particle release efficiency as the batch process (Figure 12d). It should be noted that the amount of particles released, and consequently the concentration of drug administered, relates to only a single seOF, yet due to the versatility of both seOF and microfluidics, it is possible to design an upgraded device to make an array of carrier-loaded seOF, one for any intended application. In addition, thanks to the custom adapter at the outlet, it is possible to connect a needle or a catheter to improve the delivery of the carriers to the desired site of interest.
Conclusion
Sorafenib is the standard first-line molecule-targeted drug approved for clinical treatment of HCC; however, its high toxicity and low bioavailability results in a narrow therapeutic window. To overcome these drawbacks, it is necessary to develop appropriate particle formulations able to successfully improve the SOR loading, internalization and its sustained release, thus achieving an enhanced therapeutic efficacy while mitigating the side effects.
To this aim, we have developed biodegradable and biocompatible amino-PLGA particles through an optimized emulsion solvent evaporation process for SOR delivery in HCC cell line. The particles size of 250 nm, combined with the spherical shape, the tunable surface charge and the good stability in culture medium make them a promising carrier for the hydrophobic SOR delivery. In particular, the PS particles exhibit a drug loading of 9% – that is the highest value reached compared to the most SOR loaded PLGA particles present in literature – a sustained release profile over 220 hours and a fast cellular uptake (1 hour). The particle stability and the rapid cell internalization led to a significant increase of toxicity against the hepatocellular carcinoma cells (cell viability of 17% over 168 hours at 7.5 µM), suggesting our PSs as a useful carrier to improve the SOR cytotoxic effect. Finally, as proof of concept, we demonstrated a light triggered fiber optic platform for loco-regional drug delivery, relying on particles loaded seOFs enabling advanced platforms to be easily integrated into needles and nanoendoscopes featuring highly localized drug release triggered by light.
In perspective, such drug release systems might provide new insight for the design of minimally invasive cancer-selective nanoplatforms and open a new scenario in clinical practice by reducing adverse effects and increasing the therapeutic efficiency compared with standard administration.
Acknowledgments
The support from projects “NANOFOTONICA PER LA LOTTA AL CANCRO- NANOCAN” POR CAMPANIA FESR 2014–2020 and “PIATTAFORME INNOVATIVE PER LA TERANOSTICA IN PATOLOGIE TUMORALI (OVERALL)” POR CAMPANIA FESR 2014–2020 is gratefully acknowledged.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Kong FH, Ye QF, Miao XY, et al. Current status of sorafenib nanoparticle delivery systems in the treatment of hepatocellular carcinoma. Theranostics. 2021;11(11):5464–5490. doi:10.7150/thno.54822
2. Villanueva A. Hepatocellular carcinoma. N Engl J Med. 2019;380(15):1450–1462. doi:10.1056/NEJMra1713263
3. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Prim. 2021;7(1):6. doi:10.1038/s41572-020-00240-3
4. Hourdequin KC, Schpero WL, McKenna DR, et al. Toxic effect of chemotherapy dosing using actual body weight in obese versus normal-weight patients: a systematic review and meta-analysis. Ann Oncol. 2013;24(12):2952–2962. doi:10.1093/annonc/mdt294
5. Krukiewicz K, Zak JK. Biomaterial-based regional chemotherapy: local anticancer drug delivery to enhance chemotherapy and minimize its side-effects. Mater Sci Eng C. 2016;62:927–942. doi:10.1016/j.msec.2016.01.063
