Back to Archived Journals » Hypoxia » Volume 5
SNPs, linkage disequilibrium, and chronic mountain sickness in Tibetan Chinese
Authors Buroker NE, Ning XH, Zhou ZN, Li K, Cen WJ, Wu XF, Zhu WZ, Scott CR, Chen SH
Received 23 July 2016
Accepted for publication 8 March 2017
Published 14 July 2017 Volume 2017:5 Pages 67—74
DOI https://doi.org/10.2147/HP.S117967
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Norman E Buroker,1 Xue-Han Ning,1,2,† Zhao-Nian Zhou,3 Kui Li,4 Wei-Jun Cen,4 Xiu-Feng Wu,3 Wei-Zhong Zhu,5 C Ronald Scott,1 Shi-Han Chen1
1Department of Pediatrics, University of Washington, 2Division of Cardiology, Seattle Children’s Hospital Research Foundation, Seattle, WA, USA; 3Laboratory of Hypoxia Physiology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China; 4Lhasa People Hospital, Lhasa, Tibet; 5Center for Cardiovascular Biology and Regenerative Medicine, University of Washington, Seattle, WA, USA
†Xue-Han Ning passed away on April 20, 2015
Abstract: Chronic mountain sickness (CMS) is estimated at 1.2% in Tibetans living at the Qinghai–Tibetan Plateau. Eighteen single-nucleotide polymorphisms (SNPs) from nine nuclear genes that have an association with CMS in Tibetans have been analyzed by using pairwise linkage disequilibrium (LD). The SNPs included are the angiotensin-converting enzyme (rs4340), the angiotensinogen (rs699), and the angiotensin II type 1 receptor (AGTR1) (rs5186) from the renin–angiotensin system. A low-density lipoprotein apolipoprotein B (rs693) SNP was also included. From the hypoxia-inducible factor oxygen signaling pathway, the endothetal Per-Arnt-Sim domain protein 1 (EPAS1) and the egl nine homolog 1 (ENGL1) (rs480902) SNPs were included in the study. SNPs from the vascular endothelial growth factor (VEGF) signaling pathway included are the v-akt murine thymoma viral oncogene homolog 3 (rs4590656 and rs2291409), the endothelial cell nitric oxide synthase 3 (rs1007311 and rs1799983), and the (VEGFA) (rs699947, rs34357231, rs79469752, rs13207351, rs28357093, rs1570360, rs2010963, and rs3025039). An increase in LD occurred in 40 pairwise comparisons, whereas a decrease in LD was found in 55 pairwise comparisons between the controls and CMS patients. These changes were found to occur within and between signaling pathways, which suggests that there is an interaction between SNP alleles from different areas of the genome that affect CMS.
Keywords: HAS, Qinghai-Tibetan Plateau, RAS, HIF, VEGF, pathways, miRNA
Introduction
Chronic mountain sickness (CMS) is a syndrome involving the loss of adaptation to life at high altitude, which occurs in some residents residing in this environment.1,2 Older age in these permanent residents has been linked to the sickness.3 The susceptibility to CMS between and within human populations at high altitudes has been shown to have great variability.4 The precise pathogenesis of CMS is not well understood, but the lack of oxygen at high altitudes is considered to be the major factor,5–8 which raises the question of why some individuals are susceptible to the sickness while others are not. At the same altitude, the prevalence of the disease is estimated to be 15% in Quechua Andeans and 1.2% in Tibetans.9 Based on the current genetic data, it has been suggested that Tibetans may be one of the oldest high-altitude adapted ethnic groups in the world with origins from the Neolithic period,10–13 which might explain the lower occurrence of the sickness in Tibetans.
Genome-wide association studies (GWAS) have identified >15 thousand single-nucleotide polymorphisms (SNPs) associated with various diseases and traits.14,15 Some of these SNPs have been examined in association with high-altitude sickness (HAS) among Han and Tibetan Chinese at the Qinghai–Tibetan Plateau in China.16–20 Recently, the association of SNPs that create potential changes in transcriptional factor binding sites (TFBS) and HAS have been examined.20,21 To this end, punitive changes in TFBS have been reported to be created by 17 SNPs among nine genes significantly associated with acute mountain sickness (AMS) in Han Chinese.20 In this study, linkage disequilibrium (LD) was used to analyze intra- and intergenic SNP alleles and alterations in TFBS. Pairwise LD was computed between the SNP alleles within a control group and an AMS group. An increase in LD was found to occur in 32 pairwise comparisons, whereas a decrease in LD was found in 22 comparisons between the groups.20 The pairwise LD changes were found within and between genes involving signaling pathways and systems indicating the interaction of SNP alleles or punitive TFBS from different areas of the genome. The current study examined the LD between 18 SNPs from the same nine genes in CMS Tibetan patients at the Qinghai–Tibetan Plateau in China since these SNPs have previously been associated with HAS.16–19
Materials and methods
Study groups
The Tibetan CMS patients sampled in the study had been hospitalized and diagnosed by physicians at the Lhasa People Hospital (Tibet, China, at 3,670 m above sea level).17–19 Healthy Tibetans were randomly selected from the Lhasa area to serve as control subjects. The average age of CMS patients was 53.6 years, whereas that of the non-CMS Tibetan controls was 30.5 years. All Tibetans sampled in the study signed an informed consent approved by the Human Ethics Committee of the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. The study was approved by the Shanghai Institutes for Biological Sciences review board.
