Back to Journals » OncoTargets and Therapy » Volume 11
Small ubiquitin-like modifier 1 modification of pyruvate kinase M2 promotes aerobic glycolysis and cell proliferation in A549 human lung cancer cells
Authors An S ![]() , Huang L, Miao P, Shi L, Shen M, Zhao X, Liu J
, Huang L, Miao P, Shi L, Shen M, Zhao X, Liu J ![]() , Huang G
, Huang G ![]()
Received 13 November 2017
Accepted for publication 18 January 2018
Published 13 April 2018 Volume 2018:11 Pages 2097—2109
DOI https://doi.org/10.2147/OTT.S156918
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ingrid Espinoza
Shuxian An,1,* Liangqian Huang,2,3,* Ping Miao,1 Liang Shi,1 Mengqin Shen,1 Xiaoping Zhao,1 Jianjun Liu,1 Gang Huang1,3,4
1Department of Nuclear Medicine, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China; 2Department of Cancer Biology and Abramson Family Cancer Research Institute, University of Pennsylvania School of Medicine, Philadelphia, PA, USA; 3Institute of Health Sciences, Shanghai Jiao Tong University School of Medicine & Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China; 4Shanghai University of Medicine and Health Sciences, Shanghai, China
*These authors contributed equally to this work
Objective: Lung cancer is the leading cause of cancer-related death worldwide. Aerobic glycolysis is considered the seventh hallmark of cancer. The M2 isoform of pyruvate kinase (PKM2) is an important rate-limiting enzyme in glycolytic pathway, and is strongly expressed in several types of cancer. Thus, understanding the underlying mechanisms of regulation of PKM2 is of great value for targeted therapy for lung cancer.
Patients and methods: Seventy-three lung adenocarcinoma patients were analyzed in our study. The expression levels of PKM2 were analyzed by immunohistochemistry on tissues. The effect of small ubiquitin-like modifier 1 (SUMO1) on PKM2 expression was investigated using Western blot assay and quantitative polymerase chain reaction. PKM2 SUMO1 modification was determined by in vitro and in vivo SUMOylation assays. 18F-deoxyglucose uptake and lactate production measurements were conducted to research the levels of glycolysis. The level of oxidative phosphorylation in cells was determined by cellular oxygen consumption rate measurements. Cell proliferation assays were carried out to confirm the growth ability of tumor cells.
Results: PKM2 was overexpressed in lung adenocarcinoma patients based on immunohistochemical staining. Patients with high PKM2 expression had reduced overall survival rate (P=0.017) and disease-free survival rate (P=0.027) compared with those with low PKM2 expression. SUMO1 promoted PKM2-dependent glycolysis. Western blotting analysis showed that SUMO1 knockdown in A549 cells led to a significant decrease in PKM2 protein expression. PKM2 could be covalently modified by SUMO1 at K336 (Lys336) site. SUMO1 modification of PKM2 at Lys-336 site increased glycolysis and promoted its cofactor functions. Moreover, PKM2 SUMO1 modification promoted the proliferation of A549 cells in vitro.
Conclusion: This information is important in elucidating a new mechanism of regulation of PKM2, and suggested that SUMO1 modification of PKM2 could be a potential therapeutic target in lung cancer.
Keywords: Pyruvate Kinase M2, SUMO1 modification, glycolysis, cell proliferation, cancer
Introduction
Lung cancer is the leading cause of cancer-related death worldwide. Adenocarcinoma is major histological subtype of lung cancer, and its incidence rate annually increases.1 An estimated 1,800,000 new lung cancer cases are diagnosed every year, accounting for about 13% of total cancer diagnoses.1 Although a great amount of effort has been made toward the diagnosis and treatment of lung cancer, the clinical outcomes are still unsatisfactory, with an overall 5-year survival rate of 16.8%.2 Therefore, identifying novel therapeutic target is of great importance to improve progression for lung cancer. Unlike normal cells, most cancer cells rely primarily on aerobic glycolysis to generate energy even in an oxygen-rich environment during malignant progression. This phenomenon is termed “the Warburg effect”.3 Pyruvate kinase (PK) is an important metabolic enzyme, which catalyzes the conversion of phosphoenolpyruvate and adenosine diphosphate to pyruvate and adenosine triphosphate (ATP) in the glycolytic pathway.4 There are 4 mammalian pyruvate kinase isoforms, PKL, PKR, PKM1 and PKM2, which are expressed in different cell types.5 PKM2 is mainly found in many different cancer cells and during embryonic development.6 Interestingly, PKM2 is expressed with different forms in normal proliferating cells and tumor cells. In normal proliferating cells, active tetrameric PKM2 is the main form, pyruvate is used to produce ATP in mitochondria through the tricarboxylic acid (TCA) cycle.7 However, in cancer cells, less active dimeric PKM2 is the main form with less pyruvate production, which lead to accumulation of various glycolytic metabolites for macromolecular biosynthesis (nucleotides, amino acids, and lipids) to support cell growth.7,8,10 Christofk et al reported that knockout of PKM2 could inhibit cell proliferation and tumorigenesis by reducing aerobic glycolysis.9 PKM2 has been reported to be overexpressed in many malignant tumors and makes essential contributions to development and metastasis.11,12 Yu et al reported that PKM2 could regulate neural invasion of and predict poor prognosis for human hilar cholangiocarcinoma.12 PKM2 as an independent prognostic factor is also overexpressed in lung cancer.13 Sun et al reported that knockdown of PKM2 suppressed tumor growth and invasion in lung adenocarcinoma.14 In addition, Guo et al showed that PKM2 knockdown combined with cisplatin significantly inhibited tumor growth in A549 lung cancer xenograft models.15 Considering the critical role of PKM2 in tumorigenesis, understanding the underlying mechanisms of regulation of PKM2 is of great value for targeted therapy for lung cancer.
Post-translational modifications (PTMs) are important mechanisms for regulating protein functions.16 PTM of PKM2, such as phosphorylation or acetylation, has been reported to regulate PKM2 function by affecting its protein stability, homodimerization, and translocation.8 SUMOylation is an important type of PTM, and involved in many significant cell processes such as transcription, protein stability, and protein subcellular localization.17 Recently, an increasing number of studies have shown the link between small ubiquitin-like modifier (SUMO) modification pathway and tumor development. Spoden et al reported that PIAS3 targeted PKM2 in vivo.18 Interestingly, PIAS is a SUMO-E3 Ligase. Thus, this suggested a link between SUMO system and PKM2. In this study, we aim to reveal the mechanism of SUMO1 modification of PKM2 and provide a new mechanism of regulation of PKM2.
Here, we show that expression of PKM2 was increased in lung adenocarcinoma patients based on immunohistochemical (IHC) analysis, and patients with high PKM2 expression had reduced overall survival rate and disease-free survival rate compared with those with low PKM2 expression. Mechanistic analysis demonstrated that SUMO1 modification affected the protein levels of PKM2, and Lys336 residue was the main SUMO1 modification site of PKM2. SUMO1 modification of PKM2 promoted aerobic glycolysis of cancer cells; furthermore, it promoted cell proliferation and tumorigenesis.
