Back to Journals » Drug Design, Development and Therapy » Volume 13
Smart Targeting To Improve Cancer Therapeutics
Authors Morales-Cruz M, Delgado Y ![]() , Castillo B, Figueroa CM, Molina AM, Torres A, Milián M
, Castillo B, Figueroa CM, Molina AM, Torres A, Milián M ![]() , Griebenow K
, Griebenow K ![]()
Received 14 June 2019
Accepted for publication 6 September 2019
Published 30 October 2019 Volume 2019:13 Pages 3753—3772
DOI https://doi.org/10.2147/DDDT.S219489
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Moraima Morales-Cruz,1,* Yamixa Delgado,2,* Betzaida Castillo,3 Cindy M Figueroa,4 Anna M Molina,1 Anamaris Torres,2 Melissa Milián,2 Kai Griebenow1,*
1Department of Chemistry, University of Puerto Rico, Río Piedras Campus, San Juan, PR, USA; 2Department of Biochemistry & Pharmacology, San Juan Bautista School of Medicine, Caguas, PR, USA; 3Department of Chemistry, University of Puerto Rico, Humacao Campus, Humacao, PR, USA; 4Department of Math and Sciences, Polytechnic University of Puerto Rico, San Juan, PR, USA
*These authors contributed equally to this work
Correspondence: Yamixa Delgado PO Box 4968 Caguas, PR 00726-4968, USA
Tel +1 787 743 3038
Fax +1 787 746 3093
Email [email protected]
Kai Griebenow 17 Ave Universidad STE 1701, San Juan, PR 00925-2537, USA
Tel +1 787 764 0000
Fax +1 787 746 3093
Email [email protected]
Abstract: Cancer is the second largest cause of death worldwide with the number of new cancer cases predicted to grow significantly in the next decades. Biotechnology and medicine can and should work hand-in-hand to improve cancer diagnosis and treatment efficacy. However, success has been frequently limited, in particular when treating late-stage solid tumors. There still is the need to develop smart and synergistic therapeutic approaches to achieve the synthesis of strong and effective drugs and delivery systems. Much interest has been paid to the development of smart drug delivery systems (drug-loaded particles) that utilize passive targeting, active targeting, and/or stimulus responsiveness strategies. This review will summarize some main ideas about the effect of each strategy and how the combination of some or all of them has shown to be effective. After a brief introduction of current cancer therapies and their limitations, we describe the biological barriers that nanoparticles need to overcome, followed by presenting different types of drug delivery systems to improve drug accumulation in tumors. Then, we describe cancer cell membrane targets that increase cellular drug uptake through active targeting mechanisms. Stimulus-responsive targeting is also discussed by looking at the intra- and extracellular conditions for specific drug release. We include a significant amount of information summarized in tables and figures on nanoparticle-based therapeutics, PEGylated drugs, different ligands for the design of active-targeted systems, and targeting of different organs. We also discuss some still prevailing fundamental limitations of these approaches, eg, by occlusion of targeting ligands.
Keywords: active targeting, drug delivery systems, EPR effect, nanoparticles, passive targeting, stimulus-responsive targeting
Introduction
The American Cancer Society estimates for 2018 more than 1.7 million of new cancer cases in the United States of America, and 1600 million cancer-related deaths with lung cancer being the primary cause of death (43%, www.cancer.org). These statistics are expected to increase in the coming decades “unless we make more progress today” (Joe Biden, Vice President at the American Association for Cancer Research Annual Meeting, 2016).
Currently, surgery, radiation therapy (RT), and chemotherapy are the principal treatment strategies against cancer. Surgery is usually recommended at an early stage of the disease and is most effective when all the cancer cells can be excised.1,2 It is also used in later stages but mostly to debulk tumors and improve quality of life. Thus, chemotherapy and RT are the most widely used interventions for the treatment of cancer.1–3 In contrast to surgery, chemotherapy and RT are mostly only capable of killing a fraction of tumor cells during each treatment regimen and typically never completely cure the disease.3 Cytotoxic anticancer drugs are used in chemotherapy to primarily kill metabolically active cells. Most normal cells do not divide as often as cancer cells and thus are proportionately less affected by these cytotoxic drugs. However, although chemotherapy and RT are employed to improve the patient’s quality of life or to prolong it, they are frequently associated with severe side-effects related to systemic toxicity due to the lack of tumor specificity.3–7 Similar to chemotherapy, RT also damages healthy cells, organs, and tissues. For example, the term “mucositis” describes one of the common adverse effects of RT and chemotherapy treatments. Mucositis may limit the patient’s ability to tolerate chemotherapy or RT, and the nutritional status may become compromised.3,4 In addition, one of the leading causes of treatment failure in cancer therapy is the phenomenon of multidrug resistance syndrome (MDRS), typically acquired during prolonged exposure to chemotherapy.8–12 MDRS is characterized by the ability of cancer cells to efflux drugs by molecular pumps, which results in reducing the therapeutic effect.12
With this in mind, in the last decade, a diverse range of drug delivery systems (DDS) has been developed to improve cancer therapies. There are two main types: targeted and non-targeted drug delivery systems. Both types of DDS have been designed at the nanoscale (in this review loosely defined as 10–1000 nm) to enable efficient transport in blood vessels, to overcome biological barriers during the transport, and to reach pathological cells.13
In this review, we will focus on some recent developments of smart targeting in cancer treatment, particularly promising data and advanced preclinical and clinical studies. We will evaluate the use of different nanoparticles (NPs) to facilitate drug delivery by both, passive and active targeting mechanisms and highlight the potential advantages of utilizing nanotechnology within the field of cancer therapy.
Passive Targeting For Enhanced Drug Tumor Accumulation By DDS
In cancer applications, passive targeting is defined as the preferential accumulation of the drug in the tumor. The accumulation and delivery of the drug are determined by the capability of the DDS to overcome biological barriers and the inherent characteristics of the DDS itself (eg, size, material, and charge).
Overcoming Physiological Barriers
The delivery of drugs from nanosized DDS offers a multitude of advantages over their delivery from larger-sized particles (eg, microparticles) or their delivery as single-molecule drugs (<10 nm). In general, nanosized drugs are better suited than larger delivery systems in the micrometer range for important delivery routes, eg, intravenous (i.v.) and oral delivery.13–16 In the case of oral delivery, the size limit for the particles to be able to cross the intestinal mucosal barrier of the gastrointestinal tract is the main restriction. Particles in the intestinal lumen within a size range of 3–10 µm cannot migrate through the lymphatic system whereas particles larger than 10 µm are not taken up in the gastrointestinal tract.17–19 NPs are also better suited for i.v. delivery in comparison with microparticles. Since the smallest capillaries in the body are 5–6 µm in diameter, the size of particles being distributed into the bloodstream must be significantly smaller than 5 µm and they must show no propensity for forming aggregates, to ensure that the particles do not cause an embolism.20,21 NPs with a diameter of >100 nm are susceptible to phagocytic clearance by the reticuloendothelial system (RES), an essential component of the immune system located in different organs of the human body (eg, liver, spleen, and lymph nodes). In this context, the effective diameter must be >10 nm and <100 nm because particles with a diameter of <10 nm are secreted readily by the kidney.22–24 In addition to the size restriction by drug administration routes and to avoid urinary excretion, the optimum particle size will depend on the specific target organ (also called primary targeting) and the purpose of the delivery (Table 1). At this point, it is important to elucidate that studies may show a discrepancy for what the optimal recommended size is for delivery to a specific organ. This discrepancy may be due to the use of different delivery materials and diverse methods to determine the particle size with potentially limited accuracy.25 One of the most challenging organs to be targeted is the brain, due to the blood–brain barrier which consists of tightly bound endothelial cells forming a lining that blocks the entrance of a lot of molecules into the brain. However, in general, the optimum particle size to deliver a drug to specific organs (eg, brain, liver, and lymph nodes) is in the nanoscale range.26–28
 |
Table 1 The Nanoparticle Size Recommended For Intravenous Delivery To Specific Organs |
Exploiting The Enhanced Permeation And Retention Effect
Small molecule drugs can enter and exit tumors and normal tissues and typically do not accumulate in them. In contrast, nano-sized DDS have been proven to be able to cause drug accumulation in tumors through exploiting the dimensions of fenestrae in tumor blood vessels and the lack of a proper lymphatic system.29,30 This effect has been referred to as “enhanced permeation and retention effect” (EPR effect).31–33 The EPR effect in tumor tissues is the consequence of rapid tumor growth. Angiogenesis, the process of growing new blood vessels, is necessary to satisfy the increased oxygen and nutrient demand of fast-growing cancers. Angiogenesis is strictly necessary once the tumor reaches a size of just over 1 mm in diameter to allow for further growth and produces blood vessels with large fenestrae, ie, pores between the endothelial cells.34 The irregular gaps of the vasculature have been shown to be increased by vascular endothelial growth factor (VEGF) and nitric oxide in the endothelia.13,35,36 In consequence, NPs can penetrate through the fenestrae into the tumor tissue, whereas tight junctions between healthy endothelial cells do not allow such penetration.
The EPR effect is also being caused by inefficient lymphatic drainage in tumors due to the absence or malfunction of lymphatic vessels.24,37 This, in consequence, leads to a high interstitial pressure (10–50 mm Hg) and enhanced interstitial space retention of NPs in the tumor.38 The EPR effect (Figure 1A) was first reported by Matsumura and Maeda in 198639 and was validated by Maeda and coworkers.32 Matsumura and Maeda found that polymer-protein conjugates of approximately 80 kDa MW accumulated preferentially in tumor tissues.39 Therefore, due to their high molecular weight, macromolecules (>40 kDa) can be internalized and retained in the tumors by the EPR effect.40–42 Many studies show an increment in drug efficacy when the system is designed to utilize the EPR effect.27,31
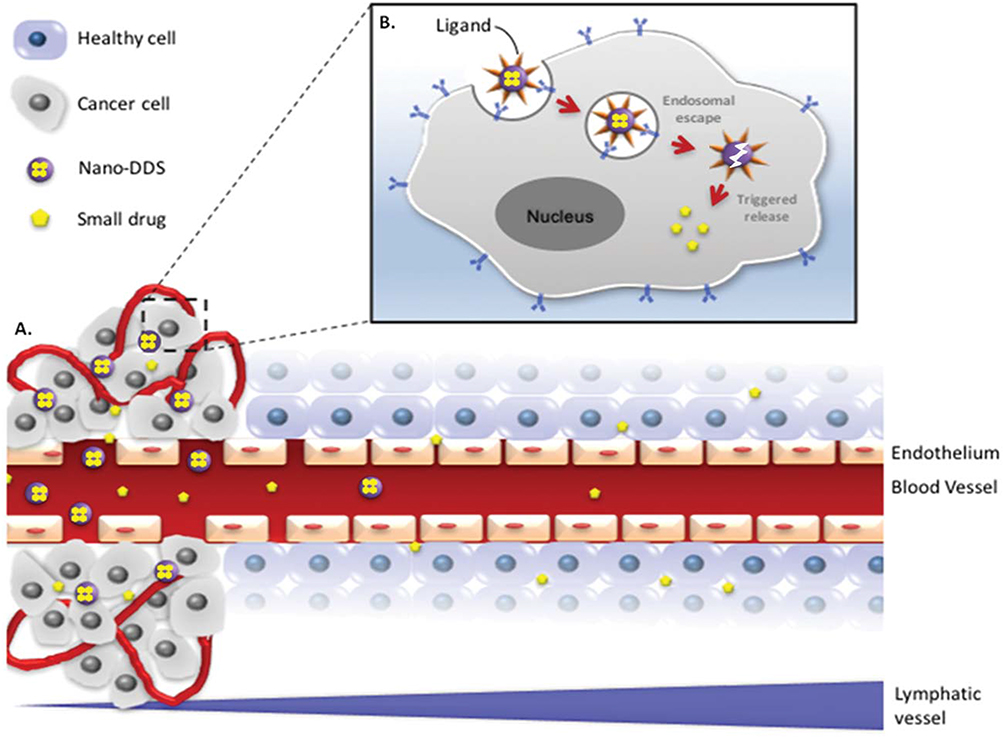 |
Figure 1 Scheme of (A) a free drug (eg, chemotherapy) versus encapsulated drug in a DDS for tumor delivery by passive targeting via the EPR effect and (B) active targeting using a ligand-mediated cellular internalization of the encapsulated drug via receptor-mediated endocytosis. The nanosize of well-designed DDS allows the drug to circulate for a longer period of time in the bloodstream to eventually extravasate and accumulate in the tumor tissue through “leaky” tumor vasculature. Decorating the nanocarriers with targeting ligands allows the specific binding to receptors overexpressed on tumor cells. |
In order to optimize the accumulation of nano-sized drugs in tumor tissues via the EPR effect, one must consider that the size of gap junctions between endothelial cells in tumor vasculature may vary from 100 to 800 nm25,29,40 unlike the tight gap junctions between endothelial cells of normal vessels (5–10 nm).43 The irregularity of “leaky” vasculature varies depending on cancer type, affected organ, and furthermore, from patient-to-patient.44 Usually, the unique pathophysiological characteristics of the tumor vasculature enable NPs of less than 400 nm to accumulate in the interstitial space in tumor tissues.17,29,31 Therefore, NPs can accumulate in tumor tissues and deposit anticancer agents passively in the interstitium or cytoplasm of tumor cells after cellular uptake.40,41,45 In contrast, conventional small anticancer drugs (<10 nm) and microparticles have a limited plasma half-life and do not take advantage of the EPR effect. The DDS size plays a significant role in passive tumor targeting in order to minimize or even overcome the toxic side effects of most current chemotherapies using small cytotoxic drugs. The basic idea is that due to the accumulation of nano-sized DDS in the tumor tissue, the local drug concentration can be higher in the tumor than in other body parts.36,40,41,46 Many scientists have designed particles in the range of 200 nm for optimum delivery to solid tumors.47–49
There are several factors which might limit the EPR effect in tumors. One of the most obvious limitations of passive targeting is that when small molecule drugs are released from DDS in cancer tissues, they can in principle diffuse out of the tumor area and interact with normal tissues. Therefore, it is important that DDS can address issues associated with the low target-site specificity. Ideally, cancer cells should do both: 1) take the DDS up preferentially and 2) the drug should only be released from the delivery device after cellular internalization. Strategies that accomplish this require much more intimate knowledge of the specific characteristics of the targeted cancer. In the next sections, we will discuss these more evolved strategies.