Sampling
Buccal brush samples were collected at the Lhasa People Hospital from 45 Tibetans diagnosed with CMS during the high occurrence period (spring and winter). Control samples consisting of 34 Tibetan unaffected natives were also collected via buccal brush. The controls were found to be in good health upon physical examination by doctors at the Lhasa People Hospital.16–19
Genotyping
The methods for genotyping each SNP have been previously outlined and reported.16–19 The genes and their SNPs used in the present study are summarized in Table 1.
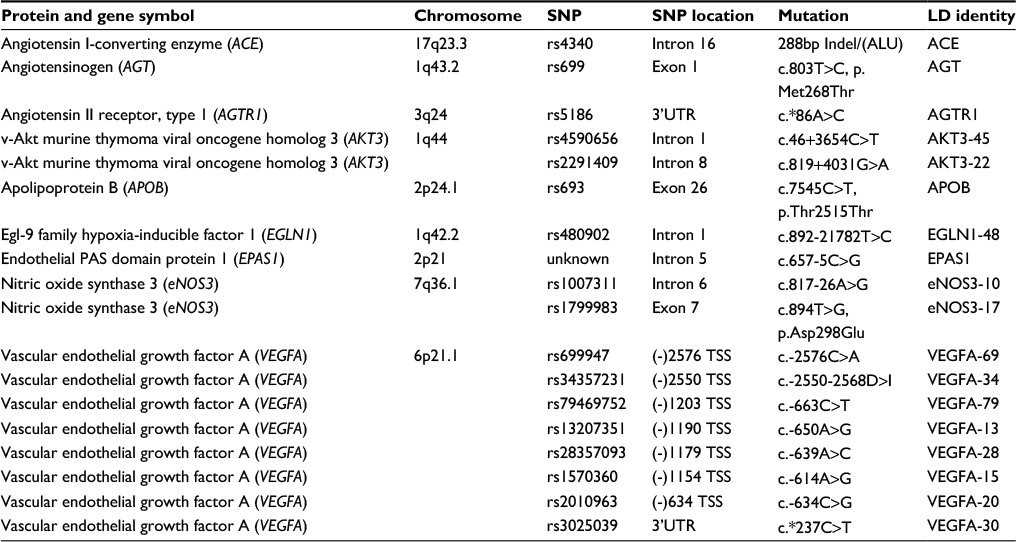 | Table 1 Genes and their SNPs that have been found to be associated with chronic mountain sickness Notes: Location of gene chromosome, SNP location in the gene, and the resulting genetic mutation as well as the LD identity using in the analysis are listed. Abbreviations: LD, linkage disequilibrium; SNP, single-nucleotide polymorphism; TSS, transcriptional start site. |
Statistical analysis
The statistical methods used to genotype samples have been previously discussed.16–19 Pairwise LD22 was computed between all SNPs from the Tibetan CMS and control groups (Figure 1). The degree of genetic linkage between the 18 SNPs in each study group was estimated as Lewontin’s coefficient D’, where no color (D’=0) indicates that LD is weak or nonexistent and the dark red (D’=1) indicates that there exists strong pairwise LD between SNPs (Figure 1). The change in LD between the control and CMS groups among all SNPs is presented in Table 2.