Patients and methods
Study population
A total of 73 patients who underwent tumor resection at Shanghai Jiaotong University affiliated Renji Hospital and Shanghai Chest Hospital from December 2007 to December 2012 were enrolled in this study. Inclusion criteria were as follows: none of the patients had received treatment before surgery; complete case records; tumor pathology of lung adenocarcinoma had been confirmed by histopathologic examination of surgical specimens; and available tissue specimen for IHC staining. This research was approved by the institutional ethics committee of Shanghai Jiao Tong University affiliated Renji Hospital and Shanghai Chest Hospital. Written informed consent was obtained from each patient according to the Declaration of Helsinki.
Immunohistochemistry
Paraffin-embedded lung cancer tissues were used for the IHC analysis. Anti-PKM2 antibody (Cell Signaling Technology, Beverly, MA, USA) was applied on 4 μm sections. Intensity of staining was scored according to the following criteria: 0 (no staining), 1 (weak staining), 2 (intermediate staining), and 3 (strong staining). In addition, 0 (0%), 1 (1%–9%), 2 (10%–49%), and 3 (50%–100%) were used to score the percentage of positive cells. Intensity of staining and percentages of cells were used to score the slides (0–9). An IHC score of <4 was considered negative, whereas an IHC score of ≥4 was considered positive. The slides were evaluated by 2 independent observers who were blinded to the clinical data.
Cell culture and transient transfection
HEK293T and A549 cell lines were purchased from the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences. All cells were maintained in (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum (FBS; Thermo Fisher Scientific). Cells were incubated at 37°C under 5% CO2. Hypoxic cells (1% O2) were placed in a sealed hypoxia chamber (Thermo Fisher Scientific). We used NEOFECT™ DNA transfection reagent (Neofect, Beijing, China) to carry out transient transfection experiments. Cells were plated in 6-well plates; 24 h after that, transient transfection experiments were carried out. Transfection reagents were diluted by opti-MEM (Thermo Fisher Scientific) for 5 min at room temperature (RT). Plasmids were also diluted by opti-MEM. Then the diluted transfection reagents were mixed with the diluted plasmids for another 30 min at RT. The mixture was added into cells; 48 h after transfection, cells were collected for the following experiments.
Transfection of siRNA
Cells were transfected with oligo small silencing RNAs using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). The sequences of siRNA oligos used in our study are as follows: SUMO1 (sense 5′-CCUUCAUAUUACCCUCUCCUU-3′, antisense 5′-AAGGAGAGGGUAAUAUGAAGG-3′), and negative control (sense 5′-UUCUCCGAGCGUGUCACGUTT-3′, antisense 5′-ACGUGACACGUUCGGAGAATT-3′).
PKM2 enzyme assay
Cells (HEK293T) were transfected with hemagglutinin (HA)-PKM2 wild type (WT) or HA-PKM2 (K336R) as indicated for 48 h. HA beads (Sigma-Aldrich-Aldrich St Louis, MO, USA) were used to immunoprecipitate HA-PKM2 (WT) or HA-PKM2 (K336R) protein; after that, indicated protein was eluted by HA peptide (Sigma). Purified HA-PKM2 or HA-PKM2 (K336R) protein was equalized by Western blotting. PK Assay Kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) was used to analyze the PK activity according to the manufacturer’s instructions.
18F-deoxyglucose (18F-FDG) uptake and lactate production measurements
Cells were transfected with indicated plasmids in 12-well plates. Approximately 48 h after transfection, the cells were collected and washed with PBS, then the collected cells were incubated in 1 mL of glucose-free DMEM containing 18F-FDG (148 kBq [4 μCi/mL]) for 1 h at 37°C. A total of 1 mL of 0.1 M NaOH was used to produce lysates. A well γ-counter was used to detect the radioactivity of lysates. At the end of the experiments, the readouts were normalized to corresponding protein amounts. Three independent experiments were performed during our study.
For lactate production measurements, cells were transfected with indicated plasmids in 12-well plates for 36 h. Then the cells were washed with PBS and cultured in serum-free DMEM for another 12 h. After that, cell supernatants were collected to measure lactate levels according to the instruction of the Lactate Assay Kit (Nanjing Jiancheng Bioengineering Institute). Data were normalized to total protein and three independent experiments were performed during our study.
Western blotting and immunoprecipitation
Cells were lysed using radio immunoprecipitation assay (RIPA) lysis solution (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, complete protease inhibitor cocktail) for 30 min on ice, then centrifuged at 15,000× g for 30 min at 4°C. Cell extracts were incubated with HA beads (Sigma) at 4°C for 3 h. After that, the beads were washed 5 times with lysed buffer (RIPA), then equal 2×SDS (sodium dodecyl sulfate) sample buffer was added to the resulting beads and boiled for 10 min at 100°C, the samples were analyzed by Western blotting. Protein samples were separated in 10% SDS-polyacrylamide gel electrophoresis and then transferred to polyvinylidene difluoride membranes. Non-fat milk 5% was used to block nonspecific background for 1 h at room temperature followed by overnight 4°C incubation with the primary antibodies. Horseradish peroxidase-conjugated secondary antibodies were used in our study.
In vitro and in vivo SUMOylation assay
A total of 200 nmol/L recombinant PKM2 was used in vitro SUMOylation assays using the SUMOylation kit (Enzo Life Sciences, Farmingdale, NY, USA) according to manufacturer’s instructions. For in vivo SUMOylation assay, HA-PKM2 (K336R) and Flag-SUMO1 were contransfected into HEK293T cells. Forty-eight hours after transfection, cells were collected, then N-ethylmaleimide (NEM)-PBS buffer (20 mM N-ethylmaleimide in phosphate-buffered saline) was used to wash the cells, HA-PKM2 (K336R) was immunoprecipitated in NEM-RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 20 mM NEM, and complete protease inhibitor cocktail) with HA beads (Sigma) at 4°C for 3 h, then SUMO1-PKM2 was detected by immunoblotting.19
Mice and in vivo small animal position emission tomography (PET) experiments
BALB/c nude mice (female, 4 to 5 weeks-old) were purchased from Ren Ji Hospital Experimental Animal Center (Shanghai, China).
Different viruses were infected into A549 cells to form stable ectopic PKM2-WT/K336R-expressing A549 cells. A total of 1×107 stably transfected cells were injected subcutaneously into nude mice. Two weeks after that, 7.4 MBq 18F-FDG was injected intravenously into tumor-bearing mice. Then the animals were anesthetized and immobilized. A 20 min PET scan was acquired using a MicroPET (Super Nova® PET/CT (PINGSENG Healthcare [KunShan] Inc, Jiangsu, China). 18F-FDG uptake by tumors was assessed by using standard uptake values.
Our study was approved by Ren Ji Institutional Animal Care and Use Committee (Shanghai, China). The procedures were performed according to the guidelines and regulations of Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University (Shanghai, China). This study was carried out in accordance with the recommendations that cover all scientific procedures involving the use of live animals.
Cellular oxygen consumption rate (OCR) measurements
We used a Seahorse XF24 Extracellular Flux Analyzer to evaluate OCR measurements. A total of 20,000 cells were plated in XF24 cell culture plates and incubated for 24 h at 37°C. After that, cells were incubated in bicarbonate-free buffered DMEM in a 37°C non-CO2 incubator for 1 h followed by XF assay at the designated time points.
