Back to Journals » Risk Management and Healthcare Policy » Volume 14
SNP Array as a Tool for Prenatal Diagnosis of Congenital Heart Disease Screened by Echocardiography: Implications for Precision Assessment of Fetal Prognosis
Authors Huang H ![]() , Cai M
, Cai M ![]() , Wang Y, Liang B
, Wang Y, Liang B ![]() , Lin N, Xu L
, Lin N, Xu L ![]()
Received 9 October 2020
Accepted for publication 30 December 2020
Published 27 January 2021 Volume 2021:14 Pages 345—355
DOI https://doi.org/10.2147/RMHP.S286001
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kent Rondeau
Hailong Huang,1,2,* Meiying Cai,1,2,* Yan Wang,1,2 Bin Liang,1,2 Na Lin,1,2 Liangpu Xu1,2
1Center for Prenatal Diagnosis, Fujian Maternity and Child Health Hospital, Affiliated Hospital of Fujian Medical University, Fuzhou 350001, Fujian Province, People’s Republic of China; 2Fujian Key Laboratory for Prenatal Diagnosis and Birth Defects, Fuzhou 350001, Fujian Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Na Lin; Liangpu Xu
Center for Prenatal Diagnosis, Fujian Maternity and Child Health Hospital, Affiliated Hospital of Fujian Medical University, No. 18 Daoshan Road, Gulou District, Fuzhou City, Fujian Province 350001, People’s Republic of China
Email [email protected]; [email protected]
Objective: This study aimed to examine the effectiveness of the SNP array for the prenatal diagnosis of congenital heart disease (CHD) screened by echocardiography.
Patients and Methods: A total of 356 pregnant women with fetal congenital heart malformations revealed by echocardiography at the Center for Prenatal Diagnosis of Fujian Maternal and Children Hospital during the period from November 2016 through July 2019 were recruited. The fetuses were assigned into three cohorts, including 142 with a single cardiac malformation, 106 with multiple cardiac malformations and 108 with cardiac and extracardiac malformations. All fetuses underwent chromosomal karyotyping and SNP array simultaneously, and the effectiveness of the SNP array for the prenatal diagnosis of CHD was evaluated.
Results: The overall prevalence of abnormal karyotypes was 9.3% among the 356 fetuses with CHD, and a higher proportion was found in fetuses with cardiac and extracardiac malformations (18.5%) than in those with single (5.6%) or multiple cardiac malformations (4.7%) (P< 0.05). Consistent with karyotype analysis, SNP array detected an additional 25 fetuses with pathogenic copy number variations (CNVs), seven with variant of unknown significance (VOUS) and seven with benign CNVs, and a lower proportion of abnormal CNV was found in fetuses with a single cardiac malformation (4.2%) than in those with multiple cardiac malformations (9.4%) or cardiac and extracardiac malformations (14.8%) (P< 0.05). Among the 33 fetuses with chromosomal abnormality, postnatal follow-up showed termination of pregnancy in 25 with pathogenic CNVs, one with VOUS, and six with normal karyotypes and SNP array findings but severe multiple malformations by ultrasonography.
Conclusion: SNP array increases the overall detection of abnormal CNVs by 9%, which improves the detection of CNVs associated with CHD. SNP array may serve as a tool for prenatal diagnosis of CHD that facilitates the discovery of pathogenic genes associated with CHD and provide valuable insights into the precision assessment of fetal prognosis during the prenatal counseling.
Keywords: congenital heart disease, SNP array, echocardiography, prenatal diagnosis
Introduction
Congenital heart disease (CHD), which consists of a wide variety of anomalies and malformations involving the heart and great vessels that develop in utero during the development of the cardiovascular system, is a problem with the cardiac structure and function that is present at birth.1 As the most common birth defect, this disorder affects approximately 1% of all live births.2 It is estimated that 1.35 million babies are born with CHD each year globally; however, the prevalence of CHD continues to increase across the world2 and, more importantly, a new group of adult CHD patients has emerged,3 which poses a great public health challenge.4 Thanks to huge technical improvements in imaging and cardiovascular surgery, surgical repair becomes possible for common cardiac defects, and both the success rate of surgery and long-term postsurgical survival rates achieve a great improvement for severe forms of CHD.5 Nevertheless, if CHD is one of phenotypes in inherited syndromes the success of surgery is not a guarantee for the normal postsurgical quality of life.6,7 A search for the cause of CHD and identification of the complicating inherited syndromes are therefore given a high priority in the currently prenatal diagnosis of CHD.8,9
Currently, the exact pathogenesis of CHD remains unclear; however, it is widely accepted that genetic and environmental factors contribute to the development of CHD.10 Increasing evidence shows that the genetic factors play a more important role in the pathogenesis of CHD among the multiple etiologic factors, including chromosomal abnormality, gene mutation and copy number variation (CNV).11–14 Chromosomal abnormality,15 notably aneuploidy, is a major cause of CHD.16 Almost a half of patients with trisomy 21 (T21) are reported to present the features of CHD, which manifest complex cardiac malformations, such as atrial septal defect (ASD), ventricular septal defect (VSD), atrial-ventricular septal defect (AVSD), and tetralogy of Fallot (TOF).17 It has been found that the majority of patients with T18 or T13 have the phenotype of CHD,18,19 and sex chromosome abnormality (Turner syndrome) or terminal deletion of 5p were reported to correlate with CHD.20,21 In addition, chromosomal microdeletions and microduplications, such as 22q11.2 microdeletion syndrome and Williams–Beuren syndrome, are also reported to contribute to the pathogenesis of CHD.22,23
Currently, the detection of CHD mainly includes karyotype analysis, fluorescence in situ hybridization (FISH), multiplex ligation dependent probe amplification (MLPA) and chromosomal microarray analysis (CMA).24 Conventional karyotyping has a low sensitivity and long duration for detection of inherited disorders,25 and FISH assay suffers problems of small probe numbers and detection of known genetic loci,26 while MLPA targets common aneuploidies and microdeletions/microduplications, which limits its clinical uses.27
Because of rapid, high-throughput and high-resolution ability, CMA has been accepted as a powerful tool for the diagnosis of developmental retardation, intellectual disability, autism and multiple congenital anomalies.28–31 Currently, CMA is classified into two categories: microarray based comparative genomic hybridization (aCGH) and single nucleotide polymorphism (SNP) array, both of which are effective to detect chromosomal microdeletions and microduplications; however, SNP array is feasible to detect not only CNVs, but also haploid and chimera.32 CMA platforms with copy number as well as SNP probes additionally provide clinically relevant information about copy neutral changes, such as uniparental disomy and will also identify ploidy changes, and such a design is feasible to identify chromosomal syndromes and single gene diseases with deletion duplication.32 In children with CHD, with or without multiple congenital anomalies or intellectual disabilities/developmental delay but normal karyotype, CMA was found to increase the diagnostic rate and improve the etiological diagnosis of CHD.32 Results from a recent prospective study showed that CMA is a reliable and high-resolution test for clinical prenatal diagnosis of CHD.33 In addition, CMA was considered a reliable and accurate prenatal approach for identifying pathogenic fetal chromosomal abnormalities associated with cardiac defects.34 However, the effectiveness of CMA for the prenatal diagnosis of CHD remains to be investigated. The purpose of this study was to examine the value of SNP array as a tool for prenatal diagnosis of CHD screened by echocardiography, which may provide implications for precision assessment of fetal prognosis.
