Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Soluble epoxide hydrolase inhibition enhances anti-inflammatory and antioxidative processes, modulates microglia polarization, and promotes recovery after ischemic stroke
Authors Yeh CF, Chuang TY, Hung YW, Lan MY ![]() , Tsai CH, Huang HX, Lin YY
, Tsai CH, Huang HX, Lin YY
Received 29 March 2019
Accepted for publication 4 July 2019
Published 15 October 2019 Volume 2019:15 Pages 2927—2941
DOI https://doi.org/10.2147/NDT.S210403
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Yu-Ping Ning
Chien-Fu Yeh,1–3,* Tung‐Yueh Chuang,4,* Yu-Wen Hung,5 Ming-Ying Lan,2,3 Ching-Han Tsai,4 Hao-Xiang Huang,4 Yung-Yang Lin1,4,6–8
1Institute of Brain Science, National Yang-Ming University, Taipei 11221, Taiwan; 2Department of Otorhinolaryngology, National Yang-Ming University, Taipei 11221, Taiwan; 3Department of Otolaryngology-Head and Neck Surgery, Taipei Veterans General Hospital, Taipei 11217, Taiwan; 4Department of Critical Care Medicine, Taipei Veterans General Hospital, Taipei 11217, Taiwan; 5Institute of Cellular and System Medicine, National Health Research Institutes, Miaoli County 35053, Taiwan; 6Institute of Physiology, National Yang-Ming University, Taipei 11221, Taiwan; 7Institute of Clinical Medicine, National Yang-Ming University, Taipei 11221, Taiwan; 8Department of Neurology, Neurological Institute, Taipei Veterans General Hospital, Taipei 11217, Taiwan
*These authors contributed equally to this work
Correspondence: Yung-Yang Lin
Department of Critical Care Medicine, Taipei Veterans General Hospital, No. 201, Sec. 2, Shipai Rd., Taipei 11217, Taiwan
Tel +886-2-2871-2121
Fax +886-2-2875-7890
Email [email protected]
Background: Ischemic stroke triggers inflammatory responses and oxidative stress in the brain, and microglia polarization affects the degree of neuroinflammation. It has been reported that the inhibition of soluble epoxide hydrolase (sEH) activity protects brain tissue. However, the anti-inflammatory and antioxidative effects of sEH inhibition in the ischemic brain are not fully understood. This study aimed to investigate the effects of a selective sEH inhibitor, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (AUDA), after ischemic stroke.
Methods: Adult male rats with middle cerebral artery occlusion (MCAO) were administered with AUDA or a vehicle. Behavioral outcome, infarct volume, microglia polarization, and gene expression were assessed.
Results: Rats treated with AUDA showed better behavioral outcomes and smaller infarct volumes after MCAO. After AUDA treatment, a reduction of M1 microglia and an increase of M2 microglia occurred at the ischemic cortex of rats. Additionally, there was an increase in the mRNA expressions of antioxidant enzymes and anti-inflammatory interleukin-10, and pro-inflammatory mediators were decreased after AUDA administration. Heme oxygenase-1 was mainly expressed by neurons, and AUDA was found to improve the survival of neurons.
Conclusion: The results of this study provided novel and significant insights into how AUDA can improve outcomes and modulate inflammation and oxidative stress after ischemic stroke.
Keywords: ischemic stroke, microglia polarization, middle cerebral artery occlusion, soluble epoxide hydrolase, anti-inflammation, antioxidant
Introduction
Stroke is a common neurological disease caused by the interruption of blood supply to the brain, and it can lead to neurological deficits.1 It is the second leading cause of death worldwide, with tremendous resulting economic and social burdens on the human population.2,3 Stroke can be classified as hemorrhagic or ischemic, wherein approximately 80% of all patients fall into the latter category.4,5 After an ischemic stroke, the mechanisms of cerebral damage are primary ischemia and secondary neuroinflammation.6 In the initial period, ischemia deprives the brain of oxygen, glucose, and other vital substances which are essential for brain cells.7,8 Subsequently, ischemic stroke leads to cerebral tissue injury by the development of inflammation. After stroke, neuroinflammation rapidly triggers microglia activation and then promotes the infiltration of circulating inflammatory cells, including monocyte/macrophages, T lymphocytes, and neutrophils.9 In the acute phase (minutes to hours), pro-inflammatory cytokines and chemokines and reactive oxygen species (ROS) are released from the injured brain tissue.10 In the subacute phase (hours to days), a high expression of matrix metalloproteinases (MMPs) and ROS promotes neuroinflammation by causing a disruption of the blood-brain barrier and neuronal death.9 Since there is a longer therapeutic time window for stroke-related cerebral inflammation than for the primary ischemic injury, there has been important experimental research in recent years focusing on such neuroinflammation.11
Acute ischemic stroke not only induces a rapid increase of ROS production but also impairs the antioxidant system. Overabundant ROS causes cellular injury, inflammation, and death via lipid peroxidation and the oxidation of protein, RNA and DNA.12 Superfluous ROS can be scavenged by some enzymatic antioxidants such as heme oxygenase-1 (HO-1), superoxide dismutases (SODs), and catalases (CATs). These enzymatic antioxidants are regulated by nuclear factor erythroid 2-related factor 2 (Nrf2), one of the critical regulators of endogenous antioxidant defense. Stimulating the Nrf2-mediated antioxidant process protects against cerebral ischemia.13,14 Thus, reducing ROS production and inducing antioxidant defense are reasonable therapeutic strategies.
Residential microglia are the primary immune cells in the brain, activated by cerebral ischemia.15 Upon activation, microglia undergo a morphologic change from the resting state of long ramifications and small soma to an activated state with a rounded amoeboid shape. Once activated, the cells develop polarized activation with distinct physiological roles: M1 and M2 polarization.16,17 M1 or the classic activation drives the microglia toward a pro-inflammatory phenotype to produce pro-inflammatory mediators such as tumor necrosis factor-α, interleukin (IL)-1β, IL-6, monocyte chemoattractant protein-1 (MCP-1), ROS, and proteolytic enzymes MMP-9, which leads to blood-brain barrier breakdown.18–22 In addition, these microglia express high levels of inducible nitric oxidase synthase (iNOS) for the production of nitric oxide.22,23 By contrast, M2 or the alternatively activated microglia execute the effect of neuroprotection by secreting neurotrophic factors and anti-inflammatory cytokines such as IL-10.24 The balance of polarization toward M1 or M2 can affect the severity of inflammation after experimental stroke, and the modulation of microglia to the M2 type has been reported to have a beneficial role in ischemic stroke.25
Epoxyeicosatrienoic acids (EETs), which are synthesized from arachidonic acid by cytochrome P450 epoxygenases, have been proven to be beneficial in several disease models. In the cardiovascular system, EETs exhibit vasodilation effects, anti-inflammatory actions and antimigratory actions on vascular smooth muscle cells.26 In brain tissue, EETs also possess an anti-inflammatory effect and can protect the brain from ischemic injury.27 In vivo, EETs are rapidly metabolized into inactive dihydroxyeicosatrienoic acids (DHET) by soluble epoxide hydrolase (sEH), thereafter decreasing its anti-inflammatory effect.26 Genetic studies of EPHX2 (which encodes the cytosolic and peroxisomal sEH) have demonstrated the association of several variants with the risk of ischemic stroke. The R287Q and G860A variants which are associated with reduced enzyme activity and protein stability were linked to lowered risk for ischemic stroke.28–30 Deletion of the sEH gene and pharmacological inhibition of sEH have both been reported to reduce infarct size after experimental ischemic stroke.31–36 In mice models with intracerebral hemorrhage (ICH) and bilateral carotid artery stenosis (BCAS), the activation of microglia was decreased by an sEH inhibitor.37,38 In our previous study, we had demonstrated that M1 monocyte/macrophages were reduced and rotarod road running time was significantly increased by an sEH inhibitor after experimental stroke.39 However, it remains unclear how the sEH inhibitor contributes to anti-inflammatory and antioxidative effects in ischemic stroke. In the present study, we used a middle cerebral artery occlusion (MCAO) model in rats to investigate whether the pharmacological inhibition of sEH by 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (AUDA) can enhance anti-inflammatory and antioxidant processes, modulate the polarization of microglia activation in the ischemic cortex and improve neurological function. The molecular mechanism underlying the AUDA-elicited beneficial process is also delineated.