6. Miyahara T, Sueoka-Aragane N, Iwanaga K, et al. Severity and predictive factors of adverse events in pemetrexed-containing chemotherapy for non-small cell lung cancer. Med Oncol. 2017;34(12):195. doi:10.1007/s12032-017-1053-8
7. Dong X, Mumper RJ. Nanomedicinal strategies to treat multidrug-resistant tumors: current progress. Nanomedicine. 2010;5(4):597–615. doi:10.2217/nnm.10.35
8. Khan MI, Hossain MI, Hossain MK, et al. Recent progress in nanostructured smart drug delivery systems for cancer therapy: a review. ACS Appl Bio Mater. 2022;5(3):971–1012. doi:10.1021/acsabm.2c00002
9. Lu Y, Luo Q, Jia X, et al. Multidisciplinary strategies to enhance therapeutic effects of flavonoids from Epimedii Folium: integration of herbal medicine, enzyme engineering, and nanotechnology. J Pharm Anal. 2023;13(3):239–254. doi:10.1016/j.jpha.2022.12.001
10. Silva JO, Fernandes RS, Lopes SCA, et al. pH-Sensitive, long-circulating liposomes as an alternative tool to deliver doxorubicin into tumors: a feasibility animal study. Mol Imaging Biol. 2016;18(6):898–904. doi:10.1007/s11307-016-0964-7
11. Feng L, Gao M, Tao D, et al. Cisplatin-prodrug-constructed liposomes as a versatile theranostic nanoplatform for bimodal imaging guided combination cancer therapy. Adv Funct Mater. 2016;26(13):2207–2217. doi:10.1002/adfm.201504899
12. Wang Y, Zhang H, Hao J, et al. Lung cancer combination therapy: co-delivery of paclitaxel and doxorubicin by nanostructured lipid carriers for synergistic effect. Drug Deliv. 2016;23(4):1398–1403. doi:10.3109/10717544.2015.1055619
13. Liu Y, Zhang H, Cui H, et al. Combined and targeted drugs delivery system for colorectal cancer treatment: conatumumab decorated, reactive oxygen species sensitive irinotecan prodrug and quercetin co-loaded nanostructured lipid carriers. Drug Deliv. 2022;29(1):342–350. doi:10.1080/10717544.2022.2027573
14. Gupta L, Sharma AK, Gothwal A, et al. Dendrimer encapsulated and conjugated delivery of berberine: a novel approach mitigating toxicity and improving in vivo pharmacokinetics. Int J Pharm. 2017;528(1–2):88–99. doi:10.1016/j.ijpharm.2017.04.073
15. Wei T, Chen C, Liu J, et al. Anticancer drug nanomicelles formed by self-assembling amphiphilic dendrimer to combat cancer drug resistance. Proc Natl Acad Sci USA. 2015;112(10):2978–2983. doi:10.1073/pnas.1418494112
16. Zhu M, Chen S, Hua L, et al. Self-targeted salinomycin-loaded DSPE-PEG-methotrexate nanomicelles for targeting both head and neck squamous cell carcinoma cancer cells and cancer stem cells. Nanomedicine. 2017;12(4):295–315. doi:10.2217/nnm-2016-0382
17. Yu L, Lin C, Zheng Z, et al. Self-assembly of pH-responsive biodegradable mixed micelles based on anionic and cationic polycarbonates for doxorubicin delivery. Colloids Surf B. 2016;145:392–400. doi:10.1016/j.colsurfb.2016.05.029
18. Hasannia M, Aliabadi A, Abnous K, et al. Synthesis of block copolymers used in polymersome fabrication: application in drug delivery. J Control Release. 2022;341:95–117. doi:10.1016/j.jconrel.2021.11.010
19. Li X, Hetjens L, Wolter N, et al. Charge-reversible and biodegradable chitosan-based microgels for lysozyme-triggered release of vancomycin. J Adv Res. 2023;43:87–96. doi:10.1016/j.jare.2022.02.014
20. Li X, Sun H, Li H, et al. Multi-responsive biodegradable cationic nanogels for highly efficient treatment of tumors. Adv Funct Mater. 2021;31(26):2100227. doi:10.1002/adfm.202100227
21. Yao X, Niu X, Ma K, et al. Graphene quantum dots-capped magnetic mesoporous silica nanoparticles as a multifunctional platform for controlled drug delivery, magnetic hyperthermia, and photothermal therapy. Small. 2017;13(2):1602225. doi:10.1002/smll.201602225
22. Wang H, Zhang M, Zhang L, et al. Near-infrared light and pH-responsive Au@carbon/calcium phosphate nanoparticles for imaging and chemo-photothermal cancer therapy of cancer cells. Dalt Trans. 2017;46(43):14746–14751. doi:10.1039/C7DT02274C
