Back to Journals » International Journal of Nanomedicine » Volume 15
Solid Lipid Nanoparticles of Mycophenolate Mofetil: An Attempt to Control the Release of an Immunosuppressant
Authors Iqbal A ![]() , Zaman M
, Zaman M ![]() , Wahab Amjad M
, Wahab Amjad M ![]() , Adnan S, Abdul Ghafoor Raja M
, Adnan S, Abdul Ghafoor Raja M ![]() , Haider Rizvi SF, Mustafa MW, Farooq U, Abbas G
, Haider Rizvi SF, Mustafa MW, Farooq U, Abbas G ![]() , Shah S
, Shah S ![]()
Received 14 April 2020
Accepted for publication 29 June 2020
Published 5 August 2020 Volume 2020:15 Pages 5603—5612
DOI https://doi.org/10.2147/IJN.S255636
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Asma Iqbal,1 Muhammad Zaman,1,2 Muhammad Wahab Amjad,3 Sharjeel Adnan,1 Maria Abdul Ghafoor Raja,3 Syed Farhan Haider Rizvi,1 Mian Waqar Mustafa,2 Umer Farooq,2 Ghulam Abbas,4 Shahid Shah4
1Faculty of Pharmacy, The University of Lahore, Lahore, Pakistan; 2Faculty of Pharmacy, University of Central Punjab, Lahore, Pakistan; 3Faculty of Pharmacy, Northern Border University Rafha, Rafha, Saudi Arabia; 4Faculty of Pharmaceutical Sciences, Government College University Faisalabad, Faisalabad, Pakistan
Correspondence: Muhammad Zaman
Faculty of Pharmacy, University of Central Punjab, Lahore, Pakistan
Tel +92 3006095928
Email [email protected]
Introduction: Organ transplantation is a critically important procedure, which requires immune modulation by using immunosuppressants. Development of nanoparticles is an emerging and beneficial engineering process to increase the dissolution rate of poorly soluble immunosuppressants as well as to provide controlled release for better therapeutic outcomes.
Method: Currently, the nanoprecipitation method was employed to fabricate β-cyclodextrin (βCD) facilitated mycophenolate mofetil (MMF)-loaded solid lipid nanoparticles (SLNPs). The prime objectives of the study included, improvement of the dissolution profile of poorly aqueous soluble drug and controlled release from the SLNs to provide steady state drug concentration. Drug release from the prepared SLNs was assessed in two different media, ie, acidic buffer at pH 1.2 and phosphate buffer at pH 7.2 using USP dissolution apparatus for 12 h, followed by the evaluation of drug release mechanism and pattern by applying kinetic models.
Results: Justifiably, in acidic medium, the release was found to be 12% more (68%) in comparison to that in basic medium (56%). However, in both dissolution media, drug release was independent of initial concentration (R2> 0.95) with non-Fickian type of diffusion mechanism. The outcomes of the study have exhibited that prepared formulations were in nanosized range (80– 170 nm) with a net charge of ± 23 charge on their surface. They possessed fairly uniform surface with acceptable polydispersity index (0.23± 0.09). Scanning electron microscopy (SEM) analysis illustrated that the nanoparticles had uniform particle size and shape.
Discussion: The findings show potential applications of the nanoparticles and the method for the development of SLNPs in controlled release of MMF for better therapeutic outcomes. Conclusively, the prepared SLNPs were well designed in nanosized ranges and justifying the once daily controlled release formulation dose of MMF to enhance patient compliance.
Keywords: mycophenolate mofetil, β-cyclodextrin, stearic acid, nanoparticles, controlled release, nanoprecipitation method
Introduction
Solid organ transplantation (SOT) is a lifesaving procedure for patients with end-stage organ failure. It bids life-saving treatment for diseases considered as terminal or those associated with a significant impairment in quality life of a patient. The early period of SOT saw its origination in the middle of the 20th century with the utilization of various strategies of methodological improvement in animal models. The progression of this idea including the practice of vascular anastomosis and surface approaches, is attributed to Alexis Carrel, Mathieu Jaboulay, and Julius Dorfler.1,2 The procedure was first used by Carrel, while Ullmann reported the first successful kidney transplants performed in dogs and goat xenografts.3 Jaboulay later performed the first renal xeno transplants in humans in 1906, using pig and goat as donors. Both xenografts eventually failed, resulting in the death of patients.2 Various experimental transplantations of thyroid, heart, ovary, lung, small bowel in animals and humans were also carried out at that time by Carrel and other scientists around the globe.4 According to Organ Procurement and Transplantation Network (OPTN) data, over 39,717 transplants were performed in 2019 in the United States. 112,720 people needed a lifesaving organ transplant at that time. About 20 people die each day waiting for a transplant. While 2019 was a record-breaking year for SOT, however the demand of organ donors is massive.