Patients and Methods
Ethical Statement
This study was approved by the Ethics Review Committee of Fujian Maternal and Children Hospital. All procedures were performed following the Declaration of Helsinki, as well as international and national laws, guidelines and regulations. Signed informed consent was obtained from all subjects with a detailed description of the purpose of the study.
Subjects
A total of 356 pregnant women with fetal congenital heart malformations revealed by echocardiography at the Center for Prenatal Diagnosis of Fujian Maternal and Children Hospital (Fuzhou, China) during the period from November 2016 through July 2019 were recruited. The pregnant women had gestational ages of 13 to 38 weeks (mean, 26.1 weeks) and ages of 18 to 46 years (mean, 29.4 years). Echocardiography detected 142 fetuses with a single cardiac malformation, 106 with multiple cardiac malformations and 108 with cardiac and extracardiac malformations. In this study, a single cardiac malformation was defined as presence of a single fetal cardiac malformation and no other anatomical deformities seen in prenatal ultrasonography, and multiple cardiac malformations were defined as development of multiple malformations in the heart, while cardiac and extracardiac malformations were defined as malformations involving the nervous system, urinary system, and digestive system in addition to the heart.
Karyotype Analysis
Chorionic villus, amniotic fluid and umbilical cord blood samples were collected through B-mode ultrasound-guided abdominal puncture, amniocentesis and cordocentesis, respectively. All prenatal samples were routinely cultured, mounted on slides and subjected to G-banding (additional C-banding and N-banding if required). Karyotype analysis was performed on a GSL-120 Streamlines Cytogenetic Analysis System (Leica Microsystems; Mannheim, Germany). At least 40 metaphases were counted for each case, and five karyotypes were randomly selected for analysis. The results were interpreted according to the 2016 International System for Human Cytogenomic Nomenclature (ISCN).
SNP Array
Short tandem repeat (STR) analysis was conducted before detection of the fetal samples. Approximately 10 mL of amniotic fluid was sampled and centrifuged, and the sediment was collected. Genomic DNA was extracted from amniotic fluid cells using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany), digested, amplified, purified, fragmented, labeled and hybridized to the array on the Affymetrix SNP Array 6.0 (Affymetrix; Santa Clara, CA, USA). The CytoScan HD array, includes the CNV probe and SNP probe, may detect CNV, mosaic (mosaic proportion >10%) and loss of heterozygosity (LOH). All data analyses were performed using the software Chromosome Analysis Suite (ChAS) version 3.2 (Affymetrix; Santa Clara, CA, USA), All nucleotide positions refer to the Human Genome Feb 2009 Assembly (GRCh37/hg19). The interpretation of CNV, which was classified as pathogenic, variants of uncertain significance (VOUS) and benign, was identified using online public databases, including the database of genomic variants (DGV, http://projects.tcag.ca/variation), the DECIPHER database (https://decipher.sanger.ac.uk/), the OMIM database (http://www.omim.org), the International Standards for Cytogenomic Arrays (ISCA) Consortium and Public Database (https://www.iscaconsortium.org/), the CAGdb database (http://www.cagdb.org/), the CHDWiki database and the NCBI database. The pathogenic CNVs detected by SNP array were validated using the FISH assay. Peripheral blood was sampled from the parents of the fetus with VOUS for the SNP array, and the type of CNV was identified by means of SNP array and pedigree analysis.
Statistics
All statistical analyses were performed using the statistical software SPSS version 22.0 (IBM Corporation, Armonk, NY, USA). Differences of proportions were tested for statistical significance with a chi-squared test or Fisher’s exact test, and a P-value <0.05 was considered statistically significant.
Results
Chromosomal Karyotypes
There were 33 fetuses with abnormal karyotypes detected in the 356 fetuses with CHD revealed by prenatal echocardiography, including 10 cases with T18, eight with T21, two with T13, three with abnormal chromosome number, and 10 with chromosomal structural abnormalities. There was a significant difference in the proportion of chromosomal abnormalities among the fetuses with a single cardiac malformation (5.6%), multiple cardiac malformations (4.7%) and cardiac and extracardiac malformations (18.5%) (P<0.01), and a higher proportion was found in fetuses with cardiac and extracardiac malformations than in those with single or multiple cardiac malformations (P<0.01) (Table 1).