23. Chen J, Gong M, Fan Y, et al. Collective plasmon coupling in gold nanoparticle clusters for highly efficient photothermal therapy. ACS Nano. 2022;16(1):910–920. doi:10.1021/acsnano.1c08485
24. Sztandera K, Gorzkiewicz M, Klajnert-Maculewicz B. Gold nanoparticles in cancer treatment. Mol Pharm. 2019;16(1):1–23. doi:10.1021/acs.molpharmaceut.8b00810
25. Lee KX, Shameli K, Mohamad SE, et al. Bio-mediated synthesis and characterisation of silver nanocarrier, and its potent anticancer action. Nanomaterials. 2019;9(10):1423. doi:10.3390/nano9101423
26. Miranda RR, Sampaio I, Zucolotto V. Exploring silver nanoparticles for cancer therapy and diagnosis. Colloids Surf B. 2022;210:112254. doi:10.1016/j.colsurfb.2021.112254
27. Zhi D, Yang T, Yang J, et al. Targeting strategies for superparamagnetic iron oxide nanoparticles in cancer therapy. Acta Biomater. 2020;102:13–34. doi:10.1016/j.actbio.2019.11.027
28. Zhao Q, Lin Y, Han N, et al. Mesoporous carbon nanomaterials in drug delivery and biomedical application. Drug Deliv. 2017;24(2):94–107. doi:10.1080/10717544.2017.1399300
29. Mohajeri M, Behnam B, Sahebkar A. Biomedical applications of carbon nanomaterials: drug and gene delivery potentials. J Cell Physiol. 2018;234(1):298–319. doi:10.1002/jcp.26899
30. Watermann A, Brieger J. Mesoporous silica nanoparticles as drug delivery vehicles in cancer. Nanomaterials. 2017;7(7):189. doi:10.3390/nano7070189
31. Vallet-Regí M, Schüth F, Lozano D, et al. Engineering mesoporous silica nanoparticles for drug delivery: where are we after two decades? Chem Soc Rev. 2022;51(13):5365–5451. doi:10.1039/d1cs00659b
32. Hua XW, Bao YW, Wu FG. Fluorescent carbon quantum dots with intrinsic nucleolus-targeting capability for nucleolus imaging and enhanced cytosolic and nuclear drug delivery. ACS Appl Mater Interfaces. 2018;10(13):10664–10677. doi:10.1021/acsami.7b19549
33. Xu X, Ho W, Zhang X, et al. Cancer nanomedicine: from targeted delivery to combination therapy. Trends Mol Med. 2015;21(4):223–232. doi:10.1016/j.molmed.2015.01.001
34. Dancy JG, Wadajkar AS, Connolly NP, et al. Decreased nonspecific adhesivity, receptor-targeted therapeutic nanoparticles for primary and metastatic breast cancer. Sci Adv. 2020;6(3):eaax3931. doi:10.1126/sciadv.aax3931
35. Wang Y, Qin B, Xia G, et al. FDA’s poly (lactic-co-glycolic acid) research program and regulatory outcomes. AAPS J. 2021;23(4):92. doi:10.1208/s12248-021-00611-y
36. Su Y, Zhang B, Sun R, et al. PLGA-based biodegradable microspheres in drug delivery: recent advances in research and application. Drug Deliv. 2021;28(1):1397–1418. doi:10.1080/10717544.2021.1938756
37. Galle PR, Forner A, Llovet JM, et al. EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182–236.
38. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. doi:10.1056/NEJMoa0708857
39. Galle PR. Sorafenib in advanced hepatocellular carcinoma - We have won a battle not the war. J Hepatol. 2008;49(5):871–873. doi:10.1016/j.jhep.2008.09.001
40. Zhu YJ, Zheng B, Wang HY, et al. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin. 2017;38(5):614–622. doi:10.1038/aps.2017.5
41. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised Phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163–1173. doi:10.1016/S0140-6736(18)30207-1
42. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382(20):1894–1905. doi:10.1056/NEJMoa1915745
43. Liu L, Cao Y, Chen C, et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66(24):11851–11858. doi:10.1158/0008-5472.CAN-06-1377
44. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362(9399):1907–1917. doi:10.1016/S0140-6736(03)14964-1
45. Bruix J, Sherman M. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53(3):1020–1022. doi:10.1002/hep.24199
