Back to Journals » Nature and Science of Sleep » Volume 13
Sleep-Related Hypermotor Epilepsy: Etiology, Electro-Clinical Features, and Therapeutic Strategies
Authors Wan H ![]() , Wang X, Chen Y, Jiang B, Chen Y, Hu W, Zhang K, Shao X
, Wang X, Chen Y, Jiang B, Chen Y, Hu W, Zhang K, Shao X
Received 25 July 2021
Accepted for publication 21 October 2021
Published 13 November 2021 Volume 2021:13 Pages 2065—2084
DOI https://doi.org/10.2147/NSS.S330986
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Ahmed BaHammam
Huijuan Wan,1– 3,* Xing Wang,4,* Yiyi Chen,1,2 Bin Jiang,3 Yangmei Chen,5 Wenhan Hu,6 Kai Zhang,7 Xiaoqiu Shao1,2
1Department of Neurology, Beijing Tiantan Hospital, Capital Medical University, Beijing, People’s Republic of China; 2China National Clinical Research Center for Neurological Diseases (NCRC-ND), Beijing, People’s Republic of China; 3Department of Neurology, First Affiliated Hospital, Xiamen University, Xiamen, People’s Republic of China; 4Department of Neurology, Chongqing University Central Hospital, Chongqing Emergency Medical Centre, Chongqing, People’s Republic of China; 5Department of Neurology, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China; 6Beijing Neurosurgical Institute, Capital Medical University, Beijing, People’s Republic of China; 7Department of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaoqiu Shao
Department of Neurology, Beijing Tiantan Hospital, Capital Medical University, No. 119 South 4th Ring West Road, Fengtai District, Beijing, 100070, People’s Republic of China
Tel +86-135-5203-4066
Email [email protected]
Abstract: Sleep-related hypermotor epilepsy (SHE) is a group of clinical syndromes with heterogeneous etiologies. SHE is difficult to diagnose and treat in the early stages due to its diverse clinical manifestations and difficulties in differentiating from non-epileptic events, which seriously affect patients’ quality of life and social behavior. The overall prognosis for SHE is unsatisfactory, but different etiologies affect patients’ prognoses. Surgical treatment is an effective method for carefully selected patients with refractory SHE; nevertheless, preoperative assessment remains challenging because of the low sensitivity of noninvasive scalp electroencephalogram and imaging to detect abnormalities. However, through a careful analysis of semiology, the clinician can deduce the potential epileptogenic zone. This paper summarizes the research status of the background, etiology, electro-clinical features, diagnostic criteria, prognosis, and treatment of SHE to provide a more in-depth understanding of its pathophysiological mechanism, improve the accuracy in the diagnosis of this group of syndromes, and further explore more targeted therapy plans.
Keywords: semiology, seizure onset zone, SOZ, epileptogenic zone, hypermotor, electroencephalogram
Background
In 1981, Lugaresi and Cirignotta reported five patients with frequent clusters of seizure events during sleep, clinically manifested by bizarre limb movements or dystonic-tonic posturing.1 These five patients responded effectively to carbamazepine treatment but not to other antiepileptic drugs, and their interictal and ictal scalp electroencephalogram (EEG) showed no significant abnormalities. Therefore, this syndrome was initially considered a wake-triggered sleep motor disorder and was first described with the term “hypnogenic paroxysmal dystonia” (Figure 1),” which was later changed to “nocturnal paroxysmal dystonia” (NPD) (Figure 1).2 In the subsequent decades, there have been constant reports of patients with similar clinical manifestations. Nevertheless, whether this syndrome is an epileptic condition is a subject of frequent debate. In 1990, Tinuper et al studied three patients with typical NPD who had frequent seizures during non-rapid eye movement sleep (NREM); two patients had generalized tonic-clonic seizures following typical NPD seizures, and epileptiform discharges in the frontal area were observed on ictal and interictal EEG. Therefore, they concluded that this NPD with a short duration was an epileptic seizure of deep frontal lobe origin, also known as nocturnal frontal lobe epilepsy (NFLE) (Figure 1).3 There has been disagreement about the diagnostic criteria for NFLE and difficulties in differentiating it from non-epileptic condition. A consensus meeting including experts in adult and pediatric epileptology, sleep medicine, and epidemiology was held in Bologna in 20144 to better characterize this disorder’s electro-clinical features and develop more accurate diagnostic criteria. At this meeting, the syndrome was renamed “sleep-related hypermotor epilepsy” (SHE) as a unique syndrome independent of the etiology (genetic, structural, or both) and the brain regions involved (Figure 1). The following consensus were reached: (1) seizures are predominantly experienced during sleep rather than at night, and (2) seizures can originate not only in the frontal lobe but also outside the frontal lobe, and there is no significant difference in epileptic semiology characteristics.
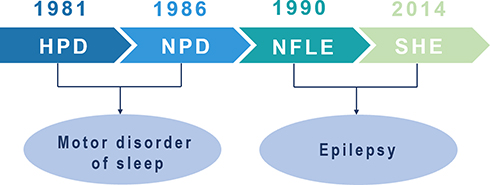 |
Figure 1 Historical background of sleep-related hypermotor epilepsy. Abbreviations: HPD, hypnogenic paroxysmal dystonia; NPD, nocturnal paroxysmal dystonia; NFLE, nocturnal frontal lobe epilepsy; SHE, sleep-related hypermotor epilepsy. |
Etiology of SHE
Most SHE cases are sporadic, with no clear etiology. The main known etiologies include genetic and structural etiologies. Structural abnormalities include focal cortical dysplasia (FCD) and acquired injuries. In terms of clinical presentation, there is no significant difference between sporadic and familial cases, that is, autosomal dominant sleep-related hypermotor epilepsy (ADSHE).4
Structural Etiology
FCD is the most common pathological manifestation in patients with surgically treated drug-resistant SHE, especially FCD type II.5,6 Previous studies have also shown that FCD, particularly small FCD type II, frequently leads to sleep-related epileptic seizures.7 Abnormal neuronal architecture within the FCD leads to abnormal synaptic connections and neurotransmitter systems, resulting in enhanced local epileptogenicity and the generation of various abnormal electrical activities. Nobili et al hypothesized that the sleep-related seizures seen in patients with FCD might be due to the specific electrical activity exhibited by the dysplastic cortex, that is, short bursts of fast discharge that can be suppressed by activity during wakefulness and are usually more sporadic during wakefulness but increase in frequency during slow-wave sleep and can be periodic. This characteristic low-amplitude fast activity has also been observed in non-lesion areas around the FCD during slow-wave sleep.8 In surgically treated patients with SHE, FCD, developmental tumors (eg, dysembryoplastic neuroepithelial tumor), brain trauma, and ganglioglioma have also been reported.5
Genetic Etiology
SHE includes sporadic and familial forms, with approximately 86% of cases being sporadic and 14% being familial.9 Although the etiology is unknown in the vast majority of patients, a small number of sporadic cases and approximately 30% of patients with familial SHE are associated with genetic mutations.9,10 As a genetic heterogeneity syndrome, the severity of epileptic seizures in SHE families varies greatly and may be related to mutations in different genes.9 We summarize the major genes that have been reported in Table 1.
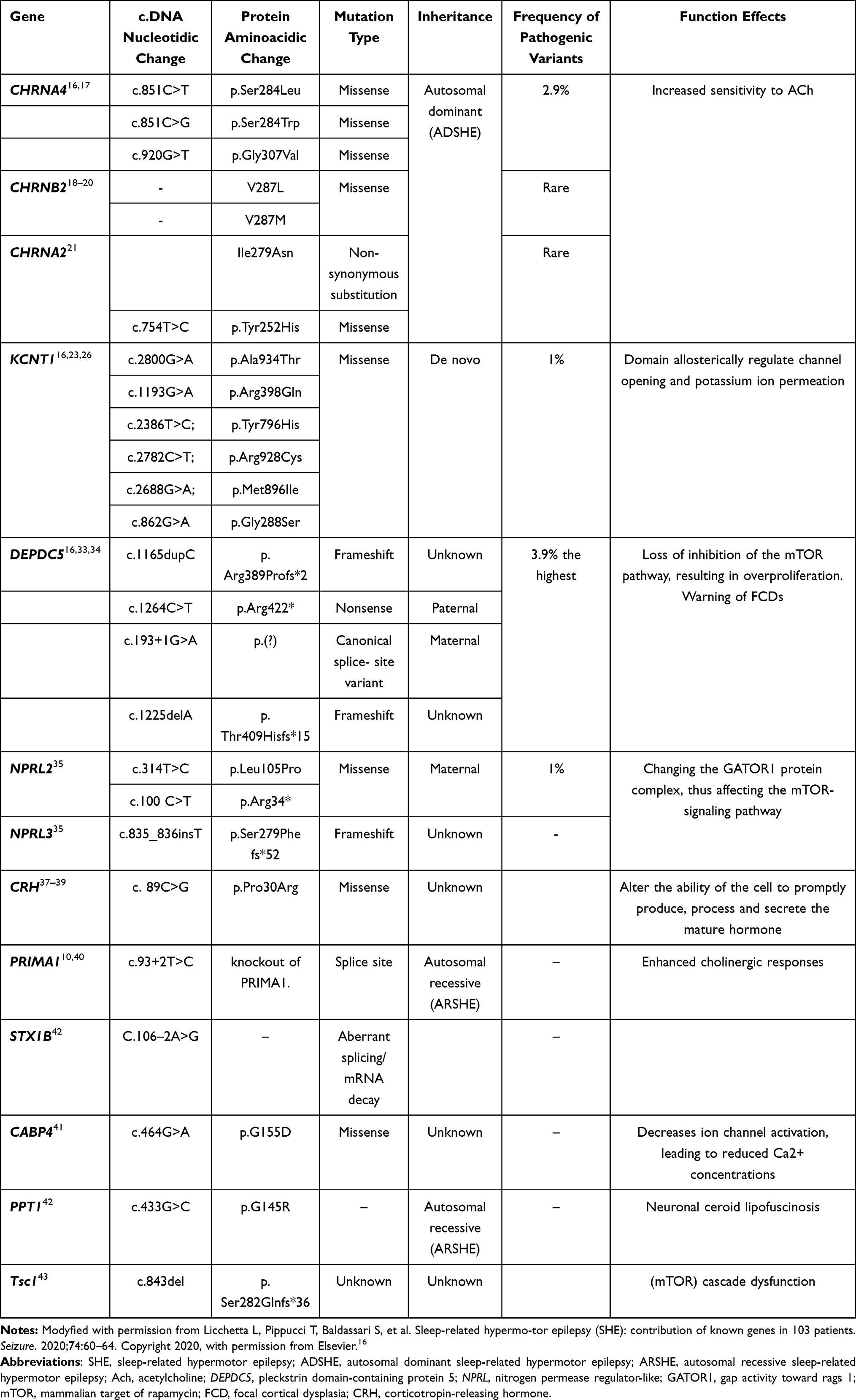 |
Table 1 Pathogenic and Likely Pathogenic Variants in SHE-Associated Genes |
In 1995, Steinlein et al reported a missense mutation in CHRNA4, which encodes the α4 subunit of the nicotinic acetylcholine receptor (nAChR) in a family of 21 patients with ADSHE and four carriers.11 Since then, mutations in other genes, such as CHRNA2 and CHRNB2, have been reported.12,13 nAChRs are widely distributed in the brain and contribute to the regulation of sleep-wake oscillations at the cortical and subcortical levels.14 However, the function of nAChRs is altered after mutations in the genes, resulting in increased sensitivity to ACh, activation of the ascending cholinergic pathway in the brainstem, and enhanced GABAergic function,15 thus creating conditions for sleep instability and the generation of epileptic seizures. Up to now, the pathogenic and likely pathogenic variants in CHRNA4 that have been identified include p.Ser284Leu, p.Ser284Trp, and p.Gly307Val,16 which have been summarized in Table 1. The table does not include the p.Ser252Leu and p.Thr26Met mutations, as well as two missense variants on the same allele of the CHRNA4 gene: c.742A>T (Ile248Phe) and c.1708G>C(Gly570Arg),17 whose variants were anecdotally implicated in inherited forms of SHE, but not confirmed in other cases. Previous studies have identified V287L and V287M mutations in the CHRNB2 gene, coding for the β2 subunit of the nAChR.18,19 Both mutations involve the same residue, located, as the CHRNA4 mutations, that cause nAChR functionality to change.18–20 Similarly, the mutation on CHRNA2, resulting in the non-synonymous substitution Ile279Asn, also increases the nAChR sensitivity, as observed in several ADSHE-linked mutations of the onα4 and β2 subunits.21 However, these mutations in CHRNB2 and CHRNA2 are rare in patients with ADSHE.18,19,21
Mutations in the KCNT1 gene encoding subunit 1 of the sodium-activated potassium channel have been identified in some patients with ADSHE and comorbid psychiatric, behavioral, and intellectual disorders.22 The pathogenic mechanism of KCNT1 mutations may be the increased firing frequency of pyramidal neurons and attenuation of the inhibition of interneuron excitability, leading to network de-inhibition and epileptic seizures.23,24 Previous studies have suggested that mutations in KCNT1 are associated with non-lesional SHE. In a recent study, four patients with SHE were reported to have mild malformations of cortical development (mMCD) and KCNT1 mutations. Three unrelated patients with magnetic resonance imaging (MRI)-negative results received a neuropathological diagnosis of FCD type I after epilepsy surgery, and one case of periventricular nodular heterotopia was detected by MRI. Studies have suggested that KCNT1 mutations are epileptogenic not only because of excitability dysregulation due to the role of this gene in controlling sodium-potassium transport but also possibly due to mMCD. Thus, epilepsy caused by pathogenic variants of KCNT1 may include both lesional and non-lesional epilepsy.25 We listed SHE-related pathogenic mutations of KCNT1 in Table 1; however, it should be noted that even in members of the same family, the same KCNT1 mutation can either cause SHE or malignant migrating focal seizures of infancy.23,26
In addition, loss-of-function (LOF) mutations in the pleckstrin (DEP) domain-containing 5 (DEPDC5) gene have been identified in some ADSHE-affected families, and patients with DEPDC5 mutations have shown a high rate of drug resistance.27 DEPDC5 was previously thought to be associated with familial focal epilepsy with variable foci.28 The mechanistic target of rapamycin (mTOR) protein has serine-threonine kinase activity; it phosphorylates several downstream proteins and regulates cell growth, proliferation, and migration.29 GTPase-activating protein activity toward the Ras-related GTPases (GATOR) complex 1 (GATOR1) is upstream of the mTOR pathway and negatively regulates mTOR complex 1 (mTORC1).30,31 The GATOR1 complex consists of three proteins: DEPDC5, nitrogen permease regulator-like 2 (NRPL2), and nitrogen permease regulator-like 3 (NRPL3). Thus, LOF mutations in the DEPDC5 gene can lead to loss of inhibition of the mTOR pathway, resulting in overproliferation, which leads to irregular growth of the lesion, anatomical disruption of the neural circuit, and consequently epilepsy. The mTOR pathway is involved in the pathogenesis of brain malformations.32