To date, hundreds of nano-sized DDS are being researched and tested (in vitro and in vivo) to improve therapeutic and diagnostic outcomes in different types of cancers. At least seven nanoparticle-based cancer therapeutic drugs are approved by the US FDA, four additional nano-sized drugs were approved in Europe and Asia, and there are other formulations in various stages in current clinical studies (Table 2).
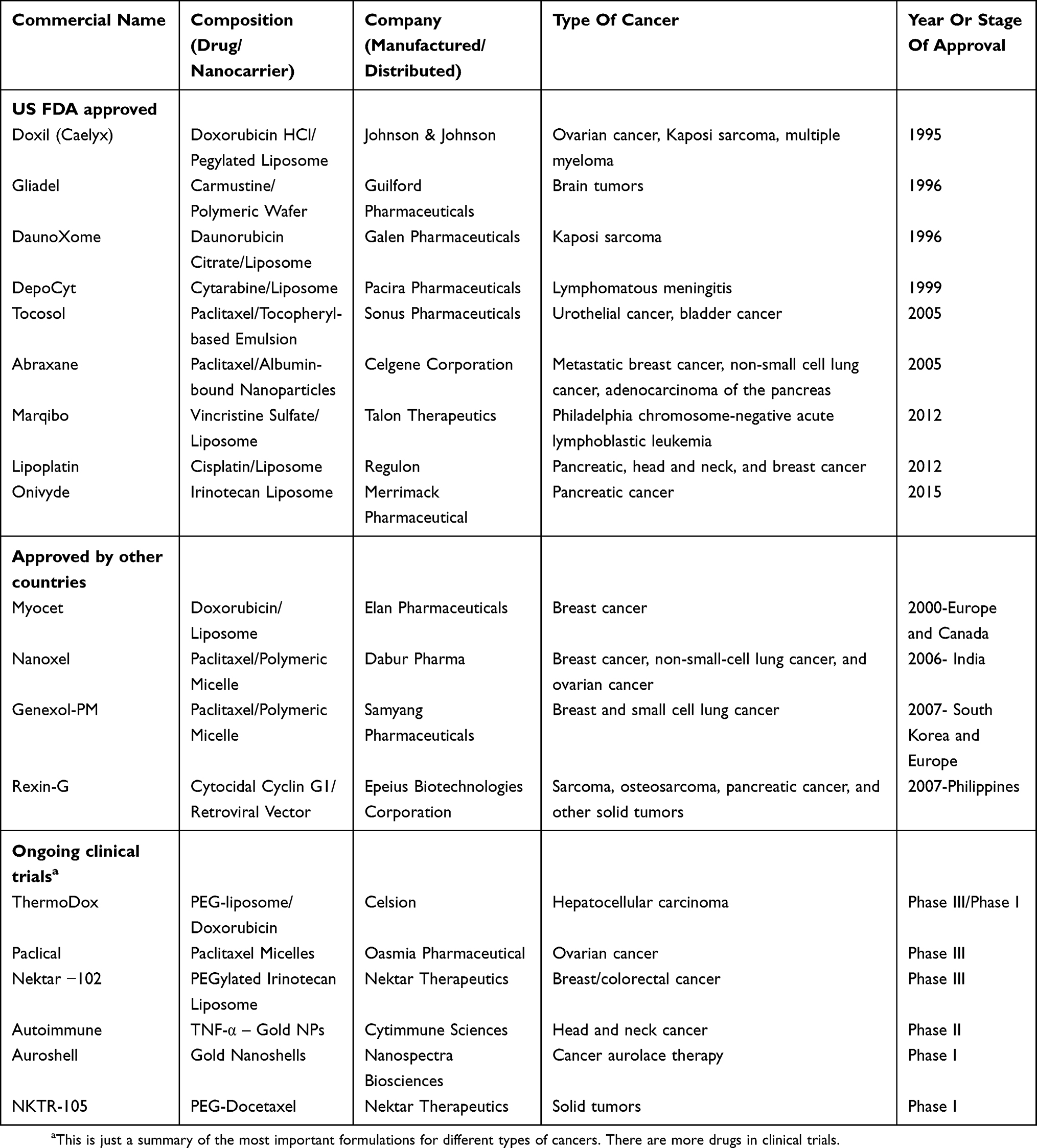 |
Table 2 Nanoparticle-Based Cancer Therapeutic Drugs For Clinical Use |
Drug delivery by exploiting the EPR effect is certainly an important starting point in improving drug efficiency and thus cancer treatment, but limitations remain with the approach. Foremost, the particle size is by far not the sole determinant of tumor specificity or lack thereof.33 There are literature data that suggest that surface charge is also an important factor to consider for DDS tumor accumulation and physiological interactions.50 Cell membranes have large negatively charged domains, which could explain the commonly reduced cellular adsorption by negatively charged nanoparticles and the effective cellular uptake of positively charged ones. Although a positively charged DDS should increase the therapeutic efficacy in vitro, molecules with a positive charge rapidly bind to vascular endothelial cells, which reduces the in vivo tumor drug accumulation.31 Likewise, highly negatively charged NPs have been shown to be taken up rapidly by the RES in the liver and spleen.42 Due to this, it is recommended to create neutral or slightly negatively charged NPs. In addition, the uptake by the target cell can be improved incorporating an active targeting modality (eg, antibodies, ligands, etc.) into the DDS; this approach will be discussed later in this review.
PEGylation As The Main Strategy For Increasing DDS Circulation Half-Life
To protect NPs from the RES and increase their circulation half-life, the most commonly used strategy is to conjugate poly-ethylene glycol (PEG) onto the nanoparticle surface to cover any undesired charge or surface properties.36 PEGylation is considered one of the best methods to stealth DDS of immunological responses.51–53 In addition, it has been shown that PEGs increase the solubility, size, molecular mass, and pharmacokinetic and pharmacodynamic properties of drugs.54–56 For example, Zhang et al encapsulated paclitaxel (PTX) into PEGylated polyphosphoester-based nanocarriers, increasing the aqueous drug solubility from <2.0 µg/mL to 4.8 mg/mL.57 In addition, Ma et al showed an increase in the therapeutic efficacy of recombinant human interleukin-11 mutein (mIL-11) when modified with PEG.55 A single-dose administration of PEGylated mIL-11 was comparable to the effect obtained with 7 to 10 consecutive daily administrations of native mIL-11.
PEG has been utilized to improve DDS based on NPs. Jain et al developed a targeted PEG-siRNA/peptide NP which demonstrated improved biophysical properties, including high serum stability and high cellular uptake.58 Zhao et al developed a biocompatible VEGF-targeted poly[bis(ε-Lys-PEI)Glut-PEG] (PLEGP) NP for triple-negative breast cancer. The authors determined that PLEGP-1800 was the most effective among others in the DDS construction leading to high cellular uptake, high tumor penetration, and downregulation of VEGF expression.59 This study showed how modulating the PEG and polymer content can influence the delivery outcome.
Due to the promising features displayed by PEGylation, some PEGylated protein drugs, summarized in Table 3, have received US FDA approval, including PEG-L-asparaginase (Oncaspar®), PEG-interferon (PEGIntron®), and PEG-granulocyte colony-stimulating factor (Neulasta®).
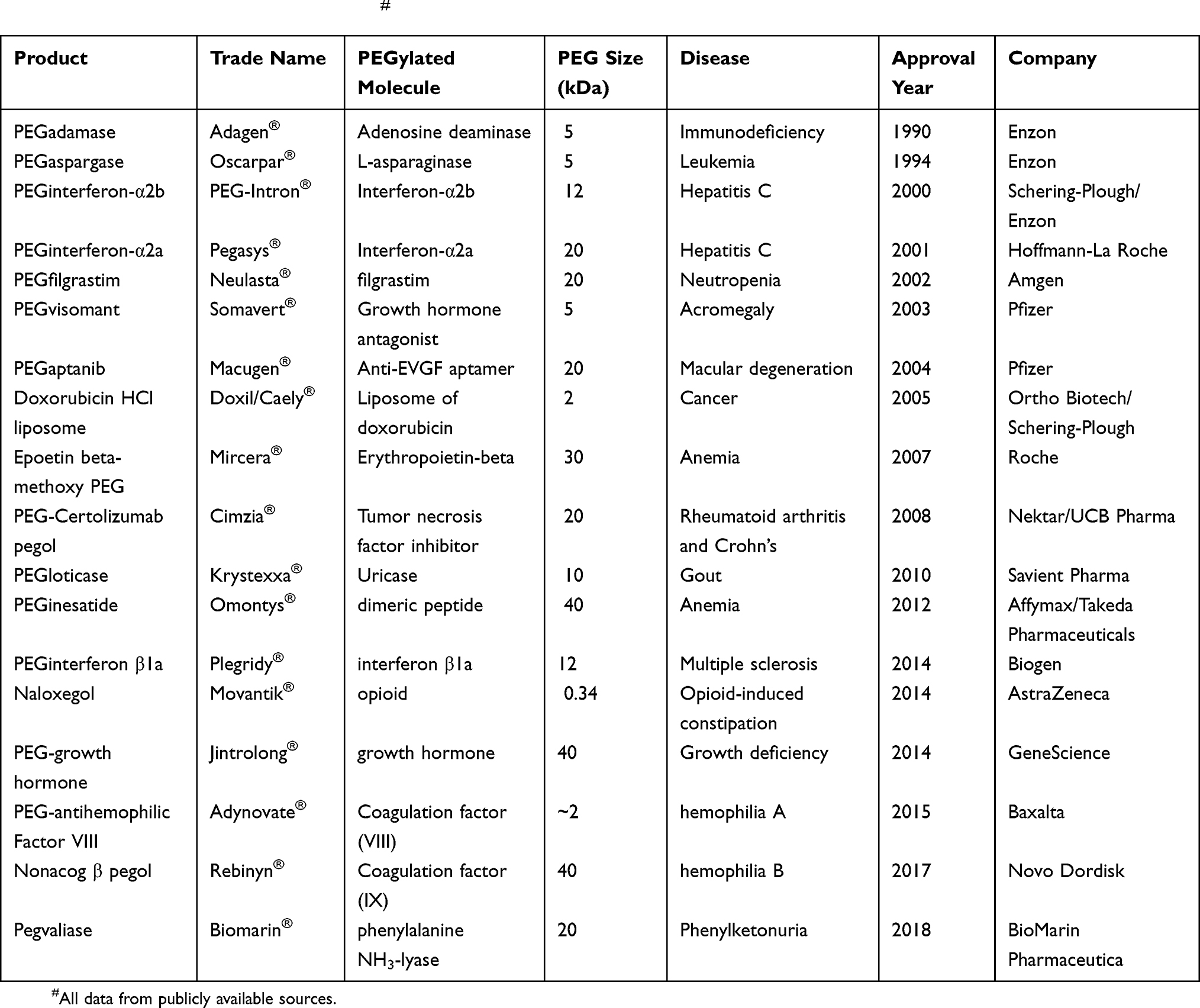 |
Table 3 FDA-Approved PEGylated Drugs# |
However, PEGylation can also impart certain disadvantages. In contrast to the general thought that PEGylation inhibits the opsonization process, some published studies have claimed that in their DDS, PEGylation actually accelerates the clearance.60,61 In another study, Mohamed et al recently reviewed different studies demonstrating an unanticipated immune response to PEGylated NPs after repeated administration and called this “the accelerated blood clearance (ABC) phenomenon”.62 In a very recent review, Shiraishi and Yokoyama analyzed different results from them and other researchers about the use of PEG and this ABC phenomenon. They concluded that PEG-liposomes and PEG-coupled to nonhuman enzymes easily exhibit the ABC phenomenon, whereas other PEG-NPs do not.63 These immunogenic responses could be deeply related to inherent characteristics of the polymer that encapsulate the drug in these PEG-conjugates than of the PEG itself. In addition, PEG-modified DDS sometimes display slower uptake into tumor cells which have coined the term “PEG dilemma”.64 To overcome this, Hatakeyama et al designed a gene delivery system modified with an enzymatically cleavable PEG-lipid which confers higher stability in systemic circulation and higher uptake in tumor cells.65 Thus, despite of some of the adverse effects PEGylation might cause, the PEG dilemma can be reduced by the using cleavable PEGs and by efficiently conjugating targeting moieties (eg, antibodies, ligands, etc.) to the PEGylated NPs,66,67 an approach referred to as active targeting discussed later in this review.