46. Wilhelm SM, Carter C, Tang LY, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–7109. doi:10.1158/0008-5472.CAN-04-1443
47. Chang YS, Adnane J, Trail PA, et al. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chemother Pharmacol. 2007;59(5):561–574. doi:10.1007/s00280-006-0393-4
48. Su GL, Altayar O, O’Shea R, et al. AGA clinical practice guideline on systemic therapy for hepatocellular carcinoma. Gastroenterology. 2022;162(3):920–934. doi:10.1053/j.gastro.2021.12.276
49. Doycheva I, Thuluvath PJ. Systemic therapy for advanced hepatocellular carcinoma: an update of a rapidly evolving field. J Clin Exp Hepatol. 2019;9(5):588–596. doi:10.1016/j.jceh.2019.07.012
50. Zhang Z, Niu B, Chen J, et al. The use of lipid-coated nanodiamond to improve bioavailability and efficacy of sorafenib in resisting metastasis of gastric cancer. Biomaterials. 2014;35(15):4565–4572. doi:10.1016/j.biomaterials.2014.02.024
51. Blanchet B, Billemont B, Cramard J, et al. Validation of an HPLC-UV method for sorafenib determination in human plasma and application to cancer patients in routine clinical practice. J Pharm Biomed Anal. 2009;49(4):1109–1114. doi:10.1016/j.jpba.2009.02.008
52. Rahiminezhad Z, Tamaddon AM, Dehshahri A, et al. PLGA-graphene quantum dot nanocomposites targeted against αvβ3 integrin receptor for sorafenib delivery in angiogenesis. Biomater Adv. 2022;137:212851. doi:10.1016/j.bioadv.2022.212851
53. Feczkó T, Piiper A, Pleli T, et al. Theranostic sorafenib-loaded polymeric nanocarriers manufactured by enhanced gadolinium conjugation techniques. Pharmaceutics. 2019;11(10):489. doi:10.3390/pharmaceutics11100489
54. Babos G, Biró E, Meiczinger M, et al. Dual drug delivery of sorafenib and doxorubicin from PLGA and PEG-PLGA polymeric nanoparticles. Polymers. 2018;10(8):895. doi:10.3390/polym10080895
55. Khan MA, Raza A, Ovais M, et al. Current state and prospects of nano-delivery systems for sorafenib. Int J Polym Mater Polym Biomater. 2018;67(18):1105–1115. doi:10.1080/00914037.2018.1429434
56. Liu J, Boonkaew B, Arora J, et al. Comparison of sorafenib-loaded poly (Lactic/Glycolic) acid and dppc liposome nanoparticles in the in vitro treatment of renal cell carcinoma. J Pharm Sci. 2015;104(3):1187–1196. doi:10.1002/jps.24318
57. Liu Y, Feng L, Liu T, et al. Multifunctional pH-sensitive polymeric nanoparticles for theranostics evaluated experimentally in cancer. Nanoscale. 2014;6(6):3231–3242. doi:10.1039/c3nr05647c
58. Kim DH, Kim MD, Choi CW, et al. Antitumor activity of sorafenib-incorporated nanoparticles of dextran/poly(DL-lactide-co-glycolide) block copolymer. Nanoscale Res Lett. 2012;7:91. doi:10.1186/1556-276X-7-91
59. Malarvizhi GL, Retnakumari AP, Nair S, et al. Transferrin targeted core-shell nanomedicine for combinatorial delivery of doxorubicin and sorafenib against hepatocellular carcinoma. Biol Med. 2014;10(8):1649–1659. doi:10.1016/j.nano.2014.05.011
60. Zhang J, Hu J, Chan HF, et al. iRGD decorated lipid-polymer hybrid nanoparticles for targeted co-delivery of doxorubicin and sorafenib to enhance anti-hepatocellular carcinoma efficacy. Biol Med. 2016;12(5):1303–1311. doi:10.1016/j.nano.2016.01.017
61. Caputo TM, Aliberti A, Cusano AM, et al. Stimuli‐responsive hybrid microgels for controlled drug delivery: sorafenib as a model drug. J Appl Polym Sci. 2021;138(14):50147. doi:10.1002/app.50147
62. Caputo TM, Cusano AM, Ruvo M, et al. Human serum albumin nanoparticles as a carrier for on-demand sorafenib delivery. Curr Pharm Biotechnol. 2021;23(9):1214–1225.
63. Giannouli M, Karagkiozaki V, Pappa F, et al. Fabrication of quercetin-loaded PLGA nanoparticles via electrohydrodynamic atomization for cardiovascular disease. Mater Today. 2018;5(8):15998–16005.