Methods
Animals
Ten-week-old male Wistar Kyoto (WKY) rats weighing 200–300 g were obtained from BioLASCO (Taipei, Taiwan). Female rats were not used to avoid any influences of sex hormones. The rats lived in clean cages with a 12-hr light/dark cycle at a constant temperature, and had free access to water and food. All animal experiments were approved by the Institutional Animal Care and Utilization Committee (IACUC) of National Yang-Ming University, Taipei, Taiwan (approval ID: 1050315), and animal care was performed in compliance with the guidelines of IACUC and the United States National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Rats were randomly assigned to sham-operated, vehicle, and AUDA treatment groups and allocation concealment was performed. The experimenters were blind to the experimental conditions and blinding was further extended through outcome assessment. All postmortem examinations were conducted blindly with pseudonymization.
Focal cerebral ischemia with MCAO
Focal cerebral ischemia with MCAO was achieved by the procedures which we previously described.40 Briefly, rats were anesthetized with Zoletil (30 mg/kg) and Xylazine (8 mg/kg) by intraperitoneal (i.p.) injection. Bilateral common carotid arteries (CCAs) were identified through a vertical incision on the midline of the neck. The temporal muscle was retracted, and the right middle cerebral artery (MCA) was exposed through a craniotomy on the right temporal bone drilled by a cutting burr. The dura matter was cut, and the MCA was permanently occluded by suture ligation between the inferior cerebral vein and the olfactory tract. Subsequently, the bilateral CCAs were occluded temporarily for 90 mins by a vessel clamp. Rats in the sham group were subjected to identical anesthesia and surgical procedure without MCAO. During MCAO, body temperature was maintained with a pad. Only rats receiving successful MCAO were included for further treatment. Rats with massive bleeding during MCAO, rats that died during or after MCAO, and rats with no neurological deficits after MCAO were excluded. The mortality rate after MCAO was less than 10%.
Administration of sEH inhibitor
Rats were randomly assigned to sham-operated, vehicle, and AUDA treatment groups. Intraperitoneal injection of AUDA (20 mg/kg, Cayman Chemicals, MI, USA) or dimethyl sulfoxide was performed every 24 hrs, beginning 2 hrs after MCAO until 1 day before the rats were sacrificed.
Animal behavioral test
Neurological function assessment was performed with the beam walk test.
The beam walk test was utilized to evaluate fine motor coordination and function, especially on the hindlimb. Rats escaped a strange cage by walking along a narrowed beam (25×1100 mm) to enter its own cage at the opposite end of the beam. A score of 7 was given when animals traversed the beam with two or less footslips; 6 was given when animals traversed the beam with less than 50% footslips; 5 was given for more than 50% but less than 100% footslips; 4 was given for 100% footslips; 3 was given for traversal with the affected limb extended and not reaching the surface of the beam; 2 was given when the animal was able to balance on the beam but not traverse it; and 1 was given when the animal could not balance on the beam. Three trials were recorded for each testing day and footslip percentage and score were used to qualify motor performance (n=6/group).
Quantification of infarct volume
At 1 and 3 days after MCAO, rats (n=6/group) were subjected to transcardial perfusion with phosphate-buffered saline (PBS) after anesthetization with Zoletil and Xylazine by intraperitoneal injection. The rats were decapitated and their brains were quickly removed from the skull. For infarction size determination, brains were sliced into 2-mm-thick coronal sections, stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma, St. Louis, MO, USA) solution for 15 mins at room temperature (RT) without light, and then fixed with 4% paraformaldehyde (PFA). The unstained region represented the infarcted area, and the non-infarcted area revealed a red color on TTC-stained sections. To perform edema correction, the infarcted area was determined by subtracting the non-infarcted area in the ipsilateral hemisphere from the contralateral hemisphere. Infarct volumes were calculated as the summation of infarcted areas on each brain slice by ImageJ software (The National Institute of Health, Bethesda, MD, USA).
Immunofluorescence staining and quantification
At 1 and 3 days after MCAO, rats (n=3/group) were subjected to transcardial perfusion with PBS, followed by 4% PFA in PBS (10 mM, pH 7.4). After anesthetization, brain tissue was removed, post-fixed in 4% PFA overnight at 4°C, and cryoprotected with 30% (w/v) sucrose in PBS for about 3 days. Then, 40-μm coronal brain cryosections were obtained using a freezing microtome (CM1900, Leica, Heidelberg, Germany) and stored in antifreeze solution (50 mM phosphate buffer, 15% glucose, 30% (v/v) ethylene glycol, 0.05% sodium azide; pH 7.4) at ‒20°C until further testing.
To characterize the microglia activation, neuron survival, and antioxidant distribution, the free-floating tissue sections were incubated in 3% normal horse serum (NHS) to bind nonspecific antigen for 1 hr at RT. The sections then were incubated with primary antibody overnight at 4°C. The following primary antibodies were used: mouse anti-CD86 (1:500; Proteintech, Rosemont, IL, USA), rabbit anti-CD206 (1:500; Abcam, Cambridge, UK), rabbit anti-neuronal nuclei (NeuN) (1:1000; Millipore, Billerica, MA, USA), and mouse anti-HO-1 (1:500; Abcam, Cambridge, UK). After conjugation with the primary antibodies, the sections were washed in PBS, incubated with the following fluor-conjugated secondary antibodies for 1 hr at RT: DyLight™ 488-conjugated secondary anti-rabbit or anti-mouse antibody and DyLight™ 594-conjugated secondary anti-rabbit antibody (1:500; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). For CD86/ionized calcium binding adapter molecule 1 (Iba-1) and CD206/Iba-1 double-labeling, the sections were incubated in goat anti-Iba-1 primary antibody (1:500, Novus, USA) overnight at 4°C. The sections were rinsed in PBS, then incubated with DyLight™ 549-conjugated secondary anti-rabbit or DyLight™ 594-conjugated secondary anti-goat antibody (1:500; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 1 hr at RT. All of the brain sections were then mounted on gelatin-coated slides. After being air-dried, these sections were placed on coverslips with Vectashield HardSet™ mounting medium with DAPI (H-1500; Vector Laboratories, Burlingame, CA, USA). Immunofluorescence was acquired by a laser confocal microscope (Zeiss LSM 700; Carl Zeiss, Germany) with four laser lines: 405, 488, 555, and 639 nm.
Microglia isolation
At 1 and 3 days after MCAO, the rats were anesthetized and subjected to transcardial perfusion with PBS. After decapitation, each rat brain was harvested and divided into two hemispheres: the ischemic and non-ischemic sides. Cerebral cortex was subjected to enzymatic digestion and mechanical dissociation as suggested by the manufacturer (Neural Dissociation Kit and gentleMACS™ Dissociator, Miltenyi Biotec, San Diego, CA, USA). PBS (10x) was added to the original Percoll (Sigma Chemical Co, St Louis, MO, USA) at a ratio of 1:9 and this solution was considered as a 100% Percoll solution. Furthermore, 30% and 70% Percoll solutions were prepared with HBSS (without phenol red) and 37% Percoll solution with HBSS (with phenol red). The cell pellet was resuspended in 4 mL of 37% Percoll solution. Subsequently, 4 mL 70% Percoll solution was added to the bottom of tube by syringe and 4 mL 30% Percoll solution was added to the top layer. The discontinuous Percoll gradient of 30%, 37%, and 70% was centrifuged at 500 g for 30 mins, and a thin white layer was observed between 37% and 70% Percoll solutions. The layer was considered to be the collection of microglia/macrophages. The white layer was then transferred and washed in three times the volume of HBSS at 600 g for 7 mins. Finally, the cell pellet was washed using PBS and the whole population of microglia/macrophages was collected.
