Back to Journals » Vascular Health and Risk Management » Volume 15
Small vessel disease to subcortical dementia: a dynamic model, which interfaces aging, cholinergic dysregulation and the neurovascular unit
Authors Caruso P, Signori R, Moretti R ![]()
Received 11 October 2018
Accepted for publication 14 January 2019
Published 7 August 2019 Volume 2019:15 Pages 259—281
DOI https://doi.org/10.2147/VHRM.S190470
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Harry Struijker-Boudier
Paola Caruso, Riccardo Signori, Rita Moretti
Department of Medical, Surgical and Health Sciences, Neurology Clinic, University of Trieste, Trieste, Italy
Background: Small vessels have the pivotal role for the brain’s autoregulation. The arteriosclerosis-dependent alteration of the brain perfusion is one of the major determinants in small vessel disease. Endothelium distress can potentiate the flow dysregulation and lead to subcortical vascular dementia (sVAD). sVAD increases morbidity and disability. Epidemiological studies have shown that sVAD shares with cerebrovascular disease most of the common risk factors. The molecular basis of this pathology remains controversial.
Purpose: To detect the possible mechanisms between small vessel disease and sVAD, giving a broad vision on the topic, including pathological aspects, clinical and laboratory findings, metabolic process and cholinergic dysfunction.
Methods: We searched MEDLINE using different search terms (“vascular dementia”, “subcortical vascular dementia”, “small vessel disease”, “cholinergic afferents”, etc). Publications were selected from the past 20 years. Searches were extended to Embase, Cochrane Library, and LILIACS databases. All searches were done from January 1, 1998 up to January 31, 2018.
Results: A total of 560 studies showed up, and appropriate studies were included. Associations between traditional vascular risk factors have been isolated. We remarked that SVD and white matter abnormalities are seen frequently with aging and also that vascular and endothelium changes are related with age; the changes can be accelerated by different vascular risk factors. Vascular function changes can be heavily influenced by genetic and epigenetic factors.
Conclusion: Small vessel disease and the related dementia are two pathologies that deserve attention for their relevance and impact in clinical practice. Hypertension might be a historical problem for SVD and SVAD, but low pressure might be even more dangerous; CBF regional selective decrease seems to be a critical factor for small vessel disease-related dementia. In those patients, endothelium damage is a super-imposed condition. Several issues are still debatable, and more research is needed.
Keywords: subcortical vascular dementia, vascular damage, small vessel disease, brain’s autoregulation
Introduction: traditional perspectives and recently acquired information
Many authors have widely questioned vascular damage for its role in causing dementia. Consensus criteria for the clinical diagnosis of the significant dementing disorders have recently been updated, both for Alzheimer Disease (AD), Parkinson disease dementia (PDD) and Lewy Body Disease (DBL), Frontotemporal Lobe Degeneration (FTLD).1–5 Previous diagnostic criteria for vascular dementia (VaD) required the presence of memory loss and the severity of cognitive impairment sufficient to adversely affect independent functioning consistent with dementia. Despite multiple attempts, no generally accepted morphologic substrates have been included in the currently used clinical diagnostic criteria for VaD.6,7
With time, different diagnostic guidelines for VaD have been described. The commonly used are the National Insititute of Neurological Disorders and Stroke Association International pour la Recherche et l’Enseignement en Neurosciences (NINDS-AIREN) guideline.8 For completion of probable vascular Dementia, all should be present:
- Dementia as impairment of memory and any other 2 or more cognitive domains, preferably established by clinical examination and documented by the neuropsychological test.
- The presence of cerebrovascular diseases as the presence of focal neurological signs and evidence of the same in the brain imaging.
- A relationship between dementia and cerebrovascular disease as one or both:
- The onset of dementia within three months following a recognized stroke;
- Abrupt deterioration on cognitive functions or fluctuation, stepwise progression of cognitive deficits.
For subcortical ischemic vascular dementia, the brain imaging would show either the Binswanger-type white matter lesions or lacunar infarcts with the absence of cortical non-lacunar territorial infarct and watershed infarcts, and other causes of white matter lesions. The temporal relationship may also be absent.8
The International Classification of disease, 10th revision (ICD-10) includes diagnostic features:
- General criteria for dementia must be met;
- Unequal distribution of deficits in higher cognitive functions, with some affected and others, relatively spared;
- Focal brain damage as at least one of the following (Unilateral spastic weakness of the limbs, unilaterally increased tendon reflexes, extensor plantar response, pseudobulbar palsy);
- Evidence from the history, examination or test of significant cerebrovascular disease, etiologically related to dementia.9
ICD-10 distinguished between:
- vascular dementia of acute onset: as it usually develops within one month, but within no longer than three months after a succession of strokes, rarely after a single broad stroke;
- multi-infarct dementia: which onset is usually gradual (within three to six months, following many minor ischemic episodes).
- Subcortical vascular dementia: with a history of hypertension and clinical examination, along with investigations showing vascular disease to be located in the deep white matter of the cerebral hemispheres, with preservation of the cerebral cortex.9
A more complex pathological description (mainly discussed below) has been employed, extending the clinical diagnosis to strategic infarct, hypoxic-ischemic dementia, venous infarct dementia, and hemorrhagic dementia.10,11
Recently, the Diagnostic and Statistical Manual of Mental disorders (5th Edition) redefined some aspects of clinical classification, and all the dementias are called Neurocognitive disorders.12 Here, the probable vascular neurocognitive disorder is described by:
- Criteria met for the major or mild neurocognitive disorder.
- Clinical features are consistent with a vascular etiology, as either the following:
- the onset of the cognitive deficits is temporally related to one or more vascular events;
- Evidence for a decline is prominent in elaborate attention (including processing speed) and frontal executive functions;
- Evidence of the presence of cerebrovascular disease from history, physical examination and neuroimaging, considering sufficient to account for neurocognitive deficits.
Possible vascular neurocognitive disorders should be diagnosed as:
- Clinical criteria supported by neuroimaging evidence of significant parenchymal injury attributed to the cerebrovascular disease.
- The neurocognitive syndrome is temporally related to one or more documented cerebrovascular events.
- Both clinical and genetic evidence of cerebrovascular disease is present.13,14
The most recent position summarizes the Standards for reporting Vascular changes on nEuroimaging (STRIVE) for a recommended standard for research with MRI, but also with CT,15 completed a structured process to develop a common advisory about terms and definitions for features visible on MRI and minimum standards for image acquisition. Signs of small vessel disease include a conventional MRI recent subcortical infarcts, white matter hyperintensities, lacunes, prominent perivascular spaces, and cerebral microbleeds, with possible consequent atrophy.
Vascular cognitive alterations are described as due to vascular dementia (VaD).16
Other include vascular cognitive disorder (VCD), while subcortical VAD (sVAD) has been employed to define a circumscribed syndrome, related to small vessel disease.17–21 Being so difficult to categorize the different vascular subtypes of dementia, today it is widely accepted to define the cognitive impairment related to vascular dementia as Vascular Cognitive Impairment (VCI).22
VCI refers to a vast and almost comprehensive spectrum of vascular brain pathologies that contribute to cognitive impairment, ranging from mild and subjective cognitive decline to overt dementia.23
The difference between VaD subtypes may depend on the anatomical distribution of the vascular insults. Usually, small artery disease is more often associated with subcortical VaD than with cortical and cortical-subcortical VaD. Subcortical VaD is homogeneous, with the small-vessel disease as the primary vascular etiology, lacunar infarcts, and ischemic white matter lesions.24
Cerebral small vessel disease (SVD) is an umbrella term covering a variety of abnormalities related to small blood vessels in the brain. SVD results from damage to the small penetrating arteries and arterioles in the brain, (9) the prevalence of SVD increases exponentially with aging: from approximately 6% to 7% at age 60 to 28% at age 80.25
Cerebral small vessel disease (SVD) is a common cause of stroke, cognitive impairment, and gait disturbances, which slowly progress to vascular Parkinsonism and dementia but can be differentiated from Alzheimer’s disease considering neuroimaging findings (it is associated with more extensive white matter lesions, less severe hippocampal atrophy and the absence of cerebral amyloid angiopathy).26,27
Substantial evidence shows that small vessel disease shares many risk factors and prognostic impacts in specific clinical settings.21 Moreover, inducing an increased blood pressure and hyperglycemia, the presence of small vessel disease confers a risk of hemorrhagic transformation in brain ischemia in 10% of cases.28–33
In the clinical practice, small vessel disease is generally associated with the typical vascular “static” risk factors, such as hypertension, hyperlipidemia, diabetes, obesity, atherosclerosis, atrial fibrillation, and insufficient physical aerobic activity.34–43 Vascular Dementia increases the morbidity, disability, and healthcare costs of the growing elderly population, decreasing their quality of life.44–46 Its prevention and treatment are critical research and clinical priorities.
Neuropsychological pattern profiles of sVaD are distinguished by poor executive function, poor planning, working memory alterations, loss of inhibition, reduced mental flexibility, multitasking procedures invalidation, and decrease speed of executive process.47 Neuroimaging standards implement the clinical definition of subcortical vascular dementia, which relies on cerebral small vessel disease. Different studies have been realized with this aim.48
Our works purposes were to detect the possible mechanisms between small vessel disease and sVaD, giving a broad vision on the topic, including pathological aspects, clinical and laboratory finding, metabolic processes and cholinergic dysfunction, pointing out the dynamic process that links small vessel disease to subcortical dementia, describing some crucial measures able to delay or even interrupt this process.
Search strategy and selection criteria
We searched MEDLINE using the search terms “vascular dementia,” “subcortical vascular dementia,” “vascular cognitive impairment,” “small vessel disease,” “cholinergic afferents,” “arteriolosclerosis” “cerebral flow regulation,” “neurovascular coupling,” “endothelium,” smooth muscle cells arteries".
Publications were selected mostly form the past 20 years, but did not exclude frequently referenced and highly regarded older publications. Researchers have been extended to EMBASE, with the same strings, to COCHRANE LIBRARY, to LILACS. All searches were done from January 1st, 1998 up to January 31st, 2018.
We have considered papers published in English, French, German and Italian. Secondary searching was performed using the bibliography of the most relevant articles (following PRISMA statement, 2009). Congress abstracts and isolated case reports were not considered. We searched the reference lists of articles identified by this search strategy and selected those we judged relevantly. Review articles and book chapters are cited to provide with additional details.
A total of 560 studies showed up, and appropriate studies (n=306) were included. The authors carefully read all the eligible articles (Figure 1).
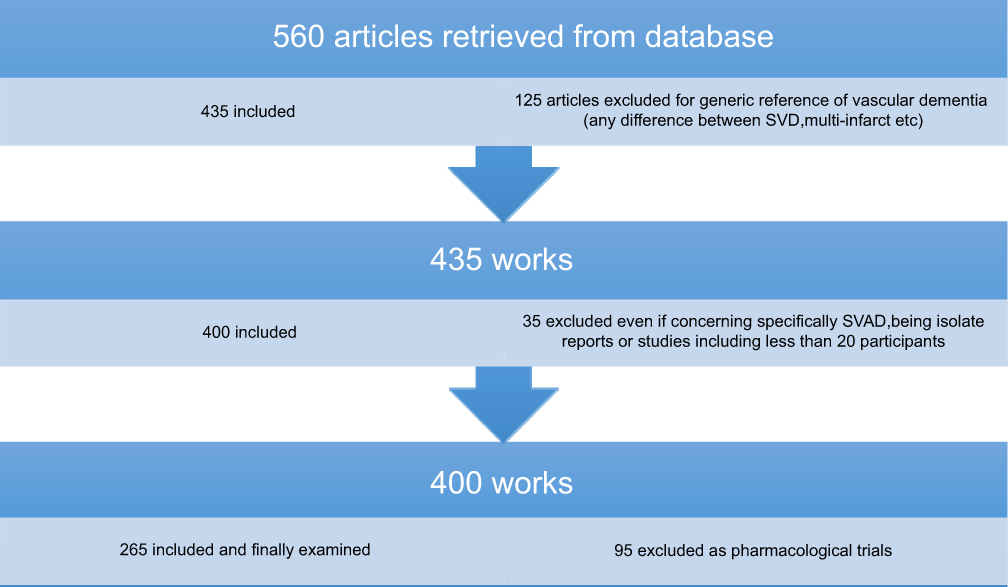 |
Figure 1 Flowchart of search strategy and selection criteria.Abbreviations: SVAD, subcortical vascular dementia; SVD, small vessel disease. |
Cerebral small vessel disease and blood pressure management
Collecting evidence suggests associations between traditional vascular risk factors, such as hypertension, diabetes, hypercholesterolemia, atherosclerosis, cardiovascular and cerebrovascular involvement, and cognitive disorders and dementia. These associations are complex and sometimes differ depending on when in life the risk factors are assessed.
Diabetes is a well-established risk factor for cognitive impairment, the mechanism of this process, however, is not fully understand. The effect of insulin acting directly on the brain is thought to increase the deposition and reduce the degradation of amyloid-ᵦprotein.49 Interestingly, Okereke et al showed that women without clinical diabetes but with higher levels of C-peptide, representing insulin secretion, had increased risk of cognitive impairment.50 Furthermore, Luchsinger et al showed that individuals with higher levels of fasting insulin, with or without clinical diabetes, exhibited a 70% higher risk of developing AD.51
Hypertension has historically been considered as one of the most critical risk factors for vascular dementia development,52–54 as one of the many causative co-factor for neurodegenerative dementia,55–58 and for white matter lesions.59 Chronic hypertension has been shown to accelerate amyloid deposition, blood-brain barrier (BBB) dysfunction, microglial cells activation, and subsequent neuronal loss and cognitive impairment in animal models.60
Rouch et al reported that patients showing a conversion from mild cognitive impairment to dementia were significantly older, more likely to have hypertension and cardiovascular diseases (Myocardial infarctions, including acute and subacute myocardial infarctions as well as older ischemic myocardial lesions). Moreover, long-standing hypertension is involved in the arterial aging process and arterial stiffness (also involved in the degenerative process).
Hypertension is a common risk factor for cerebral bleeding and hemorrhagic stroke, and it has been widely reported that there is a strict relationship between worse cognitive function and brain hemorrhage. For example, patients with Subarachnoid hemorrhage, who have no grossly evident neurological deficits after bleeding, frequently have subtle cognitive or neurobehavioral difficulties that impair their social adjustment and ability to return to their previous occupations.
On the other side, several epidemiological studies have also described low blood pressure, especially in later life as a risk factor for the development of AD, pointing to the potential risk of over treating hypertension. Indeed, the impact of hypertension on VAD has been debated and explained by several mechanisms, centering white matter damages.61,62 It has also been found that hypertension is equally distributed in non-lacunar and lacunar lesions and that many patients with lacunar stroke are normotensive.63 Thus it remains to clarify whether hypertension alone affects cognitive function or if it is the effect of higher blood pressure on the cardiovascular system that is the cause.
The Bronx Aging Study,64 followed an elderly non-demented cohort (75 years old), for up to 21 years. Dementia was diagnosed following detailed neuropsychological testing, clinical examination, and neuroimaging. Over a median follow-up of 6.7 years, 122 subjects developed dementia, of these 65 had AD. Participants with diastolic blood pressure (DBP) of <70 mmHg were twice as likely to develop AD when compared to those with a DBP >90 mmHg. The risk was higher in individuals with a persistently low diastolic blood pressure.