64. Maity S, Chakraborti AS. Formulation, physico-chemical characterization and antidiabetic potential of naringenin-loaded poly D, L lactide-co-glycolide (N-PLGA) nanoparticles. Eur Polym J. 2020;134:109818. doi:10.1016/j.eurpolymj.2020.109818
65. Wei JC, Meng F, Qu K, et al. Sorafenib inhibits proliferation and invasion of human hepatocellular carcinoma cells via up-regulation of p53 and suppressing FoxM1. Acta Pharmacol Sin. 2015;36(2):241–251. doi:10.1038/aps.2014.122
66. Carotenuto B, Ricciardi A, Micco A, et al. Smart optical catheters for epidurals. Sensors. 2018;18(7):2101. doi:10.3390/s18072101
67. Monat C, Domachuk P, Eggleton BJ. Integrated optofluidics: a new river of light. Nat Photonics. 2007;1(2):106–114. doi:10.1038/nphoton.2006.96
68. Fan X, White IM. Optofluidic microsystems for chemical and biological analysis. Nat Photonics. 2011;5(10):591–597. doi:10.1038/nphoton.2011.206
69. Sui K, Meneghetti M, Kaur J, et al. Microstructured soft fiber-based neural device for drug delivery and optical neuromodulation. Biophotonics Congr Biomed Opt. 2022;2022:
70. Nazari M, Rubio-Martinez M, Tobias G, et al. Metal-organic-framework-coated optical fibers as light-triggered drug delivery vehicles. Adv Funct Mater. 2016;26(19):3244–3249. doi:10.1002/adfm.201505260
71. Wang X, Dong X, Zhou Y, et al. Optical fiber anemometer using silver-coated fiber Bragg grating and bitaper. Sens Actuator a Phys. 2014;214:230–233. doi:10.1016/j.sna.2014.04.013
72. Yang J, Chen X, Dong X. Hot-wire anemometer based on frosted fiber Bragg grating coated with silver film. Mater Sci Eng. 2020;711:012112.
73. Caldas P, Jorge PAS, Rego G, et al. Fiber optic hot-wire flowmeter based on a metallic coated hybrid long period grating/fiber Bragg grating structure. Appl Opt. 2011;50(17):2738–2743. doi:10.1364/AO.50.002738
74. Gao R, Lu D. Temperature compensated fiber optic anemometer based on graphene-coated elliptical core micro-fiber Bragg grating. Opt Express. 2019;27(23):34011–34021. doi:10.1364/OE.27.034011
75. Silva GE, Caldas P, Santos JC, et al. All-fiber sensor based on a metallic coated hybrid LPG-FBG structure for thermal characterization of materials. In:
76. Liu Z, Wang F, Zhang Y, et al. Low-power-consumption fiber-optic anemometer based on long-period grating with SWCNT coating. IEEE Sens J. 2019;19(7):2592–2597. doi:10.1109/JSEN.2019.2891044
77. Zhang Y, Wang F, Liu Z, et al. Fiber-optic anemometer based on single-walled carbon nanotube coated tilted fiber Bragg grating. Opt Express. 2017;25(20):24521–24530. doi:10.1364/OE.25.024521
78. Liu Y, Liang B, Zhang X, et al. Plasmonic fiber-optic photothermal anemometers with carbon nanotube coatings. J Light Technol. 2019;37(13):3373–3380. doi:10.1109/JLT.2019.2916572
79. Wang F, Duan Y, Lu M, et al. Linear-response and simple hot-wire fiber-optic anemometer using high-order cladding mode. Opt Express. 2020;28(18):27028–27036. doi:10.1364/OE.399774
80. Alvi M, Yaqoob A, Rehman K, et al. PLGA-based nanoparticles for the treatment of cancer: current strategies and perspectives. AAPS Open. 2022;8(1):1–17. doi:10.1186/s41120-022-00060-7
81. Hernández-Giottonini KY, Rodríguez-Córdova RJ, Gutiérrez-Valenzuela CA, et al. PLGA nanoparticle preparations by emulsification and nanoprecipitation techniques: effects of formulation parameters. RSC Adv. 2020;10(8):4218–4231. doi:10.1039/C9RA10857B
82. Martins C, Sousa F, Araújo F, et al. Poly(lactic-co-glycolic) acid: functionalizing PLGA and PLGA derivatives for drug delivery and tissue regeneration applications. Adv Healthc Mater. 2018;7(1). doi:10.1002/adhm.201701035
83. Toy R, Peiris PM, Ghaghada KB, et al. Shaping cancer nanomedicine: the effect of particle shape on the in vivo journey of nanoparticles. Nanomedicine. 2014;9(1):121–134. doi:10.2217/nnm.13.191
84. Zhou F, Teng F, Deng P, et al. Recent progress of nano-drug delivery system for liver cancer treatment. Anticancer Agents Med Chem. 2017;17(14):1884–1897.
85. Gref R, Minamitake Y, Peracchia MT, et al. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263(5153):1600–1603. doi:10.1126/science.8128245
86. Jeong Y, Kim DH, Chung CW, et al. Doxorubicin-incorporated polymeric micelles composed of dextran-b-poly(DL-lactide-co-glycolide) copolymer. Int J Nanomedicine. 2011;6:1415–1427. doi:10.2147/IJN.S19491
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.