Luciferase reporter assay
A total of 1×104 HEK293T cells were seeded into 24-well plates and transfected with hypoxia-inducible factor 1 (HIF-1)-luc and Flag-SUMO1 or si-SUMO1 or si-negative control (si-NC), and empty vector, or PKM2-WT, or PKM2-K336R as indicated for 24 h. After that, part of cells suffer 20% O2 (normoxia), the rest of cells suffer 1% O2 (anoxia). for another 24 h, we used the Dual Luciferase Assay System (Promega, Madison, WI, USA) to analyze the cell lysates in our study. The ratio of firefly luciferase to renilla activity was calculated for each of the samples in triplicate.
Quantitative real-time polymerase chain reaction (qRT-PCR)
TRIzol Kit (Omega, Norcross, GA, USA) was used to isolate total RNA and complementary DNA (cDNA) Synthesis Kit (Takara, Otsu, Japan) was used to synthesize cDNA. SYBR Green PCR Master Mix (Takara) in a StepOnePlus RT-PCR system (Thermo Fisher Scientific) was used for qRT-PCR. The PCR cycling conditions were as follows: 95°C for 5 min, 40 cycles of 95°C for 10 s and 55°C for 20 s, 72°C for 20 s, and dissociation at 95°C for 15 s, 60°C for 1 min, and 95°C for 15 s. A 2−ΔΔCt method was used to analyze the data. All qRT-PCR reactions were performed in triplicate. Primers used in our study are as follows: PKM2-Forward Primer (GTCGAAGCCCCATAGTGAAG), PKM2-Reverse Primer (GTGAATCAATGTCCAGGCGG), GLUT1-F (CAGTTCGGCTATAACACTGGTG), GLUT1-R (GCCCCCGACAGAGAAGATG), LDHA-F (ATCTTGACCTACGTGGCTTGGA), LDHA-R (CCATACAGGCACACTGGAATCTC), PDK1-F (CTTCAGCCGCAGCTTCAGCT), PDK1-R (CAACTCTTGCCGCAGAAACA), ENO1-F (TGCCGTCTGCAAAGCTGGTG), ENO1-R (CGCATGGCTTCCCTGAAGTT).
Colony-formation assays and cell proliferation analysis
Low-melting point agarose (250 μL, 0.6%) in DMEM containing 10% FBS was poured into a 24-well plates and allowed to solidify at RT. A total of 100 cells were seeded in 250 μmL 0.4% agar with DMEM medium (10% FBS) and then plated on top of the base layer in each well. The cells were cultured for 15 days. Each well was photographed and the colony number was calculated.
A549 cells were transfected with indicated plasmids for 24 h. After that, 20,000 cells were harvested and seeded in triplicate onto 24-well plates. The cells were collected every 48 h and live cells were counted using a hemocytometer after trypan blue exclusion.
Statistical analysis
We used Graphpad Prism 5 software (GraphPad Software, San Diego, CA, USA) or SPSS 17.0 (SPSS Inc., Chicago, IL, USA) to conduct statistical analysis in our study. Overall and disease-free survival curves were calculated using Kaplan–Meier method and were compared by log-rank test. Unpaired 2-tailed t-test was used to analyze the difference between the groups. Data are expressed as mean ± SD, of 3 independent experiments. All tests of significance were 2-sided; and P-values <0.05 were considered significant.
Results
Patient characteristics
Our test subjects consisted of 40 males and 33 females. The age of patients ranged from 38 to 79 years, with mean value of 60 years. Lymph node metastasis was detected in 29 patients.
Correlation of patient characteristics with PKM2 expression
Immunohistochemical (IHC) analysis was carried to assess the expression of PKM2. Representative samples of PKM2 expression evaluated by IHC staining are shown in Figure 1A. PKM2 immunoreactivity was readily detected in the cytoplasm and was occasionally detected in the nucleus (Figure 1A). The positive expression of PKM2 was 41.1% in lung adenocarcinoma patients. To investigate the relationship between PKM2 expression and the biological characteristics of tumor tissues, we divided the patients into the following 2 categories according to immunhistochemistry of PKM2: positive PKM2 expression group (n=30) and negative PKM2 expression group (n=43). The results of the univariate analysis for each factor are show in Table 1. PKM2 expression was not associated with age and sex. However, tumor differentiation, lymph node metastasis, or TNM stage of these 2 groups were significantly different.
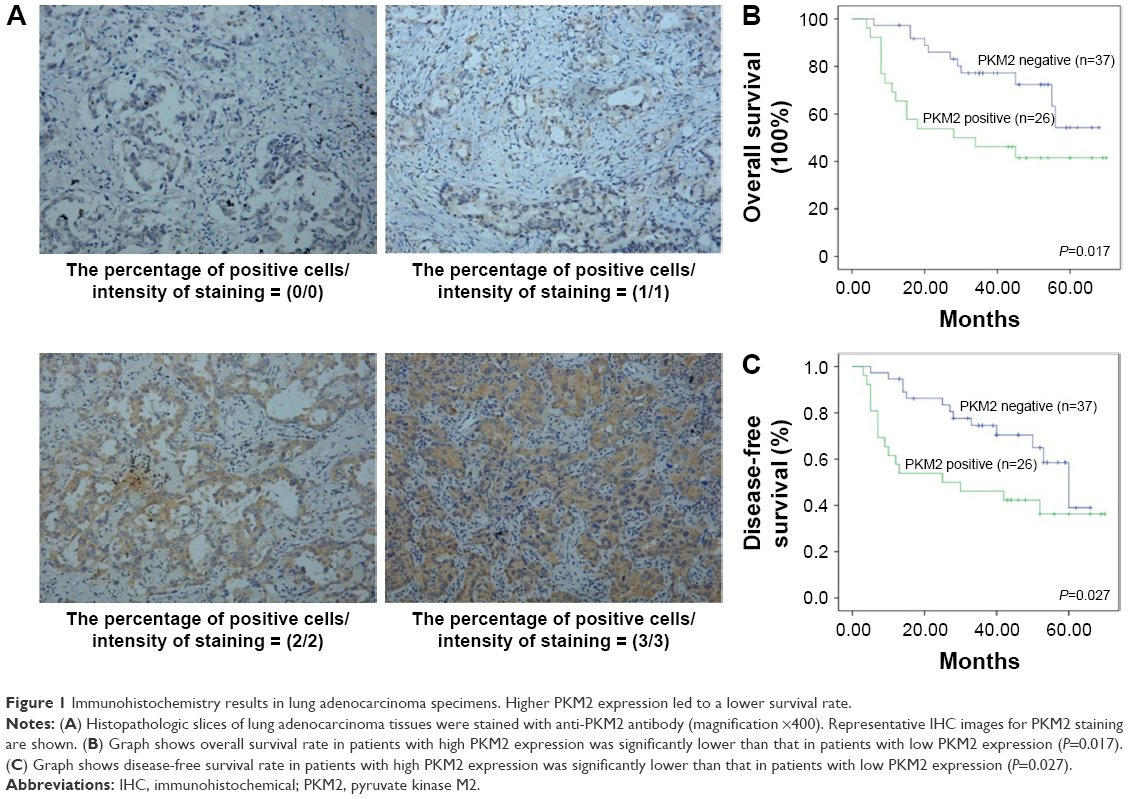 | Figure 1 Immunohistochemistry results in lung adenocarcinoma specimens. Higher PKM2 expression led to a lower survival rate. |
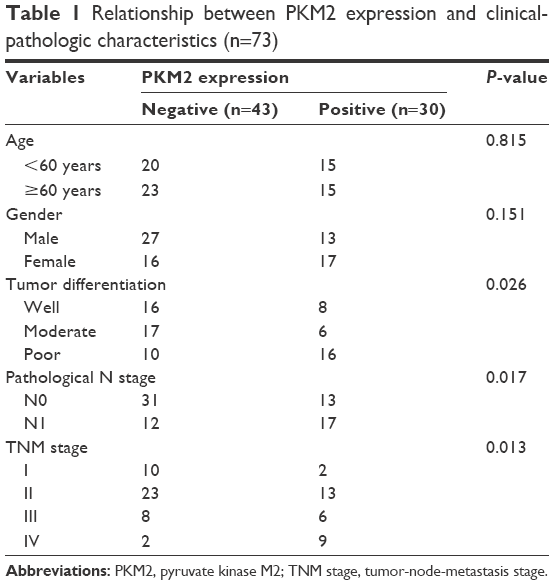 | Table 1 Relationship between PKM2 expression and clinical-pathologic characteristics (n=73) |
Correlation of high PKM2 expression with overall survival and disease-free survival in lung adenocarcinoma patients
Kaplan–Meier analysis showed that the overall survival rate was significantly lower in the positive PKM2 expression group than in the negative PKM2 expression group (P=0.017) (Figure 1B). As for disease-free survival rate, lung adenocarcinoma patients with high PKM2-expressing tumors had significantly reduced disease-free survival than patients without them (P=0.027) (Figure 1C).
