Back to Journals » Journal of Pain Research » Volume 13
Sonic Hedgehog Signaling Contributes to Chronic Post-Thoracotomy Pain via Activating BDNF/TrkB Pathway in Rats
Authors Yang Y ![]() , Wang X
, Wang X ![]() , Zhang X
, Zhang X ![]() , You S, Feng L, Zhang Y
, You S, Feng L, Zhang Y ![]() , Shi Y, Xu Y, Zhang H
, Shi Y, Xu Y, Zhang H
Received 10 January 2020
Accepted for publication 16 June 2020
Published 10 July 2020 Volume 2020:13 Pages 1737—1746
DOI https://doi.org/10.2147/JPR.S245515
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor E Alfonso Romero-Sandoval
Yitian Yang,1,* Xiaoyan Wang,2,* Xuan Zhang,3 Shaohua You,1 Long Feng,1 Yunliang Zhang,1 Yizheng Shi,1 Yuhai Xu,4 Hong Zhang1
1Anesthesia and Operation Center, The First Medical Center of Chinese PLA General Hospital, Medical School of Chinese PLA, Beijing 100853, People’s Republic of China; 2Department of Anesthesiology, The Fourth Medical Center of Chinese PLA General Hospital, Medical School of Chinese PLA, Beijing 100037, People’s Republic of China; 3Department of Anesthesiology, Tianjin Cancer Institute & Hospital, Tianjin Medical University, Tianjin 300060, People’s Republic of China; 4Department of Anesthesiology, Air Force Medical Center, Beijing 100142, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hong Zhang; Yitian Yang Email [email protected]; [email protected]
Purpose: Some patients undergoing thoracotomy may suffer from chronic post-thoracotomy pain (CPTP). Treatment of CPTP has been a clinical challenge and the underlying mechanisms of CPTP remain elusive. Recently, sonic hedgehog (Shh) signaling has been shown to be associated with various pain states but its role in the pathogenesis of CPTP is still unclear.
Methods: CPTP was induced in rats by thoracotomy. Rats were divided into CPTP group and non-CPTP group based on the mechanical withdrawal threshold (MWT). Rats were administered with Shh signaling inhibitor cyclopamine and activator smoothened agonist (SAG), and then evaluated by MWT and cold allodynia testing. The expressions of Shh signaling (Shh ligand, patched and smoothened receptor, Gli transcription factors), brain-derived neurotrophic factor (BDNF), tropomyosin-related kinase receptor B (Trk-B), phosphatidylinositol 3-kinase (PI3K) and protein kinase B (Akt) in rat T4-5 spinal cord dorsal horn (SDH) were detected by Western blotting and immunohistochemistry.
Results: The expression of Shh signaling significantly increased and the BDNF/TrkB pathway was activated in T4-5 SDH of CPTP rats. Cyclopamine attenuated hyperalgesia and down-regulated the expressions of Gil1, BDNF, p-TrkB, p-PI3K and p-Akt in CPTP rats. SAG induced hyperalgesia in non-CPTP rats and elevated the expressions of Gil1, BDNF, p-TrkB, p-PI3K and p-Akt.
Conclusion: Shh signaling may contribute to CPTP via activating BDNF/TrkB signaling pathway, and inhibition of Shh signaling may effectively alleviate CPTP.
Keywords: sonic hedgehog, chronic post-thoracotomy pain, brain-derived neurotrophic factor, spinal cord dorsal horn, PI3K, Akt
Introduction
Chronic post-thoracotomy pain (CPTP) refers to the persistent pain that occurs near the surgical incision after thoracotomy and lasts for at least 2 months.1 Epidemiological studies have shown that about 40–50% of patients still suffer from CPTP at 6 months after surgery,2,3 and 10% of them experience severe pain.4 Unfortunately, the underlying mechanism of CPTP is still unclear. Intercostal nerve injury was proposed as a major cause of CPTP,5,6 but the CPTP in most patients is not related to neuropathic pain.7 CPTP reduces the quality of life, causes atelectasis and lung infection, and even leads to anxiety and depression, but there are no effective treatments for CPTP currently. Therefore, to elucidate the pathogenesis of CPTP is particularly important for the development of effective treatment for CPTP.
Sonic hedgehog (Shh) signaling is a classical signaling pathway that controls embryonic development. It is mainly composed of Shh ligand, transmembrane patched (Ptch) and smoothened (Smo) receptors, and transcriptional factor Gli.8,9 After the Shh ligand binds to Ptch, Smo is dissociated from this complex and then activated, eventually leading to the activation of downstream Gli.10 Shh signaling is involved in the development of nervous system and regulates synaptic plasticity, and its abnormalities have been found to be related to the pathogenesis of tumors.9,11 Previous studies have shown that Shh signaling regulates nociceptive sensitization in the drosophila.12 Additionally, Shh signaling is also related to the occurrence of bone cancer pain and morphine-induced hyperalgesia and tolerance,13,14 but the role of Shh signaling in the pathogenesis of CPTP remains unclear.
As a member of the neurotrophic factor family, brain-derived neurotrophic factor (BDNF) can promote the growth and differentiation of neurons and change the neuronal excitability and synaptic plasticity.15 Previous studies have revealed that BDNF is involved in the regulation of inflammatory pain and neuropathic pain.16,17 The phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) pathway is the downstream signaling pathway of the BDNF/tropomyosin-related kinase receptor B (TrkB) pathway,18,19 and PI3K/Akt pathway is related to the BDNF-induced pain hypersensitivity.20 However, little is known about the role of BDNF/TrkB pathway in the pathogenesis of CPTP.
We hypothesize that Shh signaling is involved in the pathogenesis of CPTP via activating BDNF/TrkB pathway. In the present study, the expressions of Shh signaling and BDNF/TrkB pathway were detected in rats after thoracotomy, aiming to investigate the role of Shh signaling in the pathogenesis of CPTP.
Materials and Methods
Animals
Adult male Sprague-Dawley rats were provided by the Experimental Animal Center of the Chinese PLA General Hospital. The whole study was approved by the Institutional Animal Care Committee of the Chinese PLA General Hospital, and all the procedures were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Rats weighing 400 to 450 g were housed with a 12-h light/dark cycle and given ad libitum access to food and water at 24–26°C.