 | Figure 1 Pairwise LD for 18 SNPs in the non-CMS Tibetan Chinese group (A) compared with the CMS Tibetan Chinese group (B). The degree of genetic linkage between the 18 SNPs in each study group is estimated as Lewontin’s coefficient [D’] where no color ([D’] = 0) indicates that LD is weak or nonexistent and the dark red ([D’] = 1) indicates that there exists strong pairwise linkage disequilibrium between SNPs. Blue bar indicates tagging SNP (rs699947). Abbreviations: LD, linkage disequilibrium; SNP, single-nucleotide polymorphism; CMS, chronic mountain sickness. |
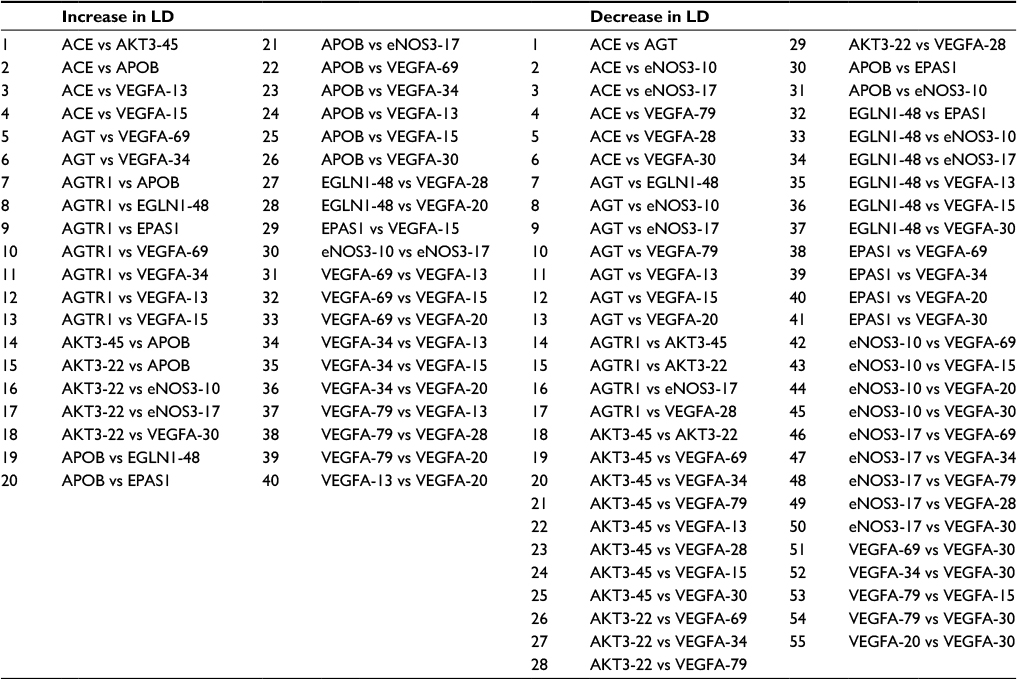 | Table 2 Changes in pairwise LD from Figure 1 between 18 SNPs in the CMS Tibetan Chinese group compared with the non-CMS Tibetan Chinese group Note: Increase and decrease in pairwise LD in the CMS group compared with the Tibetan Chinese control group are listed. Abbreviations: LD, linkage disequilibrium; SNP, single-nucleotide polymorphism; CMS, chronic mountain sickness. |
miRNA analysis
The miRdSNP23and miRBase24–28 databases were used to identify the has-miR-591 miRNA-predicted target site in association with the VEGFA (rs3025039) SNP in the 3’UTR of the VEGFA gene.
Results
Pairwise LD estimates were obtained between 18 SNPs from nine genes that had been previously found to be associated with CMS.17–19 The SNPs occur in the renin–angiotensin system (RAS) (ACE, AGT, and AGTR1), GNB3, LDL, APOB, the HIF oxygen signaling pathway EPAS1 and EGLN1), and the vascular endothelial growth factor (VEGF) signaling pathway (AKT3, eNOS3, and VEGFA) genes (Tables 1 and 2; Figures 1 and 2). In the pairwise estimates, the ACE with AKT3-45, APOB, VEGFA-13, and VEGFA-15 SNP pairs exhibit an increase in LD for the CMS study group compared with the control group and a decrease in LD in ACE with AGT, eNOS3-10, eNOS3-17, VEGFA-79, VEGFA-28, and VEGFA-30 pairs (Figure 1 and Table 2). Also, in the pairwise estimates, the AGT with VEGFA-69 and VEGFA-34 pairs exhibit an increase in LD for the CMS study group compared with the control group and a decrease in LD in AGT with EGNL1-48, eNOS3-10, VEGFA-79, VEGFA-13, VEGFA-15, and VEGFA-20 pairs (Figure 1; Table 2). In addition, in these estimates, the AGTR1 with APOB, EGLN1-48, EPAS1, VEGFA-69, VEGFA-34, VEGFA-13, and VEGFA-15 pairs exhibit an increase in LD for the CMS study group compared with the control group and a decrease in LD in AGTR1 with AKT3-45, AKT3-22, eNOS3-17, and VEGFA-28 pairs (Figure 1; Table 2). Similar results can be seen for the other pairwise LD estimates for all the SNPs (Figure 1; Table 2) including the association of SNPs between these signaling pathways and systems (Figure 2).