SOT is now possible and development in this field has become possible with the improvements in immunosuppressive pharmacological therapy. Under normal situations, when an organ is transplanted from a donor to a patient, the immune system of a recipient detects the transplant as a foreign object thus triggering a complex reaction which in due course, destroys the transplanted organ.5 To prevent this graft rejection and to uphold a successful transplant, a complex treatment of immunosuppressive medications is essential for hindering the body’s immune response. The worth and importance of adherence to these immunosuppressive agents cannot be understated. Missing dosages may lead to negative results like graft impairment, increased mortality and increased healthcare expenditures.6 The majority of SOT recipients require three immunosuppressive agents including a calcineurin inhibitor, a corticosteroid, and a mycophenolic acid (MPA) derivative such as MMF.7 In recent years MMF has become a vital alternative therapy. Calcineurin inhibitor and corticosteroids are available in once-daily formulations but mycophenolic acid derivatives are given twice a day. A sustained release formulation of MMF, capable of maintaining suitable plasma concentration of the drug for longer duration in once daily dose would be beneficial for such patients leading to improved therapeutic results. The primary purpose of developing drug delivery systems is to deliver the required amount of therapeutic agent efficiently and precisely to the target site for adequate time interval.8
MMF, amorpholinoethyl ester (prodrug) of mycophenolic acid (MPA) is an antiproliferative immunosuppressant drug. MMF belongs to biopharmaceutical classification system (BCS) class II drug having low solubility and high permeability. MMF is slightly soluble in water (43 mg/mL). Inside the body, MMF rapidly gets converted to MPA, and acts by inhibiting the proliferation of B and T lymphocytes by de novo pathway inhibition of purine synthesis. MPA acts as an inosine monophosphate dehydrogenase inhibitor (IMPDH). Inosine monophosphate dehydrogenase is an enzyme required for the de novo synthesis of purines. The proliferation of T and B cells is inhibited by MPA because these cells depend merely on this pathway for the synthesis of guanosine nucleotide. Other cells can use both the de novo pathway as well as the salvage pathway for purine formation. The latter pathway is independent of inosine monophosphate dehydrogenase and is not affected by MMF. In addition, by diminishing guanosine triphosphate (GTP), MMF can inhibit the glycosylation of adhesion molecules and the binding of activated human lymphocytes to activated human endothelial cells.9–11 MMF is transformed into MPA by carboxylesterases in the liver and intestinal wall.
Due to poor water solubility of lipophilic prodrug MMF, an excipient which acts as solubilizer and a polymer is required to make MMF soluble in any formulation. One way to enhance the solubility of lipophilic drugs is to combine these drugs with cyclodextrins, mainly β-CD forming inclusion complexes by using nanoparticles.12,13
Cyclodextrins are cyclic oligosaccharides made up of D(+)-glucopyranose units with a toroidal shape having an external hydrophilic surface and internal hydrophobic core. They usually consist of six to eight 1,4-a-glycosidic linked glucose units (α-, β-, γ-CD). These compounds are able to form inclusion complexes with lipophilic drug molecules which result in improving aqueous solubility and stability in both liquid and solid phases.14 Complexation with cyclodextrins has been reported to enhance the solubility, dissolution rate and bioavailability of poorly water soluble drugs like MMF. They are known for their ability to molecularly encapsulate a wide variety of drugs into their hydrophobic cavity without the formation of any covalent bonds.15,16 In CD-guest complex, the binding forces are present due to the hydrophobic forces, electrostatic interactions, hydrogen bonding, cavity size and the shape of the guest molecules.14,17,18 By this way they increase the aqueous solubility of less soluble drugs, resulting in an increase in the availability of drugs at the absorption site.19 The most common pharmaceutical application of cyclodextrins is to enhance the solubility, stability, and bioavailability of drug molecules.20,21 βCDs are widely used in the pharmaceutical field owing to their high aqueous solubility and ability to stabilize drug molecules.22
In this study, the objective was the development and in vitro characterization of nanoparticulate drug delivery systems based on amphiphilic βCDs that are capable of incorporating a significantly high amount of MMF with a sustained in vitro release profile and to enhance its solubility and bioavailability.
Materials and Methods
Materials
MMF was received as a generous gift from Consolidated Chemical Laboratories Pharmaceuticals. Stearic acid, βCD and Chloroform were purchased from Merck Darmstadt Germany. All ingredients used were of analytical grades.