Establishment of a Rat Model of CPTP
The rat model of CPTP was produced via thoracotomy as previously reported.21 After intraperitoneal anesthesia with sodium pentobarbital (60 mg/kg), rats were subjected to endotracheal intubation with a 16G catheter, which was connected to a rodent ventilator for mechanical ventilation (I/E: 1:2, respiratory rate: 60–70 times/min, tidal volume: 10 mL/kg). Then, a 4-cm long skin incision was made between the fourth to fifth ribs, and the muscles were separated. Subsequently, a 1.8-cm long incision was made in the pleura. The fourth and fifth ribs were separated to 1 cm with a microscopic retractor for one hour. Before closing the chest, an epidural catheter with a 10-mL syringe was inserted into the pleural cavity to extract the air. The ribs, muscles and skin were independently sutured with 3-0 non-absorbable sutures. Until the spontaneous breathing returned, the 16G catheter was removed. In the sham group, rats underwent a sham operation without rib retraction. In the control group, rats did not undergo surgical procedures.
Intrathecal Catheterization and Drug Administration
Eight days before thoracotomy, rats were inserted into the intrathecal catheter according to previously reported.22,23 After the rats were intraperitoneally anesthetized with sodium pentobarbital (60 mg/kg), a 2-cm long incision was made in the skin near the atlantooccipital space, and then the subcutaneous muscles were separated to expose the atlantooccipital membrane. A polyethylene-10 catheter (Meiweijing, Jiangsu, China) was inserted 3.3 cm from the midpoint of atlantooccipital membrane to the tail of rats and reached the T4-5 of rat spinal cord.
Shh signaling inhibitor cyclopamine (CLP) (10 μg/10 μL) (Adooq Bioscience, California, USA) or an equivalent volume of vehicle (1% DMSO) (10 μL) was administrated intrathecally in CPTP rats once daily on postoperative day (POD) 7 to 9. Subsequently, in order to remove the dead space, the catheter was rinsed with 10 μL of PBS and removed on POD 10. Shh signaling agonist smoothened agonist (SAG) (5 mg/kg) (Abcam, Cambridge, UK) or an equivalent volume of vehicle (1% DMSO) was injected intraperitoneally in rats in the non-CPTP group on POD 21. The doses were from previous studies.13,14
Mechanical Withdrawal Threshold (MWT)
After 30-min acclimation to the environment, the MWT was measured with the von-Frey filaments (VFH; Stoelting Co., Wood Dale, USA), which were used perpendicular to the dorsal skin 0.5–1 cm around the incision. The interval between each measurement was 30 seconds. The bending forces of the VFH ranged from 0.16 to 15.0 g. The intensity of VFH was increasing until a positive reaction occurred in rats. The positive reactions included contractions of muscles, escape movement, scratching the skin of the surgical area, rotation of trunk, shuddering or squealing.23,24 For avoiding tactile sensitization by the stiffer VFH, the highest threshold of MWT was assigned as 15.1 g.25 The MWT was calculated by the up-and-down method.26 The results of MWT were calculated using the formula: 50% threshold (g) = 10(X+kd)/104 [X = the value (in log units) of the final von-Frey filament, k = tabular value for the response pattern and d = the average increment (in log units) between von-Frey filaments]. Finally, rats undergoing thoracotomy and showing significant decrease in the MWT (≤4g) on POD 21 were considered to develop CPTP.21 The MWT was evaluated by the investigators who were blind to the grouping of rats.
Cold Hyperalgesia Test
Cold acetone (4°C) was dropped from the syringe to the surgical site. The number of hind limb scratching the surgical site was recorded within 1 min by the trained investigators who were blinded to the grouping, and this test was repeated 5 min later. The average of two tests was calculated for further analysis. Rats scratching at least 3 times/min were considered as having cold hyperalgesia.27
Western Blotting
Proteins were extracted from the ipsilateral T4-5 spinal cord dorsal horn (SDH), and the supernatant protein concentrations were detected with the BCA protein assay kit (Abcam). The equal number of sample proteins was separated on sodium dodecyl sulphate–polyacrylamide gels. Subsequently, they were transferred onto the polyvinylidene fluoride membrane, which was blocked with 5% non-fat milk and then incubated with primary antibodies overnight at 4°C. The primary antibodies included anti-Shh (1:2000, GeneTex, San Antonio, USA), anti-Ptch1 (1:1000, Sigma, St. Louis, USA), anti-Smo (1:2000, Abcam), anti-Gli1 (1:2000, Abcam), anti-BDNF (1:1000, GeneTex), anti-p-TrkB (1:1000, GeneTex), anti-TrkB (1:2000, GeneTex), anti-p-PI3K (1:1000, GeneTex), anti-p-Akt (1:1000, GeneTex), anti-Histone H3 (1:2000, GeneTex), and anti-GAPDH (1:10,000, GeneTex). Finally, the membranes were rinsed and the proteins were detected with the enhanced chemiluminescence method.
Immunohistochemistry
After transcardial perfusion with 4% paraformaldehyde, T4-5 SDH of rats were collected and embedded in paraffin. Subsequently, the paraffin-embedded tissues were sectioned and processed for immunohistochemistry. The sections were incubated with primary antibodies: anti-Shh (1:100, GeneTex) and anti-BDNF (1:200, Abcam), rinsed, then treated with secondary antibodies. Finally, images were captured using a digital camera.
Statistical Analysis
All data are presented as means ± standard deviation (SD). The behaviors were analyzed using repeated measures one-way analysis of variance (ANOVA) followed by the Tukey post-hoc test. The protein expressions were analyzed using Student’s t test, one- or two-way ANOVA followed by the Tukey post-hoc test. Statistical analyses were performed using SPSS version 19.0. A value of P<0.05 was considered statistically significant.