Low blood pressure was described as a predictor of increased mortality in a 5-year prospective study in Finland Zhu et al,65 found a significant correlation between low systolic blood pressure and dementia and hypothesized that hypotension could be interpreted as a precocious sign of cognitive decline. This finding was confirmed by Ruitenberg et al,66 who found that low blood pressure is related to a higher risk of developing dementia, regardless of sex or dementia-type (both AD and vascular). The risk was more evident in subjects who were permitted to continue antihypertensive drugs, suggesting that these patients become more prone to hemodynamic changes, with a higher incidence of watershed infarcts.66 Burke and colleagues firstly suggested that AD patients suffered due to pressure dysregulation, caused by cholinergic impairment.67 Guo et al,68 reported an association between increased dementia incidence and a relatively low systolic pressure, associated to a reduction of cerebral flow (CBF). The reduced CBF might be related to changes in cerebral vascular autoregulation due to the superimposing vessel degeneration, the same analysis was also reported elsewhere.69–71
Nilsson et al,72 reported that in subjects aged 80 or older, higher systolic blood pressure was associated with a better cognitive function and a lower systolic blood pressure was associated with cognitive decline and dementia. Also, many other studies reported a possible effect of low blood pressure on the cognitive decline.73–75 Guidelines published by the American Society of Hypertension and the International Society of Hypertension in 2014,76 suggest a blood pressure <140/90 mmHg for patients younger than 80 years old, the European Society has suggested a similar target for Hypertension (ESH) and by the European Society of Cardiology (ESC).77 The Guideline by the Eighth Joint National Committee (JNC8) endorses a reasonable target (<150/90 mmHg) in patients aged 80 years old.78
It appears that white matter alterations are exacerbated by a significant decrease in blood pressure.79–87 The most recently published work certified that “midlife not late-life exposure to the vascular risk factors is important for the development of dementia”.88,89
Vascular aging is associated with changes in the mechanical and structural properties of vessel walls, which leads to the loss of arterial elasticity reducing arterial compliance.90 This induces an alteration in autoregulatory capabilities of cerebral arteries, responsible of cerebral perfusion at a constant rate of blood pressure (BP). In this situation, the brain may be more vulnerable to ischemic insults when systemic blood pressure dips below a critical threshold for maintaining perfusion.91 Orthostatic hypotension is arbitrarily defined as a fall in systolic BP of 20 mmHg, or a fall in DBP of 10 mmHg on standing, but when associated with symptoms suggestive of cerebral hypoperfusion, an even smaller drop in BP may be of equal importance.92
Several studies have shown an association between severe cognitive impairment and dysregulation of postural blood pressure.93 Sudden postural hypotension can cause relatively prolonged changes in brain perfusion and may contribute to cause small lacunar events or contribute to white matter alterations and leukoaraiosis.94,95 Two recent studies found that orthostatic hypotension was more frequent in AD, with white matter disease than in AD without white matter disease.96,97 It has been speculated that a lack of capacity to regulate abnormal blood pressure, well recognized in AD, leads to chronic dysfunction of cerebral perfusion and leaves room for the development of white matter disease.98
Bytheway, in the end, some studies imply that there is a connection between cognitive impairment and hypertension, whereas others claim the opposite.
Considering other risk factors for dementia, the recent SPRINT-MIND study showed that higher urine albumin concentration was independently associated with worse cognitive functioning, including global cognitive function, executive function, and attention; lower estimated glomerular filtration rate was independently associated with worse global function and memory, assessing that there is a strict association between chronic kidney disease and poorer cognitive function. In the same paper is remarked that the conversion of patients with mild cognitive impairment to AD is lower in those with well-controlled and lower blood pressure.99
Pathological aspects of vascular cognitive impairment: the most updated vision
A review of pathologic studies shows enormous differences in the prevalence of VaD ranging from 0.03 to 85.2% with means around 11%, while in recent autopsy series from Japanese geriatric hospitals it was 23.6 to 35%. Around Europe, prevalence rates of VaD between ages 65–69 to 80+ years ranged from 2.2 to 16.3%.10
Fisher was the pioneer in describing the neuropathological details in small vessel disease. From autopsy studies, he observed that majority of the lacunes were due to segmental arterial disorganization with asymmetrical focal vascular changes leading at the loss of standard architecture of the vessels, all these lesions particularly favor the development of spontaneous hemorrhage, especially in the deeper areas of the brain.100
The vessel disorders that are most frequently associated with VaD are atherosclerosis of cerebral arteries (AS), arteriosclerosis or cerebral small vessel disease (SVD), and cerebral amyloid angiopathy (CAA).101–104 These vessel disorders frequently occur in the brains of elderly individuals and become more prevalent and severe with advancing age. There are less common forms of cerebrovascular disease and cognitive impairment, including various types of vasculitis and inherited diseases that affect vessel integrity.
SVD can result in lacunar infarcts, microinfarcts, hemorrhages, and microbleeds.
In autopsy series of elderly subjects with and without dementia, the prevalence of “pure” VaD (without concomitant cerebral pathologies) ranged from 5 to 78%, in the oldest-old from 4.5 to 46.8%.105 The majority of patients showed Alzheimer-related pathology, only part of them with AD alone, while mixed pathologies, ie, AD plus cerebrovascular or Lewy pathology, were seen in 74–93% and 9–28%, respectively. In the age group 70 to 90+, the prevalence of VaD increased from 13 to 44.8%, compared to AD (23.6–57%) and mixed dementia (2–86%). In a consecutive autopsy series of 1700 elderly demented, the AD without joint pathologies or minor CVLs, and mixed dementia increased with age. The prevalence of “pure” VaD decreased after age >80.
One of the most common causes of these striking disparities is based on the different pathological features employed in clinical classifications.106–108
Jellinger et al pointed out various clinical, pathological, and operative definitions of VaD.10
The simplest include three significant forms: multi-infarct dementia, single strategic infarct dementia, and subcortical vascular encephalopathy.
Jellinger suggested a different classification of VaD, according to primary morphologic lesions, dividing it in two broad groups: multifocal/diffuse disease (which comprises: large vessel dementia; small vessel dementia, distinguishing the subcortical infarct dementia and the cortical plus subcortical infarct dementia; hypoperfusion or hypoxic-ischemic dementia; venous infarct dementia; and hemorrhagic dementia) and focal vascular disease or strategic infarct dementia.11
Because this review is focused on small vessel disease-related dementia, also called sVaD, it is essential to describe what is known about its underlying pathological mechanisms. When discussing the microcirculation, we refer to small arteries and arterioles in the pial and lepto-meningeal circulation along with penetrating and parenchymal arteries and arterioles, pericytes, capillaries, and venules.
A semantic revision of small vessel disease
There are estimated to be approximately 47.5 million people in the world living with dementia, in up to 45% of whom SVD is a major contributing or the sole pathology.109 Markers of cerebral small vessel disease (SVD) are present on neuroimaging of almost all patients over 60. Small vessel diseases are a group of disorders that result from pathological alteration of the small blood vessels in the brain, including the small arteries, capillaries, and veins.
Cerebral small vessel disease includes a neuroimaging description which comprises different imaging changes in the white matter and subcortical grey matter, including small subcortical infarct, lacunes, white matter hyperintensities (WMHs), prominent perivascular spaces (PVS), cerebral microbleeds (CMBs) and atrophy. MRI studies have questioned the enlarged perivascular spaces (PVS), also known as Virchow-Robin spaces, as a hallmark feature of SVD. In healthy tissue, these spaces are proposed to form part of a complex brain fluid drainage system which supports interstitial fluid exchange and may also facilitate the clearance of waste products from the brain. The pathophysiological role of PVS, their function and interaction with cerebral microcirculation, has not been established yet. A possible mechanism involving impaired cerebrovascular reactivity (CVR), blood-brain barrier (BBB) dysfunction, perivascular inflammation, leading to accumulation of toxins, hypoxia and tissue damage, has been questioned.110 There is a broad consensus that PVS form a network of spaces around cerebral microvessels acting as a canal for fluid transport, the exchange between cerebrospinal fluid (CSF) and interstitial fluid (ISF) and clearance of unwanted products from the brain. In human and animal model has been proposed a brain lymphatic-like system called “para-arteriolar,” “para-venular,” “paravascular” or ‘glymphatic. Alteration of this system may induce dangerous modification of brain function because undesirable metabolic products would not be exported and nutrition would be missed. This complex brain fluid and waste clearance system appears to be necessary for the clearance of interstitial solutes from the brain and is most likely vital for maintaining brain homeostasis.111,112
Enlarged perivascular spaces (EPVS) have been related to aging, vascular risk factors (VRF) as hypertension, and other pathological conditions as cerebral small vessel disease and Alzheimer’s disease.
Has been reported that widening of PVS may indicate obstruction by protein and cell debris and thus stagnation of fluid drainage, those mechanisms can support PVS’s role in different diseases, for example increasing stroke risk particularly with lacunar rather than large vessel stroke, small vessel diseases and white matter involvement.113
The role of imaging markers of cerebral small vessel disease is essential. Cerebral white matter hyperintensities represent an essential indicator of a higher risk of all-dementia. Moreover, covert brain infarcts and cerebral microbleeds are potential risk indicators for incident dementia in the general population.
The definitions and terms of these lesions have significantly varied among studies
As stated by Roman et al,27 the term employed to define pathological features of the white matter as Binswanger’s Disease is somehow misleading.
The combination of multiple small deep infarcts and extensive white matter abnormalities has been referred to as Binswanger disease, also known as subcortical atherosclerotic encephalopathy, and the clinical findings are often classified as vascular dementia of the small artery type.
White matter hyperintensities are usually symmetrically and bilaterally distributed in the white matter including the pons and brainstem, and also occur in deep grey matter. They appear hyperintense to the healthy brain on T2 or FLAIR MRI and can be confluent. The pathology of white matter damage is still imprecise. Mainly it seems due to demyelination, loss of oligodendrocytes and axonal damage.
Other characteristics aspects are the “lacunes,” term employed by Fisher to describe a small fluid cavity in the brain, are very common and usually due to the micro-infarcts and the micro-bleeds, that are mainly located in the white matter, especially in the internal capsule and cerebellar white matter. They can also affect grey nuclei and deep white matter, such as the basal ganglia, thalamus, and brainstem white matter.
Initially, the term lacuna has been employed as a synonym for lacunar strokes, for silent brain infarct and to refer to the CSF-filled cavities on brain MRI or residual lesion of a small hemorrhage.114–119 Nowadays, it should be more appropriate the definition “lacuna of presumed vascular origin“ to replace the term“ lacuna,“ and it should be avoided the term ”lacunar infarct” to describe the ”lacuna” by definition. Lacunar infarcts are infarction areas, no larger than 15 mm; less frequently, they correspond to small hemorrhagic lesions in a reabsorption phase.120–123
Lacunes and white matter alteration share two common findings: the same pathogenic start point, arteriosclerosis, and brain damage vulnus, mainly a previous history of hypertension. Lacunes, mainly due to complete occlusion of the small arteries, lead to an infarct in a specific area (basal ganglia, capsule, pons, etc).
Moreover, there is substantial evidence in SVD of an associated hypoperfusive course, due to the altered caliber of small arteries and this is associated with incomplete ischemia of the deep white matter,124–127 accompanied by inflammation, diffuse rarefaction of myelin sheaths, axonal disruption, and astrocyte gliosis.
In small vessel disease, occlusion of deep periventricular-draining veins is also evident,128 and it has been described as a disruption of the blood-brain barrier. This leads a consequent leakage of fluid and plasma cells, which eventually might potentiate the perivascular inflammation, the demyelination process, and gliosis, accomplishing a multifactorial genesis for white matter alterations.129–131
Usually, acute small subcortical infarcts cause specific neurological symptoms, whereas cerebral small vessel disease lesions are more insidious, and referred to as silent lesions.116
Finally, concerning neuroimaging findings, cerebral microbleeds, small round and homogeneous foci of hypointensity on T2-weighted (gradient echo) MRI and susceptibility-weighted imaging have been widely described. Lipofibrohyalinosis and amyloid angiopathy are the most common vascular findings with cerebral microbleeds. Distribution of CMB are different concerning the extension of the SVD pathology, CMBs can be found in the basal ganglia, thalamus, brainstem, and cerebellum; they are related to lipofibrohyalinosis, whereas amyloid angiopathy is more frequently associated with lobar CMBs. CMBs also contribute to cognitive impairment and dementia, and neurological deficits. Several studies showed a significant relationship between CMBs and Aβ accumulation. Lobar CMBs but not deep CMBs (related to SVD) was proved to be correlated with Aβ pathology.
The burden of lacunes and deep white matter alterations (WMLs) relates to the degree of cognitive impairment, mainly when they affect the thalamus and basal ganglia.132–134 The cognitive alterations are determined by the specific point-to-point impairment, but also by the interruptions of the cortico-thalamus and thalamus-cortical networks,135,136 which can also lead to cortical atrophy, secondary to functional deafferentation.
We agree with Shi and Wardlaw,116 who have written:
common small vessel pathologies and blood-brain barrier impairment were found in clinically evident and covert cerebral small vessel disease features, suggesting that it should be regarded as a whole brain disease rather than treated separately as an individual condition.
Dynamic process for small vessel disease
Cerebral small vessel disease is a progressive disease. Lesions progress over time, and the long-term outcome and impact on brain damage vary. As far as lesions progress, cerebral small vessel disease has an uncertain long-term outcome, with a positive impact on cognition and behavior. Recent studies indicated that the strongest predictor of white matter progression is the high baseline white matter hyperintensity, with the rapid and confluent progression of lesions.137–140 Different conditions were hereafter considered.
Aging brain and vessel modifying condition
The aging of the brain is due to pathophysiological changes throughout the single lifetime.
Aging is related to vasculature changes including arteriolosclerosis and large artery stiffening, which is much more evident in proximal elastic arteries.141,142 The arterial stiffness diminishes the so-called Windkessel effect.143 Windkessel is a German term, which means air chamber, but it suggests the elastic reservoir property of vessel.144 The walls of all the large arteries which contains muscle fibers and elastin distend during systole and recoil during diastole. Since the amount of blood entering during the systolic time is larger than the de-flux of it in diastolic time, caused by peripheral resistance, the large arteries behave themselves as a capacitor. The Windkessel effects help in damping the fluctuation of blood pressure over the cardiac cycle. The Windkessel model is accompanied by the more recent theory of arterial acceleration. The onset of systole triggers a short lasting depolarization within smooth muscle cells, that spreads along the arterial tree, as a peristaltic wave; this could happen in order to amplify and distribute the pressure wave, over the arterial branches, allowing to penetrate the most profound regions.145
The most determinant effect result is an anticipated and precocious return of the so-called wave reflection, an increase of systolic and the decrease of diastolic pressure [144]. The brain usually has a high resting flow which seems to be related to the fact that heart pulsation (plus the Windkessel effect) produces a higher perfusion pressure toward the most profound small arteries of the brain.146,147
Hypertension promotes the underlying age-related mechanism of arteriolosclerosis, promoting micro-atheromatosis, which can rapidly lead to stenosis, or different degrees of vessel occlusion until the complete lumen occlusion, inducing ischemia and brain damage.148
Besides the potential effect on arterial arteriosclerosis, a fundamental role is played by subsequent hemodynamic alteration that may lead to a diminishment of blood perfusion in the brain.149,150
Like in a magic circle, the hypoperfusion, the arteriolosclerosis damage and the altered reflection on cerebral blood flow velocity perpetuates the arterial stiffness.