DEPDC5 shows the highest mutation frequency, especially in cases with a structural etiology.16 Thus, analysis of this gene is recommended even in isolated cases. The DEPDC5 variants identified include four LOF variants, namely, p.Arg389Profs*2, p.Arg422*c.193+1G>A, and p.Thr409Hisfs*15.33,34 Furthermore, variants of the genes encoding NPRL2 and NPRL3 were also confirmed, but whether these mutations are pathogenic and their role in the pathogenesis of SHE still needs to be confirmed in further studies.33–35
In addition to the aforementioned mutations, an Italian ADSHE family line study revealed a mutation in the CRH gene.36 In 2013, Sansoni et al reported a novel mutation (p.Pro30Arg) within the CRH gene cosegregating with ADSHE d detected in an Italian family. The resulting amino acid change in CRH was predicted to be pathogenic (PolyPhen-2).37,38 The mutation appears to alter the ability of the cell to promptly produce, process, and secrete the mature hormone, but the direct role of the p.Pro30Arg in the pathogenesis of SHE remains unclear.37 Mutation of the CRH gene has been confirmed in patients with ADSHE, and variations in the CRH gene detected in patients with SHE lead to altered hormone levels, suggesting that individuals with such hormonal alterations may be more susceptible to the disease.37,39
In 2015, Hildebrand et al reported a newly identified autosomal recessive pathogenic gene, the PRIMA1 gene of chromosome 14 in the splicing site (C.93+2T>C), wherein a homozygous mutation led to the skipping of the first coding exon of PRIMA1 mRNA and the deletion of the PRIMA1 gene. PRIMA1 is a transmembrane protein that anchors acetylcholinesterase (AChE) to the membrane of neurons. Reduction of this gene leads to decreased AChE, accumulation of acetylcholine and mutation of functional effects of acetylcholine receptor subunits, leading to SHE. Enhanced cholinergic response may be responsible for severe intellectual disability in this family. This is the first reported case of a recessive mutation of PRIMA1 associated with the disease but has not been confirmed in other cases.40
Chen et al performed whole-exome sequencing in four typical patients selected from a family line with seven patients. They identified the CABP4 gene, encoding calcium-binding protein 4, as a possible new pathogenic gene for ADSHE.41 Mutation of this gene mainly affects the calcium channel, leading to abnormal distribution of calcium ions inside and outside the cell, thereby inducing seizures.41 The STX1B gene encodes syntaxin-1B, which is involved in the release of glutamate and GABA. Mutations in this gene may disrupt the inherent splicing receptor site, leading to abnormal splicing and resulting in abnormal proteins or mRNA decay. STX1B gene mutation may be related to the SHE phenotype and autonomic dysfunction, characterized by peri-ictal hypotension, and testing for this gene should be considered in such patients.42 In this study, a variant of unknown significance was also detected in the PPT1 gene (encoding palmitoyl protein thioesterase 1), but whether it plays a role in SHE needs further experimental verification.42 A case report suggests that SHE may also be associated with mutations in the TSC1 gene, which plays a pathogenic role in SHE through the (mTOR) pathway.43 Recent studies in epileptic brain tissues have suggested that defective expression of circadian locomotor output cycles kaput (CLOCK), a transcription factor that regulates the circadian rhythm and mTOR pathway, may be responsible for the predisposition of epileptic seizures during sleep.44
The genetic and structural etiologies of SHE are not mutually exclusive. In particular, patients with refractory epilepsy with mutations in GATOR1-related genes combined with cortical developmental malformations can benefit from surgery. Therefore, patients with hereditary SHE are not an absolute contraindication for surgery.
Electro-Clinical Features of SHE
Clinical Features
SHE is relatively rare, with a large retrospective population-based cohort study showing its prevalence at approximately 1.8/100,000.45 According to previous reports, most patients had disease onset before the age of 20 years,7,8 with a predominance in childhood and adolescence,46 although seizures originating in adulthood have also been reported.47 Epileptic seizures occur mainly during sleep, mostly during NREM sleep, rarely during rapid eye movement sleep, and occasionally during wakefulness.3 Seizure frequency may be very high, with episodes occurring every or almost every night, usually multiple episodes per night.7 “Clusters of seizures” are a diagnostic feature but not required. As most seizures occur during sleep, sleep disruption can lead to daytime sleepiness in patients.48
The primary forms of seizures are brief (less than 2 min), individualized and stereotyped hypermotor seizures4 with sudden onset and offset.6,9,49 The clinical features are vigorous hypermotor movements (eg, hypermotor proximal joint or trunk movements, such as limb kicking, arm flinging, and body rocking),6,50 usually accompanied by vocalization, emotional facial expressions, and vegetative symptoms.50,51 Asymmetric dystonic/tonic posturing with or without head/eye deviation can also be observed in patients with SHE.46,52
In addition, patients with SHE often experience paroxysmal arousals (PAs) and minor motor events (MMEs). PAs are characterized by sudden and brief awakenings (lasting 5–10 s), sometimes accompanied by stereotyped movements, vocalizations, nervous expressions, and fearfulness.6,46 Stereotyped movements may manifest as sudden movements of the trunk and upper limbs, with the patient appearing awake but unconscious and falling asleep soon after the episode. MMEs are characterized by brief (lasting 2–4 s), stereotyped movements involving the axial musculature, limbs, and/or head.46,53 In stereoelectroencephalography (SEEG) recordings, PAs are always closely associated with epileptiform discharges.54–56 However, it is difficult to ascertain whether the MMEs should be regarded as an epilepsy event.54,57,58 By analyzing SEEG recordings in patients with SHE, Terzaghi et al found that more than two-thirds of MMEs were related to epileptiform discharges, and the variable expression of MMEs could not be attributed to the presence of epileptiform discharges.53 Current opinion considers that MMEs are probably not true epileptic manifestations but rather are motor manifestations with relation to sleep instability.56 Occasionally, patients will get out of bed and run around repetitively. These movements are called epileptic nocturnal wanderings, which are difficult to distinguish from sleepwalking.46 A video polysomnography (VPSG) study of 100 NFLE cases found that sleep-associated seizure events of varying intensity (from PAs to nocturnal wanderings) and duration may occur in a single night in a given patient.46
SHE seizures vary in complexity, ranging from sudden arousal or mild motor events46,59 to hypermotor seizures, and multiple seizure forms can be present in combinations in a single patient, even during a single night. This may be due to differences in the discharge duration within the frontal lobe and the pathways that propagate within the brain.59,60 As highlighted in the 2014 consensus conference,4 SHE does not solely originate in the frontal lobes, and seizures initiated outside the frontal lobes may also trigger hypermotor seizures or dystonic-tonic postural seizures. A SEEG study in patients with drug-resistant SHE confirmed that up to 30% of seizures were of extrafrontal origin.61 The extrafrontal epileptogenic zone (EZ) reported in several studies also includes the temporal and parietal lobes and the insula.5,56,62–66 The clinical features of SHE of the frontal and extrafrontal origins are very similar. Regardless of the origin, hypermotor seizures and complex behavior occur when ictal discharges are transmitted to structures such as the cingulate, frontal, and parietal cortices.52
Increasing number of studies have reported that some patients with SHE have cognitive deficits and behavioral and mental problems.26,67–70 Studies have also reported that SHE has negative effects on patients’ neuropsychology, with 53.3% of patients presenting with neuropsychological defects and 15% diagnosed with intellectual disabilities.71 The cognitive impairment was more significant in verbal IQ and extrafrontal and selective frontal functions.71 Furthermore, variables of clinical severity (ie, high seizure frequency, status epilepticus, and bilateral convulsive seizures, poor response to antiepileptic treatment) were negatively correlated with memory and executive functions.71
In addition to the variables of clinical severity mentioned above, it is currently believed that cognitive impairment is also related to the following factors: 1. Genetic and structural etiologies. Previous studies have shown that patients with SHE with genetic mutations have lower IQ scores than those who have do not these mutations, irrespective of the specific gene.48 Simultaneously, we also believe that the neuropsychological damage caused by different gene mutations may have different underlying mechanisms,71 which is corroborated by some clinical and basic studies, for example, KCNT1 mutations appear to be associated with more severe intellectual impairment and psychiatric symptoms;4 GATOR1 expression related gene mutations are associated with early-onset of epilepsy, cognitive decline, and sudden death in patients with epilepsy, and approximately 20% of patients may be associated with cortical dysplasia,72 whereas several studies have described mutations in the genes coding for nAChR subunits (eg, CHRNA4, CHRNA2, and CHRNB2) with particular emphasis on the impact on frontal lobe functions. Animal studies have also shown that S284L-mutation in CHRNA4 caused several important functional associations with ADSHE and its comorbidity of cognitive deficits.73–76 However, more studies are needed to further investigate the genotype-clinical correlation and their underlying mechanisms in patients with SHE. 2. Sleep fragmentation. Most patients with SHE experience frequent seizures, especially during sleep, thus frequent seizures disrupt sleep architecture. It is well known that sleep is crucial to learning and memory. Sleep reactivates neuronal learning activities, allowing the reprocessing of memories during both NREM and REM sleep, which interact during memory consolidation.77 The disruption of sleep quality caused by epilepsy (including shorter total sleep time, poorer sleep efficiency, and prolonged sleep latency) may impair executive function and memory.78 3. The onset age. Animal model studies have suggested that the cause of cognitive dysfunction due to early-onset epilepsy could be damage to neural regeneration and the reorganization of synapses, resulting in the absence of dendritic spines in hippocampal pyramidal neurons,79 thus raising the question whether there is a possibility that the younger the age of onset of SHE, the more severe the cognitive impairment will be and what the underlying mechanism; these need to be investigated in future studies.