Method
Preparation of MMF-Loaded βCD Facilitated SLNs
MMF-loaded βCD nanoparticles were prepared by using different concentrations of stearic acid and βCD (Table 1), however, after various attempts, the final formulation with optimum nanosized, least PDI and suitable release profile was selected, as described in Table 2.
 |
Table 1 Preparation of MMF-Loaded βCD Nanoparticles Using Varying Ratios of βCD and Stearic Acid to MMF |
 |
Table 2 Composition of Selected Formulation for the Preparation of MMF-Loaded βCD Nanoparticles |
Nanoprecipitation technique was first established by Fessi et al.23 The nanoparticle preparation by this method is rapid and the entire process is carried out in a single step. It involves two miscible solvents. Preferably, both polymer and drug must dissolve in the first system (the solvent), but not in the second system (the nonsolvent). Two phases were prepared in this process. The aqueous phase (Table 2) was prepared by mixing the polymer (βCD) and Tween 80 with distilled water in a beaker. The organic phase was prepared by mixing the lipid, ie stearic acid in an organic solvent (chloroform). The mixture was agitated on a hot plate magnetic stirrer for 30 min. Sonicated the organic phase in a sonicator for 30 min while adding the aqueous phase dropwise with the help of the syringe. Nanoprecipitation takes place by a rapid interaction between particles when the polymer solution is added to the nonsolvent. This results in the immediate entrapment of the drug. The emulsion was homogenized in a centrifugation machine for 30 minutes at 600 rpm. The supernatant liquid was removed from the emulsion. The emulsion was freeze-dried for 72 h and lyophilized. The resultant mass obtained was then subjected to various evaluation parameters.13
Drug-Excipients Compatibility Studies
FTIR spectroscopy studies were carried on FTIR Magma IR 7600 (Lambda Scientific Pvt Ltd, 29 Deloraine Road,Edwardstown, SA 5039, AUSTRALIA). Scanning was done between the wavelengths of 400 to 4000 cm−1. FTIR analysis provided the confirmation of compatibilities between selected ingredients. βCD, stearic acid, MMF and prepared SLNs were scanned respectively.24
Characterization of MMF-loaded βCD Facilitated Nanoparticles
The mean particle size (nm) and polydispersity index (PDI) were determined by using dynamic light scattering technique and zeta potential (mV) of prepared nanoparticles was also determined (Nano ZS, Malvern Panalytical Ltd, Malvern, Worcestershire, UK). Briefly, 300 µg of the sample was dispersed in 1 mL deionized water and ultrasonicated for 10 s at 10% amplitude before analysis.25 All the measurements were made in triplicates at 25°C and reported as mean±SD.
SEM of samples was performed using ZEISS EVO LS10 (Carl Zeiss Promenade 1007745 Jena, Germany) with a 10-kV accelerating voltage. The surface of the samples for SEM were previously made electrically conductive in a holder. A magnification of 10,000⨰ was used to observe the surface morphology of the particles.
Calibration curve of MMF was plotted as concentration vs absorbance by stock dilutions method. Different dilutions were made and examined at a wavelength of 254 nm. Tel milliliters of standard drug solution was prepared by diluting it with 90 mL acetonitrile thus giving a concentration of 10 µg/mL of the drug. Stock solutions ranging from 2 mL to 12 mL were transferred into 20 mL volumetric flask and were diluted up to the mark with acetonitrile. Thus final concentrations ranged from 2–12 µg/mL as per Beer–Lambert’s law. Absorbance of each solution was observed at 254 nm against acetonitrile as a blank or standard reference and a concentration of drug vs absorbance was plotted.
Estimation of EE is essential, because it influences the drug content and release characteristics of the drug molecule. It was calculated by using spectrophotometric method.26

Release profiles of MMF from nanoparticle formulations were determined in 900 mL of phosphate buffered solution kept at pH 7.2 as well as in acidic medium at pH 1.2 separately to observe the influence of different pH media on release of the drug.26 In a cellulose membrane dialysis tube, the prepared drug-loaded nanoparticles were placed and suspended in dissolution media to conduct the dissolution studies for 12 h. Samples of 5 mL were taken after every hour and replaced with similar volume of fresh buffer to maintain the constant volume at 37°C±0.5 and 50 rpm. The samples were filtered using a syringe filter of 0.22 µm pore size and the quantity of MMF released was quantified spectrophotometrically at 254 nm as:

Release behavior of the drug in in vitro was assessed by applying kinetic models, including first order, zero order, Higuchi and Korsmeyer–Peppas models.
The nature of the drug whether crystalline or amorphous, in pure as well as loaded onto nanoparticles was determined by XRD technique. The samples in powder form have the advantage over others such that the results are in statistically representative, volume averaged values.27–29 XRD of the samples was performed using a high-power X-ray diffractometer. The scanning rate was 4°/min. The voltage/current used was 40 kV/50 mA and the target/filter (monochromator) was copper.22
Statistical Analysis
Statistical analysis was performed using GraphPad Prism ver. 7.01. (San Diego, CA, USA) The analysis was done to observe the impact of variable ratios of βCD and stearic acid over cumulative release of MMF from MMF-loaded βCD-facilitated SLNs. Analysis of variances (ANOVA) was applied followed by Tukey’s Multiple Comparison Test with 95% of confidence interval.