Results
Thoracotomy Induced Long-Lasting Mechanical Hyperalgesia and Cold Allodynia in Rats
As reported in previous studies,21,24 our results showed thoracotomy induced long-lasting significant decrease in the MWT and increased the number of scratching around the surgical site, which persisted for at least 21 days after thoracotomy (Figure 1A and B). According to the diagnostic criteria of CPTP rats,23 the rats underwent thoracotomy and developed hyperalgesia (MWT ≤ 4 g) on POD 21 were divided into the CPTP group, while the remaining rats without hyperalgesia (MWT > 4 g) after thoracotomy were included in the non-CPTP group. Compared with the control group, rats in the sham group and non-CPTP group had decreased MWT and increased number of scratching on POD 3 and 6 (Figure 1A and B), and there was no significant difference between sham group and non-CPTP group (P > 0.05, Figure 1A and B). Ultimately, rats in the sham group and non-CPTP group did not show any mechanical hyperalgesia or cold allodynia on POD 21 (P > 0.05, Figure 1A and B). CPTP was present in 16 of 30 rats undergoing thoracotomy (53.3%), and the incidence was similar to previously reported.24 These results indicated thoracotomy produced CPTP in rats.
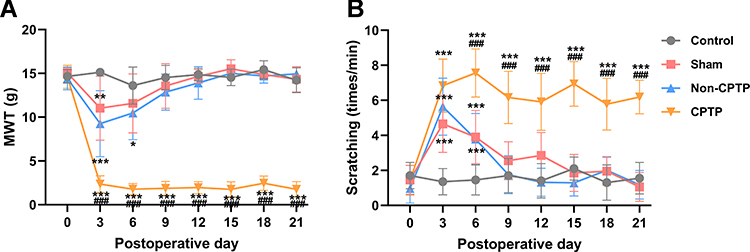 |
Figure 1 Thoracotomy induced CPTP in rats. (A) The assessment of mechanical hyperalgesia by MWT. (B) The assessment of cold allodynia by cold acetone-induced scratching; *P < 0.05, **P < 0.01 or ***P < 0.001, vs control group; ###P < 0.001, vs non-CPTP group; repeated measures one-way ANOVA; control: n= 10; sham: n=10; non-CPTP: n=14; CPTP: n=16. |
Shh Signaling and BDNF/TrkB Pathway Were Activated in CPTP Rats
The expressions of Shh-related proteins (Shh, Ptch1, Smo and Gil1) were detected in the T4-5 SDH by Western blotting. On POD 9, 15 and 21, ipsilateral T4-5 SDH of rats with hyperalgesia (MWT ≤ 4 g) after thoracotomy was collected for Western blotting. Results showed the expressions of Shh, Ptch1, Smo, and Gli1 were significantly higher than in the control group (Figure 2A and B). Moreover, the expressions of Shh, Ptch1 and Smo significantly increased in the ipsilateral T4-5 SDH of CPTP rats as compared to rats in other groups on POD 21 (P < 0.001, Figure 2C). As a nuclear transcription factor in the Shh signaling pathway, the expression of Gli1 in the nuclear extract significantly increased in CPTP rats on POD 21 (P < 0.001, Figure 2D). Additionally, immunofluorescence staining showed the expression of Shh in the ipsilateral T4-5 SDH of CPTP rats significantly increased as compared to that in the contralateral T4-5 SDH (Figure 2E). These results indicated Shh signaling was activated in CPTP rats.
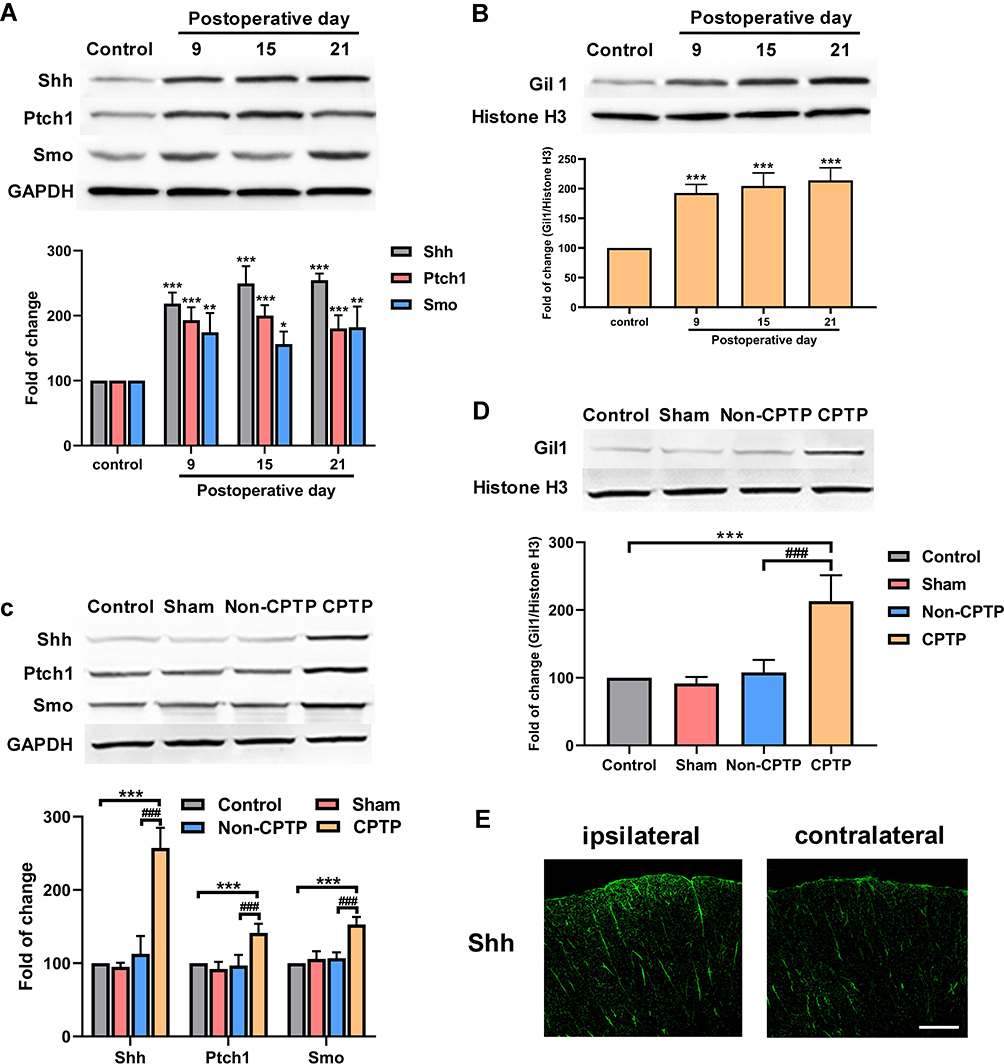 |
Figure 2 Shh signaling activation in ipsilateral T4-5 SDH of CPTP rats. (A) Results showed a time-dependent increase in the expressions of Shh, Ptch1, and Smo in the ipsilateral T4-5 SDH of rats (n=4 each group). (B) Results showed a time-dependent increase in the expression of Gil1 in the nuclear extract of the ipsilateral T4-5 SDH of rats (n=4 each group). (C) The expressions of Shh, Ptch1 and Smo were measured by Western blotting in the ipsilateral T4-5 SDH of rats on POD 21 (n=6 each group). (D) The expressions of Gil1 were measured by Western blotting in the ipsilateral T4-5 SDH of rats on POD 21 (n=6 each group). (E) The Shh expression (green) were detected by immunofluorescence staining in ipsilateral and contralateral T4-5 SDH of rats on POD 21. *P < 0.05, **P < 0.01 or ***P < 0.001, vs control group; ###P < 0.001, vs non-CPTP group; one-way ANOVA; scale bar: 100 μm. |