The reported disadvantages caused by protein or DDS PEGylation demonstrate the importance to construct and evaluate each system in the desired therapeutic application and optimize it. An improvement in the DDS efficacy in one specific application could cause a drawback in others depending on the core material used, the route of administration, its target, among others.
Active Targeting For Enhanced Tumor Cell Uptake By Steering Moieties
Tumor cells undergo rapid proliferation and uncontrolled cell growth due to their self-sufficiency in growth signals, insensitivity to anti-growth signals, apoptosis evasion, and sustained angiogenesis.68 All of these tumor hallmarks have been explored to target cancer cells. One example is the targeting of receptors overexpressed on tumor cells that respond to growth signals or facilitate nutrient uptake for DNA and protein synthesis. Such overexpressed receptors constitute excellent targets for ligands to afford docking and uptake of the delivery device by the targeted cells and thereby promoting intracellular delivery of anti-tumor agents.29,31,69 DDS are conjugated to targeting moieties to take advantage of ligand–receptor, antigen–antibody, or any other form of molecular recognition.70–72 The use of this strategy is called “active targeting”. Active tumor targeting (Figure 1B) results in more specific secondary targeting after primary targeting based on the EPR effect takes place (Figure 1A). It has been suggested that for active targeting to be effective, passive targeting must be attained first.73 While the EPR effect is considered a common phenomenon in many tumor tissues, the nature of the specific receptor overexpressed on a tumor cell is an event depending on the specific type of tumor.74–76 The moiety ligand selected (Figure 2) for the active targeting must be based on the specific cell surface properties, ie, membrane overexpressed targets of the tumor cell. To obtain an optimum and specific DDS involving active targeting, the selected targeting moiety (eg, ligand) must preferentially bind to the target receptor overexpressed by tumor cells and the target receptor must be homogeneously expressed on all target cells. Also, this targeted molecule on the membrane, ie, receptor, must be promptly recycled back on the cell membrane after uptake and mostly not released into circulation.
 |
Figure 2 From passive to active targeting by the attachment of steering molecules to the surface of the NPs for molecular recognition by cancer cell membrane. Abbreviations: FA, folic acid; HA, hyaluronic acid; Tf, transferrin; EGF, epidermal growth factor. |
It is relatively straightforward to investigate which receptors are overexpressed in a certain tumor by employing ligand-decorated chromophores for imaging.77 The same ligand can then be used to facilitate drug targeting to the tumor. Table 4 shows some examples for tumor-receptor specific delivery via small molecule ligands, such as folic acid (FA), sugar residues (eg, hyaluronic acid (HA)), peptides (eg, RGD), and proteins (eg, cytokines, lectins, and transferrin (Tf)). Other strategies include the release of a small therapeutic cargo in the tumor area or the reduction of the nanoparticle size mediated by some tumor microenvironment stimulus after nanoparticle accumulation near the blood vessels.78
 |
Table 4 Examples Of Ligands For Active Tumor Targeting |
Because active targeting can promote receptor-mediated endocytosis for increased cellular uptake, it could help overcome the reduced or slower uptake caused by PEGylation. PEGylation of DDS NPs employing carefully controlled conditions (eg, PEG chain length, shape, density, molecular weight) and incorporation of different targeting ligands are emerging as promising in DDS development for anticancer therapy.53,67,76,79,80 Bao et al designed a novel DDS consisting of the chemotherapeutic agent daunorubicin (DNR) loaded into poly(lactic-co-glycolic acid) (PLGA)-poly-L-lysine (PLL)-PEG-Tf NPs.81 Results showed that DNR-loaded NPs increased the intracellular concentration of DNR in K562 cells in vitro and induced a remarkable improvement in anticancer activity in vivo. Cruz et al covalently linked PEG molecules of various chain lengths (Mw 2,000 to 20,000 g/moL) to PLGA NP vaccines coated with various antibodies recognizing the dendritic cell-specific receptor DC-SIGN.82 These DDS were designed to study the effects of shielding on antibody–receptor interactions. Results demonstrate that binding and uptake of NPs by human DCs were affected by PEG chain length, showing that NPs coated with PEG-3,000 had the optimal chain length for antibody–receptor interactions and induction of antigen-specific T-cell responses. In another study, Su et al generated bispecific PEG-binding antibodies (PEG engagers) for targeted delivery of PEGylated nanomedicines to tumors.83 Their experiments demonstrate that pre-targeting of PEG engagers allowed subsequent accumulation and endocytosis of PEGylated nanocarriers in tumors, leading to enhanced antitumor efficacy of PEG-modified therapeutic agents in vitro and in vivo.
While ligand-selection requires specific knowledge of the type of receptor overexpressed, another type of macromolecules, aptamers, does not require such knowledge. Aptamers are nucleotides that bind to cell surface groups and can be selected from vast libraries and also allow for recursive optimization by mutational approaches.84 Similar to antibodies, they can be directed towards a wide range of targets and be useful for personalized medicine.85 While this potentially offers huge advantages, it is a tricky type of targeting because, in principle, for each patient, one or more aptamers have to be selected and it has to be assured that no unwanted cells are targeted. In how far this can be made clinically relevant remains to be seen.
It is important to mention that the aim to incorporate different targeting ligands on DDS is to increase the targetability to cancer cells, not cytotoxicity. Nevertheless, when the targetability is enhanced, this results in an increase of the intracellular drug concentration thus simultaneously enhancing therapeutic efficacy.86
Membrane Overexpressed Targets
Folic Acid Receptor (FAR)
Folic acid (FA) is one of the most frequently used tumor-targeting ligands in DDS because the FAR is a well-known tumor marker and the conjugation chemistry to therapeutic and imaging agents is well characterized.37,87–93 FA is important in the formation of new cells because it is required in one carbon metabolic reactions and consequently is essential for the synthesis of nucleotide bases.94 In fact, FARs are a family of 35–40 kDa glycoproteins which can be divided into four different isoforms (FARα, FARβ, FARγ, and FARδ). Very few places in the body express the FAR and these are typically not targeted with nanosized DDS.95 In contrast, FAR-α and FARβ have been overexpressed in lots of cancer cells, ie, ovarian, breast, lung, bladder, pancreas, prostate, colon, and kidney.96 Overall, 40% of the tumors showed FARα and 25% showed FARβ expression. In addition, FARβ is also highly expressed in activated myeloid cells of the immune system.97 This makes FA an attractive molecule to target cancer and inflammatory/autoimmune diseases. Due to the FAR overexpression in different cancer types, Morales-Cruz et al designed a smart nanosized DDS of cytochrome c modified with PEG-FA. They confirmed the FAR targeting, selective internalization, and cytotoxicity of the system on FAR-positive HeLa cells compared to FAR deficient cell lines. This DDS also showed great potential on an in vivo tumor model.98
Transferrin (Tf) Receptor
The transferrin receptor (TfR) is a cell-membrane-associated glycoprotein involved in the cellular uptake of holo-Tf (iron-bound to Tf) and the regulation of cell growth.99,100 Transferrin is a plasma protein responsible to transport iron to proliferating cells. Therefore, various studies have shown elevated levels of TfR particularly in lung, ovarian, colon, and brain cancer.101 Due to this, Tf is an important potential ligand in the active targeting of tumors. Thus, there are some promising results of TfR-targeted drugs against different types of tumors showing strong cytotoxic effects.37,102–104 For example, Saxena et al developed a DDS of a Tf-modified cytochrome c to target TfR on A549 lung cancer cells. The results suggest that this conjugate design would be effective in cancers with enhanced expression of TfR due to its nontoxic pattern to lung normal cells.105 These outcomes make TfR an important molecular target in the study of more effective cancer therapies.
Epidermal Growth Factor Receptor (EGFR)
The EGFR is a member of the family of tyrosine kinase receptors (also known as the HER family).106,107 In humans, more than 30 ligands have been identified that bind to EGFR, including EGF and EGF-like ligands, transforming growth factor (TGF)-α, and heregulins (HRGs, also known as neuregulins).74 EGFR is activated upon ligand-binding stimulating key processes involved in tumor growth and progression, including proliferation, differentiation, angiogenesis, cell motility, metastatic spread, and survival.74,107 EGFR is frequently overexpressed in many different cancers, including colon, lung, head, neck, ovarian, kidney, pancreatic, prostate, and especially breast cancer.108 Based on this, there are two classes of anti-EGFR agents that are currently approved for the treatment of cancer: monoclonal antibodies directed at the extracellular domain of the receptor and competitive inhibitors of its tyrosine kinase. Anti-EGFR and anti-VEGFR monoclonal antibodies are mostly employed to play the role of both, targeting ligand and drug (eg, Herceptin®).109 Recently, several drugs binding to EGFR have received FDA approval (ie, Kisqali® and Nerlynx®) (Table 5). Taking advantage of active targeting based on antigen-antibody recognition to promote the intracellular delivery of drugs, BESPONSA® has been designed and approved by the FDA in 2017 (Table 5). BESPONSA® (inotuzumab ozogamicin) is composed of a monoclonal antibody linked to the cytotoxic agent calicheamicin. Its mechanism of action is based on targeting of the CD22 antigen, a cell surface antigen expressed on cancer cells in almost all B-cell precursor acute lymphoblastic leukemia patients. Once attached to the CD22 antigen, the system is internalized by the cell and the cytotoxic agent released causing cell death. Additional examples in clinical trials of humanized anti-EGFR are necitumumab, zalutumumab, and nimotuzumab.37,110 In a preclinical in vivo study, Hartimath et al developed a PEGylated nimotuzumab DDS using maytansine as the drug. Their results showed higher cellular internalization due to the PEGylation versus the non-PEGylated system. However, the PEGylation has to be maintained at a low ratio (ie, ~1:3 nimotuzumab-to-PEG molar ratio) to avoid the disruption to the EGFR affinity.111
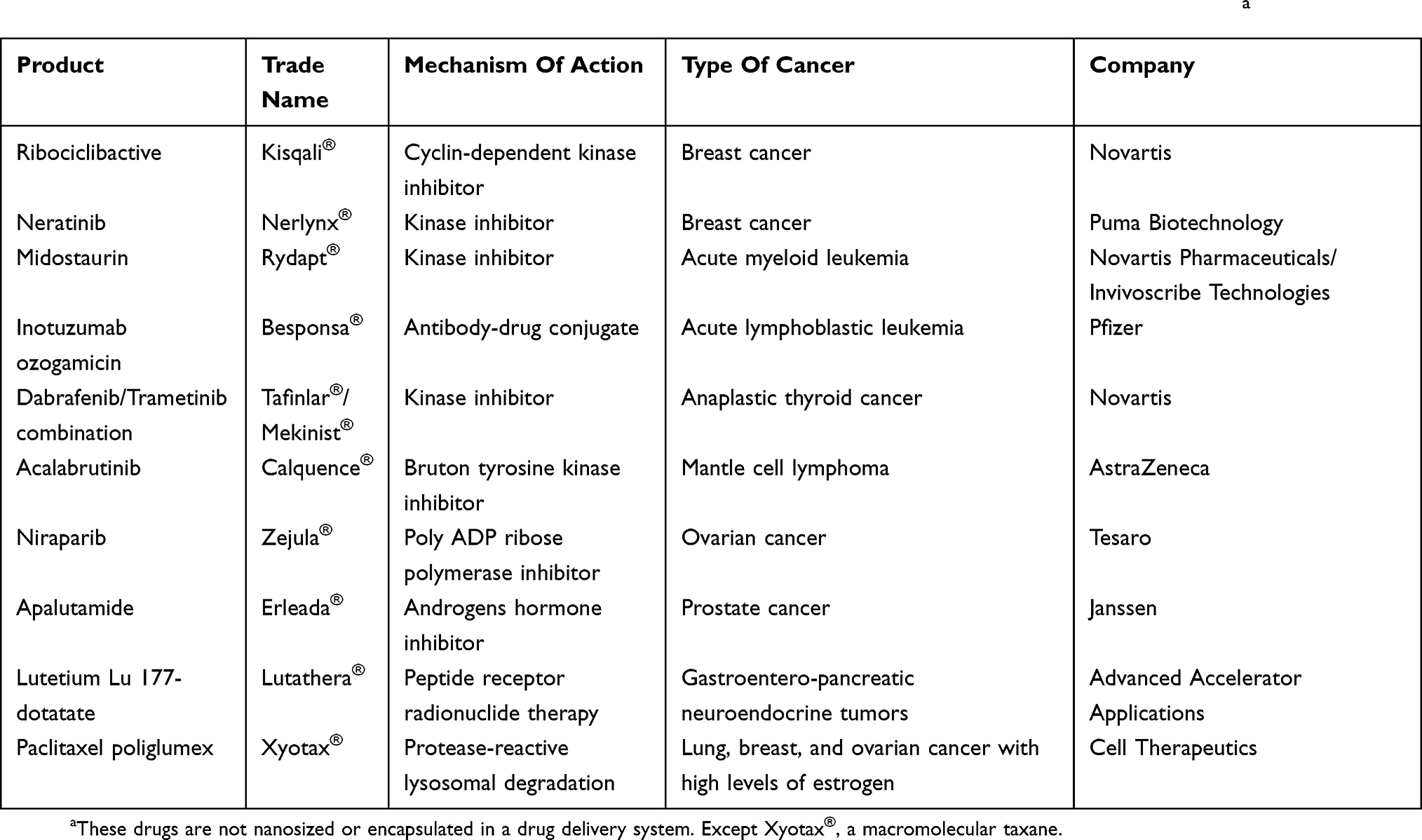 |