Seizure Semiology and Anatomoclinical Correlation
Seizure semiology refers to the physical signs and symptoms of seizures dependent on the epileptic network involved in the seizure, that is, the EZ and its propagation networks.80 Epileptic seizures can involve multiple sites sequentially and may combine multiple seizure types with a relatively fixed order of evolution.81 Epilepsies originating in a particular brain region often have a relatively characteristic clinical semiology.82 Careful generalization and search for this evidence can provide a more accurate understanding of the seizure origin and epileptic network. In the last decade, with the rapid development of SEEG, the electro-clinical features of focal epilepsy and its relationship with functional anatomy have become clearer in combination with semiology and structural and functional imaging, and anatomical-electrical-clinical data provide adequate evidence to locate the sites of origin of SHE and the epileptic networks. Although semiology is subjective in nature82 and the symptomatogenic zone is not equivalent to the EZ or seizure onset zone (SOZ),83,84 there is a strong connection between the EZ and early propagation networks;81 thus, early propagation networks can be anchored by analyzing semiology patterns and employed to find SOZ and predict the EZ.
Many patients with surgically feasible SHE have no identifiable lesions on cranial MRI.4 If, at the same time, the scalp EEG in such patients does not provide sufficient lateralization and localization information,4 ictal semiology is highly suggestive in the preoperative evaluation. After targeting the region of interest by ictal semiology, a more targeted review of MRI or its combination with functional imaging data can help identify some subtle lesions, improve the rate of positive results by MRI, and facilitate a more precise prediction of the EZ, or provide a basis for subsequent intracranial electrode implantation, enabling a significant proportion of MRI-negative patients with SHE to benefit from surgical treatment as early as possible.
Aura
An aura is a completely subjective symptom that occurs during an epileptic seizure, usually at the beginning of the seizure, and therefore often provides very useful information about the localization of the SOZ. Although most seizures in patients with SHE emerge from sleep, patients have fewer complaints about auras. Auras during wakefulness seizure events in these patients may have localization values.56 For example, abdominal auras, auditory auras, and déjà vu suggest that the seizure originates in the temporal lobe;85 the feeling of throat constriction, a choking sensation, and unpleasant somatosensory auras, such as pain, suggest that the seizure originates in the insula and operculum;5,61 and somatic sensations and complex proprioceptive sensations, such as feelings of levitation, vertigo, and falling sensations, suggest seizures originating in the parietal lobe.5,62 Cephalic auras and whole-body auras have been observed in frontal SHE; these auras can arise from the entorhinal cortex, lateral temporal neocortex, second sensory area, and supplementary sensorimotor area but often lack clear localizing value.86,87 However, these nonspecific auras were common in both frontal lobe epilepsy and the frontal SHE subgroup, thus increasing the localization yield when present.5
Seizure Semiology Patterns of Different Origins
The semiological seizure classification of hypermotor seizures was proposed and defined by Luders in 1998 and the terminology committee of the International league against epilepsy in 2001.85,88 To explore the epileptogenic networks involved in various types of hypermotor symptoms, scholars such as Rheims52 and Montavont89 classified hypermotor seizures (HMS) into three categories (Figure 2): patients with HMS type 1 have strong negative emotions (eg, fear, anger) and agitated, but relatively coordinated movements, including body rocking, pedaling or boxing, and sitting or standing up from the bed;52,89 patients with HMS type 2 have relatively mild emotions and less agitated movements, including horizontal movements or rotation of the trunk and pelvis, accompanied by tonic/dystonic postures, without leaving the bed;52,89 and patients with the mixed type of HMS mainly present with repetitive movements of the bimanual bipedal and pelvic movements, including kicking and pelvic thrust, often without leaving the bed, and these may be accompanied by strong gripping or dystonia.89 Gibbs et al analyzed the SOZ of 135 patients with SHE confirmed by a video EEG (VEEG) through preoperative anatomic-electro-clinical data (including SEEG), postoperative head MRI, and postoperative Engel’s classification and summarized four seizure semiology patterns (SPs) of SHE:5 SP1 involves early elementary motor signs and asymmetrical tonic posture, which may be combined with clonus; SP2 involves unnatural hypermotor movements, axial tonic posture, nonverbal vocalization, and facial contractions (ictal pouting); SP3 involves integrated hypermotor movements, such as pedaling behavior, which may be combined with distal limb stereotypy, speech, and manipulation behaviors; and SP4 is characterized by gestural behavior with high emotional content, including strong negative emotions, fearful facial expressions, and fight or flight behaviors, such as paroxysmal wandering, which may be combined with autonomic symptoms. In the above typing, if more than one motor symptom is exhibited, the patient is typed according to the earliest motor symptoms present.5
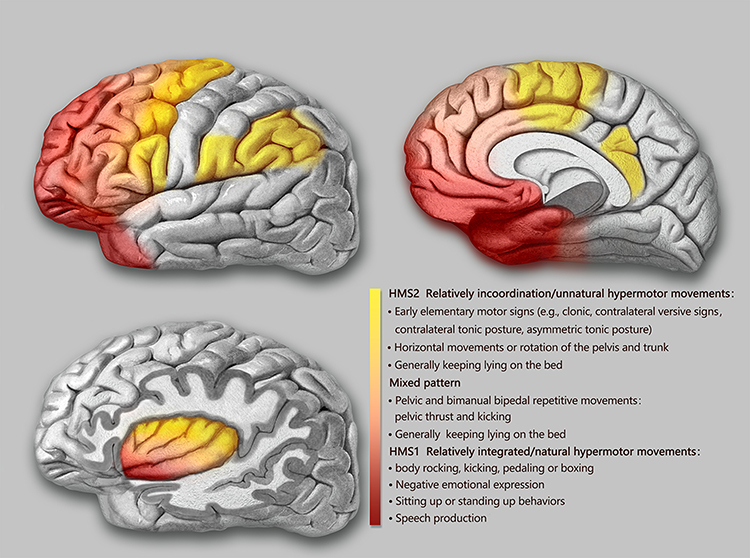 |
Figure 2 Schematic representation of the seizure semiology patterns of SHE derived from anatomical-electrical-clinical data. Abbreviation: HMS, hypermotor seizures. Notes: The seizure semiology patterns were grouped in three categories (HMS type 1 and 2 and mixed type of HMS) and depicted by three different colors (red, light red, and yellow). Although there were no strict boundaries among the color-coded groups, the specific HMS type contributed to the prediction of the EZ . |
SHE of Frontal Lobe Origin
Based on functional anatomical and electro-clinical data of the frontal lobe, the frontal lobe epileptic semiology pattern and its localization value have been extensively studied. The more posterior the EZ (ie, close to the caudal primary motor cortex of the frontal lobe), the more likely it is that elementary motor signs, such as clonus, contralateral tonic posturing, and contralateral version, will be present during seizures.90 The more anterior the EZ (ie, near the rostral side of the frontal lobe-frontal pole, orbitofrontal cortex), the more coordinated the patient’s movements will be during seizures, often accompanied by emotional responses, such as fearful facial expressions.90 Fear-like behaviors are associated with the frontal or frontotemporal paralimbic system.90 SEEG data confirmed52,89 that the SOZ suggested by different types of frontal HMS also follow the “rostrocaudal axis pattern” of frontal semiology (Figure 2). The SOZ associated with HMS type 1 is concentrated in the ventromedial frontal cortex, including the orbitofrontal cortex (OFC), rostral side of the anterior cingulate cortex (ACC), and frontal pole.52,89 Discharge during seizures can be transmitted to the temporal lobe, caudal side of the ACC, insula lobe, and transitional cortex between the prefrontal and premotor cortex. The SOZ associated with HMS type 2 is mainly located on the medial side of the premotor cortex, the caudal side of the ACC, and transition cortex between the prefrontal lobe and the premotor cortex.52,89 The SOZ associated with the mixed type of HMS is located primarily in the transitional cortex between the prefrontal and premotor cortices.89 Similarly, among the four SPs of SHE defined by Gibbs et al,5 the frontal lobe groups of SHE follow an “anteroposterior” distribution pattern, that is, the SOZ of SP1 is mainly located near the precentral sulcus, and the SOZ of SP4 is mainly located near the frontal pole and the OFC. The SOZs of SP2 and SP3 are more dispersed but follow the same “anteroposterior” trend. It is more difficult to distinguish between SP2 and SP3, and they are not as useful for localization as SP1 and SP4.5 The correlation between semiology pattern typing and the SOZ of both Gibbs5 and Rheims52 methods follows the same rule: from the rostral to the caudal side of the frontal lobe, the hypermotor symptoms range from complex to simple, from relatively coordinated to uncoordinated or unnatural hypermotor movements (often manifested as asymmetrical hypermotor movements with dystonia), with more emotional responses on the ventral side of the medial prefrontal lobe.90,91 The OFC, dorsolateral prefrontal cortex, amygdala, and ACC are key structures primarily involved in the emotional regulation circuit, and abnormalities in one of these interconnected structures or their interconnections can alter emotional regulation and increase impulsive aggression and violent tendencies.92 Using a computational model of EEG activity and nonlinear correlation analysis, the correlations between certain specific structures of the emotional network (eg, the amygdala, OFC, ACC, and temporal pole) were found to be significantly reduced in patients with frontal lobe epilepsy during the ictal phase compared to those during the background phase; in particular, the correlation between the OFC and amygdala was the most pronounced, thus leading to inhibition dissolution of negative emotions and corresponding agitated behaviors.93 This could explain the negative emotions present in patients with SHE exhibiting HMS type 1 and SP4 (Figure 2).
SHE of Temporal Lobe Origin
According to a summary of previous literature, patients with temporal SHE have a greater tendency to exhibit more integrated hypermotor behaviors (HMS type 1 or SP3 and SP4), usually of the distal limbs and accompanied by more intense negative emotions (Figure 2).5,94,95 The EZ of temporal SHE is mainly located in the anterior inferior temporal cortex (parahippocampal gyrus, anterior inferior temporal gyrus), temporal pole, and amygdala.96,97 Readings from subdural electrodes in a hypermotor patient with a temporal pole lesion confirmed that the low-voltage fast activity at the seizure’s onset began in the inferolateral anterior temporal lobe, and the patient developed hypermobility symptoms when a slow-wave rhythm appeared in the OFC, indicating that hypermotor seizures associated with temporal pole lesions are caused by the spread of discharges to the frontal lobes.91,94 The SEEG data also confirmed that the temporal pole and OFC are the most important in the HMS epileptogenic network of temporal lobe origin and that the amygdala is also involved.97 As previously mentioned, the diminished correlation between these emotional network structures in the temporal pole and the OFC results in emotion-related release signs. This could also explain the negative emotions in the semiology of patients with temporal lobe SHE.