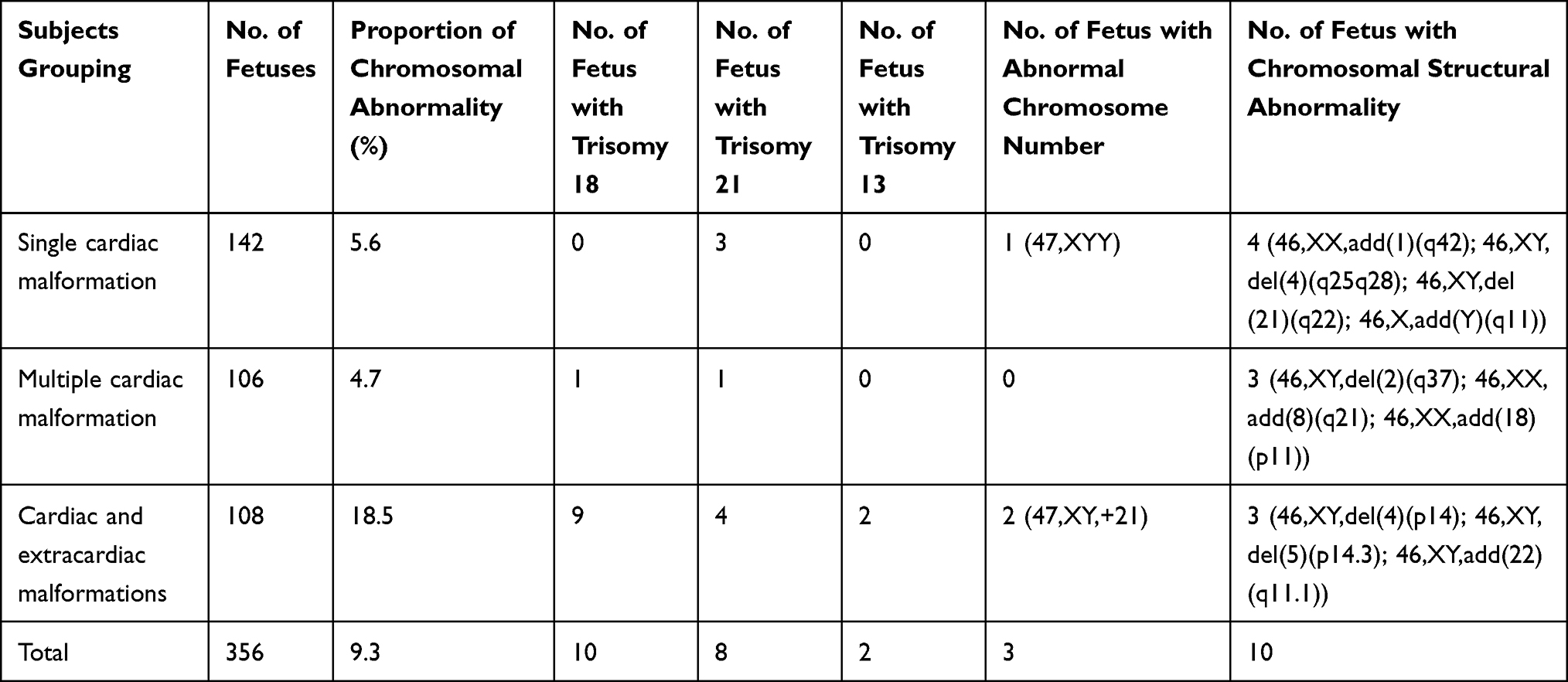 |
Table 1 Chromosomal Abnormalities Detected in Fetuses with Congenital Heart Disease Revealed by Prenatal Echocardiography |
SNP Array Findings
In addition to 33 fetuses with consistency between SNP array findings and karyotype analysis among the 356 fetuses, SNP array detected 32 fetuses with abnormal CNVs (0.4 to 6.5 Mbp in size) and seven with benign CNVs. There were 25 fetuses with pathogenic CNVs, including eight with known microdeletion/microduplication syndromes and two with uniparental disomy, and seven with VOUS, including six with copy number duplication and one with copy number loss (Table 2). There was a significant difference in the detection of abnormal CNVs among the fetuses with a single cardiac malformation (4.2%), multiple cardiac malformations (9.4%) and cardiac and extracardiac malformations (14.8%) (P<0.01), and a lower proportion was found in fetuses with single cardiac malformations than in those with multiple cardiac malformations or cardiac and extracardiac malformations (P<0.01) (Table 3).
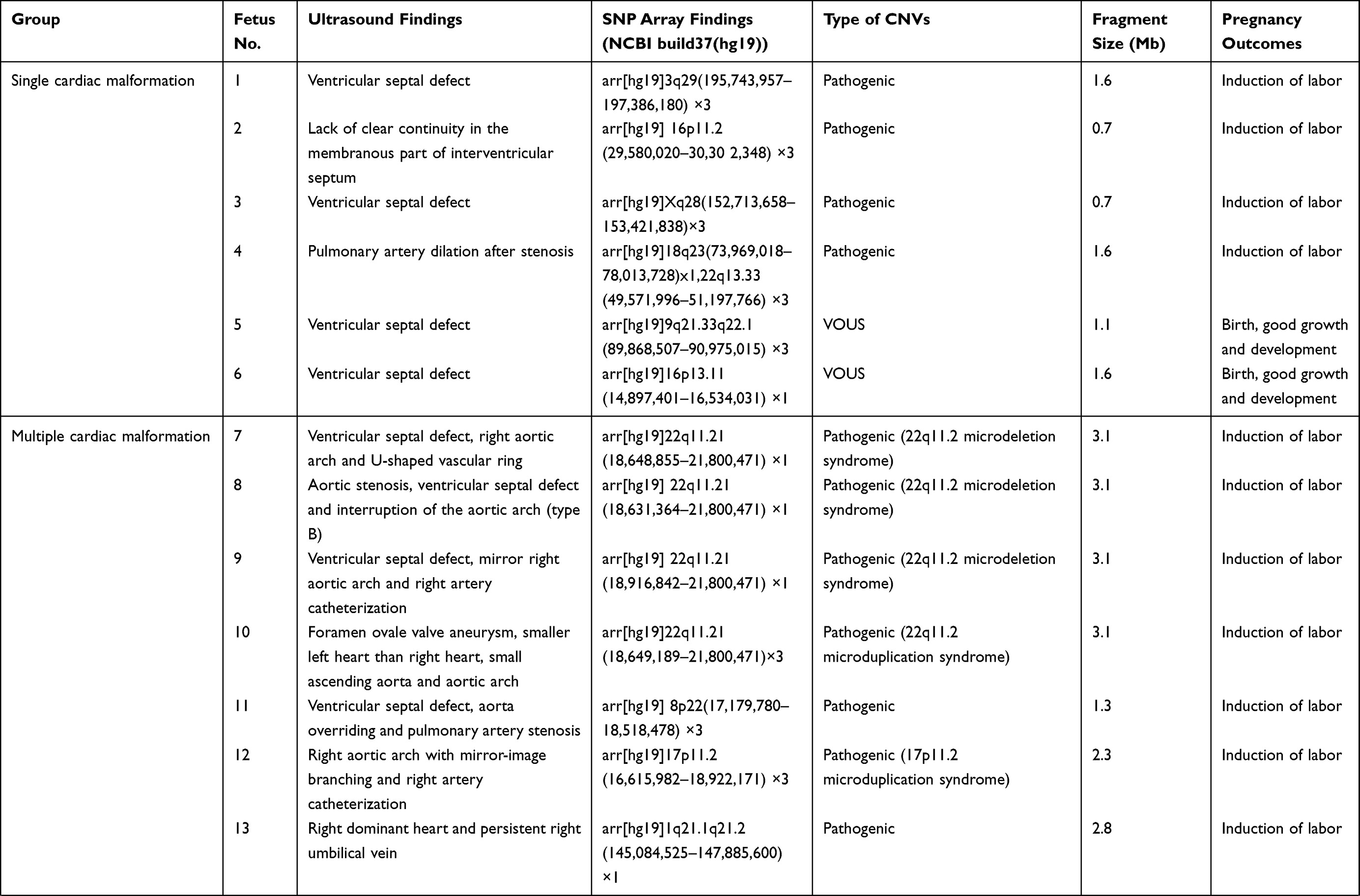 | 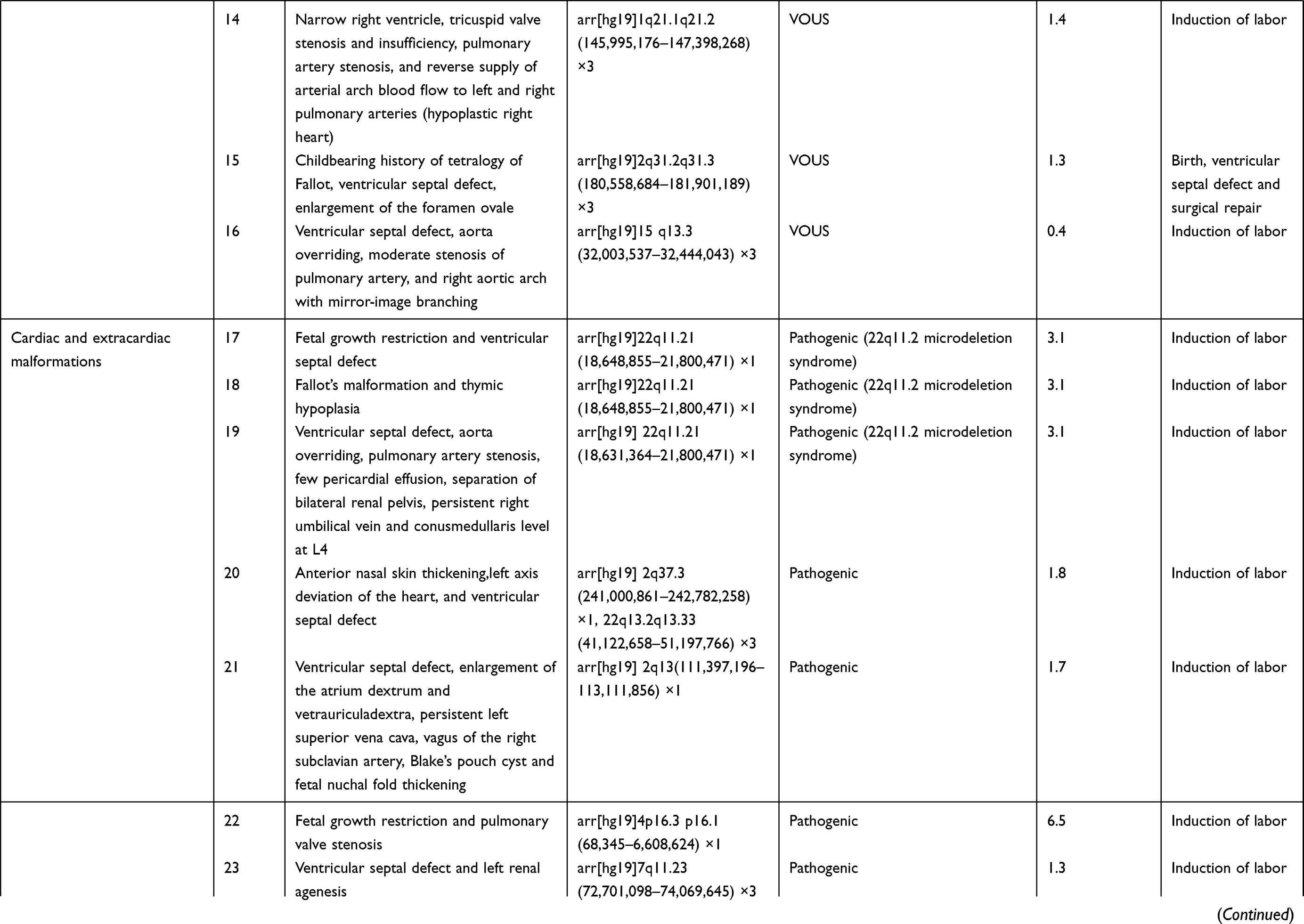 | 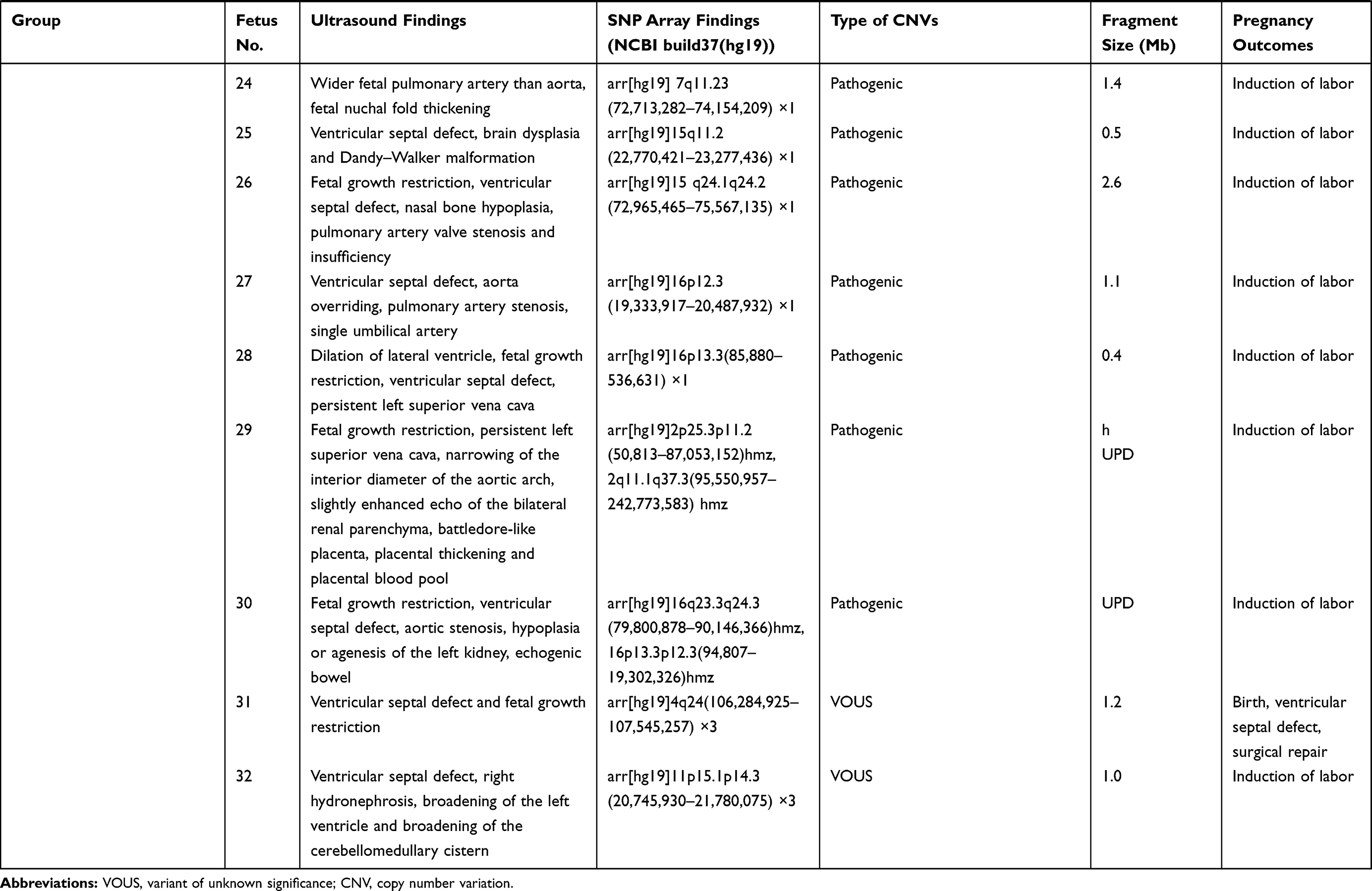 |
Table 2 SNP Array Findings in Fetuses |
 |
Table 3 Abnormal Copy Number Variations Detected in Fetuses |
Follow-up Outcomes
Among the 356 fetuses with prenatal diagnosis of CHD by echocardiography, there were seven cases lost to follow-up, and the rest were all successfully followed-up. The 33 fetuses with chromosomal abnormalities, 25 with pathogenic CNVs and one with VOUS experienced pregnancy termination, and six fetuses with normal karyotypes and SNP array findings but severe multiple malformations by ultrasonography also underwent pregnancy termination. Among the four fetuses with VOUS that continued pregnancy, the postnatal follow-up showed ventricular septal defects in two infants (the growth index became normal following surgical treatments) and good growth and development in the other two infants (Table 2).
Discussion