Table 5 Active-Targeted Drugs To Treat Different Cancer Types Approved By FDA In 2017 And 2018 (cancer.org)a |
Lectins
Lectins are proteins of non-immunological origin which preferentially recognize and bind certain carbohydrates attached to glycoproteins.112 Carbohydrate–lectin interaction-based systems have been developed to target cancer cells.37,40 The interaction of lectins with particular carbohydrates can be as specific as the interaction between those of antigen and antibody or substrate and enzyme. Therefore, lectins are used for the detection of glycoproteins on cell surfaces. Lectins mediate biological processes, such as tumor cell survival, adhesion to endothelium, or extracellular matrix, as well as tumor vascularization and other processes that are crucial for metastatic spread and growth.112 Cancer cells often express glycoproteins that are different to those of normal cells. For example, P-glycoprotein is widely expressed in many human cancers including those of the liver, pancreas, kidney, ovary, and breast.113 P-glycoprotein is one of the molecules responsible for the MDR syndrome in many types of cancer.113,114 However, if these glycoproteins could be specifically targeted, treatment would consequently be more effective. Shimomura et al conducted in vivo experiments and demonstrated how lectin could be used for active targeting and as a drug carrier in the treatment of pancreatic ductal adenocarcinoma.115
Hyaluronic Acid (HA) Receptor
HA is a linear, negatively charged polysaccharide, containing two alternating units of D-glucuronic acid and N-acetyl-D-glucosamine with a molecular weight range from 105 kDa up to 1 MDa. HA is distributed widely throughout connective, epithelial, and neural tissues. As one of the principal components of the extracellular matrix, HA is responsible for various functions within it, such as cell growth, differentiation, and migration.116 HA is the main ligand for the CD44 receptor, a multifunctional cell surface glycoprotein.117 It is well known that various tumors, such as epithelial, ovarian, colon, stomach, and acute leukemia, overexpress this receptor.118,119 Furthermore, CD44 expression is upregulated in important subpopulations of cancer cells and is recognized as a molecular marker for cancer stem cells.120,121 This makes HA a key molecule in the development of novel cancer therapies, due to there are no drugs able to kill cancer stem cells. Consequently, various anticancer therapeutics have been linked or modified with HA to increase their cellular uptake and therefore their pharmacokinetic effect on cancer cells.122,123 For example, Figueroa et al showed how pro-apoptotic protein-HA bioconjugates can reduce at least 3-fold the cell viability of CD44 overexpressing cancer cells versus normal cells.124 The ONCOFID™-S bioconjugate, which links the anticancer drug irinotecan and HA, showed a significant increment in drug efficacy in vitro and improved mice survival in ovarian cancer.125
Cell-Penetrating Peptides (CPP)
CPPs (also known as tumor-penetrating peptides) are a promising to target the nanocarriers to specific cells and penetrate cells. CPPs can be attached to the nanocarrier surface conferring the potential to translocate across the membrane via micropinocytosis.126,127 CPPs are normally short peptides of 1 to 5 kDa of less than 20 basic amino acids and can be attached to >30 kDa cargoes (eg, TAT).128 CPP-based drug strategies are considered a form of subcellular targeting due to preferential accumulation in the cell nuclei and sub-nuclear regions.129 It has been shown that CPPs are effectively internalized in lung, breast, and heart cancer cells.126–130 Hsieh et al synthesized a TAT-derived peptide that specifically targets the β-catenin signaling in breast cancer, resulting in cell growth inhibition.130 El-Sayed et al reported the conjugation of Paclitaxel and Camptothecin to a cyclic CPP containing tryptophan and arginine residues.131 Results revealed that the antiproliferative activities of the CPP-drug conjugates were less than that of the free hydrophobic drugs in the breast cancer cell line MCF-7 after 72-hr incubation. Lastly, the modification of DDS with CPP (eg, RGD peptide) made NPs capable of penetrating deeper into tumor tissue, resulting in several-fold higher therapeutic efficacy indices.71,72
Considering the targeting of CD44 receptor again, several peptides sharing sequence homology with the HA-binding domain of CD44 have been discovered for treatment and diagnosis of cancers.132–134 For example, it was found that the RP-1 peptide showed affinity and specificity to the HA-binding domain of CD44. The results demonstrate the potential of RP-1 peptide to detect gastric cancer.132,133 Furthermore, studies have shown that binding of A6-peptide to the HA-binding domain of CD44 results in the inhibition of migration, invasion, and metastasis of tumor cells, and the modulation of CD44-mediated cell signaling.134
Protein And Peptide-Based Therapeutics For Anticancer Targeting
In general, it has been shown that when DDS are modified with any of these ligands (eg, FA, HA, Tf, CPPs, and anti-EGFR) (Figure 2), therapeutic performances are improved in cellular and animal models when compared to their non-targeted counterpart. Furthermore, the use of proteins as drug carriers (as mentioned in the section “Passive targeting for enhanced drug tumor accumulation"), as targeted ligands (as discussed in the section “Membrane overexpressed targets"), or as therapeutic agents has had great impact in the development of nanomedicines for cancer therapies. The high chemical specificity, biodegradability, low toxicity, non-antigenicity, and non-mutagenic character of proteins and peptides make them attractive substitutes of small cytotoxic drugs.135,136 Highlighting this is the fact that since the early 1980s, a total of 239 therapeutic proteins and peptides have been approved for clinical use by US FDA.136,137
However, the potential of proteins has not been translated into successful clinical trials most of the time. At this point, Abraxane® is the only nanosized protein-based formulation currently approved by the FDA taking advantage of both, passive and active targeting mechanisms to treat cancer. Its mechanism of action is based on the high affinity of SA to the Gp60 receptor in endothelial cell walls.85 The use of human SA as drug carrier is based on the fact that SA is an important carrier of endogenous and exogenous hydrophobic molecules in the human circulation.138 Studies have demonstrated that the SA-bound paclitaxel formulation differs favorably from unbound paclitaxel in terms of tumor accumulation of SA, patient toleration, response rate, and extends the time to tumor progression.139 Besides Abraxane, an active-targeted NP, Rexin-G, is commercially available outside of the USA (Table 1) and there are other drug nanocarriers that incorporate an active targeting ligand in clinical trials (eg, HA-Irinotecan in phase III and RGD-PET-CT in phase II).71,140–142 Indeed, a small ligand-targeted therapeutic agent avoids deficiencies in tumor penetration sometimes shown by bigger nanoparticles (>100 nm) to cells deep within a tumor mass which can be dense and not well supported by blood vessels.79,143 Also, the anti‐metabolite methotrexate covalently bound to human SA has been evaluated, demonstrating its great potential as carrier for targeted delivery of antitumor drugs.144
Therapeutic peptides offer several advantages over protein-based therapeutics, such as less immunogenicity, lower manufacturing costs, and easier synthesis due to their simpler linear structures.145 A total of 60 peptide-based drugs are already on the market and several other therapeutic peptides are currently being evaluated in different phases of clinical trials.137
Furthermore, it has also been shown that peptides can be an alternative as drug delivery vehicles. Peptide–drug conjugates (PDCs), which are usually composed of the peptide, the small molecule drug, and a cleavable or non-cleavable linker, are an emerging technology that has resulted in many PDCs in the developmental phase of preclinical and clinical studies.146 PDCs have yet to get marketing approval from regulatory agencies.146 Zoptarelin doxorubicin, consisting of doxorubicin linked to a small peptide agonist to the luteinizing hormone-releasing hormone (LHRH) receptor, has been evaluated in phase 3 clinical trials in a number of human cancers.147. GRN1005 is a PDC composed of paclitaxel covalently linked to a peptide, angiopep-2, that targets the low-density lipoprotein receptor-related protein 1. This conjugate is in clinical trials for the treatment of advanced solid tumors in patients with brain metastases.148
A number of cancer-targeting PDCs, such as camptothecin–somatostatin conjugate,149 camptothecin–bombesin conjugate,150,151 H2009.1 peptide-polyglutamic acid–doxorubicin conjugate,152 cyclic RGD peptide–paclitaxel conjugate,153 RGD-4C peptide–doxorubicin conjugate,154,155 aptide-SN38 conjugate,156 and aptide-docetaxel conjugate,157 have been developed and are being explored in targeted cancer therapy. Tai et al prepared a PDC consisting of HER2-specific peptide conjugated to TGX-221, a potent inhibitor of PI3Kβ, a lipid enzyme that has been found to be over-activated in human cancers, such as prostate cancer.158 This PDC exhibited a much higher cellular uptake in prostate cancer cells in comparison to the parent drug, indicating its tremendous potential as a targeted therapy for prostate cancer patients.
Although PEGylation has been used to obtain a PDC with an extended half-life, their efficient tumor penetration and cost-effectiveness were adversely affected.159 Due to the need to develop a new approach that can significantly extend the half-life of PDCs while retaining the expected tumor-penetrating ability associated with the small size of PDCs, Kim et al studied the delivery of a PDC assisted by an antibody.160 They showed that the conjugation of a cotinine-labeled PDC with an anticotinine antibody resulted in a therapeutic formulation with a significantly extended circulation half-life in blood, greater penetration and accumulation within the tumor, and, ultimately, inhibition of tumor growth. It has been demonstrated that due to the low manufacturing costs, excellent cell permeability, and a high drug-loading capability exhibited by PDCs, their development is promising for the targeted delivery of chemotherapy drugs.
Stimulus-Responsive Targeting For Specific Release
An additional smart targeting strategy that takes advantage of specific characteristics of the chemical environments of the tumor interstitium or the cell interior consists in development of stimulus-responsive DDS. The DDS is designed to release or activate the drug only when triggered by specific internal or external stimuli161 (this is also known as tertiary targeting). Such stimuli include pH, proteases, redox potential, heat and light, among others (Figure 3).