SHE of Insula Lobe Origin
The insula lobe is a tissue deep in the brain covering the frontal, parietal, and temporal opercula. Because of the rich connectivity of the insula to the surrounding lobes, discharges originating in the insula have multiple propagation pathways, including the lateral sulcus, frontal lobe, and temporal lobe. Insular lobe epilepsy can mimic the symptoms of frontal, temporal, and parietal lobe epilepsy, often exhibiting a semiology of the cortices associated with the areas of discharge propagation. Therefore, it is difficult to diagnose clinically and tends to be misdiagnosed, and thus has been described as a great mimic.98 Many scholars have successively reported patients with NFLE and insular seizure onset confirmed by intracranial electrodes.64 These paroxysmal events mainly occurred during sleep and presented with symptoms of horizontal trunk hypermotor movements, asymmetric tonic/dystonic postures, and asymmetric pedaling.5,99 The SEEG recordings confirmed that ictal discharges started from the insula and hypermotor manifestations appeared when they spread to the cingulate cortex, superior frontal gyrus, supplementary motor area (SMA), and pre-SMA.64,99 Moreover, 5–40 s intervals between the ictal onset and hypermotor manifestations were observed using SEEG in these patients,99 which also suggested that hypermotor seizures in the insula are a symptom that appears after discharge diffusion. Patients with insular SHE have a greater tendency to show a semiology of HMS type 2 and mixed types or SP1 and SP2, whereas, in general, SP4 is not observed.5 However, when the SOZ or propagation networks involve the anterior insula, it can be accompanied by strong emotional responses. Ryvlin reported that three patients had hypermotor seizures of anterosuperior insular origin, confirmed by intracranial electrodes.64 Each patient also expressed fear because the ventral anterior insula is involved in emotional processing.100,101 According to the anatomic-electro-clinical data of insular lobe epilepsy by Wang et al,102 based on the Brainnetome Atlas, the semiology followed the anteroposterior “anteroventral to posterodorsal axis” distribution pattern. The semiology pattern in a group of patients with an epileptic network involving the anteroventral insular regions showed integrated gestural motor behaviors or feelings of rage or fear, whereas that in a group of patients with an epileptic network involving the posterodorsal insular regions showed non-integrated gestural motor behaviors or asymmetric tonic posture (Figure 2).102
SHE of Parietal Origin
In addition to the postcentral gyrus, the parietal cortex includes the superior parietal lobule, inferior parietal lobule, and parietal operculum.103 In previous studies, the main clinical features of parietal SHE included asymmetric “hypermotor” events and dystonia91 and were associated with an SOZ in the inferior parietal lobule, parietal operculum, or posterior cingulate cortex (PCC).91,104,105 Bartolomei et al analyzed 17 patients with epilepsy in the posterior parietal cortex (including the superior parietal lobule, inferior parietal lobule, and parietal operculum),105 of whom four patients with EZs located in the parietal operculum or inferior parietal lobule exhibited hypermotor movements and dystonic or tonic posture, tending to fit the HMS type 2 or SP1 and SP2 (Figure 2). In contrast, no hypermotor seizures were observed in patients with epilepsy in the superior parietal lobule. Contralateral dystonia, and ipsilateral hemiballic-like movements could be observed in patients with epilepsy in the inferior parietal lobule and parietal operculum. The “hemiballic-like” movement can be considered a useful clue to the localization and lateralization of the SOZ.104 The connection between the parietal operculum and premotor cortex may underlie the anatomy of hypermotor movement in patients with parietal operculum epilepsy.106
In summary, seizure SPs facilitate the prediction of SOZ localization in patients with frontal and extrafrontal SHE. Based on the intracranial electrode data and evidence of semiology, the anatomical-electrical clinical features in patients with SHE are summarized in Figure 2; however, it is worth noting that there are extensive functional connections between the cerebral cortices, and the propagation of discharges is dynamic and rapid. The mode of propagation also affects the seizures’ clinical manifestations. Differences in SP reflect the spread of seizures in different brain regions that may constitute different epileptogenic networks. Thus, although these localization trends exist in different SPs, seizure SPs cannot be completely and strictly separated from each other.
Electrophysiological Features and Imaging Manifestations of SHE
Long-term VEEG monitoring can increase the chances of detecting EEG abnormalities because the violent movements of patients with SHE during seizures lead to a large number of action artifacts in the EEG that affect judgment, or the EZ may be at relatively deep locations, such as the orbitofrontal gyrus or medial structures, so that ictal and interictal scalp EEGs often cannot provide clear diagnostic information.6,9 However, even if the EEG fails to provide clear information, the diagnosis of SHE may not be excluded.4 Gibbs retrospectively analyzed 58 patients with refractory SHE (43 in the frontal lobe and 15 extrafrontal) who underwent surgery. All patients underwent preoperative SEEG recordings and achieved freedom from seizures at 2 years of follow-up (Engel class I). The included patients were classified as having seizures of frontal or extrafrontal origin, according to the localization information acquired by SEEG.107 The seizure duration was longer in the SHE of extrafrontal origin than in that of frontal origin (mostly less than 30 s), and the mean latency between the onset of an electrographic seizure and the appearance of hypermotor manifestations of extrafrontal SHE was also longer than that of frontal SHE. During video monitoring, a latency > 5 s between the first observed action (eg, eye-opening or a mild motor event) and the appearance of hypermotor manifestations exhibited a diagnostic sensitivity of 75% and a specificity of 90% for extrafrontal SHE.107 This time interval often coincides with the patient’s duration of arousal, suggesting that it is not the hypermotor behavior itself but the recruitment and propagation of electrical activity that causes arousal in patients with SHE.56 Considering that this feature can be obtained through noninvasive video analysis, it could be a valuable clinical clue for diagnosing extrafrontal SHE.
Gibbs et al analyzed 135 patients with SHE who underwent epilepsy surgery, of whom 65% had MRI-identifiable lesions. Of these, 52% had an MRI diagnosis of “probable FCD.” Of the 47 patients with normal MRI results, the most common postoperative pathology was FCD type II (51%), followed by nonspecific gliosis (23%) and FCD type I (21%).5
Diagnostic Criteria for SHE
As mentioned above, the diagnosis of SHE is mainly based on the patient’s clinical history because of the limited value of ictal and interictal scalp EEG for the localization and lateralization of the SOZ in patients with SHE and the low positive rate of neuroimaging (Table 2).
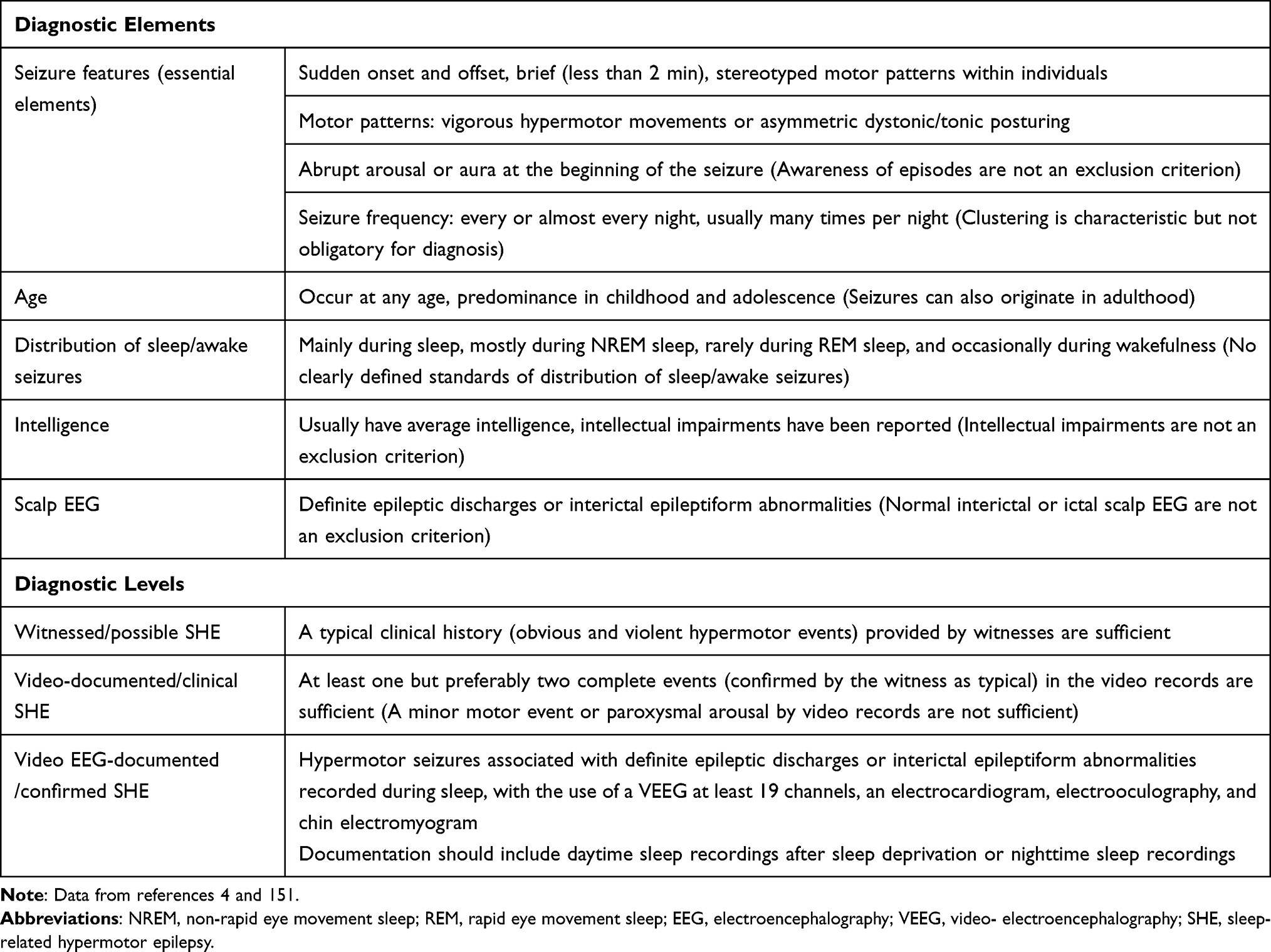 |
Table 2 Diagnostic Criteria of SHE |
The diagnosis can be divided into three levels: witnessed (possible) SHE, video-documented (clinical) SHE, and VEEG-documented (confirmed) SHE.4 In witnessed (possible) SHE, semiologically, there are obvious and violent hypermotor events, generally concordant with those documented by video analysis. Therefore, the data provided by a good clinical history are sufficient to diagnose witnessed (possible) SHE. In video-documented (clinical) SHE, at least one, but preferably two, complete events (confirmed by the witness as typical) should be documented in the video records, including the onset, evolution, and offset of the entire event. If a minor motor event or paroxysmal arousal is captured, the clinical diagnosis may not be reliable.4 In VEEG-documented (confirmed) SHE, the confirmation of a SHE diagnosis requires the use of a VEEG of at least 19 channels, an electrocardiogram, electrooculography, and chin electromyogram. Documentation should include daytime sleep recordings after sleep deprivation or nighttime sleep recordings. The diagnosis of SHE can be confirmed when hypermotor seizures associated with definite epileptic discharges or interictal epileptiform abnormalities are recorded during sleep.4 However, the proportion of seizures during sleep to those during wakefulness is not currently specified in the diagnostic criteria for SHE.
Differential Diagnosis and Overlap Between SHE and Parasomnias
Great progress has been made in the identification of these sleep-related seizures with hypermotor automatisms and/or dystonic posturing as a unique syndrome. However, the differential diagnosis between the syndrome and paroxysmal non-epileptic motor events may still be challenging. Parasomnias are undesirable behavioral events or complex motor that occur during entry to sleep, within sleep, or during sleep arousal.108 Most feature nocturnal paroxysmal motor phenomena, and their distinction from SHE can be difficult, particularly when distinguishing disorders of arousal (DOA) or REM sleep behavior disorder (RBD) from SHE. RBD is a common type of REM parasomnia characterized by the loss of normal skeletal muscle atonia with prominent dreaming and abrupt motor activity.109 Complex behaviors with violent acts (punching or kicking) or bed falls can also occur.110 RBD affects predominantly elderly men (aged ≥50 years), and episodes prevail at least 90 minutes after sleep onset (more common in the second half of the night).59 DOA are NREM parasomnias characterized by complex, seemingly purposeful, goal-directed behavioral events during NREM sleep.108 The prevalence of DOA in patients with SHE and their relatives is high, and DOA and SHE may coexist in the same patient or family members of the patient.49 Patients with sleep-related motor and behavioral disorders present to a variety of specialist outpatient centers (neurology, sleep medicine, psychiatry). The difficult diagnosis leads to significant distress for the patients, with consequences on their quality of life. It is impossible to distinguish epileptic seizures from parasomnias solely from certain semiological manifestations (eg, screaming or walking). Different core clinical features should be the first step for the differential diagnosis of SHE and parasomnias (Table 3). Distinct from patients with SHE (as described in the preceding paragraphs), those with DOA usually present with variable, complex, and non-stereotyped motor episodes.111 Core clinical features are strongly supporting a diagnosis of DOA include behaviors mimicking everyday activities associated with physical or verbal interaction with the environment (such as turning on the light, opening a door or looking out of the window, using the mobile phone, getting dressed, taking actions related to working activity, doing make-up or preparing luggage).112 Patients with parasomnia often have lower episode frequency and longer episode duration than those with SHE.113 The clinical history alone is sometimes insufficient to provide information for differential diagnosis but may still provide clues for the further development of screening programs. To obtain a systematic assessment of the diagnostic accuracy of clinical history in distinguishing SHE from parasomnias, two instruments have been devised: the Structured Interview for NFLE50 and the Frontal Lobe Epilepsy and Parasomnias scale.114 However, despite their clinical usefulness, their diagnostic accuracy is insufficient.115,116 VEEG recordings might yield an improved clinical discrimination, but not all patients with SHE exhibit EEG abnormalities, and EEG epileptiform abnormalities may also occur in some patients with RBD.109 This adds to the difficulties of successful diagnosis. Standard VEEG, used to monitor for seizures in inpatient epilepsy monitoring units, typically utilizes 19 EEG channels, electrocardiogram, and deltoids electromyography, without electrooculography and chin electromyogram. VEEG studies are not adequate for the purposes of determining sleep state and provide insufficient information regarding whether a potential sleep disorder is present due to the lack of chin electromyogram and respiratory channels. Video polysomnography was once considered the gold standard for the diagnosis of paroxysmal nocturnal events. While standard PSG typically utilizes 4–6 channels of VEEG, this is not sufficient to evaluate EEG epileptiform activity.117 In seizure monitoring (whether PSG or VEEG), the amount of seizures is often the limiting factor, as some patients have a low seizure frequency. Home-made video can be very useful in the diagnosis of major motor episodes when the patients do not recall episodes. Due to this, combined VEEG and PSG, utilizing 19-channel EEG and other physiological parameters, can assist in diagnosing suspicious nocturnal events and differentiating between the presence of a parasomnia, seizure activity, or both.4,118,119 Considering the identification of semiology is highly dependent on clinician expertise and skills, some scholars have postulated that the relative time and stage of occurrence during sleep of minor and major episodes could represent objective and simple criteria to discriminate SHE from DOA. The occurrence of at least one major event (complex arousal ambulatory movements in DOA120 and complex hypermotor seizures in SHE4) outside stage N3 sleep was highly suggestive of SHE (accuracy = 0.898, specificity = 0.949, sensitivity = 0.793), whereas the occurrence of at least one minor event (simple and rising arousal movements in DOA,120 MME and PAs in SHE4) during stage N3 sleep was highly suggestive of DOA (accuracy = 0.73, specificity = 0.723, sensitivity = 0.733).121 Despite the majority of patients being diagnosed with typical clinical history, 19-channel EEG recordings combined with PSG, and homemade video, a substantial number remain undiagnosed.