To investigate the role of BDNF/TrkB pathway in the CPTP, the BDNF expression was detected in the ipsilateral T4-5 SDH by Western blotting. Results showed the BDNF expression in the CPTP rats significantly increased as compared to naïve rats and non-CPTP rats (P < 0.001, Figure 3A). Since TrkB is a high-affinity receptor for BDNF, TrkB expression was also detected in ipsilateral T4-5 SDH. Results showed the phosphorylated TrkB (p-TrkB) significantly increased in the CPTP group (P < 0.001, Figure 3A), while the total amount of TrkB remained unchanged (P > 0.05, Figure 3A). Subsequently, the expressions of p-PI3K and p-Akt were detected, and results showed the phosphorylation of PI3K and Akt significantly increased in CPTP rats (Figure 3B). These results indicated that the upregulation of BDNF and the activation of PI3K/Akt pathway may be related to the pathogenesis of CPTP.
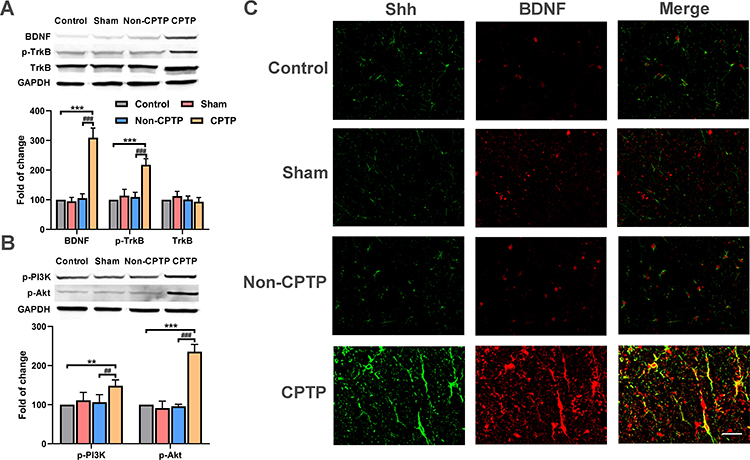 |
Figure 3 Shh signaling activated BDNF/TrkB pathway. (A) The expressions of BDNF, p-TrkB, TrkB were detected by Western blotting in ipsilateral T4-5 SDH of rats on POD 21. (B) The expressions of p-PI3K and p-Akt were measured by Western blotting in ipsilateral T4-5 SDH of rats on POD 21. (C) The expressions of Shh (red) and BDNF (green) were measured by immunofluorescence staining and Shh was colocalized with BDNF in ipsilateral T4-5 SDH of rats on POD 21 (yellow). **P < 0.01 or ***P < 0.001, vs control group; ##P < 0.01 or ###P < 0.001, vs non-CPTP group; one-way ANOVA; n=6 each group; scale bar: 50 μm. |
Additionally, to further certify the relationship between Shh and BDNF, both were detected by immunofluorescence staining. Results showed Shh and BDNF expressions significantly increased in ipsilateral T4-5 SDH of CPTP rats and co-expression of Shh and BDNF was observed (Figure 3C).
Inhibition of Shh Signaling Alleviated Hyperalgesia in CPTP Rats
Moreover, Shh signaling inhibitor cyclopamine was intrathecally injected into hyperalgesia rats (MWT ≤ 4g) on POD 7-9 (Figure 4A). Our results showed cyclopamine increased the MWT and reduced the number of scratching induced by cold acetone in CPTP rats until POD 21 (Figure 4B and C), suggesting that inhibition of Shh signaling significantly alleviated hyperalgesia in CPTP rats. Besides, the intrathecal administration of cyclopamine effectively reduced the expressions of Gil1 (P < 0.01, Figure 4D) and BDNF (P < 0.01, Figure 4E) in ipsilateral T4-5 SDH of CPTP rats on POD 21. Although the total protein expression of TrkB remained unchanged, the p-TrkB expression significantly decreased in the CPTP+CLP group (P < 0.001, Figure 4E). The expressions of p-PI3K (P < 0.05, Figure 4F) and p-Akt (P < 0.001, Figure 4F) also significantly reduced in the CPTP+CLP group. Taken together, the above results indicated inhibition of Shh signaling alleviated CPTP, suppressed BDNF/TrkB pathway and reduced the activation of PI3K/Akt pathway.