In the current study, a total of 356 fetuses with CHD detected by echocardiography received karyotype analyses, and the overall prevalence of chromosomal abnormalities was 9.3%. Among the 33 CHD fetuses with chromosomal abnormalities identified by karyotyping, there were 10 fetuses with T18, eight with T21, two with T13, three with abnormal chromosome numbers and 10 with chromosomal structural abnormalities. All 356 fetuses received SNP array simultaneously, and 33 fetuses achieved consistent findings with karyotyping results; furthermore, the SNP array detected additional 9.0% prevalence of abnormal CNV. Our data further demonstrate that in addition to abnormal chromosome numbers and chromosomal structural abnormalities, microdeletions/microduplications may contribute to the genetic etiology of CHD.35 It is suggested that the SNP array may serve as a supplement of conventional chromosomal karyotyping to increase the detection of CHD, and a further whole-genome high-resolution SNP array is recommended for fetuses with CHD but normal karyotype.36,37
In this cohort, T18 and chromosomal structural abnormality were the most common forms of chromosomal structural abnormalities among fetuses with CHD (both 30.3%), followed by T21, T13 and abnormal chromosomal numbers. The prevalence of chromosomal abnormalities was significantly greater in fetuses with CHD plus extracardiac malformations (18.5%) than in those with single cardiac malformations (5.6%) and multiple cardiac malformations (4.7%) (P<0.01); however, no significant difference was found between fetuses with single and multiple cardiac malformations (P>0.05). These findings indicate that aneuploidy and abnormality of large-fragment chromosome may cause changes in genetic materials and disrupt genetic equilibrium, thereby resulting in fetal multisystem developmental abnormalities.16
In the present study, the prevalence of abnormal CNVs was 4.2%, 9.4% and 14.8% in fetuses with a single cardiac malformation, multiple cardiac malformations, and CHD plus extracardiac malformations. Our data showed that the prevalence of abnormal CNVs was significantly higher in fetuses with cardiac and extracardiac malformations than in those with single or multiple cardiac malformations (P<0.05), and a high detection of abnormal CNVs was seen in fetuses with multiple cardiac malformations than in those with a single cardiac malformation (P<0.05), which was inconsistent with previous reports.38–40 This may be because more fetuses with CHD were enrolled in our study, and the number of subjects was comparable among groups. Compared with previous studies,38–40 this study is more persuasive, and our data demonstrate a strong correlation between fetal multiple malformations and abnormal CNVs.