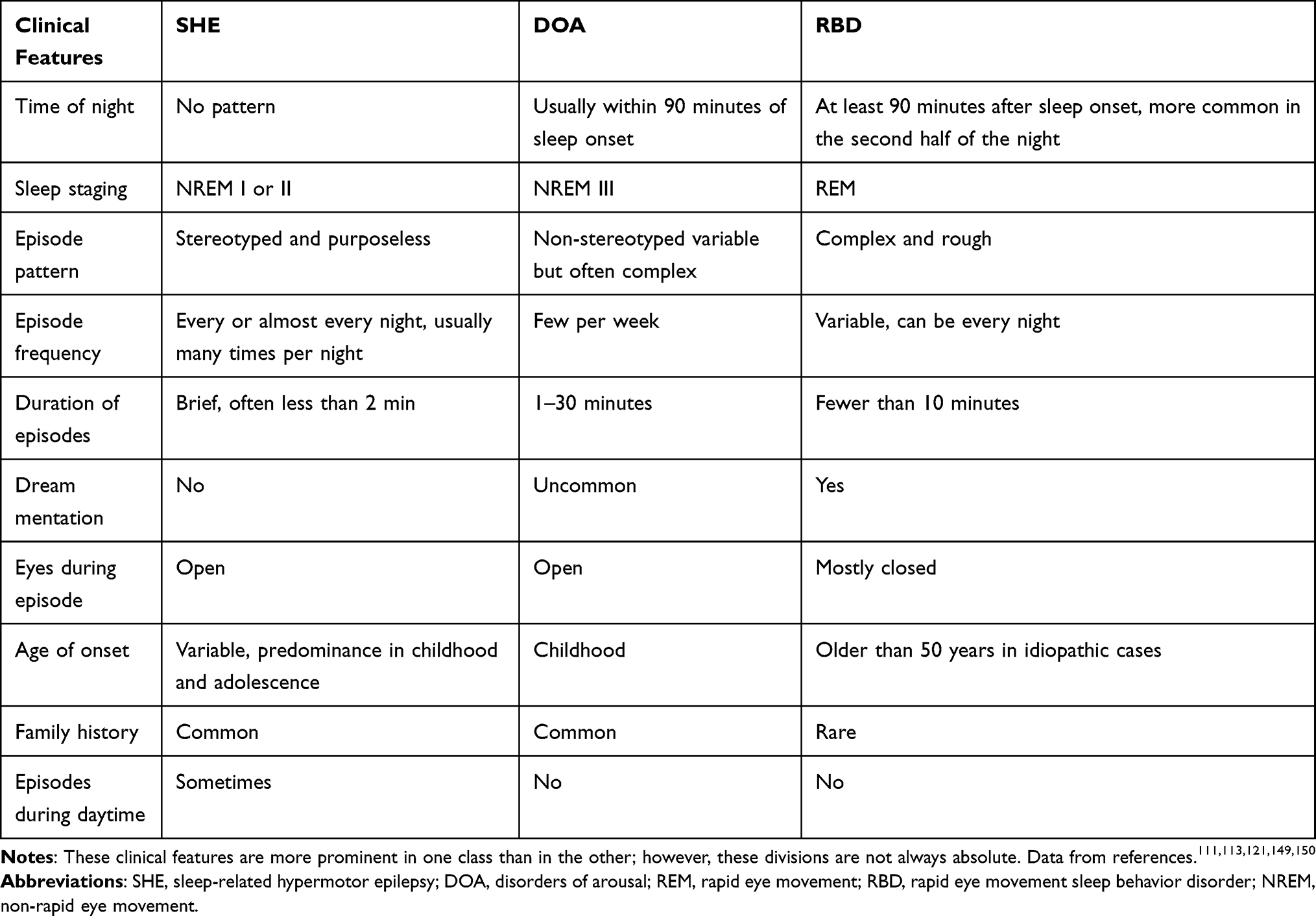 |
Table 3 Differential Diagnosis of SHE |
More and more researchers are starting to explore the possible intrinsic link between SHE and some parasomnias as they share common semiological features. It has been speculated that the similarities in motor behaviors observed with SHE and parasomnias are due to the activation of central pattern generators (CPGs).122,123 CPGs are groups of interconnected nerve cells in the spinal cord, brainstem, and other subcortical regions that activate different motor neurons in the proper sequence and intensity to generate stereotyped motor responses such as locomotion.124 In neonates, fundamental motor functions (protective reflexes like grasping and avoidance, pedaling, wandering, and cyclic leg movements) are carried out by CPGs. In adults, these motor behaviors are under the control of the neocortex. Both physiological (sleep) and pathological (seizure-related) conditions can also lead to a transient loss of control of the neocortex with a lack of CPGs inhibition.122,125,126 Thus, many scholars believe that the release of inborn fixed motor behaviors may account for the semiological overlap between SHE and some parasomnias.55,122,123 These motor patterns are already written in the brain codes (CPGs) but require a degree of activation to become visibly apparent. In general, arousal during sleep acts as a trigger releasing or facilitating an encoded inborn fixed motor behaviors.55 Numerous studies have provided support for this point of view and showed that a high number of micro-arousals was seen in both SHE and NREM parasomnias.55,127 The presence of sleep state instability is characterized by recurrent arousals that are thought to serve as the initiating source of the NREM parasomnias.128 SEEG studies have shown that epileptiform discharges would act as an internal trigger increasing micro-arousal and sleep instability and, in turn, enhance and modulate the occurrence of motor events (epileptic or not) or different types of sleep disturbances.55,56,129,130 On the other hand, the resulting sleep instability might facilitate the production of epileptic discharges in a bi-directionally influenced system and, in turn, promote seizure activity.55,56,129,130 Thus, it is not difficult to understand why the removal of the EZ after epilepsy surgery may not only eliminate major epileptic episodes but also improve sleep quality and sleep architecture (PSG documented).6,131
Several studies on the cyclic alternating pattern (CAP) also arrived at similar conclusions.55,132 CAP is a physiologic component of NREM sleep that is functionally associated with arousal oscillations, which is a measure of NREM sleep instability.128,133 CAP is composed of phase A (including sequences of K-complexes, EEG arousals, bursts of polymorphic delta or alpha waves, and vertex waves) and phase B (intervening background between two consecutive A phases).128 Phase A is the EEG marker of cerebral activation and unstable sleep phases, including cortical arousal, and for this reason, it is considered a potential trigger of somatomotor activities.134 Phase A of the CAP could not be only in favor of the co-occurrence of MMEs and epileptic discharges but also create a reciprocal facilitating effect. In earlier studies, major motor episodes (lasting between 10 and 60 s) in patients with SHE were preceded by a prolonged CAP sequence, which reflects a sustained condition of sustained arousal instability.55,135 Taken together, arousal plays a key role for the release of motor activities, ranging from physiological body movements to nocturnal seizure and NREM parasomnias. Whether the motor outcome is a normal or a pathological motor episode will depend on a number of ongoing factors such as sleep stage, the susceptibility of the patient, the nature of the putative underlying lesion, and the location and extent of the neural circuits involved.55 Mutations in the ACh receptor have been identified in some patients with ADSHE. Prior studies found increased nAChR density in the superior area of the epithalamus and cerebellum in patients with ADSHE136 and increased nAChR density in the epithalamus and interpeduncular nucleus in patients with a sleep disorder.137 The increased acetylcholine may participate regulation of sleep stability, especially micro-arousals, by activating the hyperactivation of the cholinergic pathway ascending from the brainstem.14,138 Current opinion tends to consider that the possible pathophysiologic mechanism of the intrinsic link between SHE and parasomnias is the abnormal cholinergic arousal system.
Prognosis and Treatment of SHE
Prognosis of SHE
Licchetta et al followed up 139 patients with SHE for up to 16 years, of whom 77.7% failed to reach 5 seizure-free years, thus pointing to a poor prognosis of SHE.9 The most important factor affecting remission rates is the disease’s etiology.9 Any underlying brain dysfunction, including perinatal insults, intellectual disability, abnormal neurologic examinations, structural brain abnormalities, and the presence of seizures during wakefulness, is a negative predictor of SHE prognosis.9 Although sleep-related hypermotor seizures are a hallmark feature of SHE, up to half of the patients may experience seizures during wakefulness throughout their lifetime.107 Given the greater impact on the quality of life (eg, driving limitations and social discrimination) of the presence of seizures during wakefulness in patients with SHE than that of seizures that only occur during sleep, a further analysis by Licchetta et al based on the same study population noted that the probability of the appearance of epileptic seizures in patients who only had seizures during sleep initially was 27%,139 whereas that of the appearance of seizures during wakefulness throughout their lifetime for patients who had seizures during wakefulness from the beginning was 94%.139
Treatment of SHE
Carbamazepine inhibits the SHE-associated α4β2 and α2β4 subunits of the nicotinic acetylcholine receptor,140 thereby inhibiting glutamatergic excitability and enhancing GABAergic activity in the thalamocortical system and hippocampus, thus altering neuronal excitability.141 Low doses of carbamazepine (200–1,000 mg/kg) were effective in some patients with SHE46,142 but appeared to be ineffective in at least one-third of patients.132 In uncontrolled trials, oxcarbazepine and topiramate were effective against both familial and sporadic ADSHE.143,144 Raju et al reported the significant efficacy of oxcarbazepine in eight children with NFLE who were nonresponsive to carbamazepine and other antiepileptic drugs.145 Most epilepsy cases caused by mutations in the KCNT1 gene, which encodes a sodium-activated potassium channel, are difficult to treat. Milligan et al suggested that quinidine, a partial antagonist of the KCNT1 channel, could treat ADSHE due to KCNT1 mutations.24 Quinidine has been approved by the US Food and Drug Administration for the treatment of epilepsy caused by KCNT1 mutations. However, a placebo-controlled clinical study conducted by Mullen et al confirmed the poor efficacy of quinidine on ADSHE and showed that the drug could lead to serious cardiac side effects.146 The efficacy of quinidine as an emerging drug for epilepsy treatment needs to be verified in clinical studies with large sample sizes. Lacosamide acts on slowly inactivated sodium channels and is a potentially effective adjunctive therapy for patients with refractory SHE. Samarasekera et al treated eight patients with refractory SHE with lacosamide (300–600 mg/kg) and reduced seizure frequency by more than 50% in five patients and by 25% in one patient.147 One of the pathogenic mechanisms of ADSHE is gain-of-function mutations in the α4 or β2 subunits of nAChRs, and fenofibrate, a common lipid-modulating drug, is an agonist of peroxisome proliferator-activated receptor-alpha and a ligand-activated transcription factor that negatively regulates the function of the β2-containing nAChRs. A preclinical and clinical study confirmed the efficacy of fenofibrate in transgenic mice carrying mutations in the α4-nAChR subunit homologous to those in humans and patients with drug-resistant SHE, suggesting a novel drug target for anti-epilepsy treatment.148
Gibbs et al conducted a 2-year postoperative follow-up of 127 surgically treated patients with SHE, of whom 104 (82%) had an Engel class I outcome, including 86 (68%) with an Engel class Ia outcome, and the lobar location of the SOZ showed no significant effect on postoperative outcomes.5 Surgical treatment was effective in some severe drug-resistant patients with SHE-associated with FCD.6 Genetic etiology is not a contraindication during preoperative evaluation, although there have not been many studies on the surgical outcomes of patients with known genetic mutations.4
Summary
SHE is a group of etiologically diverse clinical syndromes, and most SHEs have poor long-term control rates compared to overall remission rates in epilepsy. After a careful preoperative assessment, some drug-resistant patients with these syndromes can benefit from surgery. In recent years, anatomical-electrical-clinical data have improved our understanding of the epileptogenic network of this group of syndromes. And helped to predict potential EZs, which subsequently leads to improved chances of a good surgical outcome. SHE is characterized by the seizure pattern—the strange, abrupt hypermotor movements sometimes with high emotional content—which could be easily confused with some parasomnias or psychogenic events. The uninformative scalp EEG and MRI of some patients make an accurate diagnosis more difficult. This is especially relevant for clinicians and families less educated in epilepsy. The idea of a common semiology and genetic basis of SHE and parasomnias opens an interesting new direction for future research in this field. However, the epidemiology of SHE has not been adequately studied, and the relationship between clinical presentation homogeneity and etiologic heterogeneity has not been elucidated. Large randomized controlled studies to verify the aspects of treatment and prognosis are lacking. For this group of syndromes, we need population-based, multicenter epidemiological surveys and comprehensive analysis of data on multiple aspects, including the incidence, characteristic clinical seizure patterns, prognosis and disability rate, and improvement in the diagnosis of SHE. With the development of gene testing technology, more pathogenic mutations of ADSHE will be detected. Until now, there was no direct correlation between either known gene mutations and the disease severity or genetic result and efficacy. However, further studies will be required to establish the genotype-phenotype correlations of SHE, and further exploration of the effects of drugs and surgical treatments in patients with different etiologies and research on the disease’s etiology, especially genetic research, may provide new opportunities for targeted therapy for this group of syndromes.