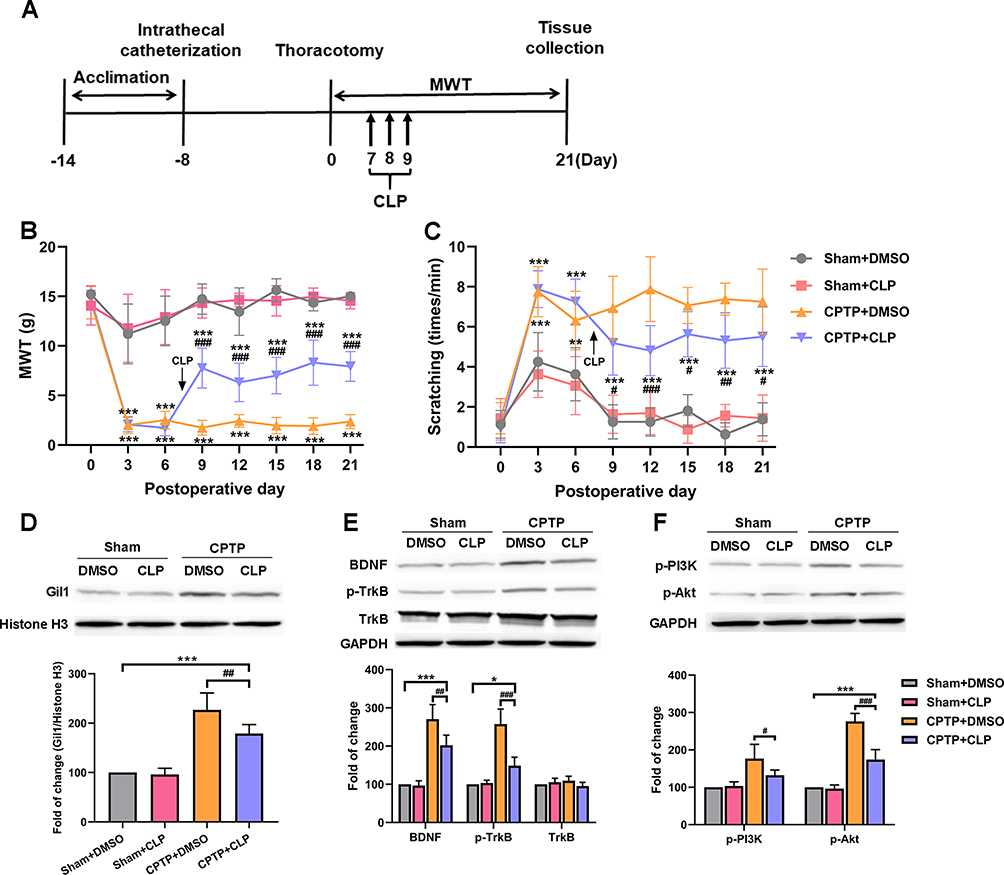 |
Figure 4 Shh signaling inhibition alleviated hyperalgesia and down-regulated BDNF/TrkB pathway in CPTP rats. (A) Cyclopamine administration. (B) Effect of cyclopamine was evaluated by MWT. (C) Effect of cyclopamine was evaluated by the number of scratching in the cold allodynia test. (D–F) After administration of cyclopamine, Western blotting showed the expression of Gil1, BDNF, p-TrkB, TrkB, p-PI3K and p-Akt in ipsilateral T4-5 SDH on POD 21. *P < 0.05, **P < 0.01 or ***P < 0.001, vs sham+DMSO group; #P < 0.05, ##P < 0.01 or ###P < 0.001, vs CPTP+DMSO group; repeated measures one-way ANOVA or two-way ANOVA; n=7 each group. |
Activation of Shh Signaling Induced Hyperalgesia in Non-CPTP Rats
To further elucidate the role of Shh signaling in CPTP, the Shh signaling agonist SAG was intraperitoneally injected into non-CPTP rats on POD 21 (Figure 5A). Our results showed SAG induced a reduction in MWT and increased the number of scratching at the surgical site in non-CPTP rats (P < 0.001, Figure 5B and C). At 6 h after SAG administration, the Gil1 expression increased significantly (P < 0.001, Figure 5D). Then, BDNF/TrkB pathway was activated, which was characterized by the elevated expressions of BDNF (P < 0.01, Figure 5E), p-TrkB (P < 0.05, Figure 5E), p-PI3K and p-Akt (P < 0.001, Figure 5F) in the non-CPTP rats after SAG administration. Based on these findings, we speculated that Shh signaling was involved in the pathogenesis of CPTP by activating the BDNF/TrkB pathway.
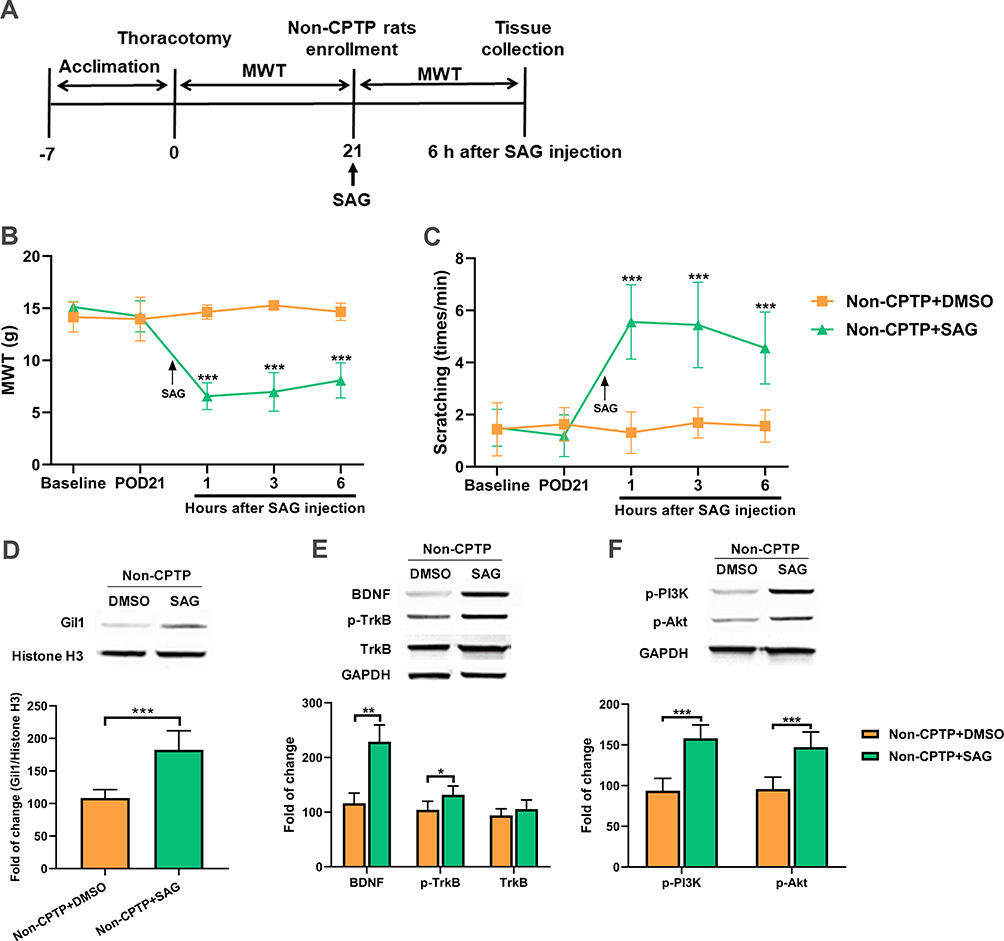 |
Figure 5 Shh signaling activation induced hyperalgesia and up-regulated BDNF/TrkB pathway in non-CPTP rats. (A) SAG administration. (B) Effect of SAG was evaluated by MWT. (C) Effect of SAG was evaluated by the number of scratching. (D–F) At 6 h after SAG administration, Western blotting showed the expressions of Gil1, BDNF, p-TrkB, TrkB, p-PI3K and p-Akt in ipsilateral T4-5 SDH on POD 21. *P < 0.05, **P < 0.01 or ***P < 0.001 vs non-CPTP+DMSO group; repeated measures one-way ANOVA or Student’s t-test; n=6 each group. |
Discussion
Our study revealed that Shh signaling was involved in the pathogenesis of CPTP via activating BDNF/TrkB pathway, and inhibition of Shh signaling effectively alleviated CPTP. To our knowledge, this study for the first time reported the role of Shh signaling in the pathogenesis of CPTP.