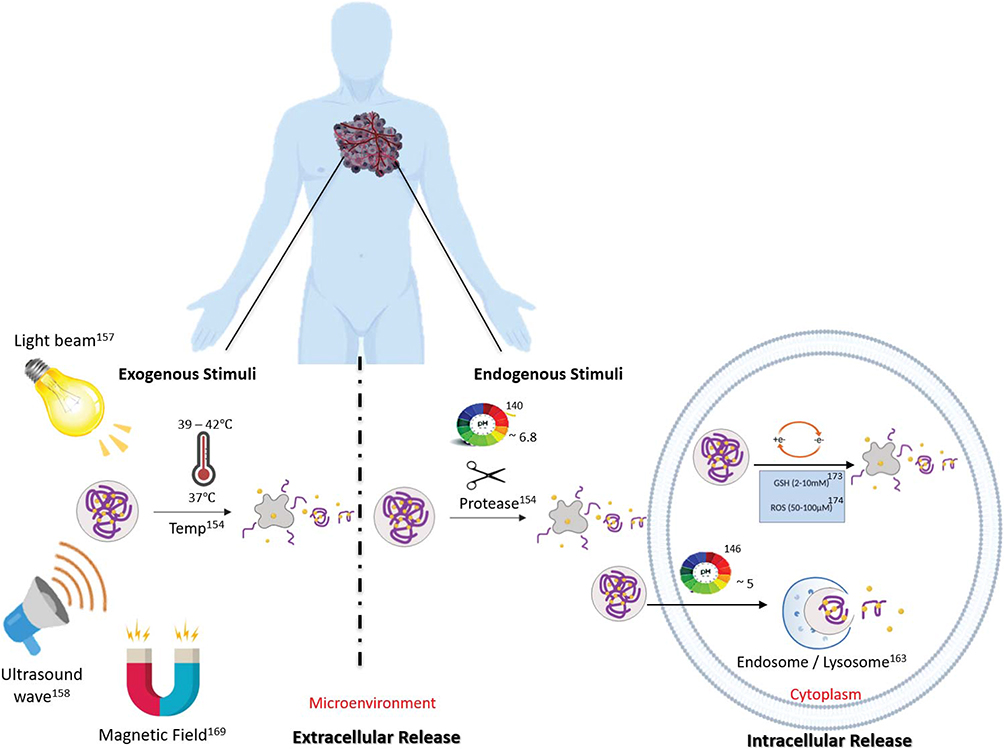 |
Figure 3 Scheme of the types of stimuli-responsive strategies utilized in the development of DDS. Drug release can in principle occur in the extracellular microenvironment or directly in the cell cytoplasm. |
Internal Stimuli
Since many cancer cells generate energy exclusively by glycolysis and are deprived of oxygen, the extracellular matrix of tumor sites frequently has a lower pH compared to normal tissues. The extracellular matrices of tumor sites frequently have an approximate pH range from 6.5 to 7.0, whereas in normal tissues and blood the typical pH is around 7.4.162,163 These circumstances of cell metabolism abnormalities and resulting low pH values are known as the Warburg effect.163,164 Exploiting the pH gradient between normal tissues and the tumor microenvironment to afford external drug release, and the low pH in cellular endosomes (~pH 5) to afford intracellular drug release, pH-sensitive NP systems have been designed to facilitate the release of anticancer drugs in a pH-controlled manner.165–168 One release mechanism is a change in the ionization of acidic or basic groups in the NP upon exposure to acidic environments. The resulting electrostatic repulsion causes the particle to swell, releasing the encapsulated drug. Studies published by Zhao et al showed that their designed PEGylated pH-sensitive NPs were long circulated in blood and highly phagocytosed by tumor cells.169 This delivery strategy resulted in several advantages including prolonged circulation, accumulation in tumors, highly efficient cellular internalization, and rapid intracellular drug release.170
The extracellular release of small drugs from DDS avoids some tumor penetration problems of nanosized DDS when tumors are very dense.24,170 However, the main problem with using extracellular stimuli is that non-specific drug leakage is likely to occur (ie, after being released small-molecule drugs can exit the tumor since they are not trapped in the interstitium). Intracellular drug release overcomes this drawback, but it is required that the DDS is taken up by the cancer cells, which typically is accomplished by decoration with receptor-targeting ligands as discussed earlier. FAR-targeted-doxorubicin-loaded polymeric micelles are one example of using intracellular stimuli once the DDS are endocytosed by the cells.167 The resulting endosome acidifies as it moves towards the lysosomes where the pH can be as low as 4. These DDS are optimized to release their drug only at a low pH. PH-sensitive nanocarriers can be constructed to be able to sense small changes in the microenvironment pH as well as the lower pH in endosomes or lysosomes.170 Zhou et al reported a pH-sensitive PEG-doxorubicin stable at pH 7.4 and activated at an endosomal acidic pH with increased circulation time and drug accumulation compared with free doxorubicin.171
Differences in the redox potential between extra- and intracellular spaces provide another opportunity for the development of a stimulus-responsive DDS. The cell cytoplasm is significantly more reducing than the outside of the cell, with a concentration of reducing species, mainly glutathione (GHS), as much as 1000 times higher.172,173 DDS that are redox-activated will exhibit intracellular drug release or activation.174 Therefore, they are only cytotoxic to the cells that internalize them. Redox-responsive DDS can be generated by using thiol-cleavable crosslinkers, which will be cleaved upon reduction by GHS. Such systems have been used to trigger the activation of pro-apoptotic proteins (eg, cytochrome c and caspase-3).98,175,176 Recently, a system composed of a Cyt c NPs stabilized by an amphiphilic copolymer through a redox-sensitive bond (FA-PEG-PLGA-S-S-Cyt c NPs) exhibited excellent stability under extracellular physiological conditions (1 µM GHS), whereas once in the intracellular reducing environment (10 mM GHS) Cyt c was released from the DDS.177 Also, smart release nanosized (passive targeting) DDS combining HA-based active and redox-responsive targeting can release up to 92% of its payload once experiencing the characteristic reductive conditions of the intracellular space. Once the NPs are internalized, they can reduce the cell viability by up to 80%.178
Another characteristic of the interstitium of tumors is that it is frequently rich in proteases (eg, membrane-type matrix metalloproteinase, MMP) and thus protease-sensitive DDS (eg, peptide-based DDS) have been developed as well. Proteases play a key role in many processes in the human body, such as cell cycle, tissue remodeling, and homeostasis. However, upregulated proteolysis is a typical hallmark of the tumor microenvironment.179 Graeser et al target the prostate-specific antigen, which is a protease overexpressed in prostate cancer, with a SA‐bound doxorubicin DDS. Then, they showed that the formulation releases the doxorubicin drug effectively due to the cleavage of the SA by the protease. Interestingly, they demonstrated that even when the SA‐bound doxorubicin DDS was ~100‐fold less active than the free doxorubicin in in vitro experiments, in an in vivo model of human prostate tumor, the DDS showed greater efficacy compared to the free doxorubicin.180 In the clinical context, Xyotax® is a pioneer protease-reactive drug in which its activity is dependent on the degradation of the poly-(L)-glutamic acid to release the paclitaxel drug.181
External Stimuli
External stimuli, such as heat, ultrasound, and light have also been exploited to afford the specific release or activation of drugs at a specific time and location. ThermoDox®, a heat-activated doxorubicin liposome, is an example of a stimulus-responsive nanomedicine in Phase III of clinical trials.182,183 In a recent study, the trial demonstrated that the ThermoDox®, designed to release targeted levels of doxorubicin into and around tumors with heat and ultrasound activation, increased doxorubicin delivery to tumors in the majority of liver cancer patients in a 10-patient trial.184
In addition, photodynamic therapy (PDT) is a novel noninvasive medical application that utilizes a photosensitizing agent (PS) to kill pathological cells. The PS is a chromophore normally derived from porphyrins which absorbs light and then produces reactive oxygen species (ROS) that kill targeted cells.183 After 1–2 days of PS administration, the tumor area is exposed to light to kill affected cells.185 This photosensitizing agent could take up to 48 to 96 hrs to successfully reach cancer cells and its body elimination up to 50 days.186 For example, patients treated with Photofrin® should avoid light for at least 1 month after administration and home equipment that emits heat (eg, hairdryer). Another limitation of this treatment is the unspecific accumulation of PS in healthy tissues near to pathological areas thus inducing adverse side effects, such as burns, swelling, and pain during treatment.186–188 To overcome this, the PS is conjugated to a DDS to increase the selectivity for tumor tissues over healthy tissues.189–191 Positive effects include accumulation of the PS in the tumor by the EPR effect and reduction of unwanted radical formation by the PS due to self-quenching prior to DDS disintegration.192,193 Internal and external stimuli can be combined within a single system to reduce self-quenching of the drug and produce the therapeutic effect preferentially at the tumor site.194
Conclusion
The present review discusses recent advances in smart-targeted delivery systems, including the use of NPs. Although stimulus-responsive strategies add tumor specificity to DDS, it is important to note that only a nanosized carrier increases the chances of the drug reaching the tumor tissue per se (See Figure 1). Both, active targeting and stimulus-responsive strategies are more efficient if the drug accumulates in the tumor tissue and is not distributed evenly throughout the body and/or eliminated from the bloodstream via renal or hepatic pathways. However, drug accumulation in the tumor tissue per se is not enough if the drug is released before reaching the target site or cannot enter the tumor cells. In summary, combining the use of passive targeting with additional targeting strategies such as active targeting and employing stimuli-responsive chemistry can potentially enhance the DDS selectivity towards cancer tissues and improve the overall therapeutic index.
Even though nanomedicine is a young science, it has demonstrated the potential to increase the therapeutic efficacy in cancer therapy when compared with the conventional chemo- and radiation-therapy by the exploitation of smart targeting. Passive targeting can significantly contribute to diminish toxic side effects by accumulation of the NPs in the tumor area taking advantage of the EPR effect. Active targeting takes advantage of different molecules overexpressed in the tumor cell to design selective NPs-based DDS that recognize the specific target. The use of stimulus-responsive DDS can prevent the premature systemic release of the drug by the design of a biologically stable NP until exposure to a specific stimulus in the tumor or cellular site. These relatively new potential nanomedicines expose the possible multifunctionality of DDS designed using different smart targeting strategies. In the future, multidrug-loaded delivery systems incorporating passive, active, and stimulus-responsive targeting, alone or in combination with chemotherapy will become a powerful tool. Carefully designed and optimized DDS will enhance the therapeutic index, reduce side effects, avoid drug resistance, and increase patient compliance and survival.
Abbreviations
ABC phenomenon, accelerated blood clearance; BBB, blood–brain barrier; CPP, cell-penetrating peptide; Cyt C, cytochrome c; MDRS, multidrug resistance syndrome; DDS, drug delivery system; DNR, daunorubicin; EGF, epidermal growth factor; EPR, enhanced permeation and retention effect; FA, folic acid; FAR, folic acid receptor; GHS, glutathione; HA, hyaluronic acid; HRG, heregulins; IL, interleukin; LHRH, luteinizing hormone-releasing hormone; MMP, membrane-type matrix metalloproteinase; NP, nanoparticle; PDC, peptide–drug conjugate; PDT, photodynamic therapy; PEG, poly(ethylene glycol); PLGA, poly(lactic-co-glycolic) acid; PLL, poly-L-lysine; PS, photosensitizing agent; PTX, paclitaxel; RES, reticuloendothelial system; RGD peptide, Arg-Gly-Asp peptide; RT, radiation therapy; ROS, reactive oxygen species; TGF, transforming growth factor; Tf, transferrin; TfR, transferrin receptor; SA, serum albumin; VEGF, vascular endothelial growth factor.
Acknowledgments
This publication was made possible by the support of the University of Puerto Rico Rio Piedras Campus and San Juan Bautista School of Medicine. The authors thank Dr. Estela Estapé for her outstanding dedication and support in the writing process of this review. Moraima Morales-Cruz and Yamixa Delgado are co-first authors.
Author Contributions
MMC and YD contributed equally in the design and selection of the topics for this article. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work. KG and YD coordinated and conceived the review article.
Disclosure
The authors report no conflict of interest in this work.
References
1. Urruticoechea A, Alemany R, Balart J, et al. Recent advances in cancer therapy: an overview. Curr Pharm Des. 2010;16:3–10. doi:10.2174/138161210789941847
2. Burstein HJ, Krilov L, Aragon-Ching JP, et al. Clinical cancer advances 2017: annual report on progress against cancer from the American Society of Clinical Oncology. J Clin Oncol. 2017;35:1341–1367.
3. Parkhill LA. Oral mucositis and stomatitis associated with conventional and targeted anticancer therapy. J Pharmacovigil. 2013;1:112. doi:10.4172/2329-6887.1000112
4. Musha A, Shimada H, Shirai K, et al. Prediction of acute radiation mucositis using an oral mucosal dose surface model in carbon ion radiotherapy for head and neck tumors. PLoS One. 2015;10:1–10.
5. Cheung-Ong K, Giaever G, Nislow C. DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol. 2013;20:648–659.
6. Pastorelli D, Soldá C. Chemotherapy-induced nausea and vomiting (CINV): the achilles heel of oncologists. Chemother. 2015;4:154.
7. Mondal J, Panigrahi AK, Khuda-Bukhsh AR. Conventional chemotherapy: problems and scope for combined therapies with certain herbal products and dietary supplements. Austin J Mol Cell Biol. 2014;1:1–10.
8. Cree IA, Charlton P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer. 2017;17:10.
9. Friberg S, Nyström AM. Nanomedicine: will it offer possibilities to overcome multiple drug resistance in cancer? J Nanobiotechnology. 2016;14:1–17.