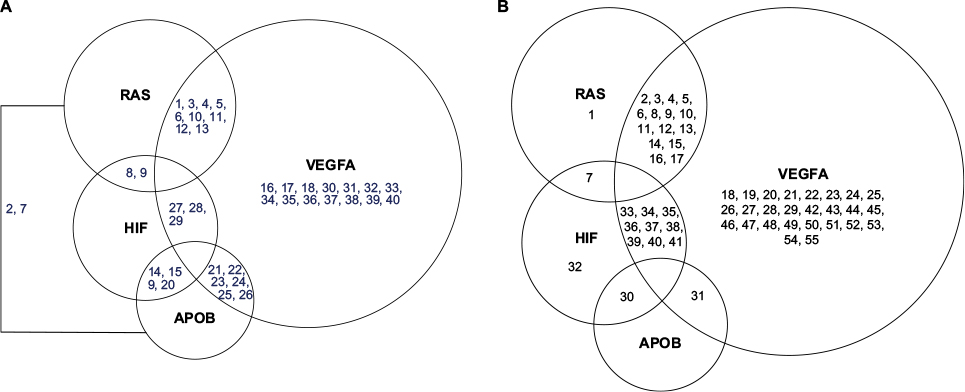 | Figure 2 Pairwise LD for 18 SNPs in the CMS Tibetan Chinese group compared with the non-CMS Tibetan Chinese groups from Table 2 is graphed between the pathways. Figure (A) represents an increase in LD in the CMS group compared with the control group, whereas figure (B) represents a decrease in LD. The numbers are the pairwise associations. Abbreviations: APOB, apolipoprotein B; HIF, hypoxia-inducible factor oxygen signaling pathway; RAS, renin–angiotension system; VEGFA, vascular endothelial growth factor signaling pathway; LD, linkage disequilibrium; SNP, single-nucleotide polymorphism; CMS, chronic mountain sickness. |
The SNPs of the AKT3, eNOS3, and VEGFA genes in the VEGF signaling pathway (a growth factor activator for angiogenesis) and the SNPs of the EGLN1 and EPAS1 genes in the HIF oxygen signaling pathway exhibit an increase in pairwise LD for the CMS group compared with the control Tibetan Chinese groups (Figure 1; Table 2) both within and between the pathways (Figure 2). Besides the SNP (rs4340) ACE-I allele, there are only two other SNPs (rs4590656 and rs3025039) AKT3-C and VEGFA-C allele, respectively, that generate potential unique hypoxia transcriptional factor binding (HIF1a:ARNT).20
A decrease in LD between SNPs for the CMS group compared with the Tibetan Chinese non-CMS group could indicate that some genes are responding to different attributes of the CMS sickness. As an example, the VEGFA-30 SNP (rs3025039) in the 3’UTR exhibits a tremendous decrease in LD in the CMS group compared with the non-CMS group with respect to the ACE, EGLN1-48, EPAS1, eNOS3-10, eNOS3-17, VEGFA-69, VEGFA-34, VEGFA-79, and VEGFA-20 SNPs (Figure 1; Table 2). Apparently, when CMS occurs among Tibetan individuals, there is less interaction between the VEGFA-30 SNP (rs3025039) and these other SNPs. A scan of the miRdSNP and miRBase databases revealed that the VEGFA (rs3025039) SNP alters the miRNA (has-miR-591)-predicted target site CA[C/T]GGTC in the 3’UTR of the gene. This SNP has been associated with several human diseases (see “Discussion” section) as well as CMS. Overall, there were 40 pairwise increases and 55 pairwise decreases in LD in the CMS group compared with the non-CMS control group (Figure 1; Table 2).
Discussion
Of the GWAS-identified disease or trait-predisposing SNPs, only 7% of these are located in protein-coding regions of the genome29,30 and the remaining 93% are located within noncoding areas31,32 such as regulatory or intergenic regions. SNPs occurring in the regulatory region of a gene are drawing more attention,33–35 where a single nucleotide change can alter the binding site of a transcriptional factor (TF), which can result in multiple consequences. In most of the cases, the SNP will neither change the TFBS interaction nor alter gene expression since a TF can usually recognize a number of different binding sites in various areas of the gene. However, in other cases, the SNP may increase or decrease the ability of TF to bind DNA which would result in allele-specific gene expression. Also, an SNP may eliminate the TFBS or generate a new binding site for another TF. In such cases, the gene would no longer be regulated by the original TF. Therefore, these functional regulatory(r) SNPs that occur in TFBS may cause changes in gene expression, phenotypes, and susceptibility to environmental exposure.35 Numerous examples and several reviews have been written about rSNPs associated with disease susceptibility.35–38 Such rSNPs occurring in the regulatory regions of genes have been associated with disease or sickness. These regulatory regions host the binding sites for transcription factors involved in gene expression.39
LD is defined as the non-random association between the alleles of two or more loci in a given population.40 LD between SNP alleles in the regulatory region of a gene has been used as a method of identifying haplotype associations relating to sickness or disease in a population.16,18 This is achieved when the levels of LD between SNP alleles within haplotypes are seen to change substantially in a sickness group when compared with a baseline population. In such cases, LD between SNP alleles can be used to identify punitive binding changes for TFs responsible for gene regulation. Such TFBS changes may result in human disease or sickness.21,41–46 In this study, LD has been considered to be the nonrandom association of SNP alleles within and between genes. Pairwise LD was computed among the 18 SNPs, and these estimates were compared between the CMS and control Tibetan Chinese group.
The ACE insertion/deletion (rs4340) polymorphism from the RAS system has been extensively studied in HAS.47–49 The ACE-I allele has been associated with superior performance in mountaineers who ascend to high altitude compared with the ACE-D allele.50,51 It has been found that mountaineers with the ACE-I/I genotype will maintain higher arterial oxygen saturation at rest and during exercise at high altitude than those having the ACE-D/D genotype.52 Perhaps the genetic reason for these findings is that the 288 bp ACE-I allele generates at least 84 punitive TFBS compared with the ACE-D allele which generates only four TFBS.20 The implications of this finding has been thoroughly discussed in an earlier paper dealing with acute mountain sickness in Han Chinese.20 In a previous work, the authors have found that there is a much higher occurrence of ACE-D allele (0.49) in the CMS patients compared with the non-CMS (0.37) Tibetan Chinese control group,17 again suggesting that the ACE-I allele preforms better among individuals in high altitude environments than the ACE-D allele. The angiotensinogen (AGT) M268T polymorphism (rs699) which is also part of the RAS system has been reported to be significantly associated with high-altitude pulmonary edema in Chinese53 and Indian54 populations. The AGTR1 polymorphism A1166C (rs5186) which is also part of the RAS system is associated with HAS and other human diseases.55–58 These three polymorphisms exhibit changes in LD between their alleles in the CMS study group compared with the non-CMS control group (Figure 1; Table 2), suggesting possible alterations in gene regulation.