Flow cytometric analysis of microglia polarization
At 3 days after MCAO, the rats (n=4/group) were anesthetized and subjected to transcardial perfusion with PBS. Following the isolation protocol, microglia/macrophages were recovered from the interphase, blocked with 0.5% bovine serum albumin and 2% NHS and stained for surface markers using fluorophore-conjugated primary antibody at 4°C for a designated time: anti-CD11b/c-PE-Cy7 (1:100; 60 mins; Biolegend, San Diego, CA, USA), anti-CD45-APC-Cy7 (1:25; 60 mins; Biolegend, San Diego, CA, USA), anti-CD86-APC (1:20; 30 mins; Miltenyi Biotec, San Diego, CA, USA), anti-CD206-FITC (1:125; 30 mins; Bioss, Boston, MA, USA). Appropriate negative and compensation controls were performed. Data were acquired on a FACSCanto II flow cytometer (BD Biosciences, San Jose, CA, USA) and analyzed using BD FACSDiva Software (BD Biosciences, San Jose, CA, USA). Ten thousand events were recorded for each sample. Cells were gated using forward and sideward scatter characteristics to exclude cell debris. Microglia were recognized as a CD11b+CD45int subpopulation. After gating microglia, the M1 and M2 type microglia were identified as CD86+ and CD206+ cells, respectively.
Reverse-transcription quantitative polymerase chain reaction (PCR)
At 1 day after MCAO, total RNA was extracted from the ischemic cortex tissue (n=4/group) and microglia (n=3/group), respectively, using E.Z.N.A.® HP Total RNA Kit (Omega Bio-tek Inc., Norcross, GA, USA). Then, they were reverse-transcribed to cDNA using a Thermo Scientific Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific Inc., Waltham, MA, USA). Reverse-transcription quantitative PCR was performed on an ABI QuantStudio 3 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using SYBR green PCR Master Mix (Vazyme, Nanjing, China) with the appropriate primers as noted in Table S1. Relative expression levels of these mRNAs were calculated as fold changes vs sham using the 2−ΔΔCT method. Tissue samples were normalized to Rpl13a and microglia were normalized to GAPDH. The expression levels of mRNAs were calculated as fold changes vs control. Melting curves were routinely performed to determine the specificity of the PCR reaction.
Statistical analyses
The statistical analyses were conducted by researchers blinded to the experimental design and conditions. Data are expressed as mean ± the standard error of the mean. Independent samples t-test or the Mann–Whitney U test was used to analyze the difference between the two groups. For multiple comparisons, one-way analysis of variance was performed followed by Tukey’s post hoc test. All statistical analyses were carried out using the SPSS 24.0 software package (SPSS, Inc, Chicago, IL, USA). Results were considered significant at P<0.05.
Results
sEH inhibitor treatment promoted functional recovery
In the beam walk test, the rats treated with AUDA had a better mean score compared with the vehicle-treated group at 1 day (6.17 vs 4.83, P=0.003) and 3 days (6.58 vs 5.92, P=0.011) after MCAO (Figure 1A). Footslips mean percentage was also improved after AUDA administration at 1 day (28.7% vs 73.9%, P=0.001) and 3 days (32.4% vs 15.0%, P=0.003) after MCAO (Figure 1B). The effect of AUDA was better at 1 day than that at 3 days after MCAO. In groups treated with vehicle, beam walk test performance at 1 day after surgery was the worst of all groups, but improved with time (all P<0.001) (Figure 1).
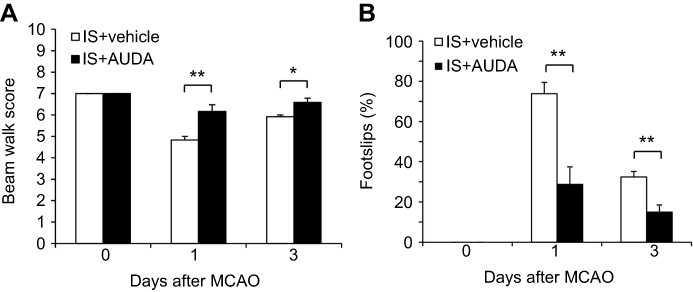 |
Figure 1 sEH inhibitor improved recovery of neurological function at 1 and 3 days after MCAO. Notes: The sensorimotor function of MCAO rats treated with or without AUDA was examined in (A) beam walk score (n=6/group) and (B) footslips percentage in beam walk test (n=6/group). Data are presented as mean ± SEM. *P<0.05, **P<0.01. Abbreviations: IS, ischemia; AUDA, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid; MCAO, middle cerebral artery occlusion; sEH, soluble epoxide hydrolase; SEM, standard error of the mean. |
sEH inhibitor reduced infarct volume after MCAO
The ischemic region revealed a pale color by the use of TTC stain, and only the cortex was shown to be involved (Figure 2A, B). At 1 day after MCAO, the mean infarct volume of the vehicle control group was 200.3 mm,3 larger than the 126.6 mm3 of the group treated with AUDA (P=0.003). At 3 days after surgery, the mean infarct volumes of the group with vehicle and AUDA treatment were 157.7 and 122.1 mm3, respectively (P=0.008). The effect of decreasing the infarct area by AUDA existed at both 1 and 3 days after MCAO. With time, the infarct size also became smaller in the group receiving vehicle treatment (P=0.005) (Figure 2C).
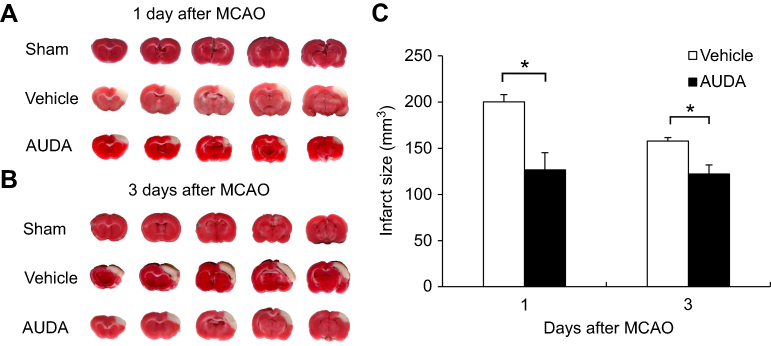 |
Figure 2 sEH inhibition decreased the infarct volume after MCAO. Notes: (A and B) Representative brain slices with TTC staining from rats of sham-operated, vehicle treatment and AUDA treatment at 1 and 3 days after MCAO. The pale region indicates the infarct area. (C) Quantification of infarct volume of rats subjected to vehicle treatment and AUDA treatment at 1 and 3 days after MCAO. The sham-operated rats revealed no infarction. n=6/group. Data are presented as mean ± SEM.*P<0.01. Abbreviations: AUDA, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid; MCAO, middle cerebral artery occlusion; sEH, soluble epoxide hydrolase; SEM, standard error of the mean; TTC, triphenyltetrazolium chloride. |
sEH inhibitor modulated the microglia polarization
Ischemic stroke triggers serious inflammatory responses. Microglia, which are major cellular contributors, polarize into either M1 classical activation or M2 alternative activation in response to brain alterations elicited by ischemic insults. Upon the further inspection of microglia with a confocal microscope, M1 type microglia/macrophages with coexpression of CD86 and Iba-1 were obviously seen in the ischemic border of rats with vehicle treatment, more so than in the ischemic region of rats with AUDA treatment at 1 and 3 days after MCAO (P=0.041 and 0.002, respectively) (Figure 3A, C). Meanwhile, there were significantly fewer M2 type microglia/macrophages with coexpression of CD206 and Iba-1 in the ischemic border of vehicle-treated rats than in the AUDA-treated rats at 3 days after MCAO (P=0.005) (Figure 3B, C). At 1 day after MCAO, CD206+/Iba-1+ double positive microglia/macrophages were rarely found in both groups. No CD86+/Iba-1+ or CD206+/Iba-1+ double positive microglia/macrophages were found in the corresponding area of the non-ischemic hemisphere. In the ischemic border area, the activated microglia presented with an amoeboid shape; however, the microglia in the non-ischemic hemisphere remained in a resting state and had small soma with long ramifications (Figure 3A, B).