SUMO1 modification promoted PKM2-dependent glycolysis via affecting PKM2 protein levels
As an important type of PTM, SUMO1 modification has been reported to be involved in glycolysis in cancer cells.20 As shown in Figure 2A and B, compared with vector control, both 18F-FDG uptake and lactate production were dramatically increased in cells overexpressed with SUMO1. On the contrary, we found that SUMO1 knockdown by specific siRNA significantly reduced the 18F-FDG uptake and lactate production in A549 (Figure 2C and D). PKM2 knockdown by specific siRNA further reduced 18F-FDG uptake and lactate production; however, no additive effect was observed when si-SUMO1 was combined with si-PKM2 (Figure 2C and D). The results showed that SUMO1 promoted glycolysis in A549, and this effect was dependent on PKM2.
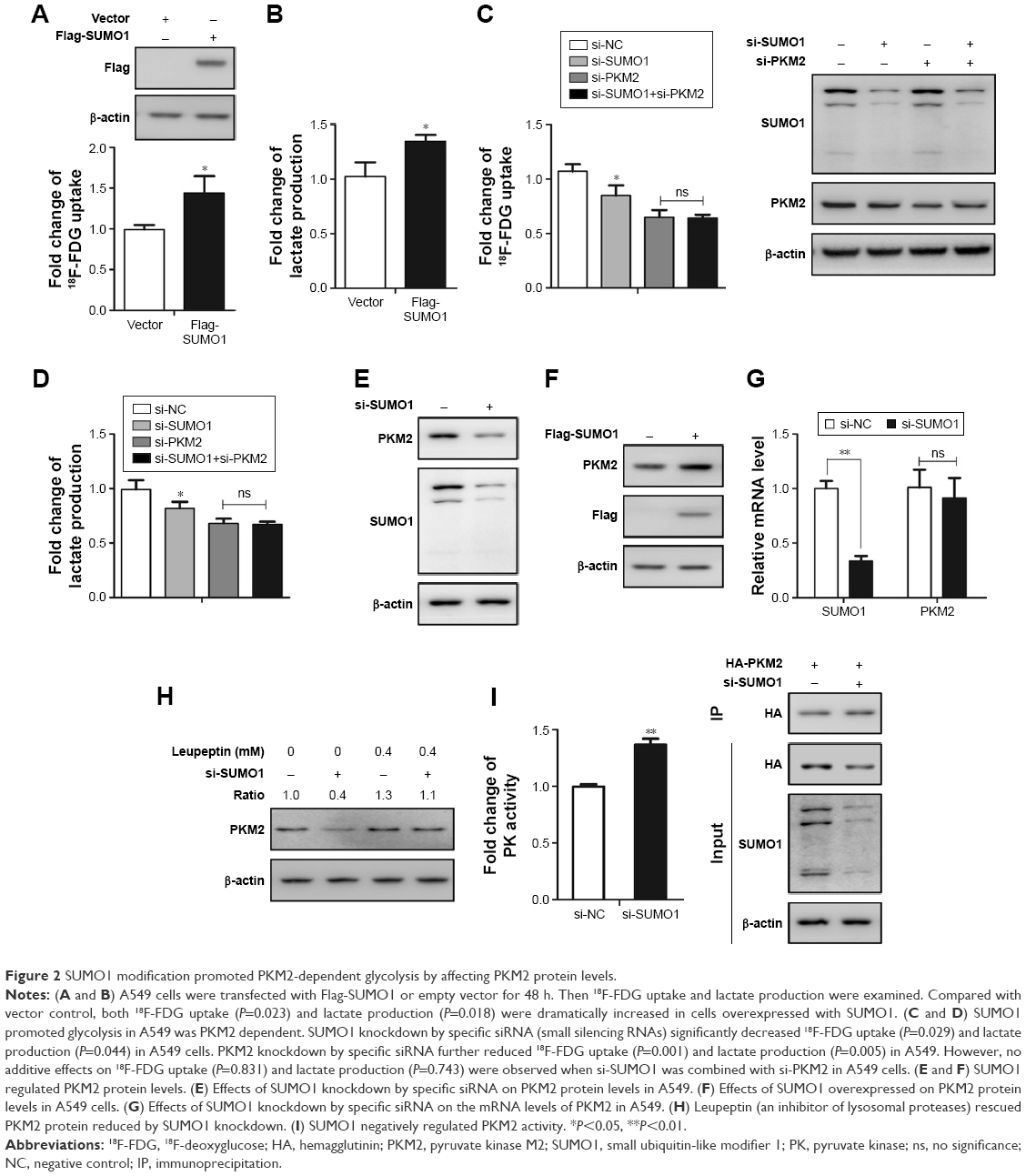 | Figure 2 SUMO1 modification promoted PKM2-dependent glycolysis by affecting PKM2 protein levels. |
As SUMO1 modification plays a critical role in protein stability, we wondered whether SUMO1 also affected PKM2 protein stability. We showed that SUMO1 knockdown by specific siRNA significantly reduced the expression level of endogenous PKM2 in A549 cells (Figure 2E). Conversely, overexpression of SUMO1 caused an increase in endogenous PKM2 proteins compared with the vector control (Figure 2F). In addition, we found that the mRNA levels of PKM2 were not affected by SUMO1 knockdown (Figure 2G). This indicated that SUMO1 played an important role in stabilizing the protein level of PKM2. A previous study showed that PKM2 was degraded by lysosomal pathway.8 We observed that Leupeptin (an inhibitor of lysosomal proteases) rescued the decrease of PKM2 protein induced by SUMO1 knockdown (Figure 2H), suggesting that SUMO1 enhanced the protein stability of PKM2 by regulating its degradation process. In addition, decreased PK activity contributes to rapidly growing embryonic and tumorigenic cells. Our results found that SUMO1 knockdown increased PK activity, suggesting that SUMO1 modification inhibited PK activity (Figure 2I).