Although the APOB polymorphism (rs693) has not been tied to HAS, it has been found to be linked to other human diseases such as dyslipidemia and higher LDL levels59 and has been shown to influence plasma levels.60,61 However, it can be seen that the rs693 APOB SNP exhibits an increase in LD between SNP alleles from the HIF, RAS, and VEGFA pathways (Figure 2; Table 2). The structural protein, apolipoprotein B is a component of LDL and can affect the blood lipid levels by regulating LDL62 and the balance of cholesterol.63,64 It evidently also has some unknown role with the components of HIF, RAS, and VEGFA pathways in CMS cases (Figure 2).
The rs3025039 VEGFA SNP affects the has-miR-591-predicted binding target CA[C/T]GGTC in the 3’UTR, where a VEGFA-C allele would eliminate the binding site for the miRNA. Strong pairwise LD exists between this SNP and the ACE (rs4340), EGNL1 (rs480902), EPAS1, eNOS3 (rs1007311 and rs1799983), and VEGFA (rs699947, rs34357231, rs79469752, and rs2010963) SNPs in the Tibetan control group but disappears in the CMS group (Figure 1). This SNP has also been associated with Behcet’s disease,65 acute myeloid leukemia,66 Kawasaki disease,67 psoriatic arthritis,68 age-related macular degeneration,69 early-stage non-small-cell lung cancer,70 and breast cancer.71
Future research should involve the ABCC9 gene, which resides on chromosome 12p and encodes the SUR2 proteins. An RNA splicing variant (SUR2A) of the gene is a cardioprotective ABC protein that serves as a subunit of sarcolemmal ATP-sensitive K+ channels and is thought to be regulated by hypoxia in high-altitude environments.72,73 Intronic SNPs between the 39 exons of the gene74 have been associated with sleep disorder,75,76 depression,76 hippocampal sclerosis of aging,77,78 blood pressure/hypertension,79,80 and Hirschsprung disease.81 It is possible that these SNPs affect BS for the TFs regulating the ABCC9 gene and thereby generate the multiple splicing variants and proteins associated with the gene.74
Conclusion
Most of the SNPs used in this pairwise LD study have been found to be associated with CMS. These alterations were discussed with regard to changes in gene regulation as a possible cause for CMS. The ACE (I/D) rs4340 SNP would be considered to be a large contributor to CMS because the ACE-I allele creates many more potential TFBS compared with the ACE-D allele. The ACE-I allele creates a punitive hypoxia-inducible factor 1: aryl hydrocarbon receptor nuclear translocator (HIF1α:ARNT) TFBS that occurs only at this location in the ACE gene. Also, a significant contribution would be the rs3025039 VEGFA SNP whose C allele eliminates the miRNA binding site for miR-591 in the 3’UTR of the gene. The interaction between SNPs within TFBS from different areas of the genome (Table 1) as indicated by LD analysis (Figure 1; Table 2) suggests that allele changes within these regulatory regions may contribute to CMS (Table 2).
Acknowledgments
The authors would like to thank Casey Guilland for the technical assistance on the manuscript. It was presented at the 66th Annual Meeting of the American Society of Human Genetics, October 17–22, 2016, Vancouver, Canada. This manuscript is dedicated to the work and memory of Xue-Han Ning.
Disclosure
The authors report no conflicts of interest in this work.