10. Holohan C, Van Schaeybroeck S, Longley DB, et al. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi:10.1038/nrc3599
11. Housman G, Byler S, Heerboth S, et al. Drug resistance in cancer: an overview. Cancers. 2014;6:1769–1792. doi:10.3390/cancers6031769
12. Yuan Y, Cai T, Xia X, et al. Nanoparticle delivery of anticancer drugs overcomes multidrug resistance in breast cancer. Drug Deliv. 2016;23:3350–3357. doi:10.1080/10717544.2016.1178825
13. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33:941–951. doi:10.1038/nbt.3330
14. Ravi K. Nano and microparticles as controlled drug delivery devices. J Pharm Pharm Sci. 2000;3:234–258.
15. Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2010;75:1–18.
16. Guo D, Xie G, Luo J. Mechanical properties of nanoparticles: basics and applications. J Phys D Appl Phys. 2014;47:013001.
17. Kim KY. Nanotechnology platforms and physiological challenges for cancer therapeutics. Nanomed Nanotechnol Biol Med. 2007;3:103–110. doi:10.1016/j.nano.2006.12.002
18. Zhang L, Yang W, Hu C, Wang Q, Wu Y. Properties and applications of nanoparticle/microparticle conveyors with adjuvant characteristics suitable for oral vaccination. Dovepress. 2018;13:2973–2987.
19. Chan JM, Valencia PM, Zhang L, et al. Polymeric nanoparticles for drug delivery. Methods Mol Biol. 2010;624:163–175.
20. Arayne MS, Sultana N, Qureshi F. Nanoparticles in delivery of cardiovascular drugs. Pak J Pharm Sci. 2007;20:340–348.
21. Ilinskaya AN, Dobrovolskaia MA. Nanoparticles and the blood coagulation system. Part II: safety concerns. Nanomedicine. 2013;8:969–981.
22. Soo Choi H, Liu W, Misra P, et al. Renal clearance of quantum dots. Nat Biotechnol. 2007;25:1165–1170.
23. Chen ZG. Small-molecule delivery by nanoparticles for anticancer therapy. Trends Mol Med. 2010;16:594–602.
24. Morachis JM, Mahmoud EA, Almutairi A. Physical and chemical strategies for therapeutic delivery by using polymeric nanoparticles. Pharmacol Rev. 2012;64:505–519.
25. Schroeder A, Heller DA, Winslow MM, et al. Treating metastatic cancer with nanotechnology. Nat Rev Cancer. 2012;12:39–50.
26. Muzykantov V, Muro S. Targeting delivery of drugs in the vascular system. Int J Transp Phenom. 2011;12:41–49.
27. Greish K. Enhanced permeability and retention of macromolecular drugs in solid tumors: a royal gate for targeted anticancer nanomedicines. J Drug Target. 2007;15:457–464.
28. Simon LC, Sabliov CM. Time analysis of poly(lactic-co-glycolic) acid nanoparticle uptake by major organs following acute intravenous and oral administration in mice and rats. Ind Biotechnol. 2013;9:19–23.
29. Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63:131–135.
30. Upreti M, Jyoti A, Sethi P. Tumor microenvironment and nanotherapeutics. Transl Cancer Res. 2013;2:309–319.
31. Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63:136–151.
32. Maeda H, Tsukigawa K, Fang J. A retrospective 30 years after discovery of the enhanced permeability and retention effect of solid tumors: next-generation chemotherapeutics and photodynamic therapy—problems, solutions, and prospects. Microcirculation. 2016;23:173–182.
33. Maeda H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Deliv Rev. 2015;91:3–6.
34. Rajabi M, Mousa S. The role of angiogenesis in cancer treatment. Biomedicines. 2017;5(2):34.
35. Islam W, Fang J, Imamura T, et al. Augmentation of the enhanced permeability and retention effect with nitric oxide-generating agents improves the therapeutic effects of nanomedicines. Mol Cancer Ther. 2018;17:2643–2653.
36. Narang AS, Varia S. Role of tumor vascular architecture in drug delivery. Adv Drug Deliv Rev. 2011;63:640–658.
37. Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148:135–146.
38. Ranganathan R, Madanmohan S, Kesavan A, et al. Nanomedicine: towards development of patient-friendly drug-delivery systems for oncological applications. Int J Nanomedicine. 2012;7:1043–1060.
39. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–6392.
40. Cho K, Wang X, Nie S, et al. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14:1310–1316.
41. Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev. 2012;64:206–212.
42. Maeda H. Tumor-selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconjug Chem. 2010;21:797–802.
43. Haley B, Frenkel E. Nanoparticles for drug delivery in cancer treatment. Urol Oncol Semin Ori Invest. 2008;26:57–64.
44. Forster J, Harriss-Phillips W, Douglass M, et al. A review of the development of tumor vasculature and its effects on the tumor microenvironment. Hypoxia. 2017;5:21–32.
45. Alexis F, Pridgen EM, Langer R, et al. Nanoparticle technologies for cancer therapy. Handb Exp Pharmacol. 2010;197:55–86.
46. Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207.
47. Tang L, Yang X, Yin Q, et al. Investigating the optimal size of anticancer nanomedicine. Proc Natl Acad Sci. 2014;111:15344–15349.
48. Biswas AK, Islam MR, Choudhury ZS, et al. Nanotechnology based approaches in cancer therapeutics. Adv Nat Sci Nanosci Nanotechnol. 2014;5:4.
49. Kwon IK, Lee SC, Han B, et al. Analysis on the current status of targeted drug delivery to tumors. J Control Release. 2012;164:108–114.
50. Honary S, Zahir F. Effect of zeta potential on the properties of nano-drug delivery systems – a review (part 1). Trop J Pharm Res. 2013;12(2):255–264.
51. Rhame K, Dagher N. Chemistry routes for copolymer synthesis containing PEG for targeting, imaging, and drug delivery purposes. Pharmaceutics. 2019;11(7):327.
52. Shan NB, Vercellotti GM, White JG, et al. Blood-nanoparticle interactions and in vivo biodistribution: impact of surface PEG and ligand properties. Mol Pharm. 2012;9(8):2146–2155.
53. Cruje C, Chithrani BD. Integration of peptides for enhanced uptake of PEGylated gold nanoparticles. J Nanosci Nanotechnol. 2015;15(3):2125–2131.
54. Mishra P, Nayak B, Dey RK. PEGylation in anti-cancer therapy: an overview. Asian J Pharm Sci. 2016;11:337–348.
55. Ma SS, Ho SH, Ma SY, et al. The pharmacokinetic and pharmacodynamic properties of site-specific pegylated genetically modified recombinant human interleukin-11 in normal and thrombocytopenic monkeys. Eur J Pharm Biopharm. 2017;119:185–191.
56. Majtan T, Bublil EM, Park I, et al. Pharmacokinetics and pharmacodynamics of PEGylated truncated human cystathionine beta-synthase for treatment of homocystinuria. Life Sci. 2018;200:15–25.
57. Zhang F, Zhang S, Pollack SF, et al. Improving paclitaxel delivery: in vitro and in vivo characterization of PEGylated polyphosphoester-based nanocarriers. J Am Chem Soc. 2015;137:2056–2066.
58. Jain A, Barve A, Zhao Z, et al. Targeted delivery of an siRNA/PNA hybrid nanocomplex reserves carbon tetrachloride-induced liver fibrosis. Adv Ther. 2019;2:1900046.
59. Zhao Z, Li Y, Shukla R, et al. Development of a biocompatible copolymer nanocomplex to deliver VEGF siRNA for triple negative breast cancer. Theranostics. 2019;9(15):4508–4524.
60. Hatakeyama H, Akita H, Harashima H. The polyethylene glycol dilemma: advantage and disadvantage of PEGylation of liposomes for systemic genes and nucleic acids delivery to tumors. Biol Pharm Bull. 2013;36:892–899.
61. Verhoef JJF, Anchordoquy TJ. Questioning the use of PEGylation for drug delivery. Drug Deliv Transl Res. 2013;3:499–503.
62. Mohamed M, Abu A, Shimizu T, et al. PEGylated liposomes: immunological responses. Sci Technol Adv Mater. 2019;20(1):710–724.
63. Shiraishi K, Yokoyama M. Toxicity and immunogenicity concerns related to PEGylated-micelle carrier systems: a review. Sci Technol Adv Mater. 2019;20(1):324–336.
64. Fang Y, Xue J, Gao S, et al. Cleavable PEGylation: a strategy for overcoming the “PEG dilemma” in efficient drug delivery. Drug Deliv. 2017;24:22–32.
65. Hatakeyama H, Akita H, Ito E, et al. Systemic delivery of siRNA to tumors using a lipid nanoparticle containing a tumor-specific cleavable PEG-lipid. Biomaterials. 2007;32(18):4306–4316.
66. Diamantis N, Banerji U. Antibody-drug conjugates - An emerging class of cancer treatment. Br J Cancer. 2016;114:362–367.
67. Yao VJ, D’Angelo S, Butler KS, et al. Ligand-targeted theranostic nanomedicines against cancer. J Control Release. 2016;240:267–286.
68. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674.
69. Jaracz S, Chen J, Kuznetsova LV, et al. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg Med Chem. 2005;13:5043–5054.
70. Steichen SD, Caldorera-Moore M, Peppas NA. A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics. Eur J Pharm Sci. 2013;48:416–427.
71. Yang J, Yang J, Wei Y, et al. Modification of IL-24 by tumor penetrating peptide iRGD enhanced its antitumor efficacy against non-small cell lung cancer. Int Inmmunopharmacol. 2019;70:125–134.
72. Yu J, Sun L, Zhou J, et al. Self-assembled tumor-penetrating peptide-modified Poly(l-γ-glutamylglutamine)–paclitaxel nanoparticles based on hydrophobic interaction for the treatment of glioblastoma. Bioconjug Chem. 2017;28:2823–2831.
73. Rosenblum D, Joshi D, Tao W, et al. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9:1410.
74. Siwak DR, Carey M, Hennessy BT, et al. Targeting the epidermal growth factor receptor in epithelial ovarian cancer: current knowledge and future challenges. J Oncol. 2010;2010:1–20.
75. Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71:409–419.
76. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12:278–287.
77. Solomon M, Liu Y, Berezin MY, et al. Optical imaging in cancer research: basic principles, tumor detection, and therapeutic monitoring. Med Princ Pract. 2011;20:397–415.
78. Li HJ, Du JZ, Du XJ, et al. Stimuli-responsive clustered nanoparticles for improved tumor penetration and therapeutic efficacy. PNAS. 2016;113:4164–4169.
79. Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188:759–768.
80. Kolate A, Baradia D, Patil S, et al. PEG — A versatile conjugating ligand for drugs and drug delivery systems. J Control Release. 2014;192:67–81.
81. Bao W, Liu R, Wang Y, et al. PLGA-PLL-PEG-Tf-based targeted nanoparticles drug delivery system enhance antitumor efficacy via intrinsic apoptosis pathway. Int J Nanomedicine. 2015;10:557–566.
82. Cruz LJ, Tacken PJ, Fokkink R, et al. The influence of PEG chain length and targeting moiety on antibody-mediated delivery of nanoparticle vaccines to human dendritic cells. Biomaterials. 2011;32:6791–6803.
83. Su YC, Burnouf PA, Chuang KH, et al. Conditional internalization of PEGylated nanomedicines by PEG engagers for triple negative breast cancer therapy. Nat Commun. 2017;8:15507.
84. Sun H, Zu Y. Aptamers and their applications in nanomedicine. Small. 2015;11:2352–2364.
85. Pérez-Herrero E, Fernández-Medarde A. Advanced targeted therapies in cancer: drug nanocarriers, the future of chemotherapy. Eur J Pharm Biopharm. 2015;93:52–79.
86. Yoo J, Park C, Yi G, Lee D, Koo H. Active targeting strategies using biological ligands for nanoparticle drug delivery systems. Cancers. 2019;11(5):640.
87. Fernández M, Javaid F, Chudasama V. Advances in targeting the folate receptor in the treatment/imaging of cancers. Chem Sci. 2017;9:790–810.
88. Srinivasarao M, Galliford CV, Low PS. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat Rev Drug Discov. 2015;14:203–219.
89. Yang Y, Li N, Nie Y, et al. Folate-modified poly(malic acid) graft polymeric nanoparticles for targeted delivery of doxorubicin: synthesis, characterization and folate receptor expressed cell specificity. J Biomed Nanotechnol. 2015;11:1628–1639.
90. Xing L, Xu Y, Sun K, et al. Identification of a peptide for folate receptor alpha by phage display and its tumor targeting activity in ovary cancer xenograft. Sci Rep. 2018;8:8426.
91. Cal P, Frade R, Chudasama V, et al. Targeting cancer cells with folic acid-iminoboronate fluorescent conjugates. Chem Commun. 2014;50:5261–5263.