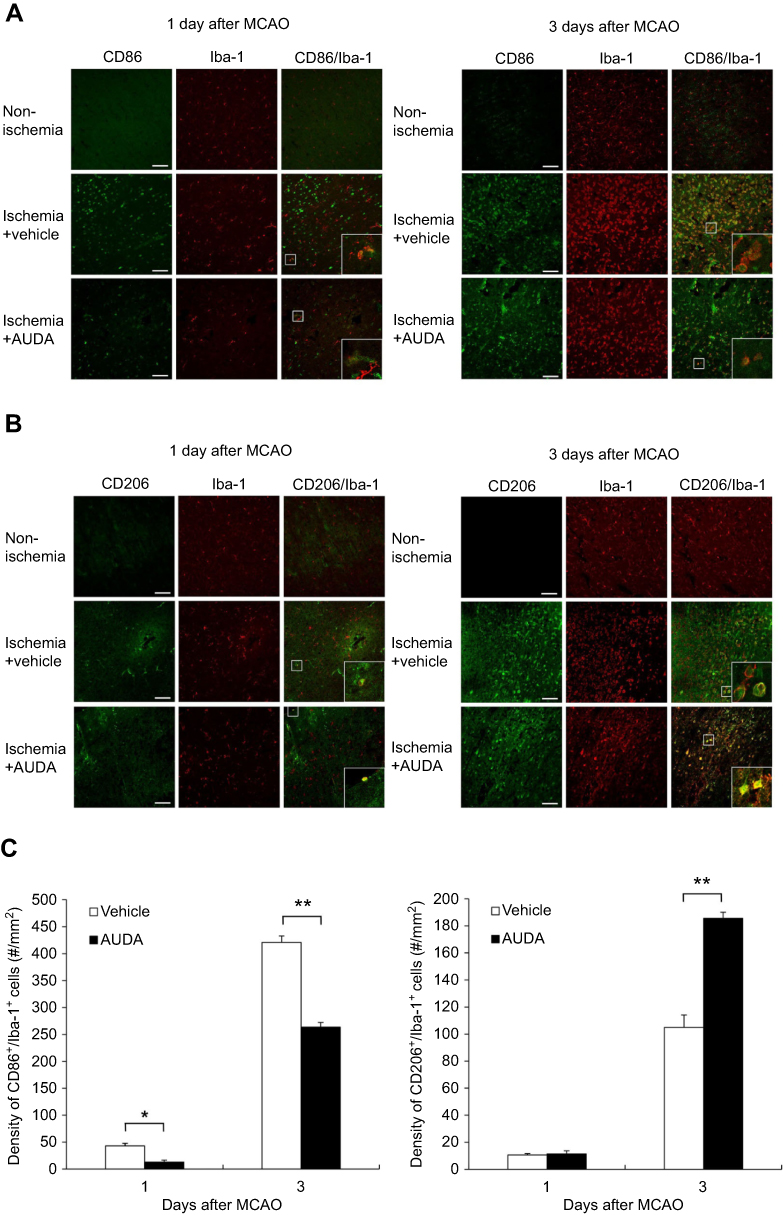 |
Figure 3 sEH inhibition modulated microglia/macrophages polarization after MCAO. Notes: (A) Representative immunofluorescent staining of CD86 (green) and Iba-1 (red) in the non-ischemic hemisphere, the ischemic border of rats in vehicle and AUDA treatment groups. Scale bar represents 100 μm. (B) Representative immunofluorescent staining of CD206 (green) and Iba-1 (red) in the non-ischemic hemisphere, the ischemic border of rats in vehicle and AUDA treatment group. Scale bar represents 100 μm. (C) Statistical analysis of CD86+/Iba-1+ or CD206+/Iba-1+ double positive microglia/macrophages in the ischemic border is expressed as cells/mm,2 n=3/group. Data are presented as mean ± SEM.*P<0.05 and **P<0.01. Abbreviations: AUDA, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid; Iba-1, ionized calcium binding adaptor molecule 1; MCAO, middle cerebral artery occlusion; sEH, soluble epoxide hydrolase; SEM, standard error of the mean. |
CD86, CD206, and Iba-1 are expressed in both activated microglia and macrophages. To further clarify the effect of AUDA on microglia polarization, flow cytometry was used to distinguish microglia from macrophages. Flow cytometry revealed the microglia polarization toward the M2 type after sEH inhibition at 3 days after surgery. In the CD11b+CD45int microglia population from the ischemic region, the percentage of CD86+ M1 microglia decreased from 51.0±4.8% to 35.6±3.4% after the administration of AUDA (P=0.041). In contrast, the percentage of CD206+ M2 microglia increased from 22.0±5.2% to 41.6±3.7% after AUDA treatment (P=0.022) (Figure 4).
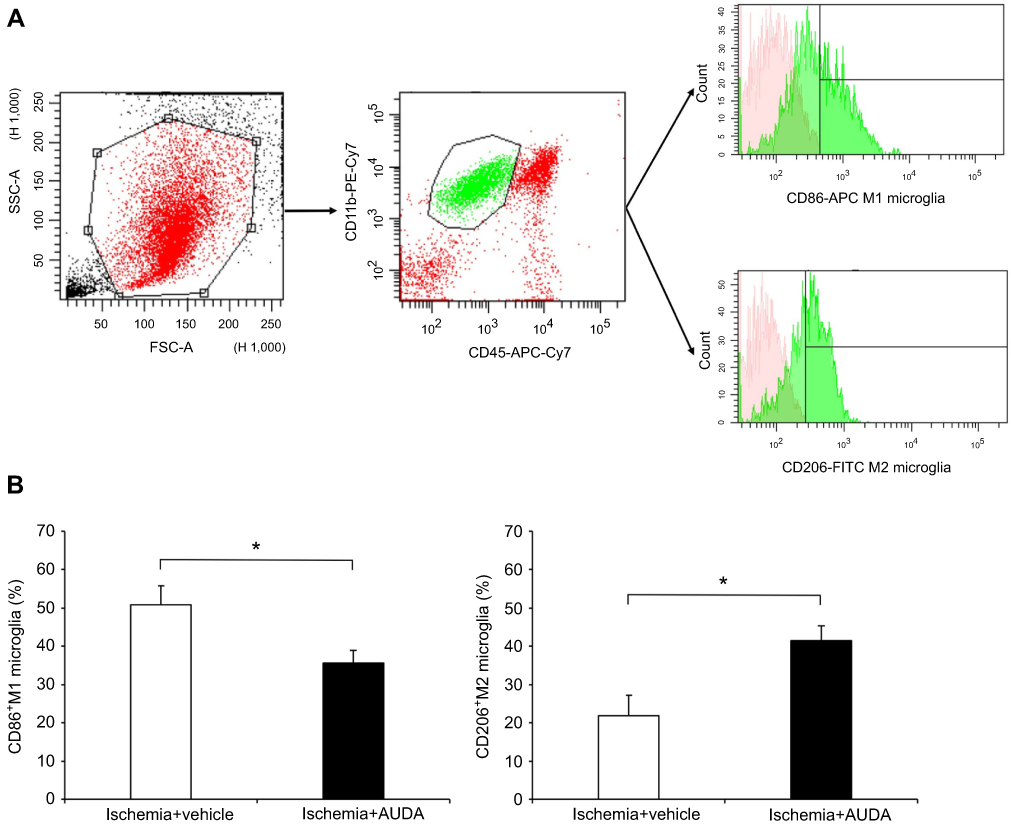 |
Figure 4 Flow cytometry analysis revealed that the sEH inhibitor suppresses M1 microglia and promotes M2 microglia polarization after MCAO. Notes: (A) Gating strategy to detect M1 microglia (CD11+CD45intCD86+) and M2 microglia (CD11+CD45intCD206+). In the histograms, the pink regions represented unstained cells (negative control). The green regions on the right of the vertical black lines represented APC-positive (M1) or FITC-positive (M2) cells. (B) The percentage of M1 and M2 microglia of rats in vehicle and AUDA treatment groups, n=4/group. Data are presented as mean ± SEM.*P<0.05. Abbreviations: APC, allophycocyanin; AUDA, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid; FITC, fluorescein isothiocyanate; MCAO, middle cerebral artery occlusion; PE, phycoerythrin; sEH, soluble epoxide hydrolase; SEM, standard error of the mean. |
sEH inhibitor regulated mRNA expression of pro- and anti-inflammatory cytokines and antioxidant enzymes
The microglia polarization shift was obvious at 3 days, but functional recovery was significant from 1 day after MCAO. Thus, in the first 24 hrs, there may exist some protective effects induced by AUDA treatment. To clarify the effect of AUDA at 1 day after MCAO, ischemic cortex tissue and microglia of the cortex were collected and analyzed by reverse-transcription quantitative PCR. In a previous study, microglia/monocytes were harvested by Percoll gradient. The authors found a slight increase but no significant difference in monocyte counts 24 hrs after MCAO surgery in ischemic brain compared with sham braincases. This means the majority of cells collected by Percoll gradient are microglia at 24 hrs after MCAO.41 The contribution of microglia in brain tissue can be demonstrated through this method at 1 day after MCAO. The mRNA expression of pro-inflammatory mediators including IL-1β, IL-6, iNOS, MCP-1, and MMP-9 was attenuated in ischemic cortex (P=0.012, 0.021, 0.044, 0.040, and 0.041, respectively). Besides, the level of mRNA of anti-inflammatory IL-10 was increased at 1 day after MCAO (P=0.040) in ischemic cortex. Upon further inspection of microglia in the ischemic area, AUDA administration decreased the mRNA level of IL-1β, IL-6, iNOS, MCP-1, and MMP-9 at 1 day after MCAO (P=0.045, 0.033, 0.041, 0.035, and 0.007, respectively). The gene expression pattern of these pro-inflammatory mediators between the cortex and microglia was similar and AUDA-regulated pro-inflammatory mediators in cortex were partially contributed by microglia (Figure 5A).