Abbreviations
ADSHE, autosomal dominant sleep-related hypermotor epilepsy; CABP, Ca(2+)-binding protein; CHRNA, gene encoding α subunit of the nicotinic acetylcholine receptor; CRH, corticotropin-releasing hormone; DEPDC5, pleckstrin (DEP) domain-containing 5; DNET, dysembryoplastic neuroepithelial tumor; EEG, electroencephalograms; NPD, nocturnal paroxysmal dystonia; EZ, epileptogenic zone; FCD, focal cortical dysplasia; GATOR, GTPase-activating protein activity toward Ras-related GTPases; HMS, hypermotor seizures; KCNT1, gene encoding subunit 1 of the sodium-activated potassium channel; LOF, loss-of-function; mMCD, mild malformations of cortical development; mTOR, mammalian target of rapamycin; nAChR, nicotinic acetylcholine receptor; NFLE, nocturnal frontal lobe epilepsy; NPD, nocturnal paroxysmal dystonia; NPRL, nitrogen permease regulator-like; NREM, non-rapid eye movement sleep; SEEG, stereoelectroencephalography; SHE, sleep-related hypermotor epilepsy; PAs, paroxysmal arousals; MME, minor motor events; OFC, orbitofrontal cortex; ACC, anterior cingulate cortex; SMA, supplementary motor area; PCC, posterior cingulate cortex; DOA, disorders of arousal; RBD, rapid eye movement sleep behavior disorder; CPGs, central pattern generators; CAP, cyclic alternating pattern; SOZ, seizure onset zone; SP, semiology pattern; VPSG, video polysomnography; CLOCK:circadian locomotor output cycles kaput.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest, financial or otherwise.
References
1. Lugaresi E. Cirignotta F Hypnogenic paroxysmal dystonia: epileptic seizure or a new syndrome? Sleep. 1981;4(2):129–138. doi:10.1093/sleep/4.2.129
2. Lugaresi E, Cirignotta F, Montagna P. Nocturnal paroxysmal dystonia. J Neurol Neurosurg Psychiatry. 1986;49(4):375–380. doi:10.1136/jnnp.49.4.375
3. Tinuper P, Cerullo A, Cirignotta F, et al. Nocturnal paroxysmal dystonia with short-lasting attacks: three cases with evidence for an epileptic frontal lobe origin of seizures. Epilepsia. 1990;31(5):549–556. doi:10.1111/j.1528-1157.1990.tb06105.x
4. Tinuper P, Bisulli F, Cross J, et al. Definition and diagnostic criteria of sleep-related hypermotor epilepsy. Neurology. 2016;86(19):1834–1842. doi:10.1212/WNL.0000000000002666
5. Gibbs SA, Proserpio P, Francione S, et al. Clinical features of sleep-related hypermotor epilepsy in relation to the seizure-onset zone: a review of 135 surgically treated cases. Epilepsia. 2019;60(4):707–717. doi:10.1111/epi.14690
6. Nobili L, Francione S, Mai R, et al. Surgical treatment of drug-resistant nocturnal frontal lobe epilepsy. Brain. 2007;130(Pt 2):561–573. doi:10.1093/brain/awl322
7. Chassoux F, Landre E, Mellerio C, et al. Type II focal cortical dysplasia: electroclinical phenotype and surgical outcome related to imaging. Epilepsia. 2012;53(2):349–358. doi:10.1111/j.1528-1167.2011.03363.x
8. Nobili L, Cardinale F, Magliola U, et al. Taylor’s focal cortical dysplasia increases the risk of sleep-related epilepsy. Epilepsia. 2009;50(12):2599–2604. doi:10.1111/j.1528-1167.2009.02169.x
9. Licchetta L, Bisulli F, Vignatelli L, et al. Sleep-related hypermotor epilepsy: long-term outcome in a large cohort. Neurology. 2017;88(1):70–77. doi:10.1212/WNL.0000000000003459
10. Kurahashi H, Hirose S, et al. Autosomal Dominant Nocturnal Frontal Lobe Epilepsy. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews((R)). Seattle (WA); 1993.
11. Steinlein O, Mulley J, Propping P, et al. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 1995;11(2):201–203. doi:10.1038/ng1095-201
12. De Fusco M, Becchetti A, Patrignani A, et al. The nicotinic receptor beta 2 subunit is mutant in nocturnal frontal lobe epilepsy. Nat Genet. 2000;26(3):275–276. doi:10.1038/81566
13. Aridon P, Marini C, Di Resta C, et al. Increased sensitivity of the neuronal nicotinic receptor alpha 2 subunit causes familial epilepsy with nocturnal wandering and ictal fear. Am J Hum Genet. 2006;79(2):342–350. doi:10.1086/506459
14. Léna C, Popa D, Grailhe R, et al. Beta2-containing nicotinic receptors contribute to the organization of sleep and regulate putative micro-arousals in mice. J Neurosci. 2004;24(25):5711–5718. doi:10.1523/JNEUROSCI.3882-03.2004
15. Marini C. Guerrini R The role of the nicotinic acetylcholine receptors in sleep-related epilepsy. Biochem Pharmacol. 2007;74(8):1308–1314. doi:10.1016/j.bcp.2007.06.030
16. Licchetta L, Pippucci T, Baldassari S, et al. Sleep-related hypermotor epilepsy (SHE): contribution of known genes in 103 patients. Seizure. 2020;74:60–64. doi:10.1016/j.seizure.2019.11.009
17. Langenbruch L, Biskup S, Young P, et al. Two mutations in the nicotinic acetylcholine receptor subunit A4 (CHRNA4) in a family with autosomal dominant sleep-related hypermotor epilepsy. Epileptic Disord. 2020;22(1):116–119.
18. Phillips H, Favre I, Kirkpatrick M, et al. CHRNB2 is the second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. Am J Hum Genet. 2001;68(1):225–231. doi:10.1086/316946
19. Duga S, Asselta R, Bonati M, et al. Mutational analysis of nicotinic acetylcholine receptor beta2 subunit gene (CHRNB2) in a representative cohort of Italian probands affected by autosomal dominant nocturnal frontal lobe epilepsy. Epilepsia. 2002;43(4):362–364. doi:10.1046/j.1528-1157.2002.39001.x
20. Labate A, Mumoli L, Fratto A, et al. Hippocampal sclerosis worsens autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) phenotype related to CHRNB2 mutation. Eur j Neurol. 2013;20(3):591–593. doi:10.1111/j.1468-1331.2012.03839.x
21. Becchetti A, Grandi L, Colombo G, et al. Nicotinic Receptors in Sleep-Related Hypermotor Epilepsy: pathophysiology and Pharmacology. Brain Sci. 2020;10:12. doi:10.3390/brainsci10120907
22. Derry C, Heron S, Phillips F, et al. Severe autosomal dominant nocturnal frontal lobe epilepsy associated with psychiatric disorders and intellectual disability. Epilepsia. 2008;49(12):2125–2129. doi:10.1111/j.1528-1167.2008.01652.x
23. Lim C, Ricos M, Dibbens L, et al. KCNT1 mutations in seizure disorders: the phenotypic spectrum and functional effects. J Med Genet. 2016;53(4):217–225. doi:10.1136/jmedgenet-2015-103508
24. Milligan C, Li M, Gazina E, et al. KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann Neurol. 2014;75(4):581–590. doi:10.1002/ana.24128
25. Rubboli G, Plazzi G, Picard F, et al. Mild malformations of cortical development in sleep-related hypermotor epilepsy due to KCNT1 mutations. Ann Clin Transl Neurol. 2019;6(2):386–391. doi:10.1002/acn3.708
26. Heron S, Smith K, Bahlo M, et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 2012;44(11):1188–1190. doi:10.1038/ng.2440
27. Picard F, Makrythanasis P, Navarro V, et al. DEPDC5 mutations in families presenting as autosomal dominant nocturnal frontal lobe epilepsy. Neurology. 2014;82(23):2101–2106. doi:10.1212/WNL.0000000000000488
28. Dibbens L, de Vries B, Donatello S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet. 2013;45(5):546–551. doi:10.1038/ng.2599
29. Laplante M. Sabatini D mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi:10.1016/j.cell.2012.03.017
30. Weckhuysen S, Marsan E, Lambrecq V, et al. Involvement of GATOR complex genes in familial focal epilepsies and focal cortical dysplasia. Epilepsia. 2016;57(6):994–1003. doi:10.1111/epi.13391
31. Baulac S, Weckhuysen S, et al. DEPDC5-Related Epilepsy. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews((R)). Seattle (WA); 1993.