References
Wu TY, Li WS, Wei LY, et al. A Preliminary Studies on the Diagnosis of Chronic Mountain Sickness in Tibetan Populations. Matsumoto: Press Committee of the 3rd World congress on Mountain Medicine and High Altitude Physiology; 1998. | ||
Leon-Velarde F, McCullough RG, McCullough RE, Reeves JT. Proposal for scoring severity in chronic mountain sickness (CMS). Background and conclusions of the CMS Working Group. Adv Exp Med Biol. 2003;543:339–354. | ||
Sime F, Monge C, Whittembury J. Age as a cause of chronic mountain sickness (Monge’s disease). Int J Biometeorol. 1975;19(2):93–98. | ||
Basang Z, Wang B, Li L, et al. HIF2A variants were associated with different levels of high-altitude hypoxia among native Tibetans. PLoS One. 2015;10(9):e0137956. | ||
West JB. The physiologic basis of high-altitude diseases. Ann Intern Med. 2004;141(10):789–800. | ||
Schoene RB. Illnesses at high altitude. Chest. 2008;134(2):402–416. | ||
Strohl KP. Lessons in hypoxic adaptation from high-altitude populations. Sleep Breath. 2008;12(2):115–121. | ||
Wilson MH, Newman S, Imray CH. The cerebral effects of ascent to high altitudes. Lancet Neurol. 2009;8(2):175–191. | ||
Mejia OM, Prchal JT, Leon-Velarde F, Hurtado A, Stockton DW. Genetic association analysis of chronic mountain sickness in an Andean high-altitude population. Haematologica. 2005;90(1):13–19. | ||
Su B, Xiao J, Underhill P, et al. Y-Chromosome evidence for a northward migration of modern humans into Eastern Asia during the last Ice Age. Am J Hum Genet. 1999;65(6):1718–1724. | ||
Su B, Xiao C, Deka R, et al. Y chromosome haplotypes reveal prehistorical migrations to the Himalayas. Hum Genet. 2000;107(6):582–590. | ||
Torroni A, Miller JA, Moore LG, et al. Mitochondrial DNA analysis in Tibet: implications for the origin of the Tibetan population and its adaptation to high altitude. Am J Phys Anthropol. 1994;93(2):189–199. | ||
Du R, Xiao C, Cavalli-Sforza LL. Genetic distances between Chinese populations calculated on gene frequencies of 38 loci. Sci China C Life Sci. 1997;40(6):613–621. | ||
Welter D, MacArthur J, Morales J, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42(Database issue):D1001–D1006. | ||
Tak YG, Farnham PJ. Making sense of GWAS: using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenetics Chromatin. 2015;8:57. | ||
Buroker NE, Ning XH, Zhou ZN, et al. VEGFA SNPs and transcriptional factor binding sites associated with high altitude sickness in Han and Tibetan Chinese at the Qinghai-Tibetan Plateau. J Physiol Sci. 2013;63(3):183–193. | ||
Buroker NE, Ning XH, Zhou ZN, et al. Genetic associations with mountain sickness in Han and Tibetan residents at the Qinghai-Tibetan Plateau. Clin Chim Acta. 2010;411(19–20):1466–1473. | ||
Buroker NE, Ning XH, Zhou ZN, et al. AKT3, ANGPTL4, eNOS3, and VEGFA associations with high altitude sickness in Han and Tibetan Chinese at the Qinghai-Tibetan Plateau. Int J Hematol. 2012;96(2):200–213. | ||
Buroker NE, Ning XH, Zhou ZN, et al. EPAS1 and EGLN1 associations with high altitude sickness in Han and Tibetan Chinese at the Qinghai-Tibetan Plateau. Blood Cells Mol Dis. 2012;49(2):67–73. | ||
Buroker NE, Ning X-H, Li K, et al. SNPs, linkage disequilibrium and transcriptional factor binding sites associated with acute mountain sickness among Han Chinese at the Qinghai-Tibetan Plateau. Int J Genom Med. 2015;3(1):120. | ||
Buroker NE, Ning X-H, Zhou A-N, et al. SNPs amd TFBS associated with high altitude sickness. Open J Blood Dis. 2013;3:85–93. | ||
Ding K, Zhou K, He F, Shen Y. LDA – a java-based linkage disequilibrium analyzer. Bioinformatics. 2003;19(16):2147–2148. | ||
Bruno AE, Li L, Kalabus JL, Pan Y, Yu A, Hu Z. miRdSNP: a database of disease-associated SNPs and microRNA target sites on 3’UTRs of human genes. BMC Genomics. 2012;13:44. | ||
Griffiths-Jones S. The microRNA Registry. Nucleic Acids Res. 2004;32(Database issue):D109–D111. | ||
Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34(Database issue):D140–D144. | ||
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36(Database issue):D154–D158. | ||
Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39(Database issue):D152–D157. | ||
Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42(Database issue):D68–D73. | ||
Pennisi E. The biology of genomes. disease risk links to gene regulation. Science. 2011;332(6033):1031. | ||
Kumar V, Wijmenga C, Withoff S. From genome-wide association studies to disease mechanisms: celiac disease as a model for autoimmune diseases. Semin Immunopathol. 2012;34(4):567–580. | ||