In our study, 25 fetuses with pathogenic CNVs were identified among the 32 fetuses with abnormal CNVs, including nine with known microdeletion/microduplication syndromes. To date, more than 40 microdeletion/microduplication syndromes have been reported to cause CHD, and most of the fetuses with these microdeletion/microduplication syndromes present developmental malformations, intellectual retardation, and cognitive impairments.41 We detected eight fetuses with microdeletion/microduplication syndromes, including six with 22q11.2 microdeletion syndrome (MIM:611,867), one with 22q11.2 microduplication syndrome (MIM:608,363) and one with 17p11.2 microduplication syndrome (MIM:610,883). Among these microdeletion/microduplication syndromes, the 22q11.2 microdeletion syndrome is most common, which has a global prevalence rate of 1/4000.42 The 22q11.2 microdeletion syndrome may present multiple clinical phenotypes, and approximately 80% patients with this disorder complicated with diverse forms of CHD.43 Genetic analysis reveals that the TBX1 gene at the 22q11.2 region is an important pathogenic gene for CHD.44 In this study, all six fetuses with the 22q11.2 microdeletion syndrome had diverse ultrasound findings, but all had cardiac malformations, including three fetuses that had complicated extracardiac malformations on ultrasonography. SNP array analysis showed a 3.1 Mb duplication at the chromosome 22q11.2 region in a fetus, which was identified as the 22q11.2 microduplication syndrome. Like the 22q11.2 microdeletion syndrome, the 22q11.2 microduplication syndrome may present intellectual deficiency, neuropsychological problems, linguistic difficulties, and common cardiac malformations.45 In this study, the fetus with the 22q11.2 microduplication syndrome had ultrasound findings of multiple cardiac malformations. SNP array analysis revealed 2.1 Mb duplication at the chromosome 17p11.2 region, and the fetus was therefore diagnosed with Potocki–Lupski syndrome (MIM: 610,883). Worldwide, the prevalence of Potocki–Lupski syndrome is 1/20,000 to 1/250,000, and this syndrome has multiple clinical manifestations, which are predominantly associated with CHD.46 In the present study, the fetus with Potocki–Lupski syndrome had ultrasound findings of multiple cardiac malformations.