Currently, the pathogenesis of CPTP remains unclear. Previously, CPTP was regarded as a type of neuropathic pain caused by intercostal nerve injury.6 Nevertheless, increasing studies show that only 23% of CPTP patients have definite neurological damage, and minimally invasive surgery with less intercostal nerve injury may also cause CPTP.5,7 This indicates CPTP has different pathological mechanisms from neuropathic pain. To further investigate the pathogenesis of CPTP, a rat model to CPTP was established by thoracotomy. In the present study, results showed 53.3% of rats developed CPTP after thoracotomy, which was similar to the clinical incidence of CPTP.2,3 Interestingly, rats had differences in pain exacerbation and relief on POD 3. Rats that had no hyperalgesia on POD 6 did not develop CPTP, which was consistent with previous findings.23,24 These results indicated that this rat model may simulate the clinical CPTP.
Shh signaling is a signaling pathway essential for the embryonic development, and its abnormalities will cause birth defects, tissue remodeling, stem cell regeneration and tumor growth.9 Additionally, Shh is a pleiotropic factor of the central nervous system, can promote cell proliferation, differentiation, and targeted growth of axons in the developing brain and spinal cord, and plays a decisive role in the differentiation of neural precursor cells into neurons or glial cells.28 Recently, emerging studies have shown that Shh signaling is closely related to the pain.12,14 Babcock et al found the Shh signaling was involved in the nociceptive sensitization in drosophila.12 Moreover, activation of Shh signaling promotes the development of bone cancer pain by regulating the excitability of sensory neurons.13 Pancreatic stellate cells may cause pancreatic cancer pain through activating Shh signaling,29 and intraperitoneal administration of Shh inhibitor cyclopamine can suppress the morphine-induced hyperalgesia and reduce morphine tolerance.14 Besides, astrocyte activation is positively related to hyperalgesia. There is evidence showing that Shh signaling can regulate Bcl-2 and Bax to affect astrocyte apoptosis,30 which may be one of mechanisms by which Shh signaling regulates nociceptive sensitization. In the present study, our results showed that the expressions of Shh signaling molecules significantly increased in the SDH of CPTP rats. Furthermore, inhibition of Shh signaling significantly alleviated CPTP in rats. Studies have shown that activation of Shh signaling by SAG can induce thermal hyperalgesia in naïve mice,14 and our results also indicated that SAG could also induce hyperalgesia in non-CPTP rats. The above results indicate that activation of Shh signaling contributes to CPTP.
Accumulating studies have shown that BDNF plays a crucial role in promoting pain transmission in chronic pain.17 The increased BDNF expression in the spinal cord can be observed in case of neuropathic pain and inflammatory pain.31,32 However, the role of BDNF in the pathogenesis of pain is still controversial. Although a majority of studies have indicated that increased expression of BDNF in the spinal cord induces pain hypersensitivity,31–33 some studies also reported that BDNF exerted limited pronociceptive effect and even antinociceptive effect on the pain.34–36 Previous studies indicated that Shh signaling activation in the cavernous and sciatic nerves up-regulated BDNF expression.10 Liu et al found that Shh signaling activation significantly elevated BDNF expression in the spinal cord of morphine-induced hyperalgesia rats, and administration of K252, an antagonist of BDNF, attenuated the thermal allodynia induced by SAG.14 These indicate that Shh signaling can activate the BDNF expression. In the present study, our results showed that BDNF expression also significantly increased in SDH of CPTP rats after Shh activation, and the expressions of BDNF and p-TrkB markedly decreased after inhibition of Shh signaling, which was accompanied by the relief of hyperalgesia and cold allodynia in CPTP rats. Besides, the Shh signaling activation in non-CPTP rats also significantly increased BDNF expression. These results suggest that Shh signaling may affect CPTP by regulating the BDNF/TrkB signaling pathway.