Aging also interferes with the brain autoregulation property. Cerebral vasoregulation is a dynamic process that redistributes cerebral blood flow (CBF) toward the brain areas in need of increased perfusion. Vasoreactivity reflects the ability of microvasculature to adapt to a changing environment, which causes fluctuating metabolic demands (such as oxygen and glucose delivery and blood pressure variations). As we already reported, chronic hypertension and hypotension alter CBF autoregulation and may affect the autoregulatory range.
“Autoregulation” most commonly refers to CBF adaptation to acute and chronic changes in arterial BP and perfusion pressure. Pressure autoregulation maintains fairly-stable perfusion over the range of mean systemic pressures 60–150 mmHg.151–153
Age-related endothelial dysfunction and risk factors such as hypertension and DM set the stage for altered neurovascular coupling and a regional decline in vasomotor capacity; all those factors reshape the sigmoidal physiologic curve of the adaptive system into “a straight line.”144
There is a strong association between hemodynamic and structural alterations of the cerebral arteries and cognitive impairment, suggesting that decreased cerebral blood flow, diffuse brain microvascular disease, increased arterial rigidity (resulting in decreased arterial compliance and putative myocardial impairment) are functionally linked to dementia. Several studies, based on transcranial Doppler ultrasounds examination of CBF, have reported lower mean flow velocity and decreased cerebrovascular reactivity in the middle cerebral artery in demented patients compared to controls.154–157
Moreover, it has been documented marked cholinergic denervation of small arteries in the brain.158The lack of muscarinic cholinergic receptors in the penetrating vessels might lead to a vital blood flow dysregulation, with scare response to blood pressure modifications and have as a consequence a loss of efficacy in vasomotor coupling response.159–161
Finally, the aging effect involves also oxidative stress at an early stage. The reactive oxygen species (ROS) produced during mitochondrial oxidative phosphorylation are associated with damage to DNA, lipids, and proteins. The accumulation of mitochondrial DNA mutations induces impairment of mitochondrial function. Mutations in mitochondrial DNA and oxidative stress both contribute to neuronal aging and neurodegeneration.162,163 All those changes contribute to neuronal loss. The effect of aging does not occur to the same extent in all brain regions. For example, compared to cerebral areas, the cerebellum is protected from aging effects. Usually, the prefrontal cortex is most affected and the occipital least. Frontal and temporal lobes are most affected in men compared with the hippocampus and parietal lobes in women.164
Metabolic and regional vulnerability
Petito et al,165 reported that after 10 mins of transient global ischemia in the rat brain, the initial changes occur in perineuronal spaces and subcortical white matter, preceding neuronal death by several days.166 The authors demonstrated the selective death of the gray matter oligodendrocytes one day after ischemia, suggesting that oligodendroglia might be more neurons in the cerebral cortex and thalamus, the damage is absent or minimal. These results appear concordant with other in vitro findings.167,168 Moreover, oligodendroglia cell death precedes neural necrosis after focal ischemia. Furukawa et al,169 described the time course of microglial activation after ischemia, with regional variability, probably due to differential selectivity across brain regions, just as what described by Gehrman et al170. They observed that the most significant and precocious microglial activation occurs in the hippocampus, soon after ischemia induction, and this decreases after 48 h. Afterward, microglial activation is slowly evident in the white matter and cortex, with an increasing trend, mostly at 48–72 h after ischemia induction. Rupalla et al,171 found that in adult mice, microglial activation increased from days 2 to 30 and days 2 to 4 after ischemia in the thalamus and cortex, respectively. Furukawa et al,169 hypothesized two subsets of microglia activation: Ml activation that induces the formation of oxidative metabolites, proteases, and cytokines after ischemia, and M2 activation that antagonizes inflammation.172 The authors demonstrated that the extent of white matter damage might be associated with more extensive activation of Ml microglia, implying that blocking this process could reduce brain damage,173 (following Yuan et al161).
Chronic cerebral hypoperfusion has been proposed as vascular risk factors for white matter injury.174 Different experimental models which studied the hypoperfusion of the corpus callosum with a stabilized drop of 52–64% of the original flow at 30 days after the ligation.175 The following white matter alteration revealed axonal damage and diffuse demyelination, indicated by an increased amyloid precursor protein expression and encephalitogenic peptide elevation.176 Oligodendrocyte degeneration was due to apoptotic cell death, with caspase 3 RNA detection, associated to an elevation of matrix- metalloprotease 2 (MMP-2) expression and microglial activation.177 More recent data confirmed that glial changes in chronic cerebral hypoperfusion could induce white matter damage, region-specific mild astrogliosis, marked microglial activation and increased oligodendrocyte density and diffuse myelin damage.178 It has been demonstrated that astrocytes react to ischemia concerning the length and severity of the insult. In the early ischemic period, the astrocytes respond with proliferation and, following persistent hypoperfusion, with degeneration and death.179 Interestingly, in the white matter and the frontal cortex of demented patients, the disintegration and the regression of astrocytes has been demonstrated and related to ischemia.180,181
When the CBF is not stopped, but it is reduced or not well matched to the energy demands of the tissue, chronic brain injuries occur in different areas, standing on a different degree of vulnerability,182 As we documented before, the neurocentric vision of the brain is no longer supported. It is quite clear that the normal functions of the brain depend on functional interactions among different components in the neurovascular unit, which includes neurons, oligodendrocytes, microglia, and astrocytes and vascular cells (endothelial cells, pericytes, and smooth muscle cells) and the basal lamina matrix within brain vasculature.183 Therefore, possibly through glutamate signaling,184 astrocytes respond to the metabolic changes of neurons. Astrocytes monitor neuronal activity and contribute to the regulation of blood flow modifying capillary permeability, by stretching out their endfeet to the microvessel and establishing a proximal connection with the capillary. The combined effect might lead to instant regulation of CBF according to neuronal activity, in the so-called neurovascular coupling.185
Moreover, endothelial cells are receiving growing interest. As pointed out by Iadecola,186 in the resting brain, CBF varies in proportion to the energy consumption of each brain region. Each increase in neuronal activity leads to increases in CBF in the activated areas, permitting a flow response to map brain functions,187 The evidence is increasing on pial arterioles: signals generated by the neuronal activity, deep in the brain, have to be conveyed to upstream arterioles, remote from the area of activation, to increase flow efficiently. Vascular mapping and fMRI demonstrated that during somatosensory activation vascular responses are first seen in the deep cortical lamina, and then, more superficially, suggesting a retrograde propagation of the vascular response.188 A possible scenario for the transmission and coordination of the vascular response is described by Iadecola,186 as follows: activation-induced increases in extracellular potassium triggers hyperpolarization of capillary endothelial cells and pericytes.189
The hyperpolarization propagates upstream and reaches smooth muscle cells in penetrating arterioles producing relaxation. At the same time, metabolic modifications (reduced viscosity, increased deformability of blood cells) on the endothelium of feeding arterioles increment the smooth muscle cell relaxation (the so-called flow-mediated vasodilation). In upstream pial arterioles remote from the site of activation, there is vasodilation, by propagation from arteriole downstream and acting as a local flow-mediated and myogenic response.
Moreover, there are also regional differences in the basal regional metabolic response.
A recent in vivo-study,190 confirmed that the caudate nucleus most precociously affected brain region, followed by the putamen, insula, precentral gyrus, inferior frontal gyrus, and middle frontal gyrus, which requires, at steady state than 20% of the metabolic request, then other brain regions.191–197
Non-demented patients with lacunar infarcts do not show cerebral flow alterations in grey matter, but they exhibit a decrease of more than 30% in white matter, associated with increased oxygen extraction of more than 130% in comparison with healthy subjects. On the contrary, patients with lacunar events and cognitive impairment have a reduced metabolic rate for oxygen, about 25% and 35% lower in grey and white matter, respectively.198,199 As a conclusion, an incongruity between the brain oxygen supply and its consumption has been described due to small vessel disease; this altered ratio causes an impaired neurovascular coupling and determines an altered vasomotor reactivity.200–205 Tak et al,200 by employing spectroscopy confirmed a reduced blood flow and low extraction of metabolic oxygen in the frontal lobes, during the resting state in subjects with small vessel disease. These changes have been observed not only in subcortical, profound regions,200–202 but also in frontal cortical areas. This was unexpected, and not fully understood, apart from the apparent speculation that cortical damage may be related to axonal dying back or Wallerian degeneration.206
Another novel aspect of the work by Tak et al,200 is the finding that in small vessel disease, the fractional change of oxygen extraction is increased during activation tasks (lower than average, in resting state). However, the oxygen extraction is not sufficient to guarantee a proper activity, due to the decrease of blood flow (mainly due to smooth muscle alterations and arterial stiffness) and to the pre-existent low metabolic oxygen rate. The temporary augmentation of oxygen extraction implies a further loss of the metabolic reserve,207 leaving the brain tissue more vulnerable to hemodynamic changes. Moreover, the clinical severity of white matter pathology is inversely related to glucose metabolism in the dorsolateral frontal lobe. Chui explained this finding with the hypothesis that the long neural fibers that connect the prefrontal-cortical areas to the periventricular brain regions are dependent on small, terminal arteries that are affected by arteriosclerosis.208
Cholinergic dysregulation in small vessel disease
A cholinergic deficit in the brain and cerebrospinal fluid of patients with vascular dementia has been widely reported. Also, cholinergic therapies have shown promising effects on cognitive improvement in those patients, even if its precise mechanisms are not well-known jet.209,210
Wilkinson et al,211 reported that patients with VAD treated with Donepezil (an acetylcholinesterase inhibitor) showed benefits in cognition and global function compared to placebo. Moreover, a significant improvement of dementia severity and slowed functional deterioration was seen.
The safety profile of donepezil has been reported in possible and probable VAD and AD; donepezil may be well tolerated, even in a relatively frail population of elderly patients with considerable comorbidities.212 Albeit, its role in VaD treatment is still debated. O’Brien and Thomas in 2015 suggested that cholinesterase inhibitors and memantine should not be used in patients with vascular dementia. Actually, in a different randomized controlled trial, a small but significant benefit of cholinesterase inhibitors on cognition was seen in vascular dementia, but the benefits on global functioning, activities of daily living, and behavior have not been consistently reported. Finally, the possible side-effects of those drugs must be taken into account.213,214
Data suggest that the cholinergic system and cerebral blood flow are linked reciprocally. An anti-inflammatory pathway in which the efferent vagus nerve signals suppress pro-inflammatory cytokine release and inhibit inflammation has been questioned.
The concept of the “cholinergic anti-inflammation pathway” has been first proposed by Tracey et al; these findings are based on the knowledge that acetylcholine (Ach) released from cholinergic axon terminals can interact with α7 nicotinic Ach receptors (nAChRs) on vicinal immune cells.215,216 The nicotinic receptors then translate the cholinergic signal into the suppression of cytokine release, involved in the inflammatory cascade.217,218
Cholinergic agents include ACh precursors, which increase the synthesis of ACh; nicotinic or M1 muscarinic agonists, which directly stimulate cholinergic receptors or allosterically modulate AChR,219 and synaptic AChE inhibitors, which prevent the degradation of ACh.
Data from literature showed that both in animal models and postmortem studies there is reduced CSF ACh concentrations in patients with Binswanger or multi-infarct dementia.220,221 Animal models of sVaD (stroke-prone rats) show significant reductions in ACh levels in the cortex, hippocampus, and cerebrospinal fluid.222–225 Furukawa et al,226 investigated the role of ACh in the development of hypoxia-ischemia lesions using in vivo animal models. They demonstrated that muscarinic receptor antagonism with atropine has a selective effect of worsening the ischemic damage, but only in the CA1 region. Selective alpha-7- nicotinic AChR antagonists exacerbate changes in the cortex and CAl and CA3 areas, while non-selective nicotinic AChR antagonists have a detrimental effect on the hippocampus but not in all the cortical areas examined.227 It is accepted that AChR stimulation reduces hypoxic-ischemic brain damage in rat pups and reduces the cytokine production as a consequence of hypoxia-ischemia damage.228–230
Elsewhere a loss of cholinergic neurons in 40% of VaD patients was reported, accompanied by reduced ACh activity in the cortex, hippocampus, and striatum.231
Changes in cerebral blood flow can affect control cholinergic networks, and a proper cholinergic function is mandatory to regulate regional brain blood flow.232,233 In primates, the central cholinergic system has three main components: the projections from the basal forebrain, innervating the hippocampus, cortical regions, and some subcortical nuclei; the projections from the brainstem which innervate the thalamus and the midbrain, as well as the brainstem; finally, the striatal and accumbens interneurons.234,235
The forebrain cholinergic bundle is supplying both the medial and lateral pathways loops around the anterior corpus callosum and the frontal horns of the ventricles. It is well-accepted the parasympathetic innervation of the circle of Willis, and pial vessels are principally mediated by Ach.236 Ach produces significant in vitro arterial relaxation per se and by increasing the synthesis of vasodilator endothelium agents, via the nitric oxide synthase,237 and via the GABA interneurons.238–240
The stimulation of the Nucleus Basalis of Meynert results in increased blood flow throughout the cerebral cortex in experimental animals.241 On stimulation, perivascular cortical afferents release Ach into endothelial M5 muscarinic receptor.242,243 M5 receptors are highly expressed in blood vessel walls. Yamada et al,222 prepared knockout mice (M5-/-) and found that compared to wild-type mice, these animals lose the ability to dilate cerebral arteries but could still regulate extra-cerebral flow; it has been suggested that the neural network from the Meynert nucleus may influence the small arteries also indirectly.
Age-related impairment of vascular autoregulation is also due to a low-level functioning of the autonomic nervous system, with direct and endothelium-mediated altered baroreflex activity.244–246
Small vessel disease, due to the primary localization of the white matter hyperintensities, may affect the integrity of the adjacent medial cholinergic pathway.247 Post-mortem studies in CADASIL demonstrated that cortical cholinergic projections from the Meynert could be affected as a consequence of white matter lesions. Bohnen et al,247 demonstrated in vivo that the severity of the periventricular white matter lesions is associated with lower AchE activity, in the middle-aged and elderly subjects without dementia, as a result of cortical cholinergic deafferentation.