In addition, we detected the uniparental disomy in two fetuses with CHD using the SNP array. A case was found to bear loss of heterozygosity on chromosome 2, and pedigree analysis showed maternal-derived uniparental disomy on chromosome 2. It has been shown that the imprinted gene on chromosome 2 correlates with fetal growth restriction, developmental retardation, cardiac malformations, hypospadias and oligohydramnios.47 Another fetus was detected with 19.2 Mb loss of heterozygosity at the p13.3-p12.3 region of the chromosome 16, and pedigree analysis confirmed the diagnosis of maternal-derived uniparental disomy. The typical intrauterine features of maternal-chromosomal uniparental disomy 16 include fetal growth restriction, cardiac malformations and urogenital abnormalities.48 Detection of uniparental disomy using a SNP array may facilitate further understandings on the genetic etiology of cardiac malformations.
Currently, the laboratory identification and interpretation of CNVs remain a great challenge in clinical practices. It is reported that CMA may detect 1.1% to 6% VOUS.49 In this study, seven fetuses were detected with VOUS among 356 fetuses with CHD, with a 1.9% detection rate of VOUS, which was similar to previous studies.34,39,50 Among the seven fetuses with CHD that were identified with VOUS, two were found to bear susceptibility loci for neurocognitive disorders, including a copy number loss of 1.4 Mb at the p13.11 region of the chromosome 16 and a copy number gain of 1.4 Mb at the q21.1-q21.2 region of the chromosome 1. Although pedigree analysis showed this mutation was inherited from the mother with a normal phenotype; however, the frequency of susceptibility loci for neurocognitive disorders is less than 1% in healthy populations.51 Therefore, the susceptibility locus for neurocognitive disorders was defined as a VOUS in this study. As a rare CNV, chromosome 16p13.11 microdeletion syndrome (MIM: 610,543) has an increased risk of complications in newborns, including bradykinesia, facial deformity, microcephaly, gastroesophageal reflux disease and CHD.52 There is little knowledge on the link between the chromosome 16p13.11 microdeletion syndrome and CHD until now, and therefore, the fetal phenotypes may provide insights into the understanding of the phenotype of the chromosome 16p13.11 microdeletion syndrome. The prevalence of the chromosome 1q21.1 microduplication syndrome (MIM: 612,475) is estimated to be 0.03% in adults, and the clinical manifestations of this disorder, which is of a high heterogeneity, include dysgnosia, developmental retardation, specific learning disabilities, autism, schizophrenia, and heart abnormalities and deformities.53 CHD is the major feature of the chromosome 1q21.1 microdeletion syndrome (MIM: 612,474),54 and is also reported in infants with the chromosome 1q21.1 microduplication syndrome.55 Previous studies have shown a correlation between the 1q21.1 microduplication and CHD, which leads to nonsyndromic coronary heart disease.56
In this cohort, a fetus was detected with a 0.4 Mb copy number duplication at the q13.3 region of the chromosome 15, and the score for the triple dose effect was one at the 15q13.3 recurrent region in the ClinGen database, with penetrance of 5% to 10%. Therefore, this duplication was also defined as a VOUS in this study. The parents of two fetuses refused to receive further genetic testing, and two other fetuses were identified to bear new CNVs through detection of their parental samples; therefore, these four fetuses were considered to bear VOUS. Although these VOUS contained genes that had indefinite roles in the alteration of cardiac structures or functions; however, the contribution of these VOUS to the disease phenotype cannot be excluded. Further studies are required to demonstrate the clinical significance of these VOUS. The improvements in the resolution and experimental procedures of CMA may result in the presence of more and more VOUS, which will increase the difficulty in prenatal counseling and the couple’s anxiety. Recently, next-generation sequencing, as a novel genetic tool to detect single-gene mutations and CNVs, may provide more comprehensive information for prenatal genetic diagnosis in fetuses with CHD screened by ultrasound, and provide better assessments of fetal prognosis.57
Genetic testing determines the decisions for termination of pregnancy.58 Among the 33 fetuses with CHD that were identified with chromosomal abnormalities recruited in this study, termination of pregnancy was decided among the 25 fetuses with pathogenic CNVs and one with VOUS. In addition, termination of pregnancy was decided in six fetuses with normal chromosomal karyotypes and normal SNP array findings, but showing severe multiple deformities on ultrasound. Among the four fetuses with VOUS that continued pregnancy, the postnatal follow-up showed ventricular septal defects in two infants (the growth index became normal following surgical treatments), and good growth and development in the other two infants. It is therefore considered that SNP array may provide a precision assessment of fetal prognosis and risk of disease recurrence, and provide valuable insights into the decisions to continue pregnancy during the prenatal counseling.
In summary, the results of the present study demonstrate that the SNP array shows consistent findings with the chromosomal karyotype analysis, and increases the overall detection of abnormal CNVs by 9%. Such an increase improves the detection of CNVs associated with CHD, which further confirms that CNVs are an important pathogenic factor of CHD. It is therefore concluded that the SNP array as a tool for prenatal diagnosis of CHD facilitates the discovery of CHD-associated pathogenic genes and provides valuable insights into the precision assessment of fetal prognosis during the prenatal counseling.