Binding of BDNF to TrkB activates multiple signaling pathways, including MAPK/ERK, PLC, and PI3K/Akt.18,19,37,38 PI3K/Akt signaling pathway plays an essential regulatory role in the development of pain.20 Studies have indicated that the expressions of p-Akt and p-mTOR increase in rats with inflammation-induced pain, which are mainly found in the neurons of the I, III, and IV layers of SDH.39 Pezet et al40 found that intrathecal administration of LY294002 (PI3K inhibitor) inhibited the increase of p-Akt expression in the SDH, exerting an analgesic effect. In the neuropathic pain rats, the p-Akt expression increased in the L4-6 DRG, and pretreatment of PI3K or Akt inhibitors dampened mechanical hyperalgesia and thermal allodynia induced by nerve damage.41 These suggest that activation of PI3K/Akt pathway is involved in the pathogenesis of chronic pain. In the present study, our results showed that the expressions of p-PI3K and p-Akt significantly increased in the SDH of CPTP rats, which was consistent with the change in the BDNF expression. Inhibition of Shh signaling decreased BDNF expression, ultimately reducing the expressions of p-PI3K and p-Akt. These results indicate that PI3K/Akt signaling pathway is involved in the pathogenesis of CPTP. Therefore, we speculate that Shh signaling activates the BDNF/TrkB signaling pathway in the spinal cord, which then activates the downstream PI3K/Akt signaling pathway and is responsible for CPTP. However, in this study, we did not further validate whether BDNF upregulation could activate PI3K/Akt pathway in CPTP rats. This was one of limitations in the present study. Therefore, more evidence is needed to elucidate the relationship between BDNF and PI3K/Akt pathway in the pathogenesis of CPTP.
Conclusion
In conclusion, our results suggest that Shh signaling contributes to CPTP via activating BDNF/TrkB pathway. Inhibition of Shh signaling can alleviate CPTP and is expected to become a new potential therapeutic target for CPTP.
Abbreviations
Shh, sonic hedgehog; CPTP, chronic post-thoracotomy pain; MWT, mechanical withdrawal threshold; CLP, cyclopamine; SAG, smoothened agonist; BDNF, brain-derived neurotrophic factor; TrkB, tropomyosin-related kinase receptor B; PI3K, phosphatidylinositol 3-kinase; Akt, protein kinase B; SDH, spinal cord dorsal horn; Ptch, patched receptor; Smo, smoothened receptors; POD, postoperative day; VFH, the von-Frey filaments.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Wildgaard K, Ravn J, Kehlet H. Chronic post-thoracotomy pain: a critical review of pathogenic mechanisms and strategies for prevention. Eur J Cardiothorac Surg. 2009;36(1):170–180. doi:10.1016/j.ejcts.2009.02.005
2. Bayman EO, Brennan TJ. Incidence and severity of chronic pain at 3 and 6 months after thoracotomy: meta-analysis. J Pain. 2014;15(9):887–897. doi:10.1016/j.jpain.2014.06.005
3. Gottschalk A, Ochroch EA. Clinical and demographic characteristics of patients with chronic pain after major thoracotomy. Clin J Pain. 2008;24(8):708–716. doi:10.1097/AJP.0b013e318174badd
4. Kehlet H, Jensen TS, Woolf CJ. Persistent postsurgical pain: risk factors and prevention. Lancet. 2006;367(9522):1618–1625. doi:10.1016/S0140-6736(06)68700-X
5. Landreneau RJ, Wiechmann RJ, Hazelrigg SR, Mack MJ, Keenan RJ, Ferson PF. Effect of minimally invasive thoracic surgical approaches on acute and chronic postoperative pain. Chest Surg Clin N Am. 1998;8(4):891–906.
6. Mitchell RL. The lateral limited thoracotomy incision: standard for pulmonary operations. J Thorac Cardiovasc Surg. 1990;99(4):
7. Steegers MA, Snik DM, Verhagen AF, van der Drift MA, Wilder-Smith OH. Only half of the chronic pain after thoracic surgery shows a neuropathic component. J Pain. 2008;9(10):955–961. doi:10.1016/j.jpain.2008.05.009
8. He W, Cui L, Zhang C, Zhang X, He J, Xie Y. Sonic hedgehog promotes neurite outgrowth of primary cortical neurons through up-regulating BDNF expression. Neurochem Res. 2016;41(4):687–695. doi:10.1007/s11064-015-1736-5
9. Tukachinsky H, Petrov K, Watanabe M, Salic A. Mechanism of inhibition of the tumor suppressor patched by Sonic Hedgehog. Proc Natl Acad Sci U S A. 2016;113(40):E5866–E5875. doi:10.1073/pnas.1606719113
10. Hashimoto M, Ishii K, Nakamura Y, Watabe K, Kohsaka S, Akazawa C. Neuroprotective effect of sonic hedgehog up-regulated in Schwann cells following sciatic nerve injury. J Neurochem. 2008;107(4):918–927. doi:10.1111/j.1471-4159.2008.05666.x
11. Hill SA, Blaeser AS, Coley AA, et al. Sonic hedgehog signaling in astrocytes mediates cell type-specific synaptic organization. Elife. 2019;8. doi:10.7554/eLife.45545
12. Babcock DT, Shi S, Jo J, Shaw M, Gutstein HB, Galko MJ. Hedgehog signaling regulates nociceptive sensitization. Curr Biol. 2011;21(18):1525–1533. doi:10.1016/j.cub.2011.08.020
13. Liu S, Lv Y, Wan XX, et al. Hedgehog signaling contributes to bone cancer pain by regulating sensory neuron excitability in rats. Mol Pain. 2018;14:1744806918767560. doi:10.1177/1744806918767560
14. Liu S, Yao JL, Wan XX, et al. Sonic hedgehog signaling in spinal cord contributes to morphine-induced hyperalgesia and tolerance through upregulating brain-derived neurotrophic factor expression. J Pain Res. 2018;11:649–659. doi:10.2147/JPR.S153544
15. Edelbrock AN, Alvarez Z, Simkin D, et al. Supramolecular nanostructure activates TrkB receptor signaling of neuronal cells by mimicking brain-derived neurotrophic factor. Nano Lett. 2018;18(10):6237–6247. doi:10.1021/acs.nanolett.8b02317