Post-mortem sVaD studies revealed lower choline acetyltransferase (ChAT) activity compared with controls,248 and sVAD patients have lower CSF concentrations of Ach.223–225,249
There are some clinical data which confirm a reduction of the number of cholinergic neurons in the Nucleus Basalis of Meynert in multi-infarct dementia, but not in sVaD.250–252 Though, the number of muscarinic cholinergic receptors is markedly reduced in mixed dementia patients,253 and in small vessel disease-related dementia. The cholinergic impairment is not mediated by a primary loss of the cholinergic neurons of Nucleus Basalis of Meynert,254,255 but rather it is a consequence of the secondary cholinergic deficits, due to the indirect less efficacious endothelium mediation through the nitric oxide synthase and the GABA interneurons.237,238 The two mediators seem less efficient to influence the small arteries contraction also due to the significant coexistent alterations of the endothelium.256–259
The pathological cascade of events, which occurs as a consequence of all the pathological alterations described, determines a decrease of the vascular tone, with a release of the blood-brain barrier permeability, with internal vascular remodeling and with vascular rarefactions. As a result, hypo-perfusion at rest occurs in the brain, combined with an impairment in the moment-to-moment control of CBF, associated to a decrease of adaptive vascular responses and a diminishment of the neurovascular coupling and auto-regulation system.257
More recent studies have suggested an important role for the autonomic nervous system in the maintenance of cerebral perfusion.260–262 It has been suggested that cholinesterase inhibitors modulate cerebral vascular functions, due to the possible role of the cholinergic fibers in cerebral flow regulation.263 Moreover, an active cholinergic vasodilatory reflex in the cerebrovascular system has significant clinical implications for the use of acetylcholinesterase inhibitors in the potential adjuvant treatment for ischemic strokes.264 Moreover, the same authors demonstrated that increasing cholinergic activity might also improve cerebral perfusion and autoregulation in demented patients.265 Once more, the distribution of muscarinic receptor subtypes may play a key role in how vasodilation is affected: in skeletal muscle, M3 receptors are dominant and are responsible for the vasodilatory response to acetylcholine.266 On stimulation, perivascular cortical afferents release Ach onto endothelial M5 muscarinic receptor,267,268 highly expressed in blood vessel walls.267 Hamner et al,261 in a limited, but well-conducted study demonstrated in healthy volunteers that cholinergic control of the cerebral vasculature might be active at low frequencies, lower than 0.05 Hz when the sympathetic nervous system appears to play a role in the cerebral auto-regulation. At these low frequencies, even the myogenic mechanisms appear to play any role; to surprise, the correspondence between cholinergic and sympathetic cerebrovascular regulation above 0.05 Hz is striking, suggesting that the cerebral circulation engages different mechanisms to protect against rising and falling pressures.
It has been demonstrated that the endothelial cells of brain arterioles promote and activates the neuronal signaling, the synaptic transmission, oligodendrocyte and white matter regulation, neurogenesis while inhibiting the activation of the microglia.269,270
Moreover, the cholinergic impairment for artery dysregulation observed in small vessel disease could also derive from the deafferentation of the basal forebrain cholinergic system to the subcortical structures.271,272 These findings are associated with a reduced synaptophysin immunoreactivity.273 In animal models, a reduction of vasopressin and histamine has been confirmed as a result of the deafferentation of the neural tracts from the supra-optic and tuberomammillary nuclei to the basal forebrain,274 with a possible self- potentiating reduction of perfusion.
The presence of lacunes and white matter lesions affecting the deep neural networks located between frontal cortical areas and basal ganglia/thalamus and anterior cingulate cortex,197,275 may account for secondary cholinergic basal-forebrain de-afferentation. Neural activity is typically accompanied by increased blood flow, with vascular dilatation propagated in a retrograde manner to upstream arterioles, located outside the activated area.276 The altered “retrograde vasodilatation system” (which occurs in small vessel disease-related dementia) could be a possible factor that may worsen vessel dysregulation.
Small vessel disease: yin and yang in subcortical vascular dementia
Small vessel disease and white matter abnormalities are seen frequently with aging.
The most relevant questions are: does the coexistence of small vessel disease and white matter alterations do represent a biological variant of healthy aging, or is small vessel disease the predictor of future dementia?
The answers are ambiguous.226
Animal models and human clinical studies showed that white matter alterations are associated with cognitive and behavioral disruptions. Though, as clearly stated by De Silva and Faraci,:118 “the associations between vascular disease and cognition are based primarily on the temporal relationship between these endpoints.”
What is lacking is a solid demonstration of the existence of a causative determinant, testifying the relation between subcortical networks disruption (due to small vessel disease) and the clinical definition of subcortical vascular dementia.
What emerges from literature are some statements: first of all, vascular and endothelium changes have been related with age; the changes can be accelerated by different vascular risk factors (hypertension, hemodynamic changes, diabetes227,228); secondly, vascular function changes can be heavily influenced by genetic and epigenetic factors,:269 they directly influence the endothelium resistance. These changes induce a progressive decrease in CBF which is thought to be one of the key element for the progression from small vessel disease towards subcortical vascular dementia.277 Aging is the most important non-modifiable risk factor for the development of white matter hyperintensities and their progression towards confluent white matter damage, for endothelium injuries, for their progression and the increased blood-brain barrier permeability.278–280 Moreover, the subcortical vascular dementia might be characterized by focal, even subtle neurological signs (such as slight motor hemiparesis, bulbar signs, gait imbalance, hypokinesia, etc.281,282). Another important key point is that the early cognitive syndrome for subcortical vascular dementia is defined by a dis-executive syndrome, with decreased abilities in goal formulation, set-shifting, and execution, associated to major problems in the retrieval of information.283–286 Similarly, behavior disruption is even more typical, with marked emotional lability, emotional bluntness, 287 and apathy.288,289 What has been described as a typical neuropsychological profile of subcortical vascular dementia can be attributed to damage to the prefrontal-frontal subcortical cortical networks.290–292
Tak et al,200 found significant alterations of the hemodynamic and metabolic responses also in the frontal cortical areas of patients suffering from small vessel disease. This can be related to axonal dying back or Wallerian degeneration,206 but it is also supported by a metabolic imbalance, due to an increased fractional change of oxygen extraction, during activation tasks, not sufficient to compensate for the constant decrease of CBF and the low metabolic oxygen rate. The increased oxygen extraction implies a progressive loss of the metabolic reserve.207
Different studies pointed out that even in the healthy aging brain, some cognitive, age-related modalities have been related to frontal lower metabolism.293,294 The ongoing process or sudden precipitating events may transform the standard aging-related subcortical alteration in dementia.
The cerebral small vessel disease has been traditionally distinguished from a neuropsychological perspective from subcortical vascular dementia.294 A well-conducted study,295 stated that lacunar infarctions and deep white matter hyperintensities seen on MRI did not correlate with neuropsychological impairments and they should be considered as “epiphenomena that morphologically characterize cerebral small vessel disease, but do not, in and themselves, indicate cognitive impairment.” Nonetheless, different studies indicated that lacunes and white matter hyperintensities were associated with dis-executive syndrome,296,297 and when lacunar events can be thoroughly studied, neuropsychological dis-executive functions can be found out in more than 55% of cases, concluding that the process from small vessel disease to subcortical vascular dementia is ongoing and its incidence is still under-recognized, in clinical and research studies.298
These conclusions are in line with those derived by more consistent studies and even recent studies.299,300 These studies indicated that most of their participants, affected by small vessel disease, demonstrated cognitive performances, below one standard deviation from the normative mean scores in executive functions. These data suggest therefore that small vessel disease could be considered as a fore-runner condition of subcortical dementia, in due time course.
Finally, the pattern progression from small vessel disease to subcortical dementia has been debated by two more recent works. Nitkunan et al,301 verified that the cognitive and executive dys-functions are related to the brain volume decline, which is progressive with time course. More sophisticated studies,302,303 suggested that the progression pattern of brain structural changes in patients with small vessel disease is consistent with the topography of selective cortical atrophy (mainly in frontal, perisylvian and posterior cingulate regions).304–307 The atrophy extends further towards the parietal and medial temporal regions, when patients manifested the definite signs of subcortical vascular dementia, exhibiting additional atrophy in the lateral head and inferior body of the hippocampus.
Taken together, all the data as mentioned earlier indicate that patients with small vessel disease manifest cognitive and behavioral impairments. They are consistent with subcortical frontal networks disruption and their evolution towards subcortical vascular dementia is consistent with the progression of the cortical thinning of prefrontal, frontal areas and with an evident shape alteration of the lateral head and inferior body of the hippocampus. Still debated aspects are time course, precipitating factors, and individual variability.308
Conclusions
At the end of this review, there are some solid conclusions and many points that require further studies. The positive aspects are:
- Small vessel disease and the related dementia are two unambiguous nosographic entities that deserve attention for their relevance and impact in clinical practice.
- Hypertension might be a historical problem of patients who suffer from small vessel disease and small vessel disease-related dementia, but at the diagnosis of small vessel disease, patients are more vulnerable to brisk pressure lowering
- CBF regional selective decrease seems to be one of the critical factors for the progression from small vessel disease to small vessel disease-related dementia.
- The arterioles are the targets of small vessel disease and sVaD, with degeneration of the smooth muscle layer, replacement by hyaline fibrosis, leading to a subtotal luminal occlusion. Patients suffering from small vessel pathology are therefore more sensitive to any pressure changes. Endothelium damage is a super-imposed, not irrelevant, condition.309
- Acetylcholine intimately regulates CBF through the parasympathetic innervation of the circle of Willis and pial vessels. It causes significant arterial relaxation by promoting the synthesis of vasodilator agents; a secondary acetyl- cholinergic impaired regulation characterizes small vessel disease.
- These aspects determine the sensitivity of elderly patients with dementia to hypotension and each clinical condition that may lead to hypotension might accelerate underlying degenerative processes.
The most relevant still under debate-aspects are:
- The modality of changing from normal aging small vessel disease towards dementia
- The influence of cells, other than neurons and astrocytes, including microglia, oligodendrocytes, perivascular and meningeal macrophages in vessel control and endothelium degeneration
- The impact of aging and the age-gene interactions in lipohyalinosis and endothelium disruption, caused, promoted or potentiated by hypoperfusion and metabolic disruption.
Acknowledgments
The authors thank Joy Bates, PhD for her assistance in editing the text and express their gratitude to Claudio Tiribelli, MD PhD, full professor, for his revision of the text, in order to implement its clarity and its rigor.
Disclosure
The authors declare that they have no actual or potential conflicts of interest in relation to this article.
References
1. Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 2010;9(11):1118–1127. Epub 2010 Oct 9. doi: 10.1016/S1474-4422(10)70223-4
2. Sorbi S, Hort J, Erkinjuntti T, et al. EFNS-ENS guidelines on the diagnosis and management of disorders associated with dementia. Eur J Neurol. 2012;19(9):1159–1179. doi: 10.1111/j.1468-1331.2012.03784.x.
3. Emre M. Clinical features, pathophysiology and treatment of dementia associated with Parkinson’s disease. Handb Clin Neurol. 2007;83:401–419. doi: 10.1016/S0072-9752(07)83018-1.
4. McKeith IG1, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology. 2005;65(12):1863–1872. Epub 2005 Oct 19.
5. Seltman RE, Matthews BR. Frontotemporal lobar degeneration: epidemiology, pathology, diagnosis and management. CNS Drugs. 2012;26(10):841–870. doi: 10.2165/11640070-000000000-00000.
6. Chui H. Vascular dementia, a new beginning: shifting focus from clinical phenotype to ischemic brain injury. Neurol Clin. 2000;18(4):951–978.
7. Román GC, Goldstein M. A population-based study of dementia in 85-year-olds. N Engl J Med. 1993;329(1):
8. Román GC, Tatemichi TK, Erkinjuntti T,et al. Vascular dementia: diagnostic criteria for research studies. Reports of the NINDS-AIREN international workshop. Neurology. 1993; 43(2):250–260.
9. World Health Organization for vascular dementia. The ICD-10 Classification of Mental and Behavioral Disorders. Diagnostic Criteria for Research, Geneva. Switzerland. World Health Organization; 1993:48–50.
10. Jellinger KA. Pathology and pathogenesis of vascular cognitive impairment - a critical update. Front Aging Neurosci. 2013;5:17. doi: 10.33897fnagi.2013.00017
11. Jellinger KA. The enigma of vascular cognitive disorder and vascular dementia. Acta Neuropathol. 2007;113:349–388.
12. American Psychiatric Association. Major or Mild Vascular Neurocognitive Disorders. Diagnostic and Statistical Manual of Mental Disorders.
13. Pohjasvaara T, Mantyla R, Ylikoski R, Kaste M, Erkiniunnti T. Comparison of different clinical criteria (DSM.III, ADDTC, ICD-10, NINDS-AIREN, DSM-IV) for the diagnosis of vascular dementia. Stroke. 2003;31: 2952–2957. doi:10.1161/01.STR.31.12.2952.
14. Sinha P, Bharath S, Chandra SR. DSM-5 in vascular dementia. Comparison with other diagnostic criteria in a retrospective study. EC Neurol. 2015;2 (3):
15. Wardlaw JM, Smith EF, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013;12:822–832. doi:10.1016/S1474-4422(13)70124-8
16. Garcia JH, Brown GG. Vascular dementia: neuropatholgic alterations and metabolic brain changes. J Neurol Sci. 1992;109:121–131.
17. Román GC, Sachdev P, Royall DR, et al. Vascular cognitive disorder: a new diagnostic category updating vascular cognitive impairment and vascular dementia. J Neurol Sci. 2004;226:81–87. doi:10.1016/j.jns.2004.09.016
18. Brown MM, Markus HS, Oppenheimer S. Stroke Medicine. New York: Taylor and Francis. 2006: 81–83.
19. Tomimoto H. Subcortical vascular dementia. Neurosci Res. 2011;71:193–199 doi:10.1016/j.neures.2011.07.1820
20. Thal DR, Grinberg LT, Attems J. Vascular dementia: different forms of vessel disorders contribute to the development of dementia in the elderly brain. Exp Gerontol. 2012;47:816–824. doi:10.1016/j.exger.2012.05.023
21. Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9:689–701. doi:10.1016/S1474-4422(10)70104-6
22. Bowler JV. Vascular cognitive impairment. J Neurol Neurosurg Psychiatr. 2005;76(5):35–44. doi:10.1136/jnnp.2005.082313
23. Erkinjunnti T, Gauthier S. Diagnosing vascular cognitive impairment and dementia. In: Wahlund LO, Erkinjunnti T, Gauthier S, editors. Concepts and Controversies in Vascular Cognitive Impairment in Clinical Practice. Cambridge: Cambridge University Press; 2009:3–9.
24. Ying H, Jianping C, Jianqing Y, Shanquan Z. Cognitive variations among vascular dementia subtypes caused by small-, large-, or mixed-vessel disease. Arch Med Sci. 2016;12(4):747–753. doi:10.5114/aoms.2016.60962
25. Patel B, Markus HS. Magnetic resonance imaging in cerebral small vessel disease and its use as a surrogate disease marker. Int J Stroke. 2011;6(1):47–59. doi:10.1111/j.1747-4949.2010.00552.x
26. Erkinjunnti T, Inzitari D, Pantoni L, et al. Resaerch criteria for subcortical vascular dementia in clinical trials. J Neur Transm Suppl. 2000;59:23–30
27. Roman GC, Erkinjunnti T, Wallin A, Pantoni L, Chui HC. Subcortical ischemic vascular dementia. Lancet Neurol. 2002;1:426–436.