Lysine-336 residue was a primary SUMO1 modification site of PKM2 protein
In that SUMO1 potentially controlled the stability of PKM2 proteins, and promoted glycolysis via PKM2, we hypothesized that PKM2 could be modified by SUMO1. Thus, we hypothesized that PKM2 could be modified by SUMO1. First, we performed in vitro SUMOylation assays and found that PKM2 protein was a substrate for modification by SUMO1 (Figure 3A). To further support the conjugation of PKM2 to SUMO1, we performed in vivo SUMOylation. HEK293T cells were transfected with HA-PKM2 in the presence or absence of Flag-SUMO1. Total cell extracts were immunoprecipitated with anti-HA antibody. We identified a slow moving immunoreactive band, which was recognized by both anti-Flag and anti-HA antibodies when supplied with SUMO1 (Figure 3B). These results supported the possibility that PKM2 was modified by SUMO1 in vivo. Then we used SUMOsp 2.021 to predict potential SUMOylation sites of PKM2 protein. In silico analysis indicated that lysine-336 (K336) residue was the most likely to be a SUMO1 modification site of PKM2 (Figure 3C). Mutation of lysine-336 to arginine largely abolished PKM2 SUMO1 modification in vitro, suggesting lysine-336 was the primary SUMOylation site of PKM2 protein (Figure 3D). In accordance with the results of in vitro, the higher molecular mass was most clearly absent in the K336R mutant in the presence of Flag-SUMO1 (Figure 3E). In vivo SUMOylation analysis further demonstrated that WT but not K336R mutant of PKM2 protein was modified by SUMO1. Taken together, these results suggested that K336 residue was a primary SUMO1 modification site of PKM2 protein.
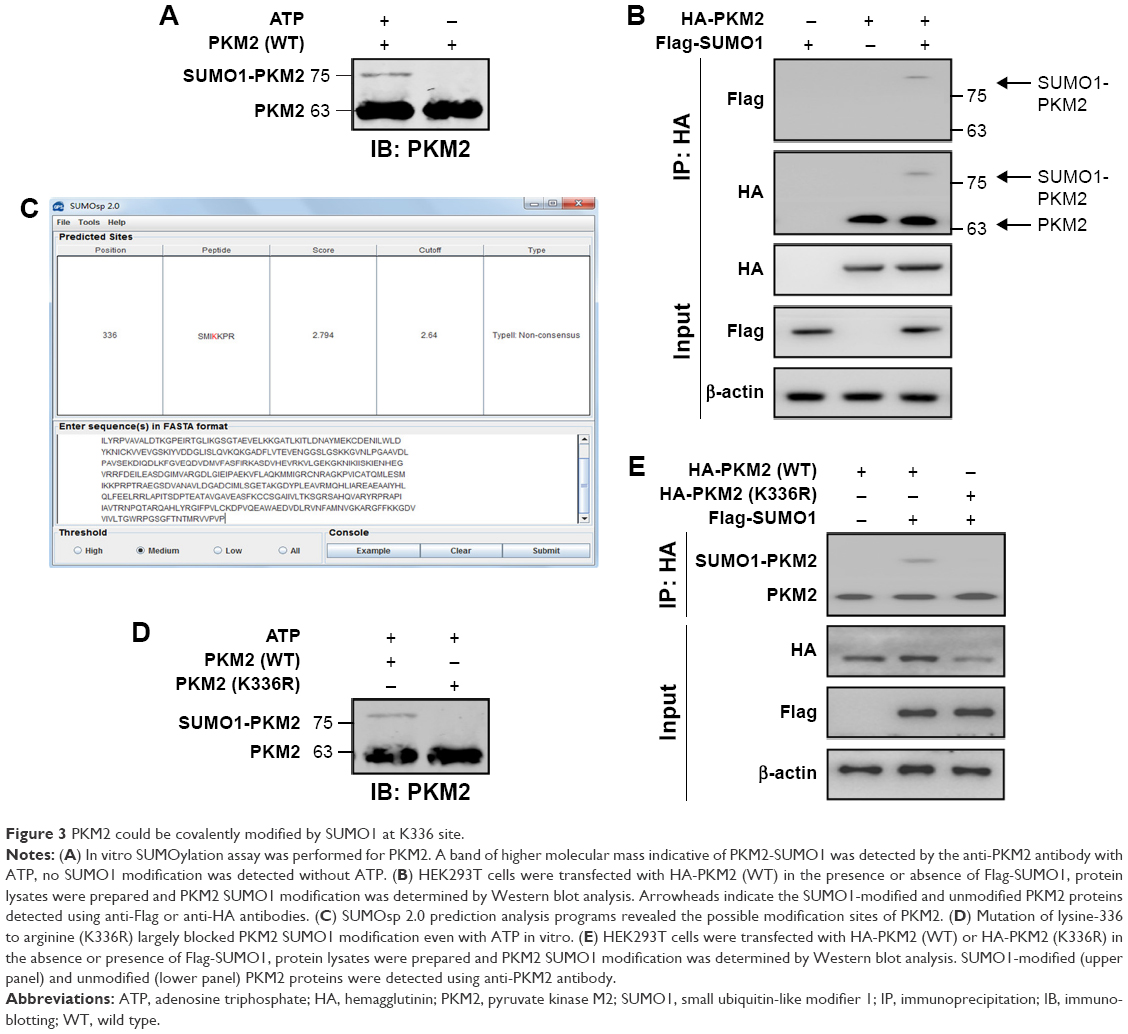 | Figure 3 PKM2 could be covalently modified by SUMO1 at K336 site. |
SUMO1 modification of PKM2 at Lys-336 site increased glycolysis and promoted its cofactor functions
Next, we analyzed whether SUMO1-modified PKM2 increased glycolysis through Lys-336 site. A549 cells were transfected with Flag-SUMO1 or si-SUMO1 or si-NC, and empty vector, or PKM2-WT, or PKM2-K336R as indicated (Figure 4A and B). In the presence of PKM2-WT, compared with empty vector and si-NC, the 18F-FDG uptake and lactate production with Flag-SUMO1 were significantly increased, whereas the values of 18F-FDG uptake and lactate production with si-SUMO1 were much reduced, indicating that SUMO1 modification of PKM2 promoted glycolysis of cancer cells. In contrast, in the presence of PKM2-K336R, the values of 18F-FDG uptake and lactate production were much reduced. Compared with expression of PKM2-WT with empty vector, expression of the K336R mutant PKM2 suppressed values of 18F-FDG uptake and lactate production to similar levels under conditions of cotransfection with empty vector, Flag-SUMO1, si-SUMO1, or si-NC. This result suggested that SUMO1 modification at the K336 residue of PKM2 increased glycolysis of cancer cells. To further support the idea that glycolysis was regulated by PKM2 Lys-336 residue SUMO1 modification, we performed 18F-FDG PET imaging on mice bearing human lung cancer xenografts. In vivo PET imaging displayed that cancer xenograft derived from cells that expressed K336R mutant PLM2 had significantly less glucose uptake than that derived from ells that expressed wild-type PKM2 (Figure 4C). This result suggested that SUMO1 modification of PKM2 at the Lys336 residue played a vital role in PKM2-mediated glucose metabolism. On the other hand, basal OCR reflecting oxidative phosphorylation was substantially higher in cancer cells that expressed K336R mutant PKM2 than those that expressed wild-type PKM2 (Figure 4D). Moreover, K336R mutant PKM2 resulted in a higher PK enzyme activity (Figure 4E). Taken together, these data suggested that SUMO1 modification of PKM2 at Lys-336 residue shunted glucose from oxidative phosphorylation metabolism to glycolysis.