Acknowledgments
We are grateful to Prof. Yuan Lin for her kind assistance during the study period. This study was funded by the grants from the Natural Science Foundation of Fujian Province (grant nos. 2017J01238 and 2018J01235).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tanner K, Sabrine N, Wren C. Cardiovascular malformations among preterm infants. Pediatrics. 2005;116(6):e833–e838. doi:10.1542/peds.2005-0397
2. Liu Y, Chen S, Zühlke L, et al. Global birth prevalence of congenital heart defects 1970–2017: updated systematic review and meta-analysis of 260 studies. Int J Epidemiol. 2019;48(2):455–463. doi:10.1093/ije/dyz009
3. Bouma BJ, Mulder BJ. Changing landscape of congenital heart disease. Circ Res. 2017;120(6):908–922. doi:10.1161/CIRCRESAHA.116.309302
4. Rossano JW. Congenital heart disease: a global public health concern. Lancet Child Adolesc Health. 2020;4(3):168–169. doi:10.1016/S2352-4642(19)30429-8
5. Meller CH, Grinenco S, Aiello H, et al. Congenital heart disease, prenatal diagnosis and management. Arch Argent Pediatr. 2020;118(2):e149–e161. doi:10.5546/aap.2020.eng.e149
6. Simmons MA, Brueckner M. The genetics of congenital heart disease… understanding and improving long-term outcomes in congenital heart disease. Curr Opin Pediatr. 2017;29(5):520–528. doi:10.1097/MOP.0000000000000538
7. Oster ME, Knight JH, Suthar D, Amin O, Kochilas LK. Long-term outcomes in single-ventricle congenital heart disease. Circulation. 2018;138(23):2718–2720. doi:10.1161/CIRCULATIONAHA.118.036821
8. Kovacevic A, Simmelbauer A, Starystach S, et al. Counseling for prenatal congenital heart disease-recommendations based on empirical assessment of counseling success. Front Pediatr. 2020;8:26. doi:10.3389/fped.2020.00026
9. Hopkins MK, Dugoff L, Kuller JA. Congenital heart disease: prenatal diagnosis and genetic associations. Obstet Gynecol Surv. 2019;74(8):497–503. doi:10.1097/OGX.0000000000000702
10. Gelb BD, Chung WK. Complex genetics and the etiology of human congenital heart disease. Cold Spring Harb Perspect Med. 2014;4(7):a013953. doi:10.1101/cshperspect.a013953
11. Blue GM, Kirk EP, Giannoulatou E, et al. Advances in the genetics of congenital heart disease: a clinician’s guide. J Am Coll Cardiol. 2017;69(7):859–870. doi:10.1016/j.jacc.2016.11.060
12. Williams K, Carson J, Lo C. Genetics of congenital heart disease. Biomolecules. 2019;9(12):E879. doi:10.3390/biom9120879
13. Edwards JJ, Gelb BD. Genetics of congenital heart disease. Curr Opin Cardiol. 2016;31(3):235–241. doi:10.1097/HCO.0000000000000274
14. Yuan S, Zaidi S, Brueckner M. Congenital heart disease: emerging themes linking genetics and development. Curr Opin Genet Dev. 2013;23(3):352–359. doi:10.1016/j.gde.2013.05.004
15. Calcagni G, Unolt M, Digilio MC, et al. Congenital heart disease and genetic syndromes: new insights into molecular mechanisms. Expert Rev Mol Diagn. 2017;17(9):861–870. doi:10.1080/14737159.2017.1360766
16. Wimalasundera RC, Gardiner HM. Congenital heart disease and aneuploidy. Prenat Diagn. 2004;24(13):1116–1122. doi:10.1002/pd.1068
17. Paladini D, Tartaglione A, Agangi A, et al. The association between congenital heart disease and down syndrome in prenatal life. Ultrasound Obstet Gynecol. 2000;15(2):104–108. doi:10.1046/j.1469-0705.2000.00027.x
18. Kosiv KA, Gossett JM, Bai S, Collins RT. Congenital heart surgery on in-hospital mortality in trisomy 13 and 18. Pediatrics. 2017;140(5):e20170772. doi:10.1542/peds.2017-0772
19. Cooper DS, Riggs KW, Zafar F, et al. Cardiac surgery in patients with trisomy 13 and 18: an analysis of the society of thoracic surgeons congenital heart surgery database. J Am Heart Assoc. 2019;8(13):e012349. doi:10.1161/JAHA.119.012349
20. Kim HK, Gottliebson W, Hor K, et al. Cardiovascular anomalies in turner syndrome: spectrum, prevalence, and cardiac MRI findings in a pediatric and young adult population. AJR Am J Roentgenol. 2011;196(2):454–460. doi:10.2214/AJR.10.4973
21. Taylor MJ, Josifek K. Multiple congenital anomalies, thymic dysplasia, severe congenital heart disease, and oligosyndactyly with a deletion of the short arm of chromosome 5. Am J Med Genet. 1981;9(1):5–11. doi:10.1002/ajmg.1320090103
22. Goldmuntz E. 22q11.2 Deletion syndrome and congenital heart disease. Am J Med Genet C Semin Med Genet. 2020;184(1):64–72. doi:10.1002/ajmg.c.31774
23. Ko JM. Genetic syndromes associated with congenital heart disease. Korean Circ J. 2015;45(5):357–361. doi:10.4070/kcj.2015.45.5.357
24. Bravo-Valenzuela NJ, Peixoto AB, AraujoJúnior E. Prenatal diagnosis of congenital heart disease: a review of current knowledge. Indian Heart J. 2018;70(1):150–164. doi:10.1016/j.ihj.2017.12.005
25. Killen SA, Mouledoux JH, Kavanaugh-McHugh A. Pediatric prenatal diagnosis of congenital heart disease. Curr Opin Pediatr. 2014;26(5):536–545. doi:10.1097/MOP.0000000000000136
26. Connor JA, Hinton RB, Miller EM, Sund KL, Ruschman JG, Ware SM. Genetic testing practices in infants with congenital heart disease. Congenit Heart Dis. 2014;9(2):158–167. doi:10.1111/chd.12112
27. Nagy O, Szakszon K, Biró BO, et al. Copy number variants detection by microarray and multiplex ligation-dependent probe amplification in congenital heart diseases. J Biotechnol. 2019;299:86–95. doi:10.1016/j.jbiotec.2019.04.025
28. Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. doi:10.1016/j.ajhg.2010.04.006
29. Werling AM, Grünblatt E, Oneda B, et al. High-resolution chromosomal microarray analysis for copy-number variations in high-functioning autism reveals large aberration typical for intellectual disability. J Neural Transm (Vienna). 2020;127(1):81–94. doi:10.1007/s00702-019-02114-9
30. Hu T, Zhang Z, Wang J, et al. Chromosomal aberrations in pediatric patients with developmental delay/intellectual disability: a single-center clinical investigation. Biomed Res Int. 2019;2019:9352581. doi:10.1155/2019/9352581
31. Jang W, Kim Y, Han E, et al. Chromosomal microarray analysis as a first-tier clinical diagnostic test in patients with developmental delay/intellectual disability, autism spectrum disorders, and multiple congenital anomalies: a prospective multicenter study in Korea. Ann Lab Med. 2019;39(3):299–310. doi:10.3343/alm.2019.39.3.299
32. Wu XL, Li R, Fu F, et al. Chromosome microarray analysis in the investigation of children with congenital heart disease. BMC Pediatr. 2017;17(1):117. doi:10.1186/s12887-017-0863-3
33. Wang Y, Cao L, Liang D, et al. Prenatal chromosomal microarray analysis in fetuses with congenital heart disease: a prospective cohort study. Am J Obstet Gynecol. 2018;218(2):
34. Zhu X, Li J, Ru T, et al. Identification of copy number variations associated with congenital heart disease by chromosomal microarray analysis and next-generation sequencing. Prenat Diagn. 2016;36(4):321–327. doi:10.1002/pd.4782
35. Goldmuntz E, Paluru P, Glessner J, et al. Microdeletions and microduplications in patients with congenital heart disease and multiple congenital anomalies. Congenit Heart Dis. 2011;6(6):592–602. doi:10.1111/j.1747-0803.2011.00582.x
36. Lin CZ, Qi BR, Hu JS, Huang YD, Huang XQ. Chromosome 15q13 microduplication in a fetus with cardiac rhabdomyoma: a case report. Mol Cytogenet. 2019;12(1):24. doi:10.1186/s13039-019-0437-1
37. Lin CZ, Qi BR, Hu JS, Huang XQ. A fetus with kabuki syndrome 2 detected by chromosomal microarray analysis. Int J Clin Exp Pathol. 2020;13(2):302–306.