28. Neumann-Haefelin T, Hoelig S, Berkefeld J, et al. Leukoaraiosi is a risk factor for symptomatic intracererbal hemorrhage after trombolysis for acute stroke. Stroke. 2006;37:2463–2466. doi:10.1161/01.STR.0000239321.53203.ea
29. Palumbo V, Boulanger JM, Hill MD, Inzitari D, Buchan AM; for the CASES Invstigators. Leukoaraiosis and intracerebral hemorrhage after trombolysis in acute stroke. Neurology. 2007;68:1020–1024. doi:10.1212/01.wnl.0000257817.29883.48
30. Launer LJ, Masaki K, Petrovitch H, Foley D, Havlik RJ. The association between midlife blood pressure levels and late-life cognitive function. The Honolulu-Asia Aging Study. JAMA. 1995;274:846–1851.
31. Smith EE, Rosand J, Knudsen KA, Hylek EM, Greenberg SM. Leukaraiosis is assocaited with arfarin-related hemorrhage following ischemic stroke. Neurology. 2002;59:193–197.
32. Streifler JY, Elasziw M, Benavente OR, et al. For the North American Symtpomatic Carotid Endarterectomy Trial Group. Prognostic importance of leukoaraiosis in patients with sympotmatic internal carotid stenosis. Stroke. 2002;33:1651–1655.
33. Lin R, Stevensson L, Gupta R, Lytle B, Krieger D. Chronic ischemic white amtter disease is a risk factor for nonfocal neurologic injury after total aortic arch replacement. J Thor Cardiovasc Surg. 2007;133:1059–1065.
34. O’Donnell MJ, Xavier D, Liu L, et al. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): a case- control study. Lancet. 2010;376:112–123.
35. Iadecola C, Park L, Capone C. Threats to the mind: aging, amyloid, and hypertension. Stroke. 2009;40:40–44.
36. Moretti R, Torre P, Antonello RM, Manganaro D, Vilotti C, Pizzolato G. Risk factors for vascular dementia: hypotension as a key point. Vasc Health Risk Manag. 2008;4(2):2432–2444.
37. Skoog I, Nilsson L, Palmertz B. A population-based study of dementia in 85-year olds. NEJM. 1993;328:153–158.
38. Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am J Public Health. 1998;88(9):1337–1342.
39. Posner HB, Tang MX, Luchsinger J, Lantigua R, Stern Y, Mayeux R. The relationship of hypertension in the elderly to AD, vascular dementia, and cognitive function. Neurology. 2002;58:1175–1181.
40. Prince MJ, Bird AS, Blizard RA, Mann AH. Is the cognitve function of older aptients affectd by antihypertensive tratement? Results from 54 months of the Medica Research Council’s trial of hypertension in older adults. BMJ. 1996;312:801–805.
41. Hofman A, Ott A, Bretler MM, et al. Atherosclerosis, apolipoproteinE, and prevalence of dementia and Alzheimer’s Dementia in the Rotterdam Study. Lancet. 1997;349:151–154.
42. Tzourio C, Anderson C, Chapman N, et al., Effects of the blood pressure lowering with perindopril and indapamide therapy on dementia and cognitive decline in aptients with cerebrovascualr disease. Arch Intern Med. 2003;163:1069–1075.
43. Forette F, Seux ML, Staessen JA, et al. The prevention of dementia with antihypertensive treatment: new evidence from the systolic hypertension in Europe (syst-Eur) study. Arch Intern Med;2002;162:20146–22052.
44. Knopman DS, Parisi JE, Boeve BF, et al. Vascular dementia in a population-based autopsy study. Arch Neurol. 2003;60(4):569–575.
45. Fitzpatrick AL, Kuller LH, Lopez OL, Kawas CH, Jagust W. Survival following dementia onset: alzheimer’s disease and vascular dementia. J Neurol Sci. 2005;229-230:43–49. Epub 2004 Dec 23.
46. Sicras A, Rejas J, Arco S, et al. Prevalence, resource utilization and costs of vascular dementia compared to Alzheimer’s dementia in a population setting. Dement Geriatr Cogn Disord. 2005;19(5–6):305–315. Epub 2005 Mar 22.
47. Ramirez-Gomez C, Zheng C, Reed B, et al. Neuropsychological profiles differentiate Alzheimer disease from subcortical ischemic vascular dementia in an autopsy-defined cohort. Dement Ag Cogn Disord. 2017;44(1–2):1–11. Doi:10.1159/000477344.
48. Kalaria RN, Erkinjuntti T. Small vessel disease and subcortical vascular dementia. J Clin Neurol 2006;2(1):1–11.
49. Coker LH, Shumaker SA. Type 2 diabetes mellitus and cognition: an understudied issue in women’s health. J Psychosom Res. 2003;54:129–139.
50. Okereke O, Hankinson SE, Hu FB, Grodstein F. Plasma C peptide level and cognitive function among older women without diabetes mellitus. Arch Intern Med. 2005;165:1651–1656.
51. Luchsinger JA, Tang MX, Shea S, Mayeux R. Hyper-insulinemia and risk of Alzheimer disease. Neurology. 2004;63:1187–1192.
52. Morris MC, Scherr PA, Hebert LE, Glynn RJ, Bennett DA, Evans DA. Association of incident Alzheimer disease and blood pressure measured from 13 years before to 2 years after diagnosis in a large community study. Arch Neurol. 2001;58:1640–1646.
53. Moretti R, Esposito F, Torre P, Antonello RM, Bellini G. Hypotension in subcortical vascular dementia, a new risk factor. Wasn’t it hypertension?. In: De La Monte S, editor. Alzheimer’s Disease Pathogenesis-Core Concepts, Shifting Paradigms and Therapeutic Targets. Rijeka: InTech; 2011. doi:10.5772/18358
54. Kennelly SP, Lawlor BA, Kenny RA. Blood pressure and dementia - a comprehensive review. Ther Adv Neurol Disord. 2009;2(4):241–260. doi: 10.11177/1756285609103483
55. Whitmer RA, Sidney S, Selby J, Johnsoton SC, Yaffe K. Midlife cardiovascular risk factors and risk of dementia in late life. Neurology. 2005;64:277–281.
56. Yamada M, Kasagi F, Sasaki H, Masunari N, Mimori Y, Suzuki G. Association between dementia and midlife risk factors: the radiation effects research foundation adult health study. J Am Ger Soc. 2003;51:410–414.
57. Kivipelto M, Helkala EL, Laakso MP, et al. Apolipoprotein E epsilo4 allele, elevated midlife total cholesterol level and high midlife systolic blood pressure are independent risk factors for late life Alzheimer disease. Ann Intern Med. 2002;137:149–155.
58. Lindsay J, Laurin D, Verrault R, et al. Risk factors for Alzheimer’s disease: a prospective analysis form the Canadian Study of health and aging. Am J Epidemiol. 2002;156:445–453.
59. van Dijck EJ, Breteler M, Schmidt R, et al. The association between blood pressure, hypertension, and cerebral white matter lesions. Hypertension. 2004;44:625–650.
60. Kruyer A, Soplop N, Strickland S, Norris EH. Chronic hypertension leads to neurodegeneration in the TgSwDI mouse model of Alzheimer’s disease. Hypertension. 2015;66(1):175–182. Epub 2015 May 4. doi: 10.1161/HYPERTENSIONAHA.115.05524
61. Verhaaren BF, Vernooij MW, de Boer R, et al. High blood pressure and cerebral white matter lesion progressions in the general population. Hypertension. 2013;61(6):1354–1359. doi:10.1161/HYPERTENSIONAHA.111.00430
62. Venkat P, Chopp M, Chen J Models and mechanisms of vascular dementia. Exp Neurol. 2015;272:97–108
63. Jackson CA, Hutchinson A, Dennis MS, et al. Differing risk factor profiles of ischemic stroke subtypes: evidence for a distinctive lacunar arteriopathy. Stroke. 2010;41:624–629.
64. Verghese J, Lipton RB, Hall CB, Kuslansky G, Katz MJ. Low blood pressure and the risk of dementia in very old individuals. Neurology. 2003;61:1667–1672.
65. Zhu L, Viitanen M, Guo Z, Winblad B, Fratiglioni L. Blood pressure reduction, cardiovascular disease, and cognitive decline in the mini-mental state examination in a community population of normal very old people: a three year follow-up. J Clin Epidem. 1998;51(5):385–391.
66. Ruitenberg A, Skoog I, Ott AM, et al. Blood pressure and risk of dementia: results from the Rotterdam Study and the Gothenburg H-70 study. Dementia and Ger Cog Dis. 2001;12:33–39.
67. Burke WJ, Coronado PG, Schmitt CA, Gillespie KM, Chung HD. Blood pressure regulation in Alzheimer’s disease. J Auton Nerv Syst. 1994;48:65–71.
68. Guo Z, Fratiglioni L, Zhu L, Fastbom J, Winblad B, Viitanen M. Occurrence and progression of dementia in a community population aged 75 years and older: relationship of antihyperthensive medication use. Arch Neurol. 1999;59:991–996.
69. Brown WD, Frackowiak RSJ. Cerebral blood flow and metabolism studies in multi-infarct dementia. Alzh Dis Assoc Dis. 1991;5:131–143.
70. Guo Z, Vintanen M, Winblad B, Fratiglioni L. Low blood pressure and incidence of dementia in a very old sample: dependent on initial cognition. J Am Ger Soc. 1994;47:723–726.
71. Mattila K, Haavisto M, Rajala S, Heikinheimo R. Blood pressure and five year survival in the very old. Br Med J (Clin Res Ed). 1988;296:887–889.
72. Nilsson SE, Read S, Berg S, Johansson B, Melander A, Lindblad U. Low systolic blood pressure is associated with impaired cognitive function in the oldest old: longitudinal observations in a population-based sample 80 years and older. Aging Clin Exp Res. 2007;19(1):41–47.
73. Qiu C, Von Strauss E, Fastblom J, Winblad B, Fratiglion L. Low blood pressure and risk of dementia in the Kungsholmen project: a 6-year follw-up study. Arch Neurol. 2003;60:223–228.
74. In’t Veld BA, Ruitenberg A, Hofman A, Stricker BH, Bretrler MM. Antihypertensive drugs and incidence of dementia: the Rotterdam Study. Neurobiol Aging. 2001;22:407–412.
75. Bilazarian S. Hypertension guidelines: clear as mud. theHeart.org. 2014. Available from: http://www.medscape.com/viewarticle/819629. Accessed February 12, 2014.
76. Weber MA, Schiffrin EL, White WB, et al. Clinical practice guidelines for the management of hypertension in the community: a statement by the American Society of Hypertension and the International Society of Hypertension. J Hypertens. 2014;32:3–15.
77. ESH/ESC Task Force for the Management of Arterial Hypertension. 2014 practice guidelines for the management of arterial hypertension of the European Society of Hypertension (ESH) and the European Society of Cardiology (ESC): ESH/ESC task force for the management of arterial hypertension. J Hypertens. 2013;31:1925–1938.
78. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311:507–520.
79. Murthy VL, Shah RV, Rubenfire M, Brook RD. A comparison of the treatment implications of American Society of Hypertension and International Society of Hypertension 2013 and Eighth Joint National Committee Guidelines: an analysis of National Health and Nutrition Examination Survey. Hypertension. 2014;64:275–280.
80. Rakugi H. DB 02-2 Strict control of hypertension in the frail elderly: is it beneficial? J Hypertens. 2016;34 Suppl 1 – ISH 2016 Abstract Book: e190.
81. Kario K, Matsuo T, Kobayashi H, Imiya M, Mastuo M, Shimada K. Nocturnal fall of blood pressure and silent cerebrovascular damage in elderly hypertensive patients: advanced silent cerebrovascular damage in extreme dippers. Hypertension. 1996;27:130–135.
82. Nakamura K, Oita J, Yamaguchi T. Nocturnal blood pressure dip in stroke survivors: a pilot study. Stroke. 1995;26:1373–1378.
83. Watanabe N, Imai Y, Nagai K, et al. Nocturnal blood pressure and silent cerebrovascular lesions in elderly Japanese. Stroke. 1996;27:1319–1327.
84. Chamorro A, Pujol J, Saiz A, et al. Periventricular white matter lucencies in patients with lacunar stroke: a marker of too high or too low blood pressure? Arch Neurol. 1997;54:1284–1288.
85. De Buyzere M, Clement DL, Duprez D. Chronic low blood pressure: a review. Cardiovasc Drugs Ther. 1998.12:29–35.
86. Matsubayashi K, Okumiya K, Wada T, et al. Postural dysregulation in systolic blood pressure is associated with worsened scoring on neurobehavioral function tests and leukoaraiosis in the older elderly living in a community. Stroke. 1997;28:2169–2173.
87. Kohara K, Jiang Y, Igase M, et al. Postprandial hypotension is associated with asymptomatic cerebrovascular damage in essential hypertensive patients. Hypertension. 1999;33:565–568.
88. Gottesman R, Schneider A, Zhou Y ,et al. Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA. 2017. doi:10.1001/jama.2017-3090.
89. Wise J. Vascular risk factors show link to development of Alzheimer’s. BMJ. 2017;357:11847. Doi: 10.1136/bmj.J1847.
90. Jani B, Rajkumar C. Ageing and vascular ageing. Postgrad Med J. 2006;82(968):357–362.
91. de la Torre JC. Vascular basis of Alzheimer’s pathogenesis. Ann N Y Acad Sci. 2002;977:196–215.
92. Mathias CJ, Kimber JR. Postural hypotension: causes, clinical features, investigation, and management. Annu Rev Med. 1999;50:317–336.
93. Roriz-Filho JS, Bernardes Silva Filho SR, Rosset I, Roriz-Cruz M. Postural blood pressure dysregulation and dementia: evidence for a vicious circle and implications for neurocardiovascular rehabilitation. In: Halliday JT ,editor. Cardiac Rehabilitation. New York: Novascience Publisher Inc.; 2009:1–37. ISBN: 987-1-60741-918-1.
94. Londos E, Passant U, Gustafson L. Blood pressure and drug treatment in clinically diagnosed LBD and AD. Arch Gerontol Ger. 2000;30:35–46
95. Kaufman H, Biaggioni I. Autonomic failure in neurodegenerative disorders. Sem Neurol. 2003;23(4):351–363.
96. Puisieux F, Monaca P, Deplanque D, et al. Relationship between leuko—araiosis and blood pressure variability in the elderly. Eur Neur. 2001;46:115–120.
97. Burke WJ, Coronado PG, Schmitt CA, Gillespie KM, Chung HD. Blood pressure regulation in AD. J Auton Nerv Syst. 1994;48:65–71
98. Englund E. A White Matter Disease in Dementia. A Study with Special Reference to AD [thesis]. Lund: Lund University; 1988:7–43.
99. Weiner DE, Gaussoin SA, Nord J, et al. Cognitive function and kidney disease: baseline data from the Systolic Blood Pressure Intervention Trial (SPRINT). Am J Kidney Dis. 2017; 70(3): 357–367
100. Fisher CM. The arterial lesions underlying lacunes. Acta Neuropathol. 1969;12:1–15. doi:10.1007/BF00685305
101. Pantoni L, Garcia JH, Gutierrez JA. Cerebral white matter is highly vulnerable to ischemia. Stroke. 1996;27:1641–1647.