92. Assaraf Y, Leamon C, Reddy J. The folate receptor as a rational therapeutic target for personalized cancer treatment. Drug Resist Updat. 2014;17:89–95.
93. Fernández M, Javaid F, Chudasama V. Advances in targeting the folate receptor in the treatment/imaging of cancers. Chem Sci. 2018;4:790–810.
94. Zwicke GL, Ali Mansoori GA, Jeffery CJ. Utilizing the folate receptor for active targeting of cancer nanotherapeutics. Nano Rev. 2012;3:18496.
95. Shi H, Guo J, Li C, et al. A current review of folate receptor alpha as a potential tumor target in non-small-cell lung cancer. Drug Des Devel Ther. 2015;9:4989–4996.
96. Shen J, Hu Y, Putt KS, et al. Assessment of folate receptor alpha and beta expression in selection of lung and pancreatic cancer patients for receptor targeted therapies. Oncotarget. 2018;9:4485–4495.
97. Shen J, Putt KS, Visscher DW, et al. Assessment of folate receptor-beta expression in human neoplastic tissues. Oncotarget. 2015;6:14700–14709.
98. Morales-Cruz M, Cruz-Montañez A, Figueroa CM, et al. Combining stimulus-triggered release and active targeting strategies improves cytotoxicity of cytochrome c nanoparticles in tumor cells. Mol Pharm. 2016;13:2844–2854.
99. Kawamoto M, Horibe T, Kohno M, et al. A novel transferrin receptor-targeted hybrid peptide disintegrates cancer cell membrane to induce rapid killing of cancer cells. BMC Cancer. 2011;11:359.
100. Daniels TR, Bernabeu E, Rodríguez JA, et al. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim Biophys Acta Gen Subj. 2012;1820(3):291–317. doi:10.1016/j.bbagen.2011.07.016
101. Shen Y, Li X, Dong D, et al. Transferrin receptor 1 in cancer: a new sight for cancer therapy. Am J Cancer Res. 2018;8:916–931.
102. Shimosaki S, Nakahata S, Ichikawa T, et al. Development of a complete human IgG monoclonal antibody to transferrin receptor 1 targeted for adult T-cell leukemia/lymphoma. Biochem Biophys Res Commun. 2017;485:144–151. doi:10.1016/j.bbrc.2017.02.039
103. Daniels-Wells T, Widney D, Leoh L, et al. Efficacy of an anti-transferrin receptor 1 antibody against AIDS-related non-hodgkin lymphoma: a brief communication. J Immunother. 2015;38:307–310. doi:10.1097/CJI.0000000000000092
104. Leoh L, Kim YK, Candelaria P, et al. Efficacy and mechanism of antitumor activity of an antibody targeting transferrin receptor 1 in mouse models of human multiple myeloma. J Immunol. 2018;200:3485–3494. doi:10.4049/jimmunol.1700787
105. Saxena M, Delgado Y, Sharma RK, et al. Inducing cell death in vitro in cancer cells by targeted delivery of cytochrome c via a transferrin conjugate. PLoS One. 2018;13:e0195542. doi:10.1371/journal.pone.0195542
106. Heinemann V, Stintzing S, Kirchner T, et al. Clinical relevance of EGFR- and KRAS-status in colorectal cancer patients treated with monoclonal antibodies directed against the EGFR. Cancer Treat Rev. 2009;35:262–271. doi:10.1016/j.ctrv.2008.11.005
107. Creixell M, Bohórquez A, Torres-Lugo M, et al. EGFR-targeted magnetic nanoparticle heaters kill cancer cells without a perceptible temperature rise. ACS Nano. 2011;5:7124–7129. doi:10.1021/nn202666w
108. Thorne AH, Zanca C, Furnari F. Epidermal growth factor receptor targeting and challenges in glioblastoma. Neuro Oncol. 2016;18:914–918. doi:10.1093/neuonc/nov319
109. Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol. 2006;33:369–385. doi:10.1053/j.seminoncol.2006.04.003
110. Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immuno therapy. Nat Rev Immunol. 2010;10:317–327. doi:10.1038/nri2744.
111. Hartimath SV, Alizadeh E, Solomon VR, et al. Preclinical evaluation of 111 in-labeled PEGylated maytansine nimotuzumab drug conjugates in EGFR-positive cancer models. J Nucl Med. 2019;17:118.
112. Poiroux G, Barre A, Van Damme EJM, et al. Plant lectins targeting O-glycans at the cell surface as tools for cancer diagnosis, prognosis and therapy. Int J Mol Sci. 2017;18:1232. doi:10.3390/ijms18061232
113. Binkhathlan Z, Lavasanifar A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: current status and future perspectives. Curr Cancer Drug Targets. 2013;13:326–346. doi:10.2174/15680096113139990076
114. Nobili S, Landini I, Mazzei T, et al. Overcoming tumor multidrug resistance using drugs able to evade P-glycoprotein or to exploit its expression. Med Res Rev. 2012;32:1220–1262. doi:10.1002/med.20239
115. Shimomura O, Oda T, Tateno H, et al. A novel therapeutic strategy for pancreatic cancer: targeting cell surface glycan using rBC2LC-N lectin-drug conjugate (LDC). Mol Cancer Ther. 2018;17:183–195. doi:10.1158/1535-7163.MCT-17-0232
116. Sherman L, Sleeman J, Herrlich P, et al. Hyaluronate receptors: key players in growth, differentiation, migration and tumor progression. Curr Opin Cell Biol. 1994;6:726–733. doi:10.1016/0955-0674(94)90100-7
117. Misra S, Hascall VC, Markwald RR, Ghatak S. Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front Immunol. 2015;6:201.
118. De la Motte CA, Drazba JA. Viewing hyaluronan: imaging contributes to imagining new roles for this amazing matrix polymer. J Histochem Cytochem. 2011;59(3):252–257. doi:10.1369/0022155411413817
119. Shigeishi H, Higashikawa K, Takechi M. Role of receptor for hyaluronan-mediated motility (RHAMM) in human head and neck cancers. J Cancer Res Clin Oncol. 2014;140:1629–1640. doi:10.1007/s00432-014-1653-z
120. Chen C, Zhao S, Karnad A, Freeman JW. The biology and role of CD44 in cancer progression: therapeutic implications. J Hematol Oncol. 2018;11(1):64. doi:10.1186/s13045-018-0605-5
121. Yin T, Wang G, He S, et al. Human cancer cells with stem cell-like phenotype exhibit enhanced sensitivity to the cytotoxicity of IL-2 and IL-15 activated natural killer cells. Cell Immunol. 2016;300:41–45. doi:10.1016/j.cellimm.2015.11.009
122. Cadete A, Alonso MJ. Targeting cancer with hyaluronic acid-based nanocarriers: recent advances and translational perspectives. Nanomedicine. 2016;11:2341–2357. doi:10.2217/nnm-2016-0233
123. Edelman R, Assaraf YG, Levitzky I, et al. Hyaluronic acid-serum albumin conjugate-based nanoparticles for targeted cancer therapy. Oncotarget. 2017;8:24337–24353. doi:10.18632/oncotarget.15363
124. Figueroa CM, Morales-Cruz M, Surarez BN, et al. Induction of cancer cell death by hyaluronic acid-mediated uptake of cytochrome c. J Nanomed Nanotechnol. 2015;6:316.
125. Montagner IM, Merlo A, Carpanese D, et al. Drug conjugation to hyaluronan widens therapeutic indications for ovarian cancer. Oncoscience. 2015;2:373–381. doi:10.18632/oncoscience.150
126. Raucher D, Ryu JS. Cell-penetrating peptides: strategies for anticancer treatment. Trends Mol Med. 2015;21:560–570. doi:10.1016/j.molmed.2015.06.005
127. Feni L, Neundorf I. The current role of cell-penetrating peptides in cancer therapy. Adv Exp Med Biol. 2017;1030:279–295.
128. Taha A, Selma MH, Jawad A, et al. Improvement of cancer therapy by TAT peptide conjugated gold nanoparticles. J Clust Sci. 2019;30(2):403–414. doi:10.1007/s10876-019-01497-9
129. Gronewold A, Horn M, Neundorf I. Design and biological characterization of novel cell-penetrating peptides preferentially targeting cell nuclei and subnuclear regions. Beilstein J Org Chem. 2018;14:1378–1388. doi:10.3762/bjoc.14.116
130. Hsieh TH, Hsu CY, Tsai CF, et al. A novel cell-penetrating peptide suppresses breast tumorigenesis by inhibiting β-catenin/LEF-1 signaling. Sci Rep. 2016;6:19156. doi:10.1038/srep19156
131. El-Sayed NS, Shizari AN, Sajid MI, et al. Synthesis and antiproliferative activities of conjugates of paclitaxel and camptothecin with a cyclic cell-penetrating peptide. Molecules. 2019;24(7):E1427. doi:10.3390/molecules24071427
132. Li W, Jia H, Wang J, et al. A CD44-specific peptide, RP-1, exhibits capacities of assisting diagnosis and predicting prognosis of gastric cancer. Oncotarget. 2017;8:30063–30076.
133. Zhang D, Jia H, Wang Y, et al. A CD44 specific peptide developed by phage display for targeting gastric cancer. Biotechnol Lett. 2015;37(11):2311–2320. doi:10.1007/s10529-015-1896-z
134. Finlayson M. Modulation of CD44 activity by A6-peptide. Front Immunol. 2015;6:135. doi:10.3389/fimmu.2015.00135
135. Nitta SK, Numata K. Biopolymer-based nanoparticles for drug/gene delivery and tissue engineering. Int J Mol Sci. 2013;14:1629–1654. doi:10.3390/ijms14011629
136. Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today. 2015;20(1):122–128. doi:10.1016/j.drudis.2014.10.003
137. Usmani SS, Bedi G, Samuel JS, et al. THPdb: database of FDA-approved peptide and protein therapeutics. PLoS One. 2017;12:e0181748. doi:10.1371/journal.pone.0181748
138. Tao C, Chuah YJ, Xu C, et al. Albumin conjugates and assemblies as versatile bio-functional additives and carriers for biomedical applications. J Mater Chem B. 2019;7:357–367. doi:10.1039/c9tb00665f
139. Gradishar WJ, Krasnojon D, Cheporov S, et al. Albumin-bound paclitaxel (ab-pac) versus docetaxel for first-line treatment of metastatic breast cancer (MBC): final overall survival (OS) analysis of a randomized phase II trial. J Clin Oncol. 2017;29(27):275. doi:10.1200/jco.2011.29.27_suppl.275
140. ClinicalTrials.gov. Trial of FOLF(HA)Iri versus FOLFIRI in mCRC (FOLF(HA)iri). Available from: http://clinicaltrials.gov/show/NCT01290783. Accessed June 30, 2019.
141. Gibbs P, Clingan PR, Ganju V, et al. Hyaluronan-irinotecan improves progression-free survival in 5-fluorouracil refractory patients with metastatic colorectal cancer: a randomized phase II trial. Cancer Chemother Pharmacol. 2011;67:153–163. doi:10.1007/s00280-010-1303-3
142. Gordon E, Hall F. Rexin-G, a targeted genetic medicine for cancer. Expert Opin Biol Ther. 2010;10:819–832. doi:10.1517/14712598.2010.481666
143. Waite CL, Roth CM. Nanoscale drug delivery systems for enhanced drug penetration into solid tumors: current progress and opportunities. Crit Rev Biomed Eng. 2012;40:21–41. doi:10.1615/CritRevBiomedEng.v40.i1
144. Burger AM, Hartung G, Stehle G, et al. Pre-clinical evaluation of a methotrexate-albumin conjugate (MTX-HSA) in human tumor xenografts in vivo. Int J Cancer. 2001;92:718–724. doi:10.1002/1097-0215(20010601)92:5<718::aid-ijc1257>3.0.co;2-d
145. Kaur K, Singh I, Kaur P, et al. Food and drug administration (FDA) approved peptide drugs. Asian J Res Biol Pharm Sci. 2015;3:75–88.
146. Ma L, Wang C, He Z, et al. Peptide-drug conjugate: a novel drug design approach. Curr Med Chem. 2017;25:3373–3396.
147. Lau JL, Dunn MK. Therapeutic peptides: historical perspectives, current development trends, and future directions. Bioorg Med Chem. 2018;26:2700–2707. doi:10.1016/j.bmc.2017.06.052
148. Kurzrock R, Gabrail N, Chandhasin C, et al. Safety, pharmacokinetics, and activity of GRN1005, a novel conjugate of angiopep-2, a peptide facilitating brain penetration, and paclitaxel, in patients with advanced solid tumors. Mol Cancer Ther. 2012;11:308–316. doi:10.1158/1535-7163.MCT-11-0824-T
149. Sun L-C, Mackey LV, Luo J, et al. Targeted chemotherapy using a cytotoxic somatostatin conjugate to inhibit tumor growth and metastasis in nude mice. Clin Med Oncol. 2008;2:491–499.