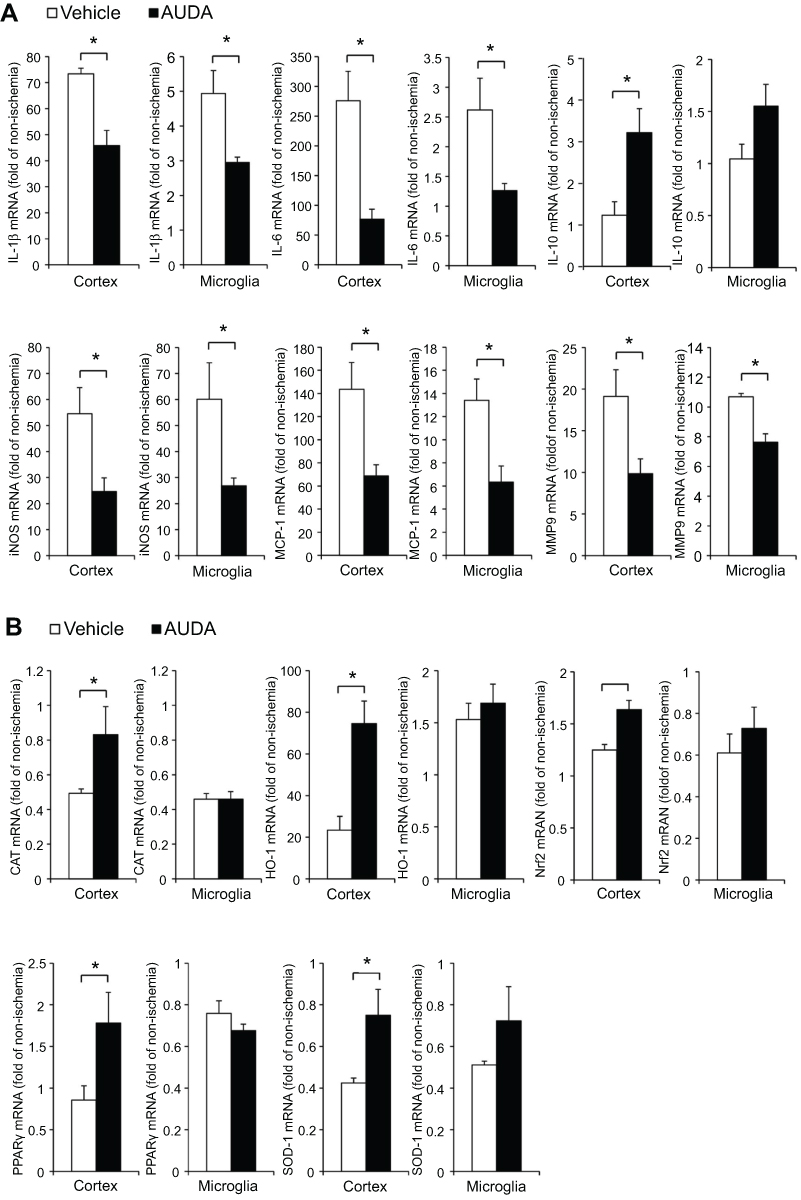 |
Figure 5 Inhibition of sEH regulated mRNA expression of pro- and anti-inflammatory cytokines and antioxidant mediators at 1 day after MCAO. Notes: (A) AUDA-regulated mRNA expression of pro- and anti-inflammatory cytokines (IL-1β, IL-6, IL-10, iNOS, MCP-1, MMP-9). (B) AUDA-regulated mRNA expression of antioxidant mediators (CAT, HO-1, Nrf2, PPARγ, SOD-1). Reverse-transcription polymerase chain reaction was performed using total RNA extracted from ischemic cortex tissue (n=4/group) and microglia (n=3‒4/group). Data are presented as mean ± SEM. *P<0.05. Abbreviations: AUDA, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid; CAT, catalase; HO-1, heme oxygenase-1; IL, interleukin; iNOS, inducible nitric oxidase synthase; MCAO, middle cerebral artery occlusion; MCP-1, monocyte chemoattractant protein-1; MMP-9, matrix metalloproteinase-9; Nrf2, nuclear factor erythroid 2-related factor 2; PPARγ, peroxisome proliferator-activated receptor-γ; sEH, soluble epoxide hydrolase; SEM, standard error of the mean; SOD-1, superoxide dismutase-1. |
After AUDA treatment, the mRNA level of antioxidant enzymes including CAT, HO-1, and SOD-1 was upregulated in ischemic cortex at 1 day after MCAO (P=0.033, 0.016, and 0.012, respectively). The critical transcription factor for controlling antioxidant enzymes, Nrf2, was also upregulated in ischemic cortex at 1 day after MCAO (P=0.011). In rats receiving AUDA treatment, peroxisome proliferator-activated receptor-γ (PPAR-γ) was upregulated in ischemic cortex at 1 day after MCAO (P=0.032) (Figure 5B). However, these five antioxidant-associated gene expressions were not altered by AUDA at 1 day after MCAO in the microglia of ischemic cortex (Figure 5B). Thus, microglia were not the target cells for the AUDA-regulated antioxidant effect. Among the antioxidant-associated genes in cortex tissue, HO-1 accounted for the most upregulated gene after AUDA treatment. Coexpression of HO-1 and Iba-1 in microglia was rare and there was no difference between vehicle and AUDA-treated groups (P=0.274) (Figure 6A, C). Double-labeling immunofluorescence showed that HO-1 was colocalized with NeuN-positive neurons. More neuronal cells with coexpression of HO-1 and NeuN were found in rats receiving AUDA treatment than in rats receiving vehicle treatment at 1 day after MCAO (P=0.012) (Figure 6B, C). NeuN gene expression in the ischemic area was significantly increased in AUDA-treated rats compared with the vehicle group (P=0.049) (Figure 6D).