102. Schmidt R, Schmidt H, Haybaeck J, et al. Heterogeneity in age-related white matter changes. Acta Neuropathol. 2011;122:171–185. doi:10.1007/s00401-011-0851-x
103. Hommet C, Mondon K, Constans T, et al. Review of cerebral microangiopathy and Alzheimer’s disease: relation between white matter hyperintensities and microbleeds. Dement Geriatr Cogn Disord. 2011;32:367–378. doi:10.1159/000335568
104. Munoz DG, Hastak SM, Harper B, et al. Pathologic correlates of increased signals of the centrum ovale on magnetic resonance imaging. Arch Neurol. 1993;50: 492–497.
105. Jellinger KA, Attems J. Prevalence and pathology of vascular dementia in the oldest-old. J Alzheimers Dis. 2010;21:1283–1293. doi:10.3233/JAD-2010-100603
106. Ince P. Acquired forms of vascular dementia. In: Kalimo H, editor. Cerebrovascular Diseases. Basel: ISN Neuropath Press; 2005:316–323.
107. Hachinski V, Iadecola C, Petersen RC, et al. National Institute of Neurological Disorders and Stroke-Canadian Stroke-Network Vascular cognitive impairment harmonization standards. Stroke. 2006;37:2220–2241. doi:10.1161/01.STR.0000237236.88823.47
108. Attems J, Jellinger K, Thal DR, Van Nostrand W. Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol. 2011;37:75–93. doi:10.1111/j.1365-2990.2010.01137.x
109. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi:10.1126/science.1072994
110. Jiménez-Balado J, Riba-Llena I, Garde E, et al. Prevalence of hippocampal enlarged perivascular spaces in a sample of patients with hypertension and their relation with vascular risk factors and cognitive function. J Neurol Neurosurg Psychiatry. 2018;89:1–6. doi:10.1136/jnnp-2017-316724.
111. Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018. doi:10.1038/nrneurol.2017.188
112. Abbott NJ, Pizzo ME, Preston JE, Janigro D, Thorne RG. The role of brain barriers in fluid movement in the CNS: is there a ‘glymphatic’ system? Acta Neuropathol. 2018;135(3):387–407. doi:10.1007/s00401-018-1812-4
113. Huijts M, Duits A, Staals J, et al. Basal ganglia enlarged perivascular spaces are linked to cognitive function in patients with cerebral small vessel disease. Curr Neurovasc Res. 2014;11:136–141. doi:10.2174/1567202611666140310102248
114. Potter GM, Marlborough FJ, Wardlaw JM. Wide variation in definition, detection and description of lacunar lesions on imaging. Stroke. 2011;42:359–366. doi:10.1161/STROKEAHA.110.594754
115. Potter GM, Doubal FN, Jackson CA, et al. Counting cavitating lacunes underestimates the burden of lacunar infarction. Stroke. 2010;41: 267–272. doi:10.1161/STROKEAHA.109.566307
116. Shi Y, Wardlaw JM. Update on cerebral small vessel disease. A dynamic whole-brain disease. Stroke Vasc Neurology. 2016;1:
117. Wardlaw JM, Doubal F, Armitage P, et al. Lacunar stroke is associated with diffuse blood brain barrier dysfunction. Ann Neurol. 2009;65:194–202. doi:10.1002/ana.21549
118. De Silva TM, Faraci FM. Microvascular dysfunction and cognitive impairment. Cell Mol Neurobiol. 2016;36(2):241–258. Doi:10.1007/s10571-015-0308-1.
119. Joutel A, Faraci FM. Cerebral small vessel disease; insights and opportunities from mouse models of collagen IV related small vessel disease and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke. 2014;45:1215–1221. doi:10.1161/STROKEAHA.113.002878
120. Lammie AG. Small vessel disease. In: Kalimo H., editor. Cerebrovascular Diseases. Basel: ISN Neuropath Press; 2005:85–91.
121. Rosenberg GA. Neurological diseases in relation to the blood- brain barrier. J Cereb Blood Flow Metab. 2012;32:1139–1151. doi:10.1038/jcbfm.2011.197
122. Pantoni L. Pathophysiology of age-related cerebral white matter changes. Cerebrovasc Dis. 2002;13(2):7–10. doi:10.1159/000049143
123. Englund E. White matter pathology of vascular dementia. In: Chui E editor. Vascular Dementia. London: M. Dunitz; 2004:117–130.
124. Englund EA, Person B. Correlations between histopathologic white matter changes and proton MR relaxation times in dementia. Alzheimer Dis Assoc Disord. 1987;1:156–170. doi:10.1097/00002093-198701030-00008
125. Román GC Senile dementia of the binswanger type: a vascular form of dementia in the elderly. JAMA. 1987;258:1782–1788. doi:10.1001/jama.1987.03400130096040
126. Moody DM, Brown WR, Challa VR, Anderson RL. Periventricular venous collagenosis: association with leukoaraiosis. Radiology. 1995;194:469–476. doi:10.1148/radiology.194.2.7824728
127. Vinters HV, Ellis WG, Zarow C, et al. Neuropathological substrate of ischemic vascular dementia. J Neuropathol Exp Neurol. 2000;59:931–945. doi:10.1093/jnen/59.11.931
128. Garcia JH, Lassen NA, Weiller C, Sperling B, Nakagawara J. Ischemic stroke and incomplete infarction. Stroke. 1996;27:761–765.
129. Dalkara T, Alarcon-Martinez L. Cerebral micro-vascular signaling in health and disease. Brain Res. 2015;1623:3–17. doi: 10.1016/j.brainres.2015.03.047
130. Giannakopoulos P, Gold G, Kowaru E, et al. Assessing the cognitive impact of Alzheimer disease pathology and vascular burden in the aging brain: the Geneva experience. Acta Neuropathol. 2007;113:1–12. doi:10.1007/s00401-006-0144-y
131. Launer LJ, Hughes TM, White LR. Microinfarcts, brain atrophy, and cognitive function: the Honolulu Asia Aging Study Autopsy Study. Ann Neurol. 2011;70:774–780. doi:10.1002/ana.22520
132. Smallwood A, Oulhaj A, Joachim C, et al. Cerebral subcortical small vessel disease and its relation to cognition in elderly subjects: a pathological study in the Oxford Project to Investigate Memory and Ageing (OPTIMA) cohort. Neuropathol Appl Neurobiol. 2012;38:337–343. doi:10.1111/j.1365-2990.2011.01221.x
133. Kramer JH, Reed BR, Mungas D, Weiner MW, Chui H Executive dysfunction in subcortical ischaemic vascular disease. J Neurol Neurosurg Psychiatr. 2002;72:217–220. doi:10.1136/jnnp.72.2.217
134. Burton E, Ballard C, Stephens S, et al. Hyperintensities and fronto-subcortical atrophy on MRI are substrates of mild cognitive deficits after stroke. Dement Geriatr Cogn Disord. 2003;16:113–118. doi:10.1159/000070684
135. Tullberg M, Fletcher E, DeCarli C, et al. White matter lesions impair frontal lobe function regard- less of their location. Neurology. 2004;63:246–253. doi:10.1212/01.WNL.0000130530.55104.B5
136. Gold G, Kovari E, Herrmann FR, et al. Cognitive consequences of thalamic, basal ganglia, and deep white matter lacunes in brain aging and dementia. Stroke. 2005;36:1184–1188. doi:10.1161/01.STR.0000166052.89772.b5
137. Van Der Veen PH, Muller M, Vinken KL, et al. Longitudinal relationship between cerebral small vessel disease and cerebral blood flow. The second manifestations of arterial disease-magnetic resonance study. Stroke. 2015;46:1233–1238 doi:10.1161/STROKEAHA.114.008030
138. Gouw AA, van Der Flier WM, Fazekas F et al. Progression of white matter hyperintensities and incidence of new lacunes over a 3-year period: the leukoaraiosis and disability study. Stroke. 2008;39:1414–1420. doi:10.1161/STROKEAHA.107.498535
139. Schmidt R, Seiler S, Loitfelder M. Longitudinal change of small vessel disease related brain abnormalities. J Cerebr Blood Flow Metab. 2016;36:26–39. doi:10.1038/jcbfm.2015.72
140. Munoz-Maniega S, Chappell FM, Valdes-Henrandez ML, et al. Integrity of normal appearing white matter: influence of age, visible lesion burden and hypertension in patients with small-vessel disease. J Cerebr Blood Flow Metab. 2016. Doi: 10.1177/027168x16635657.
141. Nichols WW, O’Rourke MF McDonald’s Blood Flow in Arteries: Theoretical, Experimental and Clinical Principles,
142. Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises. Part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107:139–146. doi:10.1161/01.CIR.0000048892.83521.58
143. Ganong MD, William F. Review of Medical Physiology,
144. Volpe M, Scuteri A. Management of risk factors in vascular dementia: hypertension. Hot Topics Neurol Psychiatry. 2008;1:17–22.
145. Najjar SS, Scuteri A, Lakatta EG. Arterial aging: is it an immutable cardiovascular risk factor? Hypertension. 2005;46:454–462. doi:10.1161/01.HYP.0000177474.06749.98
146. O’Rourke MF, Safar ME. Relationship between aortic stiffening and microvascular disease in brain and kidney: cause and logic of therapy. Hypertension. 2005;46:200–204. doi:10.1161/01.HYP.0000184108.12155.6b
147. Cervós-Navarro J, Matakas F, Roggendorf W, Christmann U. The morphology of spastic intracerebral arterioles. Neuropathol Appl Neurobiol. 1978;4:369–379. doi:10.1111/j.1365-2990.1978.tb01349.x
148. Laurent S, Katsahian S, Fassot C, et al. Aortic stiffness is an independent predictor of fatal stroke in essential hypertension. Stroke. 2003;34:1203–1206. doi:10.1161/01.STR.0000065428.03209.64
149. Baumbach GL, Siems JE, Heistad DD. Effects of local reduction in pressure on distensibility and composition of cerebral arterioles. Circ Res. 1991;68:338–351. doi:10.1161/01.RES.68.2.338
150. Liao D, Cooper L, Cai J, et al. The prevalence and severity of white matter lesions, their relationship with age, ethnicity, gender, and cardiovascular disease risk factors: the ARIC Study. Neuroepidemiology. 1997;16:149–162 doi:10.1159/000368814
151. Paulson OB, Strandgaard S, Edvinson L. Cerebral autoregulation. Cerebrovasc Brain Metab Rev. 1990;2:161–192.
152. Dawson SL, Blake MJ, Panerai RB, Potter JF. Dynamic but not static cerebral autoregulation is impaired in acute ischemic stroke. Cerebrovasc Dis. 2000;10:126–132. doi:10.1159/000016041
153. Novak V. Cognition and Hemodynamics. Curr Cardiovasc Risk Rep. 2012;6(5):380–396. doi:10.1007/s12170-012-0260-2
154. Shim Y, Yoon B, Shim DS, Kim W, An JY, Yang DW. Cognitive correlates of cerebral vasoreactivity on transcranial doppler in older adults Journal of Stroke and Cerebrovascular Diseases, 2015:1262–1269 doi:10.1016/j.jstrokecerebrovasdis.2015.01.031
155. Roher AE, Garami Z, Tyas SL, et al. Transcranial Doppler ultrasound blood flow velocity and pulsatility index as systemic indicators for Alzheimer’s disease. Alzheimers Dement. 2011;7(4):445–455. doi:10.1016/j.jalz.2010.08.229
156. Wallin A, Blennow K, Gottfries CG. Neurochemical abnormalities in vascular dementia. Dementia. 1989;13:658–667.
157. Szilagy AX, Nemeth A, Martini E, et al. Serum and CSF cholinesterase activity in various kind of dementia. Eur Arch Psych Neur Sci. 1987;236:309–311. doi:10.1007/BF00380958
158. Sakurada I, Alufuzoff I, Winblad B, et al. Substance P like immunoreactivity, choline acetyltransferase activity and cholinergic muscarinic receptors in AD and MID. Brain Res. 1990;521(1–29):329–332. doi:10.1016/0006-8993(90)91561-t
159. Serrador JM, Freeman R. Enhanced cholinergic activity improves cerebral blood flow during orthostatic stress. Frontiers in Neurol. 2017;8: 103. Doi:10.3389/fneur.2017.00103.
160. Tune L, Brandt J, Frost JJ, et al. Physstigmine in AD: effects on cognitive functioning, cerebral glucose metabolism analyzed by single photon emission tomography. Acta Psych. Scand Suppl. 1991;366:61–65. doi:10.1111/j.1600-0447.1991.tb03111.x
161. Scremin OU, Sonneschein RR, Rubinstein EH. Cholinergic cerebral vasodilatation in the rabbit: absence of concomitant metabolic activation. J Cereb Blood Flow Metab. 1982;2(2):241–247. doi:10.1038/jcbfm.1982.24
162. Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging, 2001;18(9):685–716. doi:10.2165/00002512-200118090-00004
163. Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–795. doi:10.1038/nature05292
164. Raz N. The ageing brain: structural changes and their implications for cognitive ageing. In: Dixon R, Bäckman L, Nilsson L, editors. New Frontiers in Cognitive Ageing. Telangana, India: Oxford University Press; 2004:115–134.
165. Petito C. Trasnformation of postisichemic perineuronal glial cells. I. Electron microscope studies. J Cerebr Blood Flow Metabol. 1986;6:616–624. doi:10.1038/jcbfm.1986.109
166. Petito CK, Olarte JP, Roberts B, Nowak TS, Pulsinelli WA. Selective glial vulnerability following transient global ischemia in rat brain. J Neuropath Exp Neurol. 1998;57(3):231–238. doi:10.1097/00005072-199803000-00004
167. Irving EA, Nicoll J, Graham DI, Dewar D. Increased tau immunoreactivity in oligodendrocytes following human stroke and head injury. Neurosci Lett. 1996;213:189–192. doi:10.1016/0304-3940(96)12856-1
168. Irving EA, Yatsushiro K, McCullough J, Dewar R. Rapid alteration of tau in oligodendrocytes after focal ischemic injury in the rat: involvement of free radical. J Cererb Blood Flow Metab. 1997;17:612–622.
169. Furukawa S, Sameshima H, Yang L, Hariskuma M, Ikenoue T. Regional differences of microglial accumulation within 72 hrs of hypoxia-ischemia and the effect of acetylcholine receptor agonist on brain damage and microglial activation in newborn rats. Brain Res. 2014;1562:52–58. doi:10.1016/j.brainres.2014.03.028
170. Gehrmann J, Bonnekoh P, Miyazawa T. Immunoistochemical study of an early microglial activation in ischemia. J Cer Blood Flow Metab. 1992;12:257–269. doi:10.1038/jcbfm.1992.36
171. Rupalla K, Allegrin PR, Saver D. Time course of microglia activation and apoptosis in various brain regions after permanent focal cerebral ischemia in mice. Acta Neuropathol. 1998;96:172–178.