32. Lim KC. Crino PB Focal malformations of cortical development: new vistas for molecular pathogenesis. Neuroscience. 2013;252:262–276. doi:10.1016/j.neuroscience.2013.07.037
33. Baldassari S, Picard F, Verbeek N, et al. Correction: the landscape of epilepsy-related GATOR1 variants. Genetics Med. 2019;21(8):1896. doi:10.1038/s41436-018-0325-9
34. Pippucci T, Licchetta L, Baldassari S, et al. Contribution of ultrarare variants in mTOR pathway genes to sporadic focal epilepsies. Ann Clin Translational Neurol. 2019;6(3):475–485. doi:10.1002/acn3.722
35. Ricos M, Hodgson B, Pippucci T, et al. Mutations in the mammalian target of rapamycin pathway regulators NPRL2 and NPRL3 cause focal epilepsy. Ann Neurol. 2016;79(1):120–131. doi:10.1002/ana.24547
36. Combi R, Dalprà L, Ferini-Strambi L, et al. Frontal lobe epilepsy and mutations of the corticotropin-releasing hormone gene. Ann Neurol. 2005;58(6):899–904. doi:10.1002/ana.20660
37. Sansoni V, Forcella M, Mozzi A, et al. Functional characterization of a CRH missense mutation identified in an ADNFLE family. PLoS One. 2013;8(4):e61306. doi:10.1371/journal.pone.0061306
38. Adzhubei I, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi:10.1038/nmeth0410-248
39. Ferini-Strambi L, Sansoni V, Combi R. Nocturnal frontal lobe epilepsy and the acetylcholine receptor. neurologist. 2012;18(6):343–349. doi:10.1097/NRL.0b013e31826a99b8
40. Hildebrand M, Tankard R, Gazina E, et al. PRIMA1 mutation: a new cause of nocturnal frontal lobe epilepsy. Ann Clin Translational Neurol. 2015;2(8):821–830. doi:10.1002/acn3.224
41. Chen Z, Wang C, Zhuo M, et al. CABP4Exome sequencing identified a novel missense mutation c.464G>A (p.G155D) in Ca-binding protein 4 () in a Chinese pedigree with autosomal dominant nocturnal frontal lobe epilepsy. Oncotarget. 2017;8(45):78940–78947. doi:10.18632/oncotarget.20694
42. Peres J, Antunes F, Zonjy B, et al. Sleep-related hypermotor epilepsy and peri-ictal hypotension in a patient with syntaxin-1B mutation. Epileptic Disorders. 2018;20(5):413–417. doi:10.1684/epd.2018.0996
43. Mastrangelo M, Commone C, Greco C, et al. TSC1 as a Novel Gene for Sleep-Related Hypermotor Epilepsy: a Child with a Mild Phenotype of Tuberous Sclerosis. Neuropediatrics. 2021;52(2):146–149. doi:10.1055/s-0041-1722881
44. Li P, Fu X, Smith N, et al. Loss of CLOCK Results in Dysfunction of Brain Circuits Underlying Focal Epilepsy. Neuron. 2017;96(2):387–401.e6. doi:10.1016/j.neuron.2017.09.044
45. Vignatelli L, Bisulli F, Giovannini G, et al. Prevalence of nocturnal frontal lobe epilepsy in the adult population of Bologna and Modena, Emilia-Romagna region, Italy. Sleep. 2015;38(3):479–485. doi:10.5665/sleep.4514
46. Provini F, Plazzi G, Tinuper P, et al. Nocturnal frontal lobe epilepsy. A clinical and polygraphic overview of 100 consecutive cases. Brain. 1999;122(Pt 6):1017–1031. doi:10.1093/brain/122.6.1017
47. Scheffer I, Bhatia K, Lopes-Cendes I. et al. Autosomal dominant nocturnal frontal lobe epilepsy. A distinctive clinical disorder. Brain;1995. 61–73. doi:10.1093/brain/118.1.61
48. Menghi V, Bisulli F, Tinuper P, et al. Sleep-related hypermotor epilepsy: prevalence, impact and management strategies. Nat Sci Sleep. 2018;10:317–326. doi:10.2147/NSS.S152624
49. Bisulli F, Vignatelli L, Naldi I, et al. Increased frequency of arousal parasomnias in families with nocturnal frontal lobe epilepsy: a common mechanism? Epilepsia. 2010;51(9):1852–1860. doi:10.1111/j.1528-1167.2010.02581.x
50. Bisulli F, Vignatelli L, Naldi I, et al. Diagnostic accuracy of a structured interview for nocturnal frontal lobe epilepsy (SINFLE): a proposal for developing diagnostic criteria. Sleep Med. 2012;13(1):81–87. doi:10.1016/j.sleep.2011.09.003
51. Jobst B, Siegel A, Thadani V, et al. Intractable seizures of frontal lobe origin: clinical characteristics, localizing signs, and results of surgery. Epilepsia. 2000;41(9):1139–1152. doi:10.1111/j.1528-1157.2000.tb00319.x
52. Rheims S, Ryvlin P, Scherer C, et al. Analysis of clinical patterns and underlying epileptogenic zones of hypermotor seizures. Epilepsia. 2008;49(12):2030–2040. doi:10.1111/j.1528-1167.2008.01675.x
53. Terzaghi M, Sartori I, Mai R, et al. Sleep-related minor motor events in nocturnal frontal lobe epilepsy. Epilepsia. 2007;48(2):335–341. doi:10.1111/j.1528-1167.2006.00929.x
54. Nobili L, Sartori I, Terzaghi M, et al. Intracerebral recordings of minor motor events, paroxysmal arousals and major seizures in nocturnal frontal lobe epilepsy. Neurol Sci. 2005;26(Suppl 3):s215–9. doi:10.1007/s10072-005-0490-x
55. Parrino L, Halasz P, Tassinari C, et al. CAP, epilepsy and motor events during sleep: the unifying role of arousal. Sleep Med Rev. 2006;10(4):267–285. doi:10.1016/j.smrv.2005.12.004
56. Gibbs S, Proserpio P, Terzaghi M, et al. Sleep-related epileptic behaviors and non-REM-related parasomnias: insights from stereo-EEG. Sleep Med Rev. 2016;25:4–20. doi:10.1016/j.smrv.2015.05.002
57. Sforza E, Montagna P, Rinaldi R, et al. Paroxysmal periodic motor attacks during sleep: clinical and polygraphic features. Electroencephalogr Clin Neurophysiol. 1993;86(3):161–166. doi:10.1016/0013-4694(93)90003-E
58. Nobili L, Francione S, Mai R, et al. Nocturnal frontal lobe epilepsy: intracerebral recordings of paroxysmal motor attacks with increasing complexity. Sleep. 2003;26(7):883–886.
59. Nobili L, Proserpio P, Combi R, et al. Nocturnal frontal lobe epilepsy. Curr Neurol Neurosci Rep. 2014;14(2):424. doi:10.1007/s11910-013-0424-6
60. Nobili L, Cossu M, Mai R, et al. Sleep-related hyperkinetic seizures of temporal lobe origin. Neurology. 2004;62(3):482–485. doi:10.1212/01.WNL.0000106945.68292.DC
61. Proserpio P, Cossu M, Francione S, et al. Epileptic motor behaviors during sleep: anatomo-electro-clinical features. Sleep Med. 2011;12(Suppl 2):S33–8. doi:10.1016/j.sleep.2011.10.018
62. Gibbs SA, Figorilli M, Casaceli G, et al. Sleep Related Hypermotor Seizures with a Right Parietal Onset. J Clin Sleep Med. 2015;11(8):953–955. doi:10.5664/jcsm.4952
63. Nobili L, Francione S, Cardinale F, et al. Epileptic nocturnal wanderings with a temporal lobe origin: a stereo-electroencephalographic study. Sleep. 2002;25(6):669–671.
64. Ryvlin P, Minotti L, Demarquay G, et al. Nocturnal hypermotor seizures, suggesting frontal lobe epilepsy, can originate in the insula. Epilepsia. 2006;47(4):755–765. doi:10.1111/j.1528-1167.2006.00510.x
65. Ferri L, Bisulli F, Mai R, et al. A stereo EEG study in a patient with sleep-related hypermotor epilepsy due to DEPDC5 mutation. Seizure. 2017;53:51–54. doi:10.1016/j.seizure.2017.10.022
66. Dobesberger J, Ortler M, Unterberger I, et al. Successful surgical treatment of insular epilepsy with nocturnal hypermotor seizures. Epilepsia. 2008;49(1):159–162. doi:10.1111/j.1528-1167.2007.01426.x
67. Steinlein O, Hoda J, Bertrand S, et al. Mutations in familial nocturnal frontal lobe epilepsy might be associated with distinct neurological phenotypes. Seizure. 2012;21(2):118–123. doi:10.1016/j.seizure.2011.10.003
68. Conti V, Aracri P, Chiti L, et al. Nocturnal frontal lobe epilepsy with paroxysmal arousals due to CHRNA2 loss of function. Neurology. 2015;84(15):1520–1528. doi:10.1212/WNL.0000000000001471
69. Picard F, Pegna A, Arntsberg V, et al. Neuropsychological disturbances in frontal lobe epilepsy due to mutated nicotinic receptors. Epilepsy Behav. 2009;14(2):354–359. doi:10.1016/j.yebeh.2008.11.003
70. Wood A, Saling M, Fedi M, et al. Neuropsychological function in patients with a single gene mutation associated with autosomal dominant nocturnal frontal lobe epilepsy. Epilepsy Behav. 2010;17(4):531–535. doi:10.1016/j.yebeh.2010.01.168
71. Licchetta L, Poda R, Vignatelli L, et al. Profile of neuropsychological impairment in Sleep-related Hypermotor Epilepsy. Sleep Med. 2018;48:8–15. doi:10.1016/j.sleep.2018.03.027
72. Baldassari S, Picard F, Verbeek N, et al. The landscape of epilepsy-related GATOR1 variants. Genetics Med. 2019;21(2):398–408. doi:10.1038/s41436-018-0060-2
73. Fukuyama K, Fukuzawa M, Okubo R, et al. Upregulated Connexin 43 Induced by Loss-of-Functional S284L-Mutant alpha4 Subunit of Nicotinic ACh Receptor Contributes to Pathomechanisms of Autosomal Dominant Sleep-Related Hypermotor Epilepsy. Pharmaceuticals. 2020;13:4.
74. Fukuyama K, Fukuzawa M, Shiroyama T, et al. Pathogenesis and pathophysiology of autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant alpha4 subunit of nicotinic ACh receptor. Br J Pharmacol. 2020;177(9):2143–2162. doi:10.1111/bph.14974
75. Fukuyama K, Fukuzawa M, Shiroyama T, et al. Pathomechanism of nocturnal paroxysmal dystonia in autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant α4 subunit of nicotinic ACh receptor. Biomed Pharmacotherapy. 2020;126:110070. doi:10.1016/j.biopha.2020.110070
76. Fukuyama K, Fukuzawa M, Okada M. Upregulated and Hyperactivated Thalamic Connexin 43 Plays Important Roles in Pathomechanisms of Cognitive Impairment and Seizure of Autosomal Dominant Sleep-Related Hypermotor Epilepsy with S284L-Mutant α4 Subunit of Nicotinic ACh Receptor. Pharmaceuticals. 2020;13(5):548.
77. Schonauer M, Alizadeh S, Jamalabadi H, et al. Decoding material-specific memory reprocessing during sleep in humans. Nat Commun. 2017;8:15404. doi:10.1038/ncomms15404
78. Chakravarty K, Shukla G, Poornima S, et al. Effect of sleep quality on memory, executive function, and language performance in patients with refractory focal epilepsy and controlled epilepsy versus healthy controls - A prospective study. Epilepsy Behav. 2019;92:176–183. doi:10.1016/j.yebeh.2018.12.028
79. Ampuero E, Dagnino-Subiabre A, Sandoval R, et al. Status epilepticus induces region-specific changes in dendritic spines, dendritic length and TrkB protein content of rat brain cortex. Brain Res. 2007;1150:225–238. doi:10.1016/j.brainres.2007.02.089
80. Bartolomei F, Lagarde S, Wendling F, et al. Defining epileptogenic networks: contribution of SEEG and signal analysis. Epilepsia. 2017;58(7):1131–1147. doi:10.1111/epi.13791
81. Chauvel P. McGonigal A Emergence of semiology in epileptic seizures. Epilepsy Behav. 2014;38:94–103. doi:10.1016/j.yebeh.2013.12.003
82. Tufenkjian K. Luders HO Seizure semiology: its value and limitations in localizing the epileptogenic zone. J Clin Neurol. 2012;8(4):243–250. doi:10.3988/jcn.2012.8.4.243