Hindorff LA, Sethupathy P, Junkins HA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106(23):9362–9367. | ||
Kumar V, Westra HJ, Karjalainen J, et al. Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS Genet. 2013;9(1):e1003201. | ||
Knight JC. Functional implications of genetic variation in non-coding DNA for disease susceptibility and gene regulation. Clin Sci (Lond). 2003;104(5):493–501. | ||
Wang X, Tomso DJ, Liu X, Bell DA. Single nucleotide polymorphism in transcriptional regulatory regions and expression of environmentally responsive genes. Toxicol Appl Pharmacol. 2005;207(2 Suppl):84–90. | ||
Chorley BN, Wang X, Campbell MR, Pittman GS, Noureddine MA, Bell DA. Discovery and verification of functional single nucleotide polymorphisms in regulatory genomic regions: current and developing technologies. Mutat Res. 2008;659(1–2):147–157. | ||
Prokunina L, Alarcon-Riquelme ME. Regulatory SNPs in complex diseases: their identification and functional validation. Expert Rev Mol Med. 2004;6(10):1–15. | ||
Buckland PR. The importance and identification of regulatory polymorphisms and their mechanisms of action. Biochim Biophys Acta. 2006;1762(1):17–28. | ||
Sadee W, Wang D, Papp AC, et al. Pharmacogenomics of the RNA world: structural RNA polymorphisms in drug therapy. Clin Pharmacol Ther. 2011;89(3):355–365. | ||
Buroker NE. Regulatory SNPs and transcriptional factor binding sites in ADRBK1, AKT3, ATF3, DIO2, TBXA2R and VEGFA. Transcription. 2014;5(4):e964559. | ||
Lewontin RC, Kojima K. The evolutionary dynamics of complex polymorphisms. Evolution. 1960;14(4):458–472. | ||
Buroker NE. VEGFA rSNPs, transcriptional factor binding sites and human disease. J Physiol Sci. 2014;64(1):73–76. | ||
Buroker NE. AKT3 rSNPs, transcritional factor binding sites and human disease. Open J Blood Dis. 2013;3:116–129. | ||
Buroker NE. ATF3 rSNPs, transcriptional factor binding sites and human etiology. Open J Genet. 2013;3:253–261. | ||
Buroker NE. ADRBK1 (GRK2) rSNPs, transcriptional factor binding sites and cardiovascular disease in the black population. J Cardiol Dis. 2014;2(1):62-67. | ||
Buroker NE. TBXA2R rSNPs, transcriptional factor binding sites and asthma in Asians. Open J Pediatrics. 2014;4:148–161. | ||
Buroker NE. ADRBD1 (GRK2), TBXA2R and VEGFA rSNPs in KLF4 and SP1 TFBS exhibit linkage disequilibrium. Open J Genet. 2014;4:183–189. | ||
Costerousse O, Allegrini J, Lopez M, Alhenc-Gelas F. Angiotensin I-converting enzyme in human circulating mononuclear cells: genetic polymorphism of expression in T-lymphocytes. Biochem J. 1993;290(Pt 1):33–40. | ||
Danser AH, Schalekamp MA, Bax WA, et al. Angiotensin-converting enzyme in the human heart. Effect of the deletion/insertion polymorphism. Circulation. 1995;92(6):1387–1388. | ||
Zhu X, Bouzekri N, Southam L, et al. Linkage and association analysis of angiotensin I-converting enzyme (ACE)-gene polymorphisms with ACE concentration and blood pressure. Am J Hum Genet. 2001;68(5):1139–1148. | ||
Montgomery HE, Marshall R, Hemingway H, et al. Human gene for physical performance. Nature. 1998;393(6682):221–222. | ||
Tsianos G, Eleftheriou KI, Hawe E, et al. Performance at altitude and angiotensin I-converting enzyme genotype. Eur J Appl Physiol. 2005;93(5–6):630–633. | ||
Woods DR, Pollard AJ, Collier DJ, et al. Insertion/deletion polymorphism of the angiotensin I-converting enzyme gene and arterial oxygen saturation at high altitude. Am J Respir Crit Care Med. 2002;166(3):362–366. | ||
Qi Y, Niu W, Zhu T, Zhou W, Qiu C. Synergistic effect of the genetic polymorphisms of the renin-angiotensin-aldosterone system on high-altitude pulmonary edema: a study from Qinghai-Tibet altitude. Eur J Epidemiol. 2008;23(2):143–152. | ||
Stobdan T, Ali Z, Khan AP, et al. Polymorphisms of renin--angiotensin system genes as a risk factor for high-altitude pulmonary oedema. J Renin Angiotensin Aldosterone Syst. 2011;12(2):93–101. | ||
Liu Y, Zhuoma C, Shan G, et al. A1166C polymorphism of the angiotensin II type 1 receptor gene and essential hypertension in Han, Tibetan and Yi populations. Hypertens Res. 2002;25(4):515–521. | ||
Jones A, Dhamrait SS, Payne JR, et al. Genetic variants of angiotensin II receptors and cardiovascular risk in hypertension. Hypertension. 2003;42(4):500–506. | ||
Yan C, Zhan J, Feng W. Gene polymorphisms of angiotensin II type 1 receptor and angiotensin-converting enzyme in two ethnic groups living in Zhejiang Province, China. J Renin Angiotensin Aldosterone Syst. 2005;6(3):132–137. | ||