172. Czeh M, Gressens P, Kaindl AM. The yin and yang of microglia. Dev Neurosci. 2011;33:199–209. doi:10.1159/000328989
173. Yuan TH, Yu HM. Notch signaling: key role in intrauterin infection/inflammation, embryonic development and white matter damage. J Neurosci Res. 2010;88:461–468. doi:10.1002/jnr.22229
174. Irving EA, Bentley DL, Parsons AA. Assessment of white matter injury following prolonged focal cerebral ischaemia in the rat. Acta Neuropathol. 2001;102(6):627–635.
175. Tomimoto H, Ihara M, Wakita H, et al. Chronic cerebral hypoeprfusion induces white matter lesions and loss of oligodendroglia with DNA fragmentation in the rat. Acta Neuropathol. 2003;106(6):527–534. doi:10.1007/s00401-003-0749-3
176. Wakita H, Tomimoto H, Akiguchi I, et al. Axonal damage and demyelination in the white matter after chronic cerebral hypoperfusion in the rat. Brain Res. 2002;924(1):63–70. doi:10.1016/s0006-8993(01)03223-1
177. Wakita H, Tomimoto H, Akiguchi I, Kimura J. Glial activation and white matter changes in the rat brain induced by chronic cerebral hypoperfusion: an immunoistochemical study. Acta Neuropathol. 1994;87(5):484–492.
178. Farkas E, Donka G, de Vous RAI, Mihaly A, Bari F, Luiten PGM. Experimental cerebral hypoeprfusion induces white matter injury and microglial activation in the rat brain. Acta Neuropathol. 2004;108: 57–64. doi:10.1007/s00401-004-0864-9
179. Zhang ZG, Bower L, Zhang RL, Chen S, Windham JP, Chopp M. Three dimensional measuremtn of cerebral microvascular plasma perfusion, glial fibrillary acid protein and microtubule associated P-2 immunoreactivity after embolic stroke in rats: a double fluorescent labeled laser scanning confocal microscopic study. Brain Res. 1999;844(1–2):55–66. doi:10.1016/s0006-8993(99)01886-7
180. Tomimoto H, Akiguchi I, Wakita H, Svenaga T, Nakamura S, Kimura J Regressive changes of astroglia in white matter lesions in cerebrovascular disease and AD patients. Acta Neuropathol. 1997;94(2): 146–152. doi:10.1007/s004010050686
181. Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844–866 doi:10.1016/j.neuron.2013.10.008
182. Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844–866. Doi:10.1016/jneuron.2013.10.008.
183. Lo EH, Rosenberg GA. The neuroscience unit in health and disease introduction. Stroke. 2009;40:S2–S3. doi:10.1161/STROKEAHA.108.534404
184. Filous AS, Silver J. Targeting astrocytes in CNS injury and disease: a translational research approach. Prog Neurobiol. 2016;144:173–187. Doi:10.1016/j.pneurobio.2016.03.009.
185. Cai W, Zhang K, Li P, et al. Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative disease: an ageing effect. Ageing Res Rev. 2017;34:77–87. Doi: 10.1016/jar.2016.09.006.
186. Iadecola C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron. 2017;98(1):17–42. Doi:10.1016/jneuron.2017.07030.
187. Raichle ME, Mintun MA. Brain work and brain imaging. Annu Rev Neurosci. 2006;29:449–476. doi:10.1146/annurev.neuro.29.051605.112819
188. Uhurovoa H, Kilic K, Tian P, et al. Cell-type specificity of neurovascular coupling in cerebral cortex. ELife. 2016;5:155. Doi.10.7554/elife.14315.
189. Longden TA, Dabertrand F, Koide M, et al. Capillary K+ sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat Neurosci. 2017: 275: A1489–14812.doi.10.1038/nm.4533.
190. Payabavash S, Souza LCS, Wang Y, et al. Regional ischemic vulnerability of the brain to hypoperfusion: the need for location specific CT perfusion thresholds in acute stroke patients. Stroke. 2011;42(5):1255–1260. doi: 10.1161/STROKEAHA.110.600940
191. Cheng B, Golsari A, Fiehler J, Rosenkranz M, Gerloff C, Thomalla G. Dynamics of regional distribution of ischemic lesions in middle cerebral artery trunk occlusion relates to collateral circulation.J Cereb Blood Flow Metab. 2010. doi: 10.1038/jcbfm.2010.185.
192. Dijkhuizen RM, Knollema S, van der Worp HB, et al. Dynamics of cerebral tissue injury and perfusion after temporary hypoxia-ischemia in the rat: evidence for region-specific sensitivity and delayed damage. Stroke. 1998;29:695–704
193. Garcia JH, Liu KF, Ye ZR, Gutierrez JA. Incomplete infarct and delayed neuronal death after transient middle cerebral artery occlusion in rats. Stroke. 1997;28:2303–2309.
194. Konaka K, Miyashita K, Naritomi H. Changes in diffusion-weighted magnetic resonance imaging findings in the acute and subacute phases of anoxic encephalopathy. J Stroke Cerebrovasc Dis. 2007;16:82–83. doi:10.1016/j.jstrokecerebrovasdis.2006.10.007
195. Ravens JR. Vascular changes in the human senile brain. Adv Neurol. 1978;20:487–501
196. Klassen AC, Sung JH, Stadlan EM. Histological changes in cerebral arteries with increasing age. J Neuropathol Exp Neurol. 1968;27:607–623. doi:10.1097/00005072-196810000-00006
197. Mega MS, Cummings JL. Frontal-subcortical circuits and neuropsychiatric disorders. J Neuropsychiatry Clin Neurosci. 1994;6:358–370. doi:10.1176/jnp.6.4.358
198. Yao H, Sadoshima S, Kuwabara Y, et al. Cerebral blood flow and oxygen metabolism in patients with vascular dementia of the Binswanger type. Stroke. 1990;21:1694–1699. doi:10.1161/01.STR.21.12.1694
199. Furuta A, Ishii N, Nishihara Y, Horie A. Medullary arteries in aging and dementia. Stroke. 1991;22:442–446. doi:10.1161/01.STR.22.4.442
200. Tak S, Yoon SJ, Jang J, Yoo K, Jeong Y, Ye JC. Quantitative analysis of hemodynamic and metabolic changes in subcortical vascular dementia using simulataneous near-infrared spectroscopy and FMRI measurements. Neuroimage. 2011;55:176–184. doi:10.1016/j.neuroimage.2010.11.046
201. Schroeter M, Cutini S, Wahl M, Scheid R, Yves von Cramon D. Neurovascualr coupling is impaired in cerebral microangiopathy an event related stroop study. Neuroimage. 2007;34(1):26–34. doi:10.1016/j.neuroimage.2006.09.001
202. Bar K, Boettger M, Seidler N, Mentzelh TC, Sauer H. Influence of galantamine on vasomotor reactivity in AD and vascular dementia due to microangiopathy. Stroke. 2007;38(12):3186–3192. doi:10.1161/STROKEAHA.107.492033
203. De Reuck J, Decoo D, MArchau M, Santens P, Lemahieu I, Strijckmans K. Positron emission tomography in vascular dementia. J Neurol Sci. 1998;154(1):55–61.
204. Yoshikawa T, Murase K, Oku N, et al. Statistical image analysis of cerebral blood flow in vascular dementia with small-vessel disease. J Nucl Med. 2003;44(4):505–511.
205. Yang D, Kim B, Park J, Kim S, Kim E, Sohn H. Analysis of cerebral blood flow of subcortical vascular dementia with single photon emission computed tomography: adaptation of statistical aprametric mapping. J Neurol Sci J Biomed. 2002;203:199–205. doi:10.1016/S0022-510X(02)00291-5
206. Ginsberg S, Martin L. Axonal transection in adult rat brain induces transynaptic apoptosis and persistent atrophy of target neurons. J Neurotrauma. 2002;19(1):99–109. doi:10.1089/089771502753460277
207. Heiss W, Podreka I. Role of the PET and sPECT in the assessment of ischemic cererbovascular disease. Cerebrovasc Brain Metab Rev. 1993;5(4):235–263.
208. Chui H. Subcortical ischemic vascular dementia (SIVD). Neurol Clin. 2007;25(3):717. doi: 10.1016/j.ncl.2007.04.003
209. Perry EK, Tomlinson BE, Blessed G, Bergmann K, Gibson PH, Perry RH. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. BMJ. 1978;2:1457–1459. doi:10.1136/bmj.2.6150.1457
210. Erkinjuntti T, Kurz A, Gauthier S, Bullock R, Lilienfeld S, Damaraju CV. Efficacy of galantamine in probable vascular dementia and Alzheimer’s disease combined with cerebrovascular disease: a randomized trial. Lancet. 2002;359:1283–1290. doi:10.1016/S0140-6736(02)07571-2
211. Wilkinson D, Doody R, Helme R, et al. Donepezil in vascular dementia A randomized, placebo-controlled study. NEUROLOGY. 2003;61:479–486. doi:10.1212/01.WNL.0000078943.50032.FC
212. Pratt RD, Perdomo CA, Surick IW, Ieni JR. Donepezil: tolerability and safety in Alzheimer’s disease. Int J Clin Pract. 2002;56:710–717.
213. Auchus AP, Brashear HR, Salloway S, et al. Galantamine treatment of vascular dementia: a randomized trial. Neurology. 2007; 69: 448–458. doi:10.1212/01.wnl.0000266625.31615.f6
214. Ballard C, Sauter M, Scheltens P, et al. Efficacy, safety and tolerability of rivastigmine capsules in patients with probable vascular dementia: the VantagE study. Curr Med Res Opin. 2008; 24: 2561–2574. doi:10.1185/03007990802328142
215. Borovikova LV, Ivanova S, Zhang M, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. doi:10.1038/35013070
216. Wang H, Yu M, Ochani M, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi:10.1038/nature01339
217. Conejero-Goldberg C, Davies P, Ulloa L. Alpha7 nicotinic acetylcholine receptor: a link between inflammation and neurodegeneration. Neurosci Biobehav Rev. 2008;32:693–706. doi:10.1016/j.neubiorev.2007.10.007
218. Pavlov VA, Tracey KJ. Controlling inflammation: the cholinergic anti-inflammatory pathway. Biochem Soc Trans. 2006;34:1037–1040. doi:10.1042/BST0341037
219. Maelicke A. Allosteric modulation of nicotinic receptors as a treatment strategy for Alzheimer’s disease. Dement Geriatr Cogn Disord. 2000;11 Suppl 1: 11–18. doi:10.1159/000051227
220. Tohgi H, Abe T, Kimura M, Saheki M, Takahashi S. Cerebrospinal fluid acetylcholine and choline in vascular dementia of Binswanger and multiple small infarct types as compared with Alzheimer-type dementia. J Neural Transm. 1996;103:1211–1220. doi:10.1007/BF01271206
221. Wallin A, Sjogren M, Blennow K, Davidsson P. Decreased cerebrospinal fluid acetylcholinesterase in patients with subcortical ischemic vascular dementia. Dement Geriatr Cogn Disord. 2003;16:200–207. doi:10.1159/000072803
222. Yamada M, Lamping KG, Duttaroy A, et al. Cholinergic dilatation of cerebral blood vessels is abolished in M5 muscarinic acetylcoline receptor knockout mice. Prc Natl Ac Sci USA. 2001;98:14096–14101. doi:10.1073/pnas.251542998
223. Togashi H, Matsumoto M, Yoshioka M, et al. Neurochemical profiles in cerebrospinal fluid of stroke-prone spontaneously hypertensive rats. Behav Lett. 1994;166:117–120.
224. Togashi H, Kimura S, Matsumoto M, et al. Cholinergic changes in the hippocampus of stroke-prone spontaneously hypertensive rats. Stroke. 1996;27:520–526.
225. Scremin OU, Li MG, Scremin AM, Jenden DJ. Cholinesterase inhibition improves blood flow in the ischemic cerebral cortex. Brain Res Bull. 1997;42:59–70. doi:10.1016/S0361-9230(96)00207-9
226. Furukawa S, Sameshima H, Yang L, Ikenove T. Activation of acetylcholine receptros and microglia in hypoxic-ischemic brain damage in newborn rats. Brain and Develop. 2013;35:607–613. doi:10.1016/j.braindev.2012.10.006
227. Hejmadi MV, Dajas-BAilador F, Barns SM, Jones B, Wonnacott S. Neuroprotection by nicotine against hypoxia-induced apopotosis in cortical cultures involves activation of multiple nicotinic acethylcholine receptor subtypes. Mol Cell Neurosci. 2003;24:779–786.
228. Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120. doi:10.1152/physrev.00015.2008
229. Alkondon M, Pereira EF, Eisenberg HM, Albuquerque EX. Choline and selective antagonists identify two subtypes of nicotinic acetylcholine receptors that modulate GABA release from CA1 interneurons in rat hippocampal slices. J Neurosci. 1999;19:2693–2705.
230. Alkondon M, Albuquerque EX. The nicotinic acetylcholine receptor subtypes and their function in the hippocampus and cerebral cortex. Prog Brain Res. 2004;145:109–120. doi:10.1016/S0079-6123(03)45007-3
231. Court J, Perry E, Kalaria R. Neurotransmitter control of the cerebral vasculature and abnormalities in vascular dementia. In: Erkinjuntti T, Gauthier S, editors. Vascular Cognitive Impairment. Martin Dunitz: London; 2002:167–185.
232. Roman GC, Kalaria RN. Vascular determinants of cholinergic deficits in AD and vascular dementia. Neurobiol Aging. 2006;27:1769–1785.
233. Roman GC. Brain Hypoperfusion: a critical factor in vascular dementia. Neurol Res. 2004;26:454–458 doi:10.1179/016164104225017686
234. Scarr E. Muscarinc receptors: their roles in disorders of the central nervous system and potential therapeutic targets. CNS Neurosci Ther. 2012;18:369–379. Doi 10.1111/j.1755-5949.201100249.x
235. Perry E, Walker M, Grace J, Perry R. Acetylcholine in mind: a neurotransmitter correlate of consciousness? Trends Neurosci. 1999;22:273–280
236. Vasquez J, Purve MJ. The cholinergic pathway to cerebral blood vessels. I. Morphological Studies. Pflugers Arch. 1979;379:157–163. doi:10.1007/BF00586942
237. Tong XK, Hamel E. Regional cholinergic denervation of cortical microvessels and nitric oxid synthase-containing neurons in AD. Neuroscience. 1999;92:163–175.
238. Cauli B, Tong XK, Rancillac A, et al. Cortical GABA interneurons in neurovascular coupling: relays for the subcortical vasoactive pathways. J Neurosci. 2004;24:8940–8949. doi:10.1523/JNEUROSCI.3065-04.2004
239. Vanhoutte PM. Endothelium and control of vascular function. State of the art lecture. Hypertension. 1989;13:658–667.