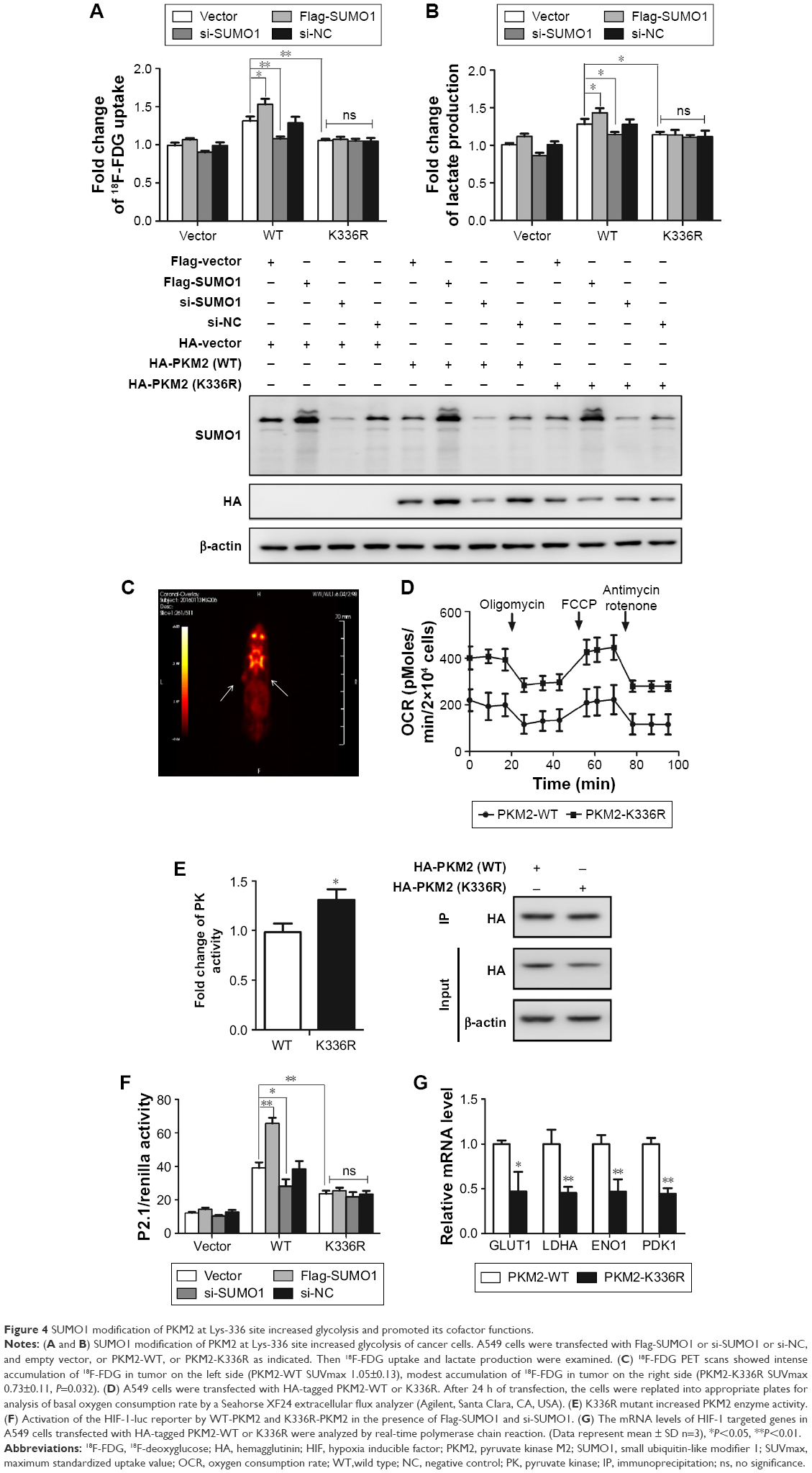 | Figure 4 SUMO1 modification of PKM2 at Lys-336 site increased glycolysis and promoted its cofactor functions. |
Besides being an important rate-limiting enzyme in aerobic glycolysis, PKM2 functioned as a transcriptional coactivator of HIF-1 in the nucleus.22,23 To test whether SUMO1-modified PKM2 affected HIF-1 transcriptional activity, a luciferase reporter containing hypoxia-response element derived from the human ENO1 gene was used for analyzing HIF-1 transcriptional activity. The 293T cells were transfected with HIF-1-luc and Flag-SUMO1 or si-SUMO1 or si-NC, and empty vector, or PKM2-WT, or PKM2-K336R as indicated (Figure 4F). In the presence of PKM2-WT, compared with empty vector and si-NC, the activity of HIF-1-luc with Flag-SUMO1 was significantly increased, whereas the activity of HIF-1-luc with si-SUMO1 was much reduced, indicating that SUMO1 modification of PKM2 controls PKM2 activity on HIF-1 transcription. On the contrary, in the presence of PKM2-K336R, the activities of HIF-1-luc were much reduced. Compared with expression of PKM2-WT with empty vector, the expression of the K336R mutant PKM2 suppressed transactivation of HIF-1-luc to similar levels under conditions of cotransfection with empty vector, Flag-SUMO1, si-SUMO1, or si-NC. This result suggests that SUMO1 modification at the K336 residue contributes to PKM2-mediated HIF-1 transcription. Then we tested HIF-1 target genes in cancer cells that expressed either wild-type or K336R mutant PKM2. The expression of HIF-1 targets consisting of GLUT1, LDHA, ENO1 and PDK1 was lower in cells that expressed K336R mutant PKM2 than those that expressed wild-type PKM2 (Figure 4G). All these findings indicated that SUMO1 modification of PKM2 at Lys-336 residue was controlling PKM2 activity on HIF-1 transcription.