Results
Preparation of MMF-Loaded βCD Facilitated SLNs
There are many known methods to prepare nanoparticles. The one adopted in this study was the nanoprecipitation method. This method enables the production of nanoparticles in the desired nano range, ie 50–500 nm. Findings of different characterization studies including zeta sizer, EE and SEM confirmed the successful fabrication of MMF-loaded βCD facilitated SLNs.
Drug-excipients Compatibility Studies
FTIR spectra of MMF, stearic acid, βCD and MMF-loaded nanoparticles were assessed and illustrated in Figure 1. It is generally performed to determine the functional groups in the ingredients and to check any possible chemical interaction between them. Since FTIR is a highly sensitive method of analysis, all spectra of ingredients and formulation were analyzed. The IR spectrum of pure drug MMF was also recorded. The stretching region of functional group C-O ranged from 1700–1775cm−1 depicting carboxylic acids. Bands between 800 and 1000 cm−1 showed C-H band of alkenes. Bands ranging from 1020–1250 cm−1 showed C-N stretch of aliphatic amines. The asymmetric stretch of N-O between the wavelengths of 1475–1550 cm−1 confirmed the presence of nitro compounds. The FTIR spectrum of formulation MMF-loaded βCD nanoparticles was recorded. The stretching region of functional group C-N stretch ranged from 1050–1100 cm−1 indicating aliphatic amines. Bands in the range of 1200–1300 cm−1 showed C-H wag confirming the presence of alkyl halides. Peaks ranging between the wavelengths of 2800–3000 cm−1 confirmed C-H stretch of alkanes. C=O stretch at 1760 cm−1 was observed that confirmed the presence of carboxylic acids. The IR spectrum of raw βCD showed a broad band that appeared at 3312 cm−1 indicating an O-H stretch of alcohols. An intense peak at 2942 cm−1 due to C-H stretch of alkanes was also seen. A strong –C-O band of carboxylic acids around 1025 cm1 was also observed in the IR spectrum. The results showed no chemical interactions between the molecules used in the process.
 |
Figure 1 FTIR spectra of (A) MMF, (B) stearic acid, (C) βCD and (D) MMF-loaded βCD facilitated SLNs, describing the chemical compatibilities of selected the ingredients. |
Characterization of MMF-Loaded βCD Facilitated Nanoparticles
Particle Size, Polydispersity and Zeta Potential
Particle size and zeta potential analyses have illustrated that average particles size was between 80 and 170 nm and surface charge ranged from ±20 to ± 23 mV respectively (Figure 2). Furthermore, the reports obtained from Malvern UK Nano ZS exhibited fairly uniform distribution of the prepared particles as polydispersity of the formulations was found in the range of 0.2–0.4.
 |
Figure 2 Zeta size and particle size distribution analysis of MMF-loaded βCD facilitated SLNs. |
Scanning Electron Microscopy (SEM)
The morphology of nanoparticles was investigated through scanning electron microscopy. SEM analysis revealed that MMF-loaded βCD facilitated SLNs were of smooth surface with regular shape. PDI findings also strengthened the SEM analysis where uniformly distributed nanoparticles could be observed (Figure 3).
 |
Figure 3 SEM micrograph of MMF-loaded βCD facilitated SLNs, illustrating nanosized particles with smooth spherical shape. |
Qualitative Analysis of MMF
A linearity graph constructed between concentration and absorbance demonstrated reasonable linearity having R2 was 0.9995, following Beer–Lambert’s law (Figure 4).
 |
Figure 4 Calibration curve for MMF, describing linearity behavior in the selected range of concentration (2–12 µg/mL) at 254 nm. |
Percentage Entrapment Efficiency (%EE)
MMF showed an EE of 98.96% with βCD showing that hydrophilic βCD had the capability to entrap a hydrophobic drug. Hydrophobic MMF can be easily incorporated into the hydrophobic inner cavity of βCD surrounding the hydrophilic outer surface thereby improving the solubility of MMF.30
In vitro Drug Release Studies
In vitro drug release profile of four formulations was studied initially in KCl at pH 1.2 dissolution medium. Studies revealed that F1 and F2 having greater amount of lipophilic polymer were able to release 30.01±6.3% and 37±5.9%, respectively in 12 h. Alternatively, F4 which contained only βCD released 100% of the drug in the initial two hours of the study. However, F3 having drug, βCD, and stearic acid, at a ratio of 1:4:2 showed satisfactory results. It was found to be accomplishing the objectives of the study, as it not only provided the sustained release, but also allowed suitable amount of drug to be released (≈70%) in studied duration to provide the therapeutic effects (Figure 5).