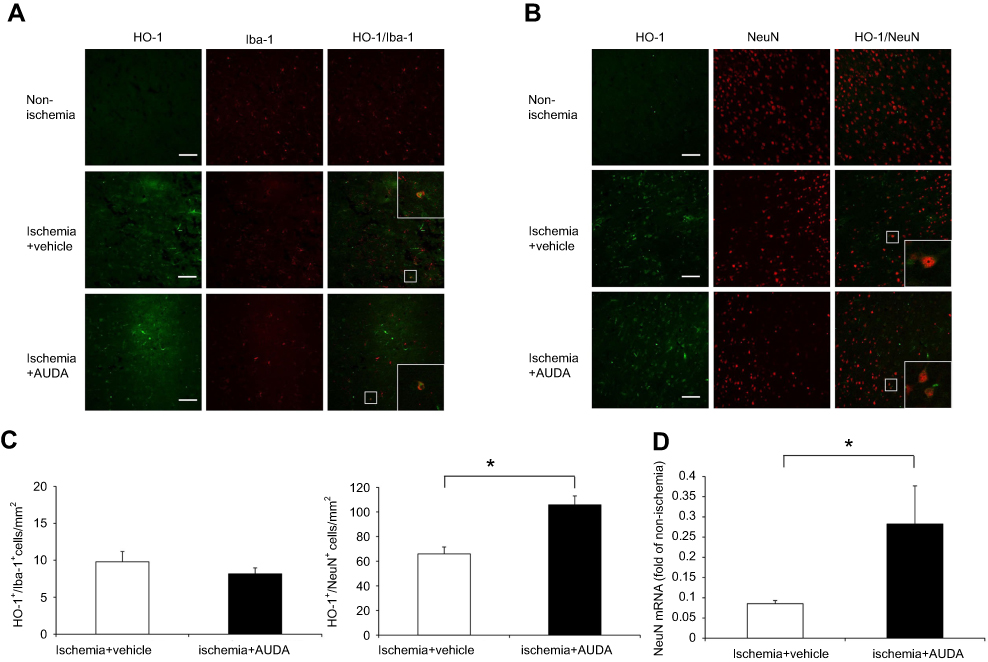 |
Figure 6 sEH inhibition modulated HO-1 and NeuN expression in neuronal cells at 1 day after MCAO. Notes: (A) Representative immunofluorescence staining of HO-1 (green) and Iba-1 (red) in the non-ischemic hemisphere, the ischemic border of rats in vehicle and AUDA treatment groups. Scale bar: 100 μm. (B) Representative immunofluorescence staining of HO-1 (green) and NeuN (red) in the non-ischemic hemisphere, the ischemic border of rats in vehicle and AUDA treatment groups. Scale bar: 100 μm. (C) Statistical analysis of HO-1+/Iba-1+ in microglia and HO-1+/NeuN+ in neuronal cells in the ischemic border expressed as cells/mm2 n=3/group. (D) The mRNA expression of NeuN in the ischemic cortex, n=4/group. Data are presented as mean ± SEM. *P<0.05.Abbreviations: AUDA, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid; HO-1, heme oxygenase-1; Iba-1, ionized calcium binding adaptor molecule 1; MCAO, middle cerebral artery occlusion; NeuN, neuronal nuclei; sEH, soluble epoxide hydrolase; SEM, standard error of the mean; SOD-1, superoxide dismutase-1. |
Discussion
In this study, the administration of AUDA in rats improved behavior outcomes and decreased the infarct size after ischemic brain injury. Moreover, AUDA modulated microglia polarization to decrease M1 type and increase M2 type microglia. In ischemic brain tissue and microglia, AUDA decreased the mRNA expression of pro-inflammatory mediators including IL-1β, IL-6, iNOS, MCP-1, MMP-9, and increased the gene expression of anti-inflammatory cytokine IL-10. The gene expression of antioxidative enzymes including HO-1, CAT, and SOD-1 was also upregulated after AUDA treatment. This antioxidative process may be initiated by the activation of transcription factor PPAR-γ and Nrf2. Furthermore, HO-1, which was expressed mainly in neurons, was increased by AUDA and more neuronal cells survived in the ischemic environment after AUDA treatment. To the best of our knowledge, this is the first study to reveal that a selective sEH inhibitor modulates microglia activation toward the M2 neuroprotective type, as well as upregulates anti-inflammatory and antioxidant genes in a short time in rats with ischemic stroke.
To date, only one previous report has described that an sEH inhibitor improved functional outcome at 1 day after ischemic stroke in a rat model.42 Our previous study has proved that an sEH inhibitor improved the performance of the rotarod test at 1 and 3 days after MCAO.39 Our present study further utilized precise behavior tests including beam walk test to evaluate the effect of the sEH inhibitor. The rotarod test is a useful test to assess motor coordination and balance changes in rodents.43 The beam walk test is a locomotor assessment and focuses on hindlimb functioning.44 Using these two tools, we confirmed that the sEH inhibitor ameliorated the behavior outcome after ischemic stroke. However, in rats without sEH inhibitor treatment, we also observed that the behavior outcome improved with time.
Previous reports indicated that an sEH inhibitor reduced infarct size at 24 hrs after MCAO.31,34,35,42 Our previous study indicated that inhibition of sEH can reduce infarct size at 3 days after MCAO.39 Our present research further revealed that the effect of the sEH inhibitor on infarct size reduction existed on both 1 and 3 days after ischemic brain injury. However, there was no difference between rats receiving AUDA at 1 day and 3 days after MCAO. The reason for this may be associated with the concept that there is an area which is not able to be rescued in an acute phase. Another reason is that self-recovery from brain injury in the young stroke animal model is fast.45 These possible factors may cause AUDA effects at 3 days to be inferior to AUDA effects at 1 day after MCAO.
Inhibition of sEH can decrease the degradation of EET into DHET and increase the level of EET, which itself possesses an anti-inflammatory effect. Liu et al, reported that the administration of 14, 15-EET, and AUDA both inhibited microglia activation, reduced infarct volume and improved behavioral function after focal ischemia.42 Most recent studies, as well as our present research, focused on the effect of sEH inhibitor, which has a more direct impact on anti-inflammation.
Ischemic brain injury activates microglia and these cells develop an M1 or M2 phenotype. M1 microglia produce pro-inflammatory cytokines and increase the level of injury. However, M2 microglia secrete anti-inflammatory cytokines and execute neuroprotection.46 Thus, driving the microglia activation to the M2 phenotype can reduce neuroinflammation. Recently, the decrease of microglia activation by pharmacological inhibition of sEH was reported in both ICH and BCAS models.37,38 Wu et al, revealed that an sEH inhibitor decreased M1 microglia and activated microglia at 1 day after ICH. However, if M2 microglia were affected, this was not mentioned and there was no conclusion regarding the modulation of microglia toward the M2 type by the sEH inhibitor.38 At 4 weeks after BCAS, Chen et al, proved that an sEH inhibitor was able to decrease M1 microglia and increase M2 microglia, and thus microglia were activated toward the M2 type.37 In our previous study, we also found M1 monocyte/macrophages were decreased by an sEH inhibitor.39 In our present study, we first discovered that the sEH inhibitor modulates microglia polarization toward the M2 neuroprotective type in rats with MCAO in the acute stage. In this study focusing on gene expression in ischemic cerebral cortex and microglia, the mRNA level of pro-inflammatory cytokines IL-1β, IL-6, iNOS, MCP-1, and MMP-9 decreased and the anti-inflammatory cytokine IL-10 increased in rats receiving MCAO and treated by AUDA. This was compatible with the consequence of the modulation of microglia polarization to decrease the proportion of M1 type microglia.
Transcriptional regulators including PPAR-γ and CCAAT/enhancer-binding protein-β (C/EBP-β) have been reported to turn on the M2 gene and promote polarization in research on macrophages.47,48 In microglia, there were no previous reports which discussed the molecular pathway in microglia polarization after sEH inhibitor treatment. Our result revealed that AUDA treatment increased the mRNA expression of C/EBP-β, but not PPAR-γ, in microglia at 3 days after MCAO (data not shown). C/EBP-β may modulate the microglia polarization toward the M2 phenotype in addition to producing anti-inflammatory cytokines and decreasing pro-inflammatory cytokines.