SUMO1-modified PKM2 promoted cell proliferation in vitro
To investigate whether SUMO1-modified PKM2 influenced the growth of cancer cells, cell-counting experiments were conducted. We found that cells that overexpressed WT PKM2 with si-NC proliferated faster than cells overexpression of K336R mutant PKM2 with si-NC (P=0.001) (Figure 5A). More importantly, the growth promotion effect was comparable between wild-type and K336R mutant PKM2 cells in the absence of SUMO1 (P=0.662) (Figure 5A). Soft agar colony-forming assays were conducted to further confirm the effect of PKM2 SUMO1 modification on the proliferation potential of cancer cells. The results showed that WT PKM2 significantly promoted the anchorage-independent growth of cancer cells compared with K336R mutant PKM2 in the presence of SUMO1 (P=0.002) (Figure 5B). However, the growth advantage was comparable between wild-type and K336R mutant PKM2 cells when SUMO1 gene was silenced by specific siRNA (P=0.901) (Figure 5B). Thus, these data suggested that PKM2 Lys-336 residue SUMO1 modification conferred tumor cell growth advantage in vitro.
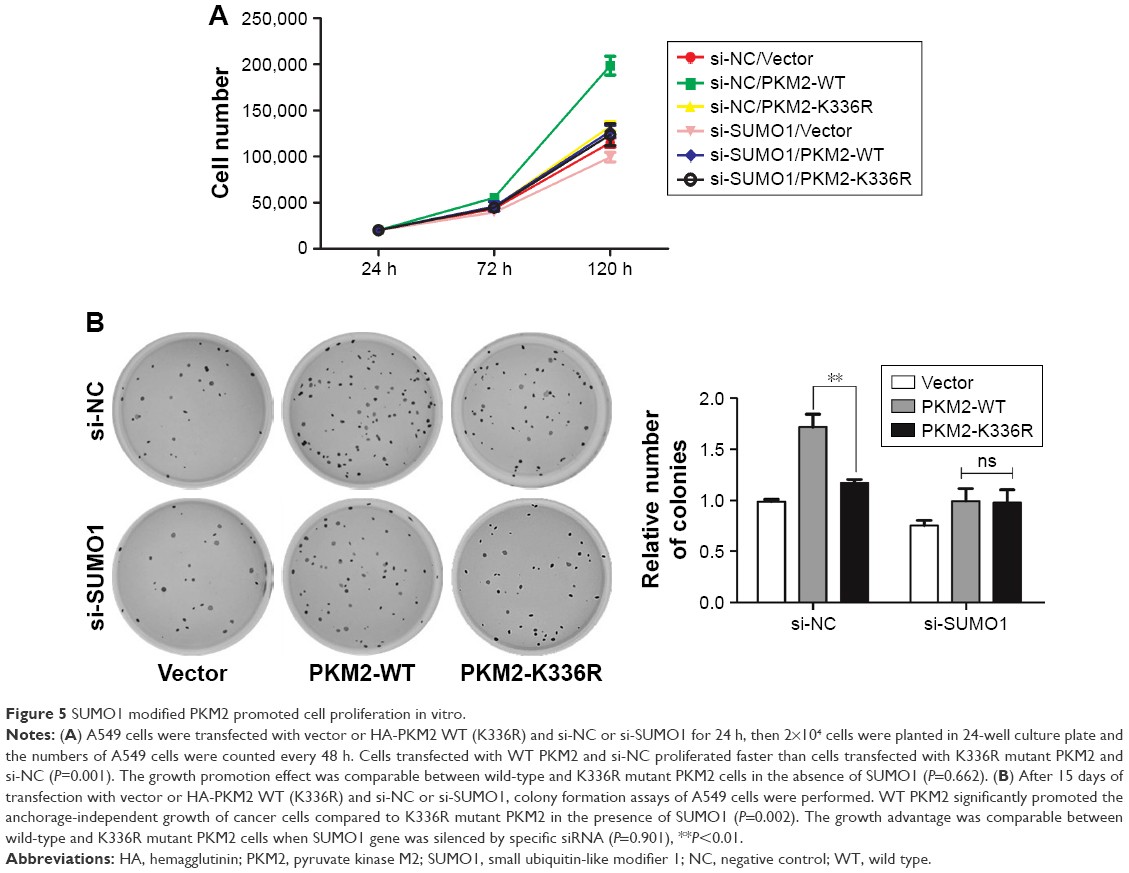 | Figure 5 SUMO1 modified PKM2 promoted cell proliferation in vitro. |
Discussion
Lung cancer is the leading cause of cancer-related death worldwide,1 among which adenocarcinoma is the major histological subtype. Although massive efforts have been made toward the diagnosis and treatment of lung cancer, the 5-year survival rate is still poor. Therefore, identifying novel therapeutic target is of great importance to facilitate targeted therapies or interventions for human lung cancer.
About 80 years ago, Otto Warburg found that most cancer cells relied primarily on aerobic glycolysis to generate energy even in an oxygen-rich environment, which is termed “the Warburg effect”.24 It is well known that aerobic glycolysis is an inefficient way to generate ATP, but it enables cancer cells to produce more important biomass, which is needed for producing a new cell.25 As the last rate-timing enzyme in glycolysis, PKM2 plays a crucial part in regulating cancer cells metabolism and tumorigenesis.4,9 Christofk et al reported that knockout of PKM2 could inhibit cell proliferation and tumorigenesis by reducing aerobic glycolysis.9 The PKM2 mRNA levels have been reported to be elevated in lung cancer, breast cancer, colon cancer, and bladder cancer.26–29
IHC results further showed that PKM2 was overexpressed in many malignant tumors and made essential contributions in development and metastasis.11,12 In this study, we found a 41.1% rate of PKM2 expression in lung adenocarcinoma using immunohistochemistry, which was in line with a previous report.13 More importantly, we found that patients with relatively high PKM2 expression had lower overall and disease-free survival rates when compared with rates in patients with low expression. Considering its relatively high levels of expression and critical role in tumors, understanding the underlying mechanisms of regulation of PKM2 may represent an effective treatment approach for human lung cancer.
PTMs as an important protein-regulating mechanism are reported to cause cancers by regulation of key factors and signaling pathways.30 PTM of PKM2, such as phosphorylation or acetylation, has been reported to regulate PKM2 function by affecting its protein stability, homodimerization, and translocation.8 SUMO1 modification is an important type of PTMs, and is involved in the regulation of tissue development and cancer progression.17 And this type of PTM manifests its effect through regulating protein stability, protein–protein interaction, and protein localization. However, how SUMO1 modification affects the role of PKM2 in tumorigenesis in lung cancer is still unknown. A previous study has reported a stimulatory role of SUMO1 on glycolysis in cancer cells.20 Consistent with this report, we found that glucose uptake and lactate production were decreased by SUMO1 knockdown, and its effect on glycolysis was independent of PKM2. Further research revealed that SUMO1 played a vital role in improving protein stability for PKM2. We speculated such regulation was most likely posttranslational because SUMO1 did not affect PKM2 mRNA expression. PKM2 has been reported to be degraded by lysosomal pathway.8 Further experiments revealed that Leupeptin (an inhibitor of lysosomal proteases) inhibited the decrease of PKM2 protein induced by SUMO1 knockdown, suggesting that SUMO1 enhanced the protein stability of PKM2 by regulating its degradation process. In the meantime, we found that PKM2 could be modified by SUMO1 at Lys336 site in vivo and in vitro.