 |
Figure 5 In vitro drug release profile of F1, F2 F3 and F4 of MMF-loaded βCD facilitated SLNs. |
In vitro release study of MMF from nanoparticles was conducted for 12 h. In PBS kept at pH 7.2 nanoparticles showed 57.74±8.3% of drug release. In KCl kept at pH 1.2 the nanoparticles showed sustained release of drug with a cumulative percentage release of 68.12±6.2% (Figure 6). The figure illustrates an initial burst release from the formulation in both media. Initially, prompt release of a fraction of the drug can be attributed to the amount of drug adsorbed or weakly bound to the large surface area of the polymer nanoparticles instead of the drug fused in polymer nanoparticles.31
 |
Figure 6 Graphical representation of in vitro drug release, conducted at acidic pH (1.2) and basic pH (7.2), showing sustained and controlled release of drug from MMF-loaded βCD facilitated SLNs, during 12 h of the studies. |
Kinetic Modeling of Release Data
Drug release pattern and mechanism are considered to be the important factors for polymeric nanocarriers of drugs. It can be assessed by kinetic modeling of release data through the application of statistical models, which suggested that release of the drug was not only sustained, but also controlled following diffusion mechanism (Table 3). Zero order and Korsmeyer–Peppas model were dominating with comparatively higher values of R2, ie 0.9514 and 0.9523 respectively.
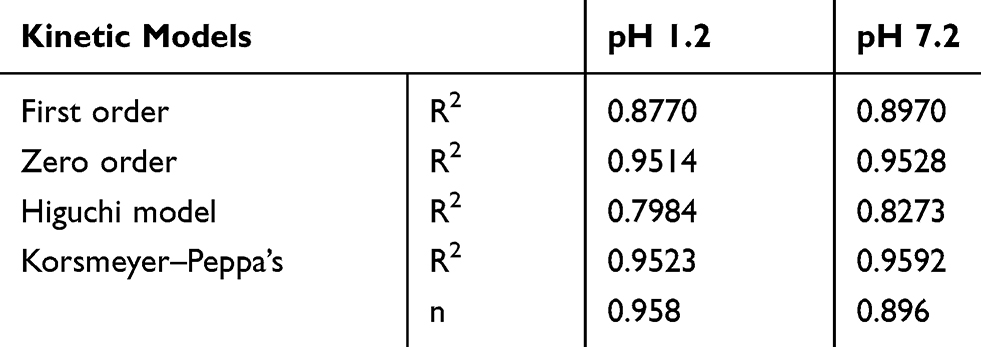 |
Table 3 Kinetic Modeling of the Release Data Obtain from in vitro Dissolution Studies |
Powder X-Ray Diffraction (XRD) Studies
X-ray diffractogram revealed that the drug (Figure 7A) was crystalline in nature, moreover the scan of formulations also showed some peaks, that might have appeared due to the attachment of some drug onto the surface of the particles (Figure 7B). Initial burst release had also exhibited such type of behavior. However, formulated SLNs without MMF exhibited some fused peaks, indicating a decrease in the crystallinity of the polymer and other ingredients upon conversion into lyophilized SLNs (Figure 7C).
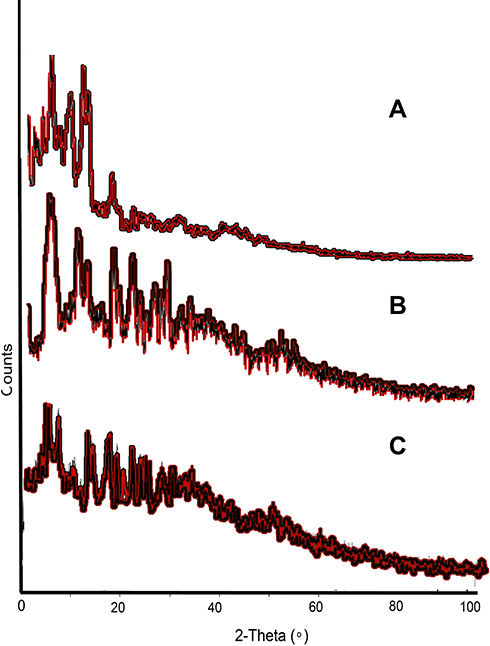 |
Figure 7 XRD diffractogram, (A) describing the crystalline nature of the drug, which has also been observed in prepared, (B) MMF-loaded βCD facilitated SLNs, while, (C) also showed some less intense peaks, attributed to crystalline βCD. |
Discussion
MMF-loaded βCD facilitated SLNs were synthesized by employing the nanoprecipitation technique. This technique is widely used for the preparation of nanoparticles. Dissolving the drug in its solvent helps to achieve the minimum possible size and then careful addition of dissolved drug into its antisolvent leads to achieving uniform nanosized particles, as reported in the current study. The nanoparticle suspension formulated in the current study was white and milky in appearance. It is well known that surface charge, shape, size are the primary parameters of nanoparticles for cellular uptake.32 The particle diameter was found in the range of 80–170 nm (Figure 2), while the zeta potential of MMF-loaded βCD assisted SLNs ranged from −20 to −23 mV. Zeta potential, being the indicator of nanoparticle’s stability, is an important parameter to be studied.33 From the literature it is evident that βCD and stearic acid both show negative zeta potentials, furthermore, Wu et al, also reported that nanosuspension containing MMF showed negative zeta potential.34,35 The observed surface charge might be the indication of stable SLNs, as the surface charge below 20 could result in particle aggregation, but in recent studies the values greater than 20 were found to yield well-dispersed and stable formulation. The reports obtained from Malvern UK Nano ZS indicated that polydispersity of the formulations was below 0.4, indicating a single homogenous nanoparticle population.36 Homogeneous formulation is the desired one for achieving uniformity in the drug content as well as in the dose of the drug, it is preferred to have the monodispersing population of the SLNs. Findings of the zeta sizer were also supported by the SEM analysis, where relatively uniform and nanosized particles were observed. Apparently, as illustrated by SEM, the particles had a smooth surface and were of spherical shape, advocating the suitability of selected ingredients as well as the selected method used for preparation. One of the advantages of observing nanoparticles with SEM rather than transmission electron microscopy is the opportunity to see the surface of the particles.36
Entrapment of the drug into a polymeric network is one of the challenges in nanotechnology, and for this purpose selection of the excipients is difficult. Stearic acid, that is used frequently in SLNs, was found to be a good choice to entrap the lipophilic drug followed by stabilizing it with the help of βCD, primarily known to be the ingredient used for solubilization and stabilization of the drugs. Combination of stearic acid and βCD made it possible to achieve EE>95%.
In vitro drug release testing is an essential analytical tool that is used to investigate and establish product behavior during the various stages of development, as well as life cycle management. If designed suitably, an in vitro release profile can reveal fundamental information regarding the dosage form and its performance and can also provide information on the release mechanism and kinetics, permitting a rational and scientific approach to drug product development. For complex dosage forms like a nanoparticulate carrier, in vitro release testing assumes greater importance. To optimize the suitable composition of formulation, different formulations with varying ratios of polymers to drug were tried and observed for drug release studies. Studies revealed that variations in the polymeric contents significantly (p<0.05) affected the release of the drug. F1 without βCD released the least amount of drug, however, its addition improved the cumulative amount of drug released (Table 4). It might be due to the solubility enhancing effect of βCD, which was confirmed in F4, where 100% of the drug was found to be released in the initial two hours of the study. Furthermore, the ratios of drug, βCD and stearic acid 1:4:2 were considered to be optimum as the stated composition showed satisfactory results. After selecting F2 as the optimum formulation, further studies were performed. It was observed that in acidic medium the release of the drug from selected formulation was somewhat greater, which might be due to the reason that acidic pH might had greater influence in degrading the stearic acid, leading to comparatively greater release of the drug (≈70%) in comparison to that obtained in basic medium (≈57%). Two main models were applied to study the release of the drug from the matrix of nanoparticles: firstly, diffusion of drug and secondly, degradation of the lipids in vivo by enzymes (eg, lipase, colipase). The initial rapid release, as seen in current study, was either due to increase in solubility or because of greater drug concentration near the particle surface. In the current study, a good control over release of the drug from SLNs was observed during 12 h. Output of kinetic modeling was advocating the controlled release of the drug which had followed the non-Fickian type of release mechanism. Drug release was found to be independent of the initial concentration of the drug available in the SLNs, as predicted by the application of zero order kinetics.
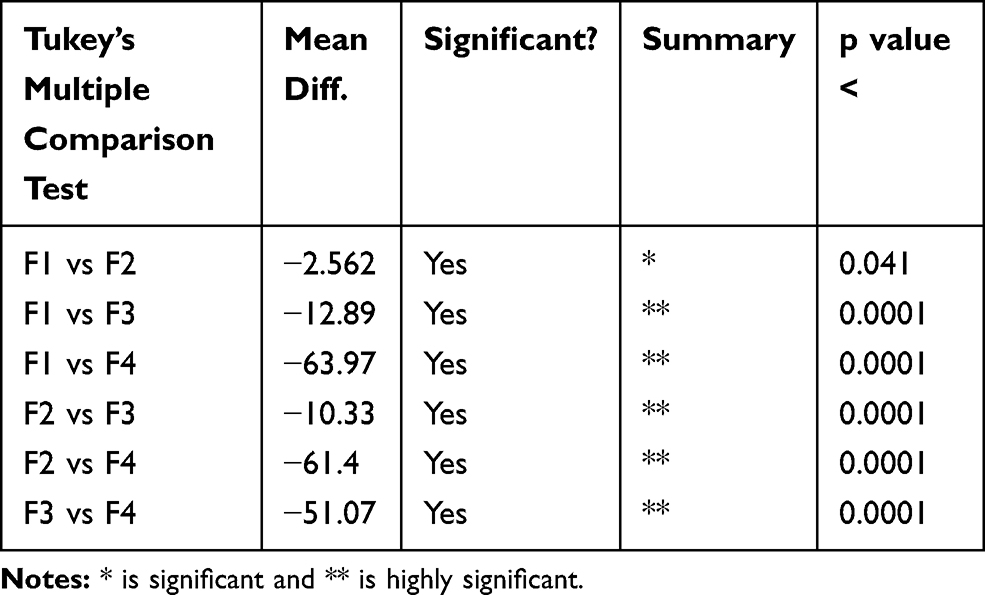 |
Table 4 Outcomes of Statistical Analysis |
Statistical Analysis
The analysis showed a significant effect of both βCD and stearic acid on the release of MMF from MMF-loaded βCD facilitated SLNs. The p values were less than 0.05 (Table 4), advocating the significance of variation in the polymeric ratios to the drug.
Conclusion
In the current study we have successfully developed the nanoparticles of MMF with βCD molecule by forming a complex using nanoprecipitation technique. The lipophilic drugs were found to be more easily released from the βCD complex. The study also provides evidence that the solubility of MMF is enhanced by the formulation of nanoparticles as a drug delivery system. The work represented a drug delivery system in which βCD acts as a carrier and solubilizer for delivery of poorly soluble MMF.
Acknowledgment
Authors were highly grateful to the CCL Pharmaceuticals Lahore, Pakistan for providing mycophenolate mofetil for the successful accomplishment of the project.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Carrel A. The transplantation of organs: a preliminary communication. 1905 [classical article]. Yale J Biol Med. 2001;74(4):239.
2. Barker CF, Markmann JF. Historical overview of transplantation. Cold Spring Harb Perspect Med. 2013;3(4):a014977. doi:10.1101/cshperspect.a014977
3. Ullman E. Tissue and organ transplantation. Ann Surg. 1914;60(2):195. doi:10.1097/00000658-191408000-00006
4. Black CK, Termanini KM, Aguirre O, Hawksworth JS, Sosin M. Solid organ transplantation in the 21st century. Ann Transl Med. 2018;6:20. doi:10.21037/atm.2018.09.68
5. Mohammed M, Mansell H, Shoker A, Wasan KM, Wasan EK. Development and in vitro characterization of chitosan-coated polymeric nanoparticles for oral delivery and sustained release of the immunosuppressant drug mycophenolate mofetil. Drug Dev Ind Pharm. 2019;45(1):76–87. doi:10.1080/03639045.2018.1518455
6. Dew MA, DiMartini AF, Dabbs ADV, et al. Rates and risk factors for nonadherence to the medical regimen after adult solid organ transplantation. Transplantation. 2007;83(7):858–873. doi:10.1097/01.tp.0000258599.65257.a6
7. Muduma G, Shupo FC, Dam S, et al. Patient survey to identify reasons for non-adherence and elicitation of quality of life concepts associated with immunosuppressant therapy in kidney transplant recipients. Patient Prefer Adherence. 2016;10:27.
8. Sepe V, Libetta C, Giuliano M, Adamo G, Dal Canton A. Mycophenolate mofetil in primary glomerulopathies. Kidney Int. 2008;73(2):154–162. doi:10.1038/sj.ki.5002653
9. He X, Smeets RL, Koenen HJ, et al. Mycophenolic acid‐mediated suppression of human CD4+ T cells: more than mere guanine nucleotide deprivation. Am J Transplant. 2011;11(3):439–449. doi:10.1111/j.1600-6143.2010.03413.x
10. Reis A, Reinhard T, Sundmacher R, Althaus C, Voiculescu A, Kutkuhn B. Mycophenolatemofetil in ocular immunological disorders. A survey of the literature with 3 case reports. Klin Monbl Augenheilkd. 1998;213(5):257–261. doi:10.1055/s-2008-1034985
11. Reis A, Spelsberg H, Reinhard T, Braunstein S, Godehardt E, Sundmacher R. Beneficial effect of preoperative mycophenoiate mofetil in murine corneal transplantation. Transplant Int. 1999;12(5):341–345. doi:10.1007/s001470050237
12. Knapp S, Bertelmann E, Hartmann C, Keipert S, Pleyer U. Intraocular availability of topically applied mycophenolate mofetil in rabbits. J Ocular Pharmacol Ther. 2003;19(2):181–192. doi:10.1089/108076803321637717
13. Bilati U, Allémann E, Doelker E. Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. Eur J Pharm Sci. 2005;24(1):67–75. doi:10.1016/j.ejps.2004.09.011
14. Bongiorno D, Ceraulo L, Ferrugia M, Filizzola F, Ruggirello A, Liveri VT. Inclusion complexes of cyclomaltooligosaccharides (cyclodextrins) with melatonin in solid phase. Arkivoc. 2005;14:118–130.
15. Mukne AP, Nagarsenker M. Triamterene-β-cyclodextrin systems: preparation, characterization and in vivo evaluation. AAPS PharmSciTech. 2004;5(1):142.
16. Fernandes CM, Vieira MT, Veiga FJB. Physicochemical characterization and in vitro dissolution behavior of nicardipine–cyclodextrins inclusion compounds. Eur J Pharm Sci. 2002;15(1):79–88. doi:10.1016/S0928-0987(01)00208-1
17. Liu L, Guo Q-X. The driving forces in the inclusion complexation of cyclodextrins. J Incl Phenom Macrocycl Chem. 2002;42(1–2):1–14. doi:10.1023/A:1014520830813
18. Schneiderman E, Stalcup AM. Cyclodextrins: a versatile tool in separation science. J Chromatogr B Biomed Sci Appl. 2000;745(1):83–102. doi:10.1016/S0378-4347(00)00057-8
19. Scarpignato C. Piroxicam-β-cyclodextrin: a GI safer piroxicam. Curr Med Chem. 2013;20(19):2415–2437. doi:10.2174/09298673113209990115
20. Hirayama F, Uekama K. Cyclodextrin-based controlled drug release system. Adv Drug Deliv Rev. 1999;36(1):125–141. doi:10.1016/S0169-409X(98)00058-1
21. Uekama K, Hirayama F, Irie T. Cyclodextrin drug carrier systems. Chem Rev. 1998;98(5):2045–2076. doi:10.1021/cr970025p
22. Baboota S, Dhaliwal M, Kohli K. Physicochemical characterization, in vitro dissolution behavior, and pharmacodynamic studies of rofecoxib-cyclodextrin inclusion compounds. Preparation and properties of rofecoxib hydroxypropyl β-cyclodextrin inclusion complex: A technical note. AAPS PharmSciTech. 2005;6(1):E83–E90.
23. Fessi HP, Puisieux F, Devissaguet JP, Ammoury N, Benita S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. International Journal of Pharmaceutics. 1989;55(1):R1–4. doi:10.1016/0378-5173(89)90281-0
24. Kang J-H, Chon J, Kim Y-I, et al. Preparation and evaluation of tacrolimus-loaded thermosensitive solid lipid nanoparticles for improved dermal distribution. Int J Nanomedicine. 2019;14:5381. doi:10.2147/IJN.S215153
25. Liaw J, Rojanasakul Y, Robinson JR. The effect of drug charge type and charge density on corneal transport. Int J Pharm. 1992;88(1–3):111–124. doi:10.1016/0378-5173(92)90308-O
26. Zur Mühlen A, Schwarz C, Mehnert W. Solid lipid nanoparticles (SLN) for controlled drug delivery–drug release and release mechanism. Eur J Pharm Biopharm. 1998;45(2):149–155. doi:10.1016/S0939-6411(97)00150-1
27. Mourdikoudis S, Pallares RM, Thanh NT. Characterization techniques for nanoparticles: comparison and complementarity upon studying nanoparticle properties. Nanoscale. 2018;10(27):12871–12934.
28. Garud A, Singh D, Garud N. Solid lipid nanoparticles (SLN): method, characterization and applications. Int Curr Pharm J. 2012;1(11):384–393. doi:10.3329/icpj.v1i11.12065
29. Loganathan S, Valapa RB, Mishra RK, Pugazhenthi G, Thomas S. Thermogravimetric analysis for characterization of nanomaterials. Thermal and Rheological Measurement Techniques for Nanomaterials Characterization. Elsevier; 2017;67–108.
30. Gao S, Sun J, Fu D, Zhao H, Lan M, Gao F. Preparation, characterization and pharmacokinetic studies of tacrolimus-dimethyl-β-cyclodextrin inclusion complex-loaded albumin nanoparticles. Int J Pharm. 2012;427(2):410–416. doi:10.1016/j.ijpharm.2012.01.054
31. Kumar S, Randhawa JK. Solid lipid nanoparticles of stearic acid for the drug delivery of paliperidone. RSC Adv. 2015;5(84):68743–68750. doi:10.1039/C5RA10642G
32. Albanese A, Tang PS, Chan WC. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu Rev Biomed Eng. 2012;14:1–16. doi:10.1146/annurev-bioeng-071811-150124
33. Vakilinezhad MA, Amini A, Dara T, Alipour S. Methotrexate and Curcumin co-encapsulated PLGA nanoparticles as a potential breast cancer therapeutic system: in vitro and in vivo evaluation. Colloids Surf B Biointerfaces. 2019;184:110515. doi:10.1016/j.colsurfb.2019.110515
34. Usui S, Healy T. Zeta potential of stearic acid monolayer at the air–aqueous solution interface. J Colloid Interface Sci. 2002;250(2):371–378. doi:10.1006/jcis.2002.8340
35. Wu X-G, Xin M, Yang L-N, Shi W-Y. The biological characteristics and pharmacodynamics of a mycophenolate mofetil nanosuspension ophthalmic delivery system in rabbits. J Pharm Sci. 2011;100(4):1350–1361. doi:10.1002/jps.22356
36. Mohammed MA. Development of Once-Daily Mycophenolate Mofetil Sustained Release Oral Nanoparticles. University of Saskatchewan; 2017.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.