240. Kocharyan A, Fernandes P, Tong XP, Vaucher E, Hamel E. Specific subtypes of cortical GABA interneurons contribute to the neurovascular coupling response to basal forebrain stimulation. J Cerebr Blood Flow Metab. 2008;28:221–231. doi:10.1038/sj.jcbfm.9600558
241. Lacombe P, Sercombe R, Vaucher E, Seylaz J. reduced cortical vasodilatory response to stimulation of the nucleus basalis of Meynert in the aged rats and evidence for a control of the cerebral circulation. Ann Ny Acad Sci. 1997;826:410–415. doi:10.1111/j.1749-6632.1997.tb48494.x
242. Elhusseiny A, Hamel E. Muscarinic but not nicotinic acetylcholine receptros mediated nitric oxide dependent dilatation in brain cortical arterioles: a possible role for the M5 receptor subtype. J Cerebr Blood Flow Metab. 2000:298–305. doi:10.1097/00004647-200002000-00011
243. Yamada M, Lamping KG, Duttaroy A, et al. Cholinergic dilation of cerebral blood vessels is abolished in M5 muscarinic acetylcholine receptor knockout mice. PNAS. 2001;98(24):14096–14101. doi:10.1073/pnas.251542998
244. Salloway S. Subcortical Vascular Dementia: Binswanger’s and CADASIL. Honolulu: American Academy of Neurology (AAN); 2003:(CD 8AC.006-2):1–29.
245. Mirski MA. Pharmacology of Blood Pressure Management during Cerebral Ischemia. Miami: American Academy of Neurology (AAN); 2005:5PC-004:456–469.
246. Wallin A, Blennow K, Gottfries CG. Neurochemical abnormalities in vascular dementia. Dementia. 1989;1:120–130.
247. Bohnen NI, Muller MLTM, Kuwabara H, Ocnstantien GM, Studentski SA. Age-associated leukoaraiosis and cortical cholinergic deafferentation. Neurology. 2009;72:1411–1416. doi:10.1212/WNL.0b013e3181a187c6
248. Szilagy AK, Nemeth A, Martini E, Lendvai B, Venter V. Serum and CSF cholinesterase activity in various kind of dementia. Eur Arch Psychiatry Neurol Sci. 1987;236:309–311.
249. Kimura S, Saito H, Minami M, et al. Pathogenesis of vascular dementia in stroke-prone spontaneously hypertensive rats. Toxicology. 2000;153:167–187. doi:10.1016/S0300-483X(00)00312-7
250. Tomimoto H, Ohtani R, Shibata M, Nakamura N, Ihara M Loss of cholinergic pathways in vascular dementia of the Binswanger type. Dement Geriatr Cogn Disord 2005; 19:282–288. doi:10.1159/000084553
251. Wang J, Zhang HY, Tang XC. Cholinergic deficiency involved in vascular dementia: possible mechanism and strategy of treatment. Acta Pharamcol Sinica. 2009;30:879–888. doi:10.1038/aps.2009.82
252. Mann DM, Yates PD, Marcyniuk B. The nucleus basalis of Meynert in multi-infarct dementia. Acta Neuropathol. 1986;71:332–337.
253. Sakurada T, Alufuzoff I, Winblad B, Nordberg A. Substance P like immunoreactivity, choline acetyltransferase activity and cholinergic muscarinic receptors in Alzheimer’s disease and multi-infarct dementia. Brain Res .1990;521:329–332. doi:10.1016/0006-8993(90)91561-t
254. Jung S, Zarow C, Mack WJ, et al. Preservation of neurons of the nucleus basalis in subcortical ischemic vascular disease. Arch Neurol. 2012;69:879–886. doi: 10.1001/archneurol.2011.2874
255. Swartz RH, Sahlas DJ, Black SE. Strategic involvement of cholinergic pathways and executive dysfunction: does location of white matter signal hyperintensities matter? J Stroke Cerebrovasc Dis. 2003;12:29–36. doi:10.1053/jscd.2003.5
256. Wardlaw JM, Smith C, Dichgans M. Mechanism of sporadic cerebral small vessel disease: insight from neuroimaging. Lancet Neurol. 2013;12:483–497. doi:10.1016/S1474-4422(13)70060-7
257. De Silva TM, Miller AA. Cerebral small vessel disease: targeting oxidative stress as a novel therapeutic strategy. Frontiers in Pharmacol. 2016. doi:10.3389/fpharm.2016.00061.
258. Chan SL, Umesalma S, Baumbach GL. Epidermal growth factor receptor is critical for angiotensin II mediated hypertrophy in cerebral arterioles. Hypertension. 2015;65:808–812. Doi: 10.1161/HYEPRTENSIONHA.114.04794;
259. Umesalma S, Houwen FK, Baumbach GL, Chan SL. Roles of ceveolin-1 in angiotensin-ii induced hypertrophy and inward remodeling of cerebral pial arterioles. Hypertension. 2016;67:623–629.
260. Zhang R, Zuckerman JH, Iwasaki K, Wilson TE, Crandall CG, Levine BD. Autonomic neural control of dynamic cerebral autoregulation in humans. Circul. 2002;106:1814–1820. doi:10.1161/01.CIR.0000031798.07790.FE
261. Hamner JW, Tan CO, Lee K, Cohen MA, Taylor JA. Sympathetic control of the cerebral vasculature in humans. Stroke. 2010;41;102–109. doi:10.1161/STROKEAHA.109.557132
262. Hamner JW, Tan CO, Tzeng YC, Taylor JA. Cholinergic control of the cerebral vasculature in humans. J Physiol. 2012;590(24):6343–6352. doi:10.1113/jphysiol.2012.245100
263. Brown WR, Thore CR. Review: cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol Appl NeuroBiol. 2011;37:56–74. doi:10.1111/j.1365-2990.2010.01139.x
264. Barrett KM, Brott TG, Brown RD Jr, et al. Enhancing recovery after acute ischemic stroke with donepezil as an adjuvant therapy to standard medical care: results of a phase IIA clinical trial. J Stroke Cerebrovasc Dis. 2011;20:177–182. doi:10.1016/j.jstrokecerebrovasdis.2010.12.009
265. Silver JM, Koumaras B, Mneg X, et al. Long term effects of rivastigmine capsules in patients with transient brain injury. Brain Inj. 2009;23:123–132. doi:10.1080/02699050802649696
266. Gericke A, Sniatechi JJ, Mayer VG, et al. Role of the M1, M3 and M5 muscarinic Ach receptors in cholinergic dilatation of small Arteries studied with gene targeted mice. Am J Physiol Heart Circ Physiol. 2011;300; H1602–H1608. doi:10.1152/ajpheart.00982.2010
267. Elhusseiny A, Hamel E. Muscarinic, but not nicotinic acetylcholine receptors mediate nitric oxid dependent dilation in brain cortical arterioles: a possible role for the M5 receptor subtype. J Cerebr Blood Flow Metab. 2000;20;298–305. doi:10.1097/00004647-200002000-00011
268. Katusic ZS, Austin SA. Endothelial nitric oxide: protector of a healthy mind. Eur Heart J. 2014;35:888–894. doi:10.1093/eurheartj/eht544
269. Joutel A. The Notch3-ECD cascade hypothesis of cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy disease. Neurol Clin Neurosci. 2015;3:1–6. doi:10.1111/ncn3.2015.3.issue-1
270. Ridder DA, Wenzel J, Muller K, et al. Brain endothelial TAK1 and NEMO safeguard the neurovascular unit. J Exp Med. 2015;212:1529–1549. doi:10.1084/jem.20150165
271. Román GC. Brain hypoperfusion: a critical factor in vascular dementia. Neurol Res. 2004; 26:454–458. doi:10.1179/016164104225017686
272. Zhan SS, Beyreuther K, Schmitt HP. Synaptophysin immunoreactivity of the cortical neuropil in vascular dementia of Binswanger type compared with the dementia of Alzheimer type and non-demented controls. Dementia. 1994;5:79–87.
273. Ishunina TA, Kamphorst W, Swaab DF. Metabolic alterations in the hypothalamus and basal forebrain in vascular dementia. J Neuropathol Exp Neurol. 2004;63(12):1243–1254. doi:10.1093/jnen/63.12.1243
274. Iadecola C, Yang G, Ebner TJ, Chen G. Local and propagated vascular responses evoked by focal synaptic activity in cerebellar cortex. J Neurophysiol. 1997;78(2):651–659. doi:10.1152/jn.1997.78.2.651
275. Cummings JL. Frontal-subcortical circuits and human behavior. Arch Neurol. 1993;50:873–880. doi:10.1001/archneur.1993.00540080076020
276. Ahtiluoto S, Polvikoski T, Peltonen M, et al. Diabetes, Alzheimer disease, and vascular dementia: a population-based neuropathologic study. Neurology. 2010;75:1195–1202. doi:10.1212/WNL.0b013e3181ed735b
277. Poggesi A, Pasi M, Pascini F, Pantoni L, Inzitari D. Circulating biologic markers of endothelial dysfunction in cerebral small vessel disease: a review. J Cerebr Blood Flow Metab. 2015. doi: 10.1038/jcbfm.2015.116
278. Gorelick PB, Scuteri A, Black SE et al. Vascular contributions to cognitive impairment and dementia a statement for healthcare professional from the American Heart Association/American Stroke Association. Stroke. 2011;42:2672–2713. doi:10.1161/STR.0b013e3182299496
279. Kivipelto M, Ngandu T, Laatikainen T, WInblad B, Soinen H, Tuomilehto J. Risk score for the prediction of dementia risk in 20 years among middle aged people: a longitudinal, population-based study. Lancet Neurol. 2006;5:735–741. doi:10.1016/S1474-4422(06)70537-3
280. Prins ND, Scheltens P. White matter hyper-intensities, cognitive impairment and dementia: an update, Nature Rev Neurol. 2015;11:157–165. doi:10.1038/nrneurol.2015.10
281. Wallin A, Blennow K, Gottfires CG. Subcortical symptoms predominate in vascular dementia. Int J Ger Psych. 1991;6:137–146. doi:10.1002/gps.930060305
282. Moretti R, Cavressi M, Tomietto P. Gait and apathy as relevant symptoms of subcortical vascular dementia. Am J Alzh Dis Other Dem. 2015;30(4):390–399. Doi:10.1177/1533317514550329.
283. Fischer P, Gaterer G, Martere A, Simanyi M, Danielcyzk W. Course characteristics in the differentiation of dementia of the Alzheimer’s type and multi-infarct dementia. Acta Psych Scand 1990;81:551–553. doi:10.1111/j.1600-0447.1990.tb05497.x
284. Cummings JL. Vascular subcortical dementias: clinical aspects. Dementia 1994;5:177–180.
285. Desmond D, Erkinjuntti T, Sano M, et al. The cognitive syndrome of vascular dementia: implications for clinical trials. Alzh Dis Ass Dis 1999;13:21–29.
286. Sachdev PS, Brodaty H, Valenzuela MJ, et al. the neuropsychological profile of vascular cognitive impairment in stroke and TIA paitents. Neurol. 2004;62(6):912-919.
287. Traykov L, Baudic S, Thibaudet MC, Rigaud AS, Samgghe A, Boller F. Neuropsychological deficit in early subcortical vascular dementia: comparison to AD. Demen Geriatr Cogn Dis. 2002;14:26–32. doi:10.1159/000058330
288. Wallin A, Milo S, Sjogren M, Pantoni L, Erkinjuntti T Classification and subtypes of vascular dementia. Int PSychoger. 2003;15:27–37. doi:10.1017/S1041610203008937
289. Moretti R, Torre P, Antonello RM, Cazzato G. Behavioral alterations and vascular dementia. Neurologist. 2006;12(81):43–47. doi:10.1097/01.nrl.0000186806.54314.e8
290. Moretti R, Signori R. Neural correlates for apathy: frontal-prefrontal and parietal cortical-subcortical circuits. Front Aging Neurosci. 2016;9:8:289. doi: 10.3389/fnagi.2106.00289.
291. Ishii N, Nashihara Y, Imamura T. Why do frontal lobe symptoms predominate in vascular dementia with lacunes? Neurology. 1986; 36:340–345. doi:10.1212/wnl.36.3.340
292. Levine B, Stuss DT, Milberg WP. Effects of aging on conditioned associative learning: process analysis and comparison with focal frontal lesions. Neupsychol. 1997;11:367–381.
293. Jorm AF, Jolley D. The incidence of dementia: a meta-analysis. Neurology. 1998:51(3):728–733.
294. Meyer JS, Xu G, Thornby J, Chowdhury MH, Quach M. Is mild cognitive impairment prodromal for vascular dementia like AD? Stroke. 2002;33(8):1981-1985.
295. Grau-Olivares M, Arboix A. Mild cognitive impairment in stroke patients with ischemic cerebral small vessel disease: a forerunner of vascular dementia. Exp Rev Neurotherpae. 2009;9:1201–1217.
296. Sabri O, Hellwig D, Schreckenberger M, et al. Correlation of neuropsychological, morphological and functional findings in cerebral microangiopathy. J Nucl Med 1998;39:147–154.
297. Prins ND, van Dijk EJ, Den Heijer T, et al. Cerebral small-vessel disease and decline in information processing speed, executive function and memory. Brain 2005; 128: 20134–22041.
298. Grau-Olivares M, Arboix A, Bartres-Faz D, Junque C. Neuropsychological abnormalities associated with lacunar infarction. J Neurol Sci. 2007;257:160–165.
299. Jokinen H, Kalska H, Ylikoski R, et al. Longitudinal cognitive decline in subcortical ischemic vascular disease-the LADIS study. Cerebrovasc Dis. 2009;27:384–391.
300. Baker JG, Williams AJ, Ionita CC, Lee-Kwen P, Ching M, Miletich RS. Cerebral small-vessel disease: cognition, mood, daily functioning, and imaging finding from a small pilot sample. Dem Ger Cogn Disord. 2012;2:169–179. doi: 10.1159/0000333482.
301. Nitkunan A, Lanfranconi S, Charlton RA, Barrick TR, Markus HS. Brain atrophy and cerebral small vessel disease. A prospective follow-up study. Stroke. 2011; 42:133–138.
302. Kim HJ, Ye BS, Yoon CW, et al. Cortical thickness and hippocampal shape in pure vascular mild cognitive impairment and dementia of subcortical type. Eur J Neur. 2014;21:744–751. doi: 10.111/ene.123769.
303. Shi Y, Tirpletton MJ, Makin SD, et al. Cerebral blood flow in small vessel disease: a systematic review and meta-analysis. J Cerebr Blood Flow Metab. 201636(10):1653-1667. doi:10.1177/0271678X166662891.
304. Maillard P, Fletcher E, Lockhart SN, et al. White matter hyperintensities and their penumbra lie along a continuum of injury in the aging brain. Stroke. 2014;45:1721–1726.
305. Maniega SM, Valdes Hernandez MC, Clayden JD, et al. White matter hyperintensities and normal-appearing white matter integrity in the aging brain. Neurobiol Aging. 2015;36:909–918.
306. Madden DJ, Bennett IJ, Burzynska A, et al. Diffusion tensor imaging of cerebral white matter integrity in cognitive aging. Biochim Biophys Acta. 2012:1822:386–400.
307. Iliff JJ, Wang M, Liao Y, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4(147):147ra111.
308. O’Brien JT, Thomas A. Vascular dementia. Lancet. 2015;386:1698–1706.
309. O’Brien JT, Thomas A. Non-Alzheimer’s dementia 3 vascular dementia. Lancet. 2015;386:1698–1706. doi:10.1016/S0140-6736(15)00463-8
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.