38. Sukenik-Halevy R, Sukenik S, Koifman A, et al. Clinical aspects of prenatally detected congenital heart malformations and the yield of chromosomal microarray analysis. Prenat Diagn. 2016;36(13):1185–1191. doi:10.1002/pd.4954
39. Wu X, Fu F, Li R, et al. Clinical value of genome-wide high resolution chromosomal microarray analysis in etiological study of fetuses with congenital heart defects. Zhonghua Fu Chan Ke Za Zhi. 2014;49(12):893–898.
40. Xia Y, Yang Y, Huang S, Wu Y, Li P, Zhuang J. Clinical application of chromosomal microarray analysis for the prenatal diagnosis of chromosomal abnormalities and copy number variations in fetuses with congenital heart disease. Prenat Diagn. 2018;38(6):406–413. doi:10.1002/pd.5249
41. Lalani SR, Belmont JW. Genetic basis of congenital cardiovascular malformations. Eur J Med Genet. 2014;57(8):402–413. doi:10.1016/j.ejmg.2014.04.010
42. Sullivan KE. Chromosome 22q11.2 deletion syndrome and DiGeorge syndrome. Immunol Rev. 2019;287(1):186–201. doi:10.1111/imr.12701
43. Huber J, Peres VC, de Castro AL. Molecular screening for 22Q11.2 deletion syndrome in patients with congenital heart disease. Pediatr Cardiol. 2014;35(8):1356–1362. doi:10.1007/s00246-014-0936-0
44. Hou HT, Chen HX, Wang XL, et al. Genetic characterisation of 22q11.2 variations and prevalence in patients with congenital heart disease. Arch Dis Child. 2020;105(4):367–374. doi:10.1136/archdischild-2018-316634
45. Di Matteo F, Bettin P, Ferrari G, et al. 22q11.2 microduplication syndrome and juvenile glaucoma. Ophthalmic Genet. 2018;39(4):532–538. doi:10.1080/13816810.2018.1484926
46. Praticò AD, Falsaperla R, Rizzo R, Ruggieri M, Verrotti A, Pavone P. A new patient with potocki-lupski syndrome: a literature review. J Pediatr Genet. 2018;7(1):29–34. doi:10.1055/s-0037-1604479
47. Skaar DA, Jirtle RL. Analysis of imprinted gene regulation. Methods Mol Biol. 2017;1589:161–183. doi:10.1007/7651_2015_264
48. Yingjun X, Zhiyang H, Linhua L, et al. Chromosomal uniparental disomy 16 and fetal intrauterine growth restriction. Eur J Obstet Gynecol Reprod Biol. 2017;211:1–7. doi:10.1016/j.ejogrb.2016.12.019
49. Breman A, Pursley AN, Hixson P, et al. Prenatal chromosomal microarray analysis in a diagnostic laboratory; experience with >1000 cases and review of the literature. Prenat Diagn. 2012;32(4):351–361. doi:10.1002/pd.3861
50. Song T, Wan S, Li Y, et al. Detection of copy number variants using chromosomal microarray analysis for the prenatal diagnosis of congenital heart defects with normal karyotype. J Clin Lab Anal. 2019;33(1):e22630. doi:10.1002/jcla.22630
51. Digilio MC, Marino B. What is new in genetics of congenital heart defects? Front Pediatr. 2016;4:120. doi:10.3389/fped.2016.00120
52. Smith AE, Jnah A. Chromosome 16p13.11 microdeletion syndrome in a newborn: a case study. Neonatal Netw. 2018;37(5):303–309. doi:10.1891/0730-0832.37.5.303
53. Dolcetti A, Silversides CK, Marshall CR, et al. 1q21.1 Microduplication expression in adults. Genet Med. 2013;15(4):282–289. doi:10.1038/gim.2012.129
54. Digilio MC, Bernardini L, Consoli F, et al. Congenital heart defects in recurrent reciprocal 1q21.1 deletion and duplication syndromes: rare association with pulmonary valve stenosis. Eur J Med Genet. 2013;56(3):144–149. doi:10.1016/j.ejmg.2012.12.004
55. Xavier J, Zhou B, Bilan F, et al. 1q21.1 microduplication: large verbal-nonverbal performance discrepancy and ddPCR assays of HYDIN/HYDIN2 copy number. NPJ Genom Med. 2018;3(1):24. doi:10.1038/s41525-018-0059-2
56. Sun G, Tan Z, Fan L, Wang J, Yang Y, Zhang W. 1q21.1 microduplication in a patient with mental impairment and congenital heart defect. Mol Med Rep. 2015;12(4):5655–5658. doi:10.3892/mmr.2015.4166
57. Mellis R, Chandler N, Chitty LS. Next-generation sequencing and the impact on prenatal diagnosis. Expert Rev Mol Diagn. 2018;18(8):689–699. doi:10.1080/14737159.2018.1493924
58. Tsai GJ, Cameron CA, Czerwinski JL, Mendez-Figueroa H, Peterson SK, Noblin SJ. Attitudes towards prenatal genetic counseling, prenatal genetic testing, and termination of pregnancy among southeast and east asian women in the United States. J Genet Couns. 2017;26(5):1041–1058. doi:10.1007/s10897-017-0084-9
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.