150. Moody T, Mantey S, Pradhan T, et al. Development of high affinity camptothecin-bombesin conjugates that have targeted cytotoxicity for bombesin receptor-containing tumor. J Biol Chem. 2004;279(22):23580−23589. doi:10.1074/jbc.M401938200
151. Moody T, Sun L, Mantey S, et al. In vitro and in vivo antitumor effects of cytotoxic camptothecin-bombesin conjugates are mediated by specific interaction with cellular bombesin. J Pharmacol Exp Ther. 2006;318(3):1265−1272. doi:10.1124/jpet.106.104141
152. Guan H, McGuire MJ, Li S, et al. Peptide-targeted polyglutamic acid doxorubicin conjugates for the treatment of αvβ6-positive cancers. Bioconjug Chem. 2008;19:1813–1821. doi:10.1021/bc800154f
153. Chen X, Plasencia C, Hou Y, Neamati N. Synthesis and biological evaluation of dimeric RGD peptide-paclitaxel conjugate as a model for integrin-targeted drug delivery. J Med Chem. 2005;48:1098–1106. doi:10.1021/jm049494r
154. Kim JW, Lee HS. Tumor targeting by doxorubicin-RGD-4C peptide conjugate in an orthotopic mouse hepatoma model. Int J Mol Med. 2004;14:529–535.
155. Burkhart DJ, Kalet BT, Coleman MP, Post GC, Koch TH. Doxorubicin-formaldehyde conjugates targeting alphavbeta3 integrin. Mol Cancer Ther. 2004;3(12):1593–1604.
156. Kim H, Lee Y, Kang S, et al. Self-assembled nanoparticles comprising aptide-SN38 conjugates for use in targeted cancer therapy. Nanotechnology. 2016;27(48). doi:10.1088/0957-4484/27/36/365202.
157. Kim H, Lee Y, Lee IH, et al. Synthesis and therapeutic evaluation of an aptide-docetaxel conjugate targeting tumor-associated fibronectin. J Control Release. 2014;178:118−124. doi:10.1016/j.jconrel.2014.01.015
158. Tai W, Shukla RS, Qin B, et al. Development of a peptide-drug conjugate for prostate cancer therapy. Mol Pharm. 2011;8:901–912. doi:10.1021/mp200125j
159. Hamley IW. PEG-peptide conjugates. Biomacromolecules. 2014;15(5):1543–1559. doi:10.1021/bm500246w
160. Kim H, Hwang D, Choi M, et al. Antibody-assisted delivery of a peptide-drug conjugate for targeted cancer therapy. Mol Pharm. 2019;16(1):165–172. doi:10.1021/acs.molpharmaceut.9b00422
161. Raza A, Rasheed T, Nabeel F, Hayat U, Bilal M, Iqbal H. Endogenous and exogenous stimuli-responsive drug delivery systems for programmed site-specific release. Molecules. 2019;24(6):1117. doi:10.3390/molecules24061117
162. Justus CR, Dong L, Yang LV. Acidic tumor microenvironment and pH-sensing G protein-coupled receptors. Front Physiol. 2013;4:354. doi:10.3389/fphys.2013.00354
163. Barar J, Omidi Y. Dysregulated pH in tumor microenvironment checkmates cancer therapy. Bioimpacts. 2013;3(4):149–162. doi:10.5681/bi.2013.036
164. Kato Y, Ozawa S, Miyamoto C, et al. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013;13:89. doi:10.1186/1475-2867-13-89
165. Dalela M, Shrivastav TG, Kharbanda S, Singh H. pH-sensitive biocompatible nanoparticles of paclitaxel-conjugated poly(styrene-co-maleic acid) for anticancer drug delivery in solid tumors of syngeneic mice. ACS Appl Mater Interfaces. 2015;7:26530–26548. doi:10.1021/acsami.5b07764
166. Ding HM, Ma YQ. Controlling cellular uptake of nanoparticles with pH-sensitive polymers. Sci Rep. 2013;3:2804. doi:10.1038/srep02804
167. Kim D, Lee ES, Park K, et al. Doxorubicin loaded pH-sensitive micelle: antitumoral efficacy against ovarian A2780/DOXR tumor. Pharm Res. 2008;25:2074–2082. doi:10.1007/s11095-007-9382-5
168. Chuang CH, Wu PC, Tsai TH, et al. Development of pH-sensitive cationic PEGylated solid lipid nanoparticles for selective cancer-targeted therapy. J Biomed Nanotechnol. 2017;13(2):192–203. doi:10.1166/jbn.2017.2338
169. Zhao G, Long L, Zhang L, et al. Smart pH-sensitive nanoassemblies with cleavable PEGylation for tumor targeted drug delivery. Sci Rep. 2017;7:3383. doi:10.1038/s41598-017-03111-2
170. Liu Y, Wang W, Yang J, et al. Sun J. pH-sensitive polymeric micelles triggered drug release for extracellular and intracellular drug targeting delivery. Asian J Pharm Sci. 2013;8:159–167. doi:10.1016/j.ajps.2013.07.021
171. Zhou L, Cheng R, Tao H, et al. Endosomal pH-activatable poly(ethylene oxide)-graft-doxorubicin prodrugs: synthesis, drug release, and biodistribution in tumor-bearing mice. Biomacromolecules. 2011;15(5):1460–1467. doi:10.1021/bm101340u
172. Lushchak VI. Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids. 2012;2012:1–26. doi:10.1155/2012/736837
173. Mortera R, Vivero-Escoto J, Slowing II, et al. Cell-induced intracellular controlled release of membrane impermeable cysteine from a mesoporous silica nanoparticle-based drug delivery system. Chem Commun. 2009;22:3219–3221. doi:10.1039/b900559e
174. Hong R, Han G, Fernández JM, et al. Glutathione-mediated delivery and release using monolayer protected nanoparticle carriers. J Am Chem Soc. 2006;128:1078–1079. doi:10.1021/ja056726i
175. Méndez J, Monteagudo A, Griebenow K. Stimulus-responsive controlled release system by covalent immobilization of an enzyme into mesoporous silica nanoparticles. Bioconjug Chem. 2012;23:698–704. doi:10.1021/bc200301a
176. Zhao M, Biswas A, Hu B, et al. Redox-responsive nanocapsules for intracellular protein delivery. Biomaterials. 2011;32:5223–5230. doi:10.1016/j.biomaterials.2011.03.060
177. Morales-Cruz M, Figueroa CM, González-Robles T, et al. Activation of caspase-dependent apoptosis by intracellular delivery of cytochrome c-based nanoparticles. J Nanobiotechnol. 2014;12:33. doi:10.1186/s12951-014-0033-9
178. Figueroa CM, Suárez BN, Molina AM, et al. Smart release nano-formulation of cytochrome C and hyaluronic acid induces apoptosis in cancer cells. J Nanomed Nanotechnol. 2017;8:427.
179. Choi KY, Swierczewska M, Lee S, et al. Protease-activated drug development. Theranostics. 2012;2:156–179. doi:10.7150/thno.4068
180. Graeser R, Chung D, Esser N, et al. Synthesis and biological evaluation of an albumin-binding prodrug of doxorubicin that is cleaved by prostate-specific antigen (PSA) in a PSA-positive orthotopic prostate carcinoma model (LNCaP). Int J Cancer. 2008;122:1145–1154.
181. Chipman S, Oldham F, Pezzoni G, et al. Biological and clinical characterization of paclitaxel poliglumex (PPX, CT-2103), a macromolecular polymer-drug conjugate. Int J Nanomedicine. 2006;1:375–383.
182. Pillai G. Nanomedicines for cancer therapy: an update of FDA approved and those under various stages of development. SOJ Pharm Pharm Sci. 2014;1:1–13.
183. Kwiatkowski S, Knap B, Przystupski D, et al. Photodynamic therapy - mechanisms, photosensitizers and combinations. Biomed Pharmacother. 2018;106:1098–1107.
184. Gray MD, Lyon PC, Mannaris C, et al. Focused ultrasound hyperthermia for targeted drug release from thermosensitive liposomes: results from a phase I trial. Radiology. 2019;291:232–238.
185. Agostinis P, Berg K, Cengel KA, et al. Photodynamic therapy of cancer: an update. CA Cancer J Clin. 2011;61:250–281.
186. Dos Santos AF, Queiroz de Almeida DR, Terra LF, et al. Photodynamic therapy in cancer treatment - an update review. J Cancer Metastasis Treat. 2019;5:25.
187. Van Straten D, Mashayekhi V, De Bruijn HS, et al. Oncologic photodynamic therapy: basic principles, current clinical status and future directions. Cancers. 2017;9(2):19.
188. Hong EJ, Choi DG, Shim MS. Targeted and effective photodynamic therapy for cancer using functionalized nanomaterials. Acta Pharm Sin B. 2016;6:297–307.
189. Liang P, Huang K, Tung C, et al. A novel photodynamic therapy-based drug delivery system layered on a stent for treating cholangiocarcinoma. Biomed Microdevices. 2018;20:3.
190. Alves L, Ferreira L, Pacheco P, et al. Pore forming channels as a drug delivery system for photodynamic therapy in cancer associated with nanoscintillators. Oncotarget. 2018;9:25342–25354.
191. Calixto G, Bernegossi J, De Freitas L, et al. Nanotechnology-based drug delivery systems for photodynamic therapy of cancer: A review. Molecules. 2016;21:342.
192. Molina AM, Morales-Cruz M, Cindy M, et al. Function as delivery system for photodynamic cancer therapy. J Nanomed Naotechnol. 2016;6:1–20.
193. Tu J, Wang T, Shi W, et al. Multifunctional ZnPc-loaded mesoporous silica nanoparticles for enhancement of photodynamic therapy efficacy by endolysosomal escape. Biomaterials. 2012;33:7903–7914.
194. Molina AM, Morales-Cruz M, Benítez M, et al. Redox-sensitive cross-linking enhances albumin nanoparticle function as delivery system for photodynamic cancer therapy. J Nanomed Nanotechnol. 2016;3.
195. Veiseh O, Sun C, Fang C, et al. Specific targeting of brain tumors with an optical/magnetic resonance imaging nanoprobe across the blood-brain barrier. Cancer Res. 2009;69:6200–6207.
196. Dong X. Current strategies for brain drug delivery. Theranostics. 2018;8:1481–1493.
197. Longmire M, Choyke PL, Kobayashi H. Clearance properties of nano-sized particles and molecules as imaging agents: considerations and caveats. Nanomedicine. 2008;3:703–717.
198. Azarmi S, Roa WH, Löbenberg R. Targeted delivery of nanoparticles for the treatment of lung diseases. Adv Drug Deliv Rev. 2008;60:863–875.
199. Rijt SH, Bein T, Meiners S. Medical nanoparticles for next generation drug delivery to the lungs. Eur Resp J. 2014;44:765–774.
200. Perry JL, Reuter KG, Luft JC, et al. Mediating passive tumor accumulation through particle size, tumor type, and location. Nano Lett. 2017;17:2879–2886.
201. Yoshida M, Takimoto R, Murase K, et al. Targeting anticancer drug delivery to pancreatic cancer cells using a fucose-bound nanoparticle approach. PLoS One. 2012;7:e39545.
202. Guarneri V, Frassoldati A, Bottini A, et al. Preoperative chemotherapy plus trastuzumab, lapatinib, or both in human epidermal growth factor receptor 2-positive operable breast cancer: results of the randomized phase II CHER-LOB study. J Clin Oncol. 2012;30:1989–1995.
203. Stish BJ, Chen H, Shu Y, et al. A bispecific recombinant cytotoxin (DTEGF13) targeting human interleukin-13 and epidermal growth factor receptors in a mouse xenograft model of prostate cancer. Clin Cancer Res. 2007;13:6486–6493.
204. Goldstein R, Hanley C, Morris J, et al. Clinical investigation of the role of interleukin-4 and interleukin-13 in the evolution of prostate cancer. Cancers (Basel). 2011;3(4):4281–4293.
205. Yau T, Dan X, Ng CC, et al. Lectins with potential for anti-cancer therapy. Molecules. 2015;20:3791–3810.
206. Luria-Pérez R, Helguera G, Rodríguez JA. Antibody-mediated targeting of the transferrin receptor in cancer cells. Bol Med Hosp Infant Mex. 2016;73:372–379.
207. Bazak R, Houri M, El Achy S, et al. Cancer active targeting by nanoparticles: a comprehensive review of literature. J Cancer Res Clin Oncol. 2015;141:769–784.
208. Padró T, Bieker R, Ruiz S, et al. Overexpression of vascular endothelial growth factor (VEGF) and its cellular receptor KDR (VEGFR-2) in the bone marrow of patients with acute myeloid leukemia. Leukemia. 2002;16:1302–1310.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.