16. Ho IHT, Liu X, Zou Y, et al. A novel peptide interfering with proBDNF-sortilin interaction alleviates chronic inflammatory pain. Theranostics. 2019;9(6):1651–1665. doi:10.7150/thno.29703
17. Sikandar S, Minett MS, Millet Q, et al. Brain-derived neurotrophic factor derived from sensory neurons plays a critical role in chronic pain. Brain. 2018;141(4):1028–1039. doi:10.1093/brain/awy009
18. Chen A, Xiong LJ, Tong Y, Mao M. Neuroprotective effect of brain-derived neurotrophic factor mediated by autophagy through the PI3K/Akt/mTOR pathway. Mol Med Rep. 2013;8(4):1011–1016. doi:10.3892/mmr.2013.1628
19. Li XT, Liang Z, Wang TT, et al. Brain-derived neurotrophic factor promotes growth of neurons and neural stem cells possibly by triggering the phosphoinositide 3-Kinase/AKT/Glycogen Synthase Kinase-3beta/beta-catenin Pathway. CNS Neurol Disord Drug Targets. 2017;16(7):828–836. doi:10.2174/1871527316666170518170422
20. Duan B, Liu DS, Huang Y, et al. PI3-kinase/Akt pathway-regulated membrane insertion of acid-sensing ion channel 1a underlies BDNF-induced pain hypersensitivity. J Neurosci. 2012;32(18):6351–6363. doi:10.1523/JNEUROSCI.4479-11.2012
21. Buvanendran A, Kroin JS, Kerns JM, Nagalla SN, Tuman KJ. Characterization of a new animal model for evaluation of persistent postthoracotomy pain. Anesth Analg. 2004;99(5):
22. Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17(6):1031–1036. doi:10.1016/0031-9384(76)90029-9
23. Zhu A, Shen L, Xu L, Chen W, Huang Y. Suppression of Wnt5a, but not Wnts, relieves chronic post-thoracotomy pain via anti-inflammatory modulation in rats. Biochem Biophys Res Commun. 2017;493(1):474–480. doi:10.1016/j.bbrc.2017.08.167
24. Wang H, Liu W, Cai Y, et al. Glutaminase 1 is a potential biomarker for chronic post-surgical pain in the rat dorsal spinal cord using differential proteomics. Amino Acids. 2016;48(2):337–348. doi:10.1007/s00726-015-2085-z
25. Hung CH, Wang JC, Strichartz GR. Spontaneous chronic pain after experimental thoracotomy revealed by conditioned place preference: morphine differentiates tactile evoked pain from spontaneous pain. J Pain. 2015;16(9):903–912. doi:10.1016/j.jpain.2015.06.006
26. Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53(1):55–63. doi:10.1016/0165-0270(94)90144-9
27. Nara T, Saito S, Obata H, Goto F. A rat model of postthoracotomy pain: behavioural and spinal cord NK-1 receptor assessment. Can J Anaesth. 2001;48(7):665–676. doi:10.1007/BF03016201
28. Marti E, Bovolenta P. Sonic hedgehog in CNS development: one signal, multiple outputs. Trends Neurosci. 2002;25(2):89–96. doi:10.1016/S0166-2236(02)02062-3
29. Han L, Ma J, Duan W, et al. Pancreatic stellate cells contribute pancreatic cancer pain via activation of sHH signaling pathway. Oncotarget. 2016;7(14):18146–18158. doi:10.18632/oncotarget.7776
30. Chen KY, Cheng CJ, Wang LC. Activation of sonic hedgehog leads to survival enhancement of astrocytes via the GRP78-dependent pathway in mice infected with angiostrongylus cantonensis. Biomed Res Int. 2015;2015:674371.
31. Ding X, Cai J, Li S, Liu XD, Wan Y, Xing GG. BDNF contributes to the development of neuropathic pain by induction of spinal long-term potentiation via SHP2 associated GluN2B-containing NMDA receptors activation in rats with spinal nerve ligation. Neurobiol Dis. 2015;73:428–451. doi:10.1016/j.nbd.2014.10.025
32. Duric V, McCarson KE. Neurokinin-1 (NK-1) receptor and brain-derived neurotrophic factor (BDNF) gene expression is differentially modulated in the rat spinal dorsal horn and hippocampus during inflammatory pain. Mol Pain. 2007;3:32. doi:10.1186/1744-8069-3-32
33. Zhou LJ, Yang T, Wei X, et al. Brain-derived neurotrophic factor contributes to spinal long-term potentiation and mechanical hypersensitivity by activation of spinal microglia in rat. Brain Behav Immun. 2011;25(2):322–334. doi:10.1016/j.bbi.2010.09.025
34. Pezet S, Cunningham J, Patel J, et al. BDNF modulates sensory neuron synaptic activity by a facilitation of GABA transmission in the dorsal horn. Mol Cell Neurosci. 2002;21(1):51–62. doi:10.1006/mcne.2002.1166
35. Zhao J, Seereeram A, Nassar MA, et al. Nociceptor-derived brain-derived neurotrophic factor regulates acute and inflammatory but not neuropathic pain. Mol Cell Neurosci. 2006;31(3):539–548. doi:10.1016/j.mcn.2005.11.008
36. Dembo T, Braz JM, Hamel KA, Kuhn JA, Basbaum AI. Primary afferent-derived BDNF contributes minimally to the processing of pain and itch. eNeuro. 2018;5(6):
37. Garraway SM, Petruska JC, Mendell LM. BDNF sensitizes the response of lamina II neurons to high threshold primary afferent inputs. Eur J Neurosci. 2003;18(9):2467–2476. doi:10.1046/j.1460-9568.2003.02982.x
38. Sun W, Zhang L, Li R. Overexpression of miR-206 ameliorates chronic constriction injury-induced neuropathic pain in rats via the MEK/ERK pathway by targeting brain-derived neurotrophic factor. Neurosci Lett. 2017;646:68–74. doi:10.1016/j.neulet.2016.12.047
39. Xu Q, Fitzsimmons B, Steinauer J, et al. Spinal phosphinositide 3-kinase-Akt-mammalian target of rapamycin signaling cascades in inflammation-induced hyperalgesia. J Neurosci. 2011;31(6):2113–2124. doi:10.1523/JNEUROSCI.2139-10.2011
40. Pezet S, Marchand F, D’Mello R, et al. Phosphatidylinositol 3-kinase is a key mediator of central sensitization in painful inflammatory conditions. J Neurosci. 2008;28(16):4261–4270. doi:10.1523/JNEUROSCI.5392-07.2008
41. Liu W, Lv Y, Ren F. PI3K/Akt pathway is required for spinal central sensitization in neuropathic pain. Cell Mol Neurobiol. 2018;38(3):747–755. doi:10.1007/s10571-017-0541-x
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.