The modulation of microglia polarization became most obvious at 3 days after MCAO; however, the improvement of behavior outcome and infarct size was significant from 1 day after MCAO. Thus, there exists other protection mechanism in the acute stage and we proved that the antioxidant process plays an important role. HO-1 is an anti-inflammatory and antioxidant enzyme that can be induced by an inflammatory trigger or stress.49 HO-1 has been reported to be beneficial in ischemic stroke, and its upregulation after sEH inhibition was also noted in diabetic spontaneously hypertensive rats and in platelet-derived growth factor-induced rat vascular smooth muscle cell proliferation.50–52 M2 microglia triggered antioxidative responses including the upregulation of HO-1.53,54 However, there has been no report describing the relationship between HO-1 and the sEH inhibitor in ischemic brain injury. In addition to HO-1, CAT and SOD1 are other antioxidant enzymes and all of these are downstream genes of Nrf2, a transcription factor regulating cellular antioxidant response.55 PPAR-γ is a transcription factor known to play an anti-inflammatory and antioxidative role. Previous studies revealed that PPAR-γ transrepresses pro-inflammatory mediators such as IL-1ß, IL-6, interferon-γ (IFN-γ), iNOS, MCP-1, and MMP-9, and transactivates anti-inflammatory cytokines including IL-10.56–59 PPAR-γ has also been reported to execute its antioxidative function by activating the gene expression of HO-1, CAT, and SOD1.60 Nrf2 and PPAR-γ are linked by a positive feedback loop that maintains their expression and their target antioxidant genes during oxidative stress.60,61 The increased expression of antioxidant enzymes and transcription factors was found in ischemic cerebral cortex but not microglia. Thus, we performed further experiments to clarify the origin of these antioxidant enzymes. Immunofluorescence staining revealed that HO-1 induced by AUDA was mainly produced by neurons and the neuron survival improved after AUDA treatment. It has been reported that the mRNA expression of NeuN is significantly reduced by ischemia.62 Based on our results, the sEH inhibitor may execute anti-inflammatory and antioxidative functions to reduce neuronal injury partially via the PPAR-γ and Nrf2 pathway in ischemic brain tissue.
Conclusion
In summary, our present report indicated that applying the sEH inhibitor AUDA in rats with ischemic stroke can reduce the infarct size and improve behavior outcome at the acute stage. Furthermore, the inhibition of sEH modulates the polarization of microglia activation toward the M2 anti-inflammatory type. AUDA also reduces the mRNA expression of inflammatory mediators, promotes antioxidative genes and alleviates neuronal injury in ischemic brain tissue. These results suggest that sEH inhibitors may be a potentially promising treatment for ischemic stroke.
Acknowledgment
The study was supported in part by Ministry of Science and Technology of Taiwan (Grant number 105-2314-B-075-069-MY3, 106-2221-E-010-009, 106-3114-B-010-00, 107-2314-B-075-015-MY2, 107-2321-B-039−004, 108-2314-B-075-015), and Taipei Veterans General Hospital (Grant numbers V106D21-001-MY2-1, V107C-044, V108B-028, V108C-048, Neurovascular research grant 28070506-A0000014).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Casals JB, Pieri NC, Feitosa ML, et al. The use of animal models for stroke research: a review. Comp Med. 2011;61(4):305–313.
2. Mergenthaler P, Meisel A. Do stroke models model stroke? Dis Model Mech. 2012;5(6):718–725. doi:10.1242/dmm.010033
3. Lee HC, Chang KC, Huang YC, et al. Readmission, mortality, and first-year medical costs after stroke. J Chin Med Assoc. 2013;76(12):703–714. doi:10.1016/j.jcma.2013.08.003
4. Sacco RL, Wolf PA, Gorelick PB. Risk factors and their management for stroke prevention: outlook for 1999 and beyond. Neurology. 1999;53(7 Suppl 4):S15–S24.
5. Beal CC. Gender and stroke symptoms: a review of the current literature. J Neurosci Nurs. 2010;42(2):80–87.
6. Woodruff TM, Thundyil J, Tang SC, Sobey CG, Taylor SM, Arumugam TV. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol Neurodegener. 2011;6(1):11. doi:10.1186/1750-1326-6-11
7. Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79(4):1431–1568. doi:10.1152/physrev.1999.79.4.1431
8. Tobin MK, Bonds JA, Minshall RD, Pelligrino DA, Testai FD, Lazarov O. Neurogenesis and inflammation after ischemic stroke: what is known and where we go from here. J Cereb Blood Flow Metab. 2014;34(10):1573–1584. doi:10.1038/jcbfm.2014.130
9. Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87(5):779–789. doi:10.1189/jlb.1109766
10. Kriz J. Inflammation in ischemic brain injury: timing is important. Crit Rev Neurobiol. 2006;18(1–2):145–157.
11. Kawabori M, Yenari MA. Inflammatory responses in brain ischemia. Curr Med Chem. 2015;22(10):1258–1277.
12. Rodrigo R, Fernandez-Gajardo R, Gutierrez R, et al. Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets. 2013;12(5):698–714.
13. Zhang R, Xu M, Wang Y, Xie F, Zhang G, Qin X. Nrf2-a promising therapeutic target for defensing against oxidative stress in stroke. Mol Neurobiol. 2017;54(8):6006–6017. doi:10.1007/s12035-016-0111-0
14. Zhang F, Wang S, Zhang M, et al. Pharmacological induction of heme oxygenase-1 by a triterpenoid protects neurons against ischemic injury. Stroke. 2012;43(5):1390–1397. doi:10.1161/STROKEAHA.111.647420
15. Kawabori M, Yenari MA. The role of the microglia in acute CNS injury. Metab Brain Dis. 2015;30(2):381–392. doi:10.1007/s11011-014-9531-6
16. Chhor V, Le Charpentier T, Lebon S, et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav Immun. 2013;32:70–85. doi:10.1016/j.bbi.2013.02.005
17. Patel AR, Ritzel R, McCullough LD, Liu F. Microglia and ischemic stroke: a double-edged sword. Int J Physiol Pathophysiol Pharmacol. 2013;5(2):73–90.
18. Becker K, Kindrick D, Relton J, Harlan J, Winn R. Antibody to the alpha4 integrin decreases infarct size in transient focal cerebral ischemia in rats. Stroke. 2001;32(1):206–211. doi:10.1161/01.str.32.1.206
19. Zheng Z, Yenari MA. Post-ischemic inflammation: molecular mechanisms and therapeutic implications. Neurol Res. 2004;26(8):884–892. doi:10.1179/016164104X2357
20. Lai AY, Todd KG. Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol. 2006;84(1):49–59. doi:10.1139/Y05-143
21. Chuang YC, Yang JL, Yang DI, Lin TK, Liou CW, Chen SD. Roles of sestrin2 and ribosomal protein S6 in transient global ischemia-induced hippocampal neuronal injury. Int J Mol Sci. 2015;16(11):26406–26416. doi:10.3390/ijms161125963
22. Yenari MA, Kauppinen TM, Swanson RA. Microglial activation in stroke: therapeutic targets. Neurotherapeutics. 2010;7(4):378–391. doi:10.1016/j.nurt.2010.07.005
23. Varnum MM, Ikezu T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch Immunol Ther Exp (Warsz). 2012;60(4):251–266. doi:10.1007/s00005-012-0181-2
24. Ponomarev ED, Veremeyko T, Weiner HL. MicroRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS. Glia. 2013;61(1):91–103. doi:10.1002/glia.22363
25. Han L, Cai W, Mao L, et al. Rosiglitazone promotes white matter integrity and long-term functional recovery after focal cerebral ischemia. Stroke. 2015;46(9):2628–2636. doi:10.1161/STROKEAHA.115.010091
26. Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev. 2012;92(1):101–130. doi:10.1152/physrev.00021.2011
27. Iliff JJ, Alkayed NJ. Soluble epoxide hydrolase inhibition: targeting multiple mechanisms of ischemic brain injury with a single agent. Future Neurol. 2009;4(2):179–199.