Then we tested whether SUMO1-modified PKM2 promoted glycolysis via Lys336 residue. And through loss-of-function analysis, we showed that Lys336 residue was critical for SUMO1-modified PKM2 to function as a key glycolytic enzyme. The findings from OCR indicated that SUMO1 modified PKM2 shunted glucose from TCA cycle to glycolysis, which was in line with the function of PKM2 as a central point of regulation the switch of glycolysis and TCA cycle.7 Besides being an important rate-limiting enzyme in aerobic glycolysis, the non-metabolic function of PKM2 is emerging in cancer. Nuclear PKM2 as a coactivator of HIF-1α plays another vital role in Warburg effect and tumorigenesis.22,23 Thus, we tested whether SUMO1 modification of PKM2 at Lys336 site affected the cofactor functions of this protein. The results showed that SUMO1-modified PKM2 significantly increased HIF-1α transcriptional activity, thus increasing the expression of its target genes, including GLUT1, LDHA, ENO1, and PDK1. GLUT1 and LDHA are essential for glucose uptake and the conversion of pyruvate to lactate.9 PDK1 inactivates pyruvate dehydrogenase, thereby shunting pyruvate away from the mitochondria.31 Our findings highlight the essential nuclear functions of SUMO1-modified PKM2 in the regulation of cancer cell metabolism.
It has been shown that PKM2 is strongly expressed in several types of cancer. Knockout of PKM2 could inhibit cell proliferation and tumorigenesis by reducing aerobic glycolysis.9 PTM of PKM2, such as phosphorylation or acetylation, has been reported to be involved in tumor proliferation in vivo and in vitro.8 Our in vitro growth assays revealed that cancer cells overexpressing wild-type PKM2 grew quicker than K336R mutant PKM2, and this growth-promotion effect was SUMO1-dependent. Taken together, our study suggested that SUMO1-modified PKM2 promoted tumor cell proliferation in vitro.
Conclusion
In this study, we found that PKM2 as a vital rate-limiting enzyme in glycolytic pathway was increased in lung adenocarcinoma patients. Patients with high PKM2 expression had reduced overall survival and disease-free survival rates compared with those with low PKM2 expression, suggesting that upregulated PKM2 expression could serve as a promising marker for tumor progression and prognosis in lung adenocarcinoma patients. Mechanistic analysis revealed that SUMO1 modification played an important role in stabilizing the protein level of PKM2 and promoting tumorigenesis in lung cancer cell lines. Thus, our results will provide a novel mechanistic basis of targeting SUMO1 modification of PKM2 in treating lung cancer.
Acknowledgments
The study was supported by research grants from National Natural Science Foundation of China (Grant No 81530053, 81471685, 81372195, 81471687, 81572719). “973” Project (2012CB932600), and Shanghai Municipal Commission of Health and Family Planning (Grant No 201440606).
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Didkowska J, Wojciechowska U, Manczuk M, Lobaszewski J. Lung cancer epidemiology: contemporary and future challenges worldwide. Ann Transl Med. 2016;4(8):150. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Israelsen WJ, Vander Heiden MG. Pyruvate kinase: function, regulation and role in cancer. Semin Cell Dev Biol. 2015;43:43–51. | ||
Noguchi T, Yamada K, Inoue H, Matsuda T, Tanaka T. The L- and R-type isozymes of rat pyruvate kinase are produced from a single gene by use of different promoters. J Biol Chem. 1987;262(29):14366–14371. | ||
Wang YH, Israelsen WJ, Lee D, et al. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. 2014;158(6):1309–1323. | ||
Wong N, De Melo J, Tang D. PKM2, a central point of regulation in cancer metabolism. Int J Cell Biol. 2013;2013:242513. | ||
Lv L, Li D, Zhao D, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell. 2011;42(6):719–730. | ||
Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC.The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth.Nature. 2008;452(7184):230–223. | ||
Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab. 2012;23(11):560–566. | ||
Zhou CF, Li XB, Sun H, et al. Pyruvate kinase type M2 is upregulated in colorectal cancer and promotes proliferation and migration of colon cancer cells. IUBMB Life. 2012;64(9):775–782. | ||
Yu G, Yu W, Jin G, et al. PKM2 regulates neural invasion of and predicts poor prognosis for human hilar cholangiocarcinoma. Mol Cancer. 2015;14:193. | ||
Huang P, Zhao X, Xiao W, Dong Y, Hu G. 18F-fluorodeoxyglucose uptake predicts PKM2 expression in lung adenocarcinoma. Oncotarget. 2017;8(24):39618–39626. | ||
Sun H, Zhu A, Zhang L, Zhang J, Zhong Z, Wang F. Knockdown of PKM2 suppresses tumor growth and invasion in lung adenocarcinoma. Int J Mol Sci. 2015;16(10):24574–24587. | ||
Guo W, Zhang Y, Chen T, et al. Efficacy of RNAi targeting of pyruvate kinase M2 combined with cisplatin in a lung cancer model. J Cancer ResClin Oncol. 2010;137(1):65–72. | ||
Yang XJ. Multisite protein modification and intramolecular signaling. Oncogene. 2005;24(10):1653–1662. | ||
Johnson ES. Protein modification by SUMO. Annu Rev Biochem. 2004;73:355–382. | ||
Spoden GA, Morandell D, Ehehalt D, et al. The SUMO-E3 ligase PIAS3 targets pyruvate kinase M2. J Cell Biochem. 2009;107(2):293–302. | ||
Yu J, Zhang SS, Saito K, et al. PTEN regulation by Akt–EGR1–ARF–PTEN axis. EMBO J. 2009;28(1):21–33. | ||
Agbor TA, Cheong A, Comerford KM, et al. Small ubiquitin-related modifier (SUMO)-1 promotes glycolysis in hypoxia. J Biol Chem. 2011;286(6):4718–4726. | ||
Ren J, Gao X, Jin C, et al. Systematic study of protein sumoylation: development of a site-specific predictor of SUMOsp 2.0. Proteomics. 2009;9(12):3409–3412. | ||
Luo W, Hu H, Chang R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145(5):732–744. | ||
Yang W, Zheng Y, Xia Y, et al. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol. 2012;14(12):1295–1304. | ||
Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13(6):472–482. | ||
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. | ||
Beer DG, Kardia SL, Huang CC, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8(8):816–824. | ||
Karnoub AE, Dash AB, Vo AP, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449(7162):557–563. | ||
Alon U, Barkai N, Notterman DA, et al. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc Natl Acad Sci U S A. 1999;96(12):6745–6750. | ||
Lee JS, Leem SH, Lee SY, et al. Expression signature of E2F1 and its associated genes predict superficial to invasive progression of bladder tumors. J Clin Oncol. 2010;28(16):2660–2667. | ||
Chen Z, Lu W. Roles of ubiquitination and SUMOylation on prostate cancer: mechanisms and clinical implications. Int J Mol Sci. 2015;16(3):4560–4580. | ||
Wheaton WW, Chandel NS. Hypoxia regulates cellular metabolism. Am J Physiol Cell Physiol. 2011;3000(3):C385–C393. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.