83. Rosenow F. Lüders H Presurgical evaluation of epilepsy. Brain. 2001;124:1683–1700. doi:10.1093/brain/124.9.1683
84. Lüders H, Najm I, Nair D, et al. The epileptogenic zone: general principles. Epileptic Disorders. 2006;1:S1–9.
85. Lüders H, Acharya J, Baumgartner C, et al. Semiological seizure classification. Epilepsia. 1998;39(9):1006–1013. doi:10.1111/j.1528-1157.1998.tb01452.x
86. Foldvary-Schaefer N, Unnwongse K. Localizing and lateralizing features of auras and seizures. Epilepsy Behav. 2011;20(2):160–166. doi:10.1016/j.yebeh.2010.08.034
87. Yin F, Ni D, Xu C, et al. Auras in intractable frontal lobe epilepsy: clinical characteristics, values, and limitations. Epilepsy Behav. 2021;115:107724. doi:10.1016/j.yebeh.2020.107724
88. Blume W, Lüders H, Mizrahi E, et al. Glossary of descriptive terminology for ictal semiology: report of the ILAE task force on classification and terminology. Epilepsia. 2001;42(9):1212–1218. doi:10.1046/j.1528-1157.2001.22001.x
89. Montavont A, Kahane P, Catenoix H, et al. Hypermotor seizures in lateral and mesial parietal epilepsy. Epilepsy Behav. 2013;28(3):408–412. doi:10.1016/j.yebeh.2013.05.030
90. Bonini F, McGonigal A, Trebuchon A, et al. Frontal lobe seizures: from clinical semiology to localization. Epilepsia. 2014;55(2):264–277. doi:10.1111/epi.12490
91. Fayerstein J, McGonigal A, Pizzo F, et al. Quantitative analysis of hyperkinetic seizures and correlation with seizure onset zone. Epilepsia. 2020;61(5):1019–1026. doi:10.1111/epi.16510
92. Davidson R, Putnam K, Larson C. Dysfunction in the neural circuitry of emotion regulation–a possible prelude to violence. Science. 2000;289(5479):591–594. doi:10.1126/science.289.5479.591
93. Bartolomei F, Trebuchon A, Gavaret M, et al. Acute alteration of emotional behaviour in epileptic seizures is related to transient desynchrony in emotion-regulation networks. Clin Neurophysiol. 2005;116(10):2473–2479. doi:10.1016/j.clinph.2005.05.013
94. Wang L, Mathews GC, Whetsell WO, et al. Hypermotor seizures in patients with temporal pole lesions. Epilepsy Res. 2008;82(1):93–98. doi:10.1016/j.eplepsyres.2008.07.005
95. Staack A, Bilic S, Wendling A, et al. Hyperkinetic seizures in patients with temporal lobe epilepsy: clinical features and outcome after temporal lobe resection. Epilepsia. 2011;52(8):1439–1446. doi:10.1111/j.1528-1167.2011.03100.x
96. Nitta N, Usui N, Kondo A, et al. Semiology of hyperkinetic seizures of frontal versus temporal lobe origin. Epileptic Disord. 2019;21(2):154–165.
97. Vaugier L, Aubert S, McGonigal A, et al. Neural networks underlying hyperkinetic seizures of “temporal lobe” origin. Epilepsy Res. 2009;86(2–3):200–208. doi:10.1016/j.eplepsyres.2009.06.007
98. Nguyen DK, Nguyen DB, Malak R, et al. Revisiting the role of the insula in refractory partial epilepsy. Epilepsia. 2009;50(3):510–520.
99. Proserpio P, Cossu M, Francione S, et al. Insular-opercular seizures manifesting with sleep-related paroxysmal motor behaviors: a stereo-EEG study. Epilepsia. 2011;52(10):1781–1791. doi:10.1111/j.1528-1167.2011.03254.x
100. Klein TA, Ullsperger M, Danielmeier C. Error awareness and the insula: links to neurological and psychiatric diseases. Front Hum Neurosci. 2013;7:14. doi:10.3389/fnhum.2013.00014
101. Kurth F, Zilles K, Fox PT, et al. A link between the systems: functional differentiation and integration within the human insula revealed by meta-analysis. Brain Struct Funct. 2010;214(5–6):519–534. doi:10.1007/s00429-010-0255-z
102. Wang H, McGonigal A, Zhang K, et al. Semiologic subgroups of insulo-opercular seizures based on connectional architecture atlas. Epilepsia. 2020;61(5):984–994. doi:10.1111/epi.16501
103. Nieuwenhuys R, Voogd J, Van Huijzen C. The Human Central Nervous System. Springer Science & Business Media; 2008.
104. Fluchere F, McGonigal A, Villeneuve N, et al. Ictal “hemiballic-like” movement: lateralizing and localizing value. Epilepsia. 2012;53(2):e41–5. doi:10.1111/j.1528-1167.2011.03362.x
105. Bartolomei F, Gavaret M, Hewett R, et al. Neural networks underlying parietal lobe seizures: a quantified study from intracerebral recordings. Epilepsy Res. 2011;93(2–3):164–176. doi:10.1016/j.eplepsyres.2010.12.005
106. Rushworth MF, Behrens TE, Johansen-Berg H. Connection patterns distinguish 3 regions of human parietal cortex. Cereb Cortex. 2006;16(10):1418–1430. doi:10.1093/cercor/bhj079
107. Gibbs SA, Proserpio P, Francione S, et al. Seizure duration and latency of hypermotor manifestations distinguish frontal from extrafrontal onset in sleep-related hypermotor epilepsy. Epilepsia. 2018;59(9):e130–e134. doi:10.1111/epi.14517
108. Sateia MJ. International Classification of Sleep Disorders-Third Edition. Chest. 2014;146(5):1387–1394. doi:10.1378/chest.14-0970
109. Schenck C. Mahowald M REM sleep behavior disorder: clinical, developmental, and neuroscience perspectives 16 years after its formal identification in SLEEP. Sleep. 2002;25(2):120–138. doi:10.1093/sleep/25.2.120
110. Fernández-Arcos A, Iranzo A, Serradell M, et al. The Clinical Phenotype of Idiopathic Rapid Eye Movement Sleep Behavior Disorder at Presentation: a Study in 203 Consecutive Patients. Sleep. 2016;39(1):121–132. doi:10.5665/sleep.5332
111. Zucconi M, Oldani A, Ferini-Strambi L, et al. Nocturnal paroxysmal arousals with motor behaviors during sleep: frontal lobe epilepsy or parasomnia? J Clin Neurophysiol. 1997;14(6):513–522. doi:10.1097/00004691-199711000-00008
112. Montini A, Loddo G, Baldelli L, et al. Sleep-Related Hypermotor Epilepsy vs Disorders of Arousal in Adults: a Step-Wise Approach to Diagnosis. Chest. 2021;160(1):319–329. doi:10.1016/j.chest.2021.01.059
113. Breen D, Högl B, Fasano A, et al. Sleep-related motor and behavioral disorders: recent advances and new entities. Movement Disorders. 2018;33(7):1042–1055. doi:10.1002/mds.27375
114. Derry C, Davey M, Johns M, et al. Distinguishing sleep disorders from seizures: diagnosing bumps in the night. Arch Neurol. 2006;63(5):705–709. doi:10.1001/archneur.63.5.705
115. Bisulli F, Vignatelli L, Provini F, et al. Parasomnias and nocturnal frontal lobe epilepsy (NFLE): lights and shadows–controversial points in the differential diagnosis. Sleep Med. 2011;12(Suppl 2):S27–32. doi:10.1016/j.sleep.2011.10.008
116. Manni R, Terzaghi M, Repetto A. The FLEP scale in diagnosing nocturnal frontal lobe epilepsy, NREM and REM parasomnias: data from a tertiary sleep and epilepsy unit. Epilepsia. 2008;49(9):1581–1585. doi:10.1111/j.1528-1167.2008.01602.x
117. Foldvary-Schaefer N, De Ocampo J, Mascha E, et al. Accuracy of seizure detection using abbreviated EEG during polysomnography. J Clin Neurophysiol. 2006;23(1):68–71. doi:10.1097/01.WNP.0000174544.86406.8D
118. Jain S, Dye T, Kedia P. Value of combined video EEG and polysomnography in clinical management of children with epilepsy and daytime or nocturnal spells. Seizure. 2019;65:1–5. doi:10.1016/j.seizure.2018.12.009
119. Bubrick E, Yazdani S, Pavlova M. Beyond standard polysomnography: advantages and indications for use of extended 10–20 EEG montage during laboratory sleep study evaluations. Seizure. 2014;23(9):699–702. doi:10.1016/j.seizure.2014.05.007
120. Loddo G, Sessagesimi E, Mignani F, et al. Specific motor patterns of arousal disorders in adults: a video-polysomnographic analysis of 184 episodes. Sleep Med. 2018;41:102–109. doi:10.1016/j.sleep.2017.08.019
121. Proserpio P, Loddo G, Zubler F, et al. Polysomnographic features differentiating disorder of arousals from sleep-related hypermotor epilepsy. Sleep. 2019;42:12.
122. Tassinari C, Rubboli G, Gardella E, et al. Central pattern generators for a common semiology in fronto-limbic seizures and in parasomnias. A neuroethologic approach. Neurological Sci. 2005;26(S3):s225–32. doi:10.1007/s10072-005-0492-8
123. Tassinari C, Cantalupo G, Högl B, et al. Neuroethological approach to frontolimbic epileptic seizures and parasomnias: the same central pattern generators for the same behaviours. Rev Neurol (Paris). 2009;165(10):762–768. doi:10.1016/j.neurol.2009.08.002
124. Grillner S. Wallen P Central Pattern Generators for Locomotion, with Special Reference to Vertebrates. Annu Rev Neurosci. 1985;8(1):233. doi:10.1146/annurev.ne.08.030185.001313
125. Howell M. Parasomnias: an updated review. Neurotherapeutics. 2012;9(4):753–775.
126. Crompton D. Berkovic S The borderland of epilepsy: clinical and molecular features of phenomena that mimic epileptic seizures. Lancet Neurol. 2009;8(4):370–381. doi:10.1016/S1474-4422(09)70059-6
127. Gaudreau H, Joncas S, Zadra A, et al. Dynamics of slow-wave activity during the NREM sleep of sleepwalkers and control subjects. Sleep. 2000;23(6):755–760. doi:10.1093/sleep/23.6.1d
128. Rodriguez C. Foldvary-Schaefer N. Clinical neurophysiology of NREM parasomnias. Handb Clin Neurol. 2019;161:397–410.
129. Nobili L. Nocturnal frontal lobe epilepsy and non-rapid eye movement sleep parasomnias: differences and similarities. Sleep Med Rev. 2007;11(4):251–254. doi:10.1016/j.smrv.2007.03.009
130. Pincherle A, Proserpio P, Didato G, et al. Epilepsy and NREM-parasomnia: a complex and reciprocal relationship. Sleep Med. 2012;13(4):442–444. doi:10.1016/j.sleep.2011.09.011
131. Zanzmera P, Shukla G, Gupta A, et al. Effect of successful epilepsy surgery on subjective and objective sleep parameters–a prospective study. Sleep Med. 2013;14(4):333–338. doi:10.1016/j.sleep.2012.11.017
132. Provini F, Plazzi G, Montagna P, et al. The wide clinical spectrum of nocturnal frontal lobe epilepsy. Sleep Med Rev. 2000;4(4):375–386. doi:10.1053/smrv.2000.0109
133. Terzano M, Parrino L, Spaggiari M. The cyclic alternating pattern sequences in the dynamic organization of sleep. Electroencephalogr Clin Neurophysiol. 1988;69(5):437–447. doi:10.1016/0013-4694(88)90066-1
134. Terzano M, Parrino L, Sherieri A, et al. Atlas, rules, and recording techniques for the scoring of cyclic alternating pattern (CAP) in human sleep. Sleep Med. 2001;2(6):537–553. doi:10.1016/S1389-9457(01)00149-6
135. Terzano M, Monge-Strauss M, Mikol F, et al. Cyclic alternating pattern as a provocative factor in nocturnal paroxysmal dystonia. Epilepsia. 1997;38(9):1015–1025. doi:10.1111/j.1528-1157.1997.tb01485.x
136. Picard F, Bruel D, Servent D, et al. Alteration of the in vivo nicotinic receptor density in ADNFLE patients: a PET study. Brain. 2006;129:2047–2060. doi:10.1093/brain/awl156
137. Nauta W. Hippocampal projections and related neural pathways to the midbrain in the cat. Brain. 1958;81(3):319–340. doi:10.1093/brain/81.3.319
138. Halasz P, Kelemen A, Szucs A. The role of NREM sleep micro-arousals in absence epilepsy and in nocturnal frontal lobe epilepsy. Epilepsy Res. 2013;107(1–2):9–19. doi:10.1016/j.eplepsyres.2013.06.021
139. Licchetta L, Vignatelli L, Zenesini C, et al. Sleep-related hypermotor epilepsy: a prediction cohort study on sleep/awake patterns of seizures. Epilepsia. 2019;60(11):e115–e120. doi:10.1111/epi.16369
140. Di Resta C, Ambrosi P, Curia G, et al. Effect of carbamazepine and oxcarbazepine on wild-type and mutant neuronal nicotinic acetylcholine receptors linked to nocturnal frontal lobe epilepsy. Eur J Pharmacol. 2010;643(1):13