28. Przybyla-Zawislak BD, Srivastava PK, Vazquez-Matias J, et al. Polymorphisms in human soluble epoxide hydrolase. Mol Pharmacol. 2003;64(2):482–490. doi:10.1124/mol.64.2.482
29. Srivastava PK, Sharma VK, Kalonia DS, Grant DF. Polymorphisms in human soluble epoxide hydrolase: effects on enzyme activity, enzyme stability, and quaternary structure. Arch Biochem Biophys. 2004;427(2):164–169. doi:10.1016/j.abb.2004.05.003
30. Zhang L, Ding H, Yan J, et al. Genetic variation in cytochrome P450 2J2 and soluble epoxide hydrolase and risk of ischemic stroke in a Chinese population. Pharmacogenet Genomics. 2008;18(1):45–51. doi:10.1097/FPC.0b013e3282f313e8
31. Zhang W, Koerner IP, Noppens R, et al. Soluble epoxide hydrolase: a novel therapeutic target in stroke. J Cereb Blood Flow Metab. 2007;27(12):1931–1940. doi:10.1038/sj.jcbfm.9600494
32. Zhang W, Otsuka T, Sugo N, et al. Soluble epoxide hydrolase gene deletion is protective against experimental cerebral ischemia. Stroke. 2008;39(7):2073–2078. doi:10.1161/STROKEAHA.107.508325
33. Zhang W, Iliff JJ, Campbell CJ, Wang RK, Hurn PD, Alkayed NJ. Role of soluble epoxide hydrolase in the sex-specific vascular response to cerebral ischemia. J Cereb Blood Flow Metab. 2009;29(8):1475–1481. doi:10.1038/jcbfm.2009.65
34. Jouihan SA, Zuloaga KL, Zhang W, et al. Role of soluble epoxide hydrolase in exacerbation of stroke by streptozotocin-induced type 1 diabetes mellitus. J Cereb Blood Flow Metab. 2013;33(10):1650–1656. doi:10.1038/jcbfm.2013.130
35. Shaik JS, Ahmad M, Li W, et al. Soluble epoxide hydrolase inhibitor trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid is neuroprotective in rat model of ischemic stroke. Am J Physiol Heart Circ Physiol. 2013;305(11):H1605–H1613. doi:10.1152/ajpheart.00471.2013
36. Zuloaga KL, Zhang W, Roese NE, Alkayed NJ. Soluble epoxide hydrolase gene deletion improves blood flow and reduces infarct size after cerebral ischemia in reproductively senescent female mice. Front Pharmacol. 2014;5:290.
37. Chen Y, Tian H, Yao E, et al. Soluble epoxide hydrolase inhibition promotes white matter integrity and long-term functional recovery after chronic hypoperfusion in mice. Sci Rep. 2017;7(1):7758. doi:10.1038/s41598-017-08227-z
38. Wu CH, Shyue SK, Hung TH, et al. Genetic deletion or pharmacological inhibition of soluble epoxide hydrolase reduces brain damage and attenuates neuroinflammation after intracerebral hemorrhage. J Neuroinflammation. 2017;14(1):230. doi:10.1186/s12974-017-1005-4
39. Yeh CF, Chuang TY, Hung YW, et al. Inhibition of soluble epoxide hydrolase regulates monocyte/macrophage polarization and improves neurological outcome in a rat model of ischemic stroke. Neuroreport. 2019;30(8):567–572. doi:10.1097/WNR.0000000000001248
40. Yeh CF, Chuang TY, Hung YW, et al. Development of a modified surgical technique for simulating ischemic cerebral cortex injury in rats. In Vivo. 2019. doi:10.21873/invivo.11588
41. Ritzel RM, Patel AR, Grenier JM, et al. Functional differences between microglia and monocytes after ischemic stroke. J Neuroinflammation. 2015;12:106. doi:10.1186/s12974-015-0329-1
42. Liu Y, Wan Y, Fang Y, et al. Epoxyeicosanoid signaling provides multi-target protective effects on neurovascular unit in rats after focal ischemia. J Mol Neurosci. 2016;58(2):254–265. doi:10.1007/s12031-015-0670-y
43. Bouet V, Freret T, Toutain J, Divoux D, Boulouard M, Schumann-Bard P. Sensorimotor and cognitive deficits after transient middle cerebral artery occlusion in the mouse. Exp Neurol. 2007;203(2):555–567. doi:10.1016/j.expneurol.2006.09.006
44. Barth TM, Jones TA, Schallert T. Functional subdivisions of the rat somatic sensorimotor cortex. Behav Brain Res. 1990;39(1):73–95.
45. Suenaga J, Hu X, Pu H, et al. White matter injury and microglia/macrophage polarization are strongly linked with age-related long-term deficits in neurological function after stroke. Exp Neurol. 2015;272:109–119. doi:10.1016/j.expneurol.2015.03.021
46. Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation. 2014;11:98. doi:10.1186/s12974-014-0139-x
47. Bouhlel MA, Derudas B, Rigamonti E, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6(2):137–143. doi:10.1016/j.cmet.2007.06.010
48. Ruffell D, Mourkioti F, Gambardella A, et al. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc Natl Acad Sci U S A. 2009;106(41):17475–17480. doi:10.1073/pnas.0908641106
49. Chen HG, Xie KL, Han HZ, et al. Heme oxygenase-1 mediates the anti-inflammatory effect of molecular hydrogen in LPS-stimulated RAW 264.7 macrophages. Int J Surg. 2013;11(10):1060–1066. doi:10.1016/j.ijsu.2013.10.007
50. Elmarakby AA, Faulkner J, Pye C, et al. Role of haem oxygenase in the renoprotective effects of soluble epoxide hydrolase inhibition in diabetic spontaneously hypertensive rats. Clin Sci (Lond). 2013;125(7):349–359. doi:10.1042/CS20130003
51. Kim HS, Kim SK, Kang KW. Differential effects of sEH inhibitors on the proliferation and migration of vascular smooth muscle cells. Int J Mol Sci. 2017;18(12):2683. doi:10.3390/ijms18122683
52. Shah ZA, Nada SE, Dore S. Heme oxygenase 1, beneficial role in permanent ischemic stroke and in Gingko biloba (EGb 761) neuroprotection. Neuroscience. 2011;180:248–255. doi:10.1016/j.neuroscience.2011.02.031
53. Schaer CA, Schoedon G, Imhof A, Kurrer MO, Schaer DJ. Constitutive endocytosis of CD163 mediates hemoglobin-heme uptake and determines the noninflammatory and protective transcriptional response of macrophages to hemoglobin. Circ Res. 2006;99(9):943–950. doi:10.1161/01.RES.0000247067.34173.1b
54. Choi KM, Kashyap PC, Dutta N, et al. CD206-positive M2 macrophages that express heme oxygenase-1 protect against diabetic gastroparesis in mice. Gastroenterology. 2010;138(7):2399–2409, 2409 e2391. doi:10.1053/j.gastro.2010.02.014
55. Chen B, Lu Y, Chen Y, Cheng J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J Endocrinol. 2015;225(3):R83–R99. doi:10.1530/JOE-14-0662
56. Crosby MB, Svenson J, Gilkeson GS, Nowling TK. A novel PPAR response element in the murine iNOS promoter. Mol Immunol. 2005;42(11):1303–1310. doi:10.1016/j.molimm.2004.12.009
57. Dworzanski T, Celinski K, Korolczuk A, et al. Influence of the peroxisome proliferator-activated receptor gamma (PPAR-gamma) agonist, rosiglitazone and antagonist, biphenol-A-diglicydyl ether (BADGE) on the course of inflammation in the experimental model of colitis in rats. J Physiol Pharmacol. 2010;61(6):683–693.
58. Giaginis C, Giagini A, Theocharis S. Peroxisome proliferator-activated receptor-gamma (PPAR-gamma) ligands as potential therapeutic agents to treat arthritis. Pharmacol Res. 2009;60(3):160–169. doi:10.1016/j.phrs.2009.02.005
59. Becker J, Delayre-Orthez C, Frossard N, Pons F. Regulation of inflammation by PPARs: a future approach to treat lung inflammatory diseases? Fundam Clin Pharmacol. 2006;20(5):429–447. doi:10.1111/j.1472-8206.2006.00425.x
60. Polvani S, Tarocchi M, Galli A. PPARgamma and oxidative stress: con(beta) catenating NRF2 and FOXO. PPAR Res. 2012;2012:641087. doi:10.1155/2012/641087
61. Lee C. Collaborative power of nrf2 and ppargamma activators against metabolic and drug-induced oxidative injury. Oxid Med Cell Longev. 2017;2017:1378175. doi:10.1155/2017/1378175
62. Zhou F, Gao S, Wang L, et al. Human adipose-derived stem cells partially rescue the stroke syndromes by promoting spatial learning and memory in mouse middle cerebral artery occlusion model. Stem Cell Res Ther. 2015;6:92. doi:10.1186/s13287-015-0114-1
Supplementary material
 |
Table S1 The detected target gene primers |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.