Wu CK, Tsai CT, Chang YC, et al. Genetic polymorphisms of the angiotensin II type 1 receptor gene and diastolic heart failure. J Hypertens. 2009;27(3):502–507. | ||
Rodrigues AC, Sobrino B, Genvigir FD, et al. Genetic variants in genes related to lipid metabolism and atherosclerosis, dyslipidemia and atorvastatin response. Clin Chim Acta. 2013;417:8–11. | ||
Benn M, Stene MC, Nordestgaard BG, Jensen GB, Steffensen R, Tybjaerg-Hansen A. Common and rare alleles in apolipoprotein B contribute to plasma levels of low-density lipoprotein cholesterol in the general population. J Clin Endocrinol Metab. 2008;93(3):1038–1045. | ||
Zhu H, Tucker HM, Grear KE, et al. A common polymorphism decreases low-density lipoprotein receptor exon 12 splicing efficiency and associates with increased cholesterol. Hum Mol Genet. 2007;16(14):1765–1772. | ||
Besseling J, Kastelein JJ, Defesche JC, Hutten BA, Hovingh GK. Association between familial hypercholesterolemia and prevalence of type 2 diabetes mellitus. JAMA. 2015;313(10):1029–1036. | ||
Disterer P, Al-Shawi R, Ellmerich S, et al. Exon skipping of hepatic APOB pre-mRNA with splice-switching oligonucleotides reduces LDL cholesterol in vivo. Mol Ther. 2013;21(3):602–609. | ||
Corsetti JP, Gansevoort RT, Bakker SJ, Sparks CE, Vart P, Dullaart RP. Apolipoprotein B attenuates albuminuria-associated cardiovascular disease in prevention of renal and vascular endstage disease (PREVEND) participants. J Am Soc Nephrol. 2014;25(12):2906–2915. | ||
Salvarani C, Boiardi L, Casali B, et al. Vascular endothelial growth factor gene polymorphisms in Behcet’s disease. J Rheumatol. 2004;31(9):1785–1789. | ||
Kim DH, Lee NY, Lee MH, Sohn SK, Do YR, Park JY. Vascular endothelial growth factor (VEGF) gene (VEGFA) polymorphism can predict the prognosis in acute myeloid leukaemia patients. Br J Haematol. 2008;140(1):71–79. | ||
Breunis WB, Biezeveld MH, Geissler J, et al. Vascular endothelial growth factor gene haplotypes in Kawasaki disease. Arthritis Rheum. 2006;54(5):1588–1594. | ||
Butt C, Lim S, Greenwood C, Rahman P. VEGF, FGF1, FGF2 and EGF gene polymorphisms and psoriatic arthritis. BMC Musculoskelet Disord. 2007;8:1. | ||
Lin JM, Wan L, Tsai YY, et al. Vascular endothelial growth factor gene polymorphisms in age-related macular degeneration. Am J Ophthalmol. 2008;145(6):1045–1051. | ||
Heist RS, Zhai R, Liu G, et al. VEGF polymorphisms and survival in early-stage non-small-cell lung cancer. J Clin Oncol. 2008;26(6):856–862. | ||
Lin GT, Tseng HF, Yang CH, et al. Combinational polymorphisms of seven CXCL12-related genes are protective against breast cancer in Taiwan. OMICS. 2009;13(2):165–172. | ||
Mohammed Abdul KS, Jovanovic S, Du Q, Sukhodub A, Jovanovic A. Mild hypoxia in vivo regulates cardioprotective SUR2A: a role for Akt and LDH. Biochim Biophys Acta.2015;1852(5):709–719. | ||
Mohammed Abdul KS, Jovanovic S, Du Q, Sukhodub A, Jovanovic A. A link between ATP and SUR2A: a novel mechanism explaining cardioprotection at high altitude. Int J Cardiol. 2015;189:73–76. | ||
Nelson PT, Jicha GA, Wang WX, et al. ABCC9/SUR2 in the brain: implications for hippocampal sclerosis of aging and a potential therapeutic target. Ageing Res Rev. 2015;24(Pt B):111–125. | ||
Allebrandt KV, Amin N, Muller-Myhsok B, et al. A K(ATP) channel gene effect on sleep duration: from genome-wide association studies to function in Drosophila. Mol Psychiatry. 2013;18(1):122–132. | ||
Parsons MJ, Lester KJ, Barclay NL, Nolan PM, Eley TC, Gregory AM. Replication of Genome-Wide Association Studies (GWAS) loci for sleep in the British G1219 cohort. Am J Med Genet B Neuropsychiatr Genet. 2013;162B(5):431–438. | ||
Nelson PT, Estus S, Abner EL, et al. ABCC9 gene polymorphism is associated with hippocampal sclerosis of aging pathology. Acta Neuropathol. 2014;127(6):825–843. | ||
Nelson PT, Wang WX, Wilfred BR, et al. Novel human ABCC9/SUR2 brain-expressed transcripts and an eQTL relevant to hippocampal sclerosis of aging. J Neurochem. 2015;134(6):1026–1039. | ||
Sato N, Nakayama T, Asai S, Soma M. A haplotype in the human Sur2 gene is associated with essential hypertension. J Hum Hypertens. 2006; 20(1):87–90. | ||
Kamide K, Asayama K, Katsuya T, et al. Genome-wide response to antihypertensive medication using home blood pressure measurements: a pilot study nested within the HOMED-BP study. Pharmacogenomics. 2013;14(14):1709–1721. | ||
Kim JH, Cheong HS, Sul JH, et al. A genome-wide association study identifies potential susceptibility loci for Hirschsprung disease. PLoS One. 2014;9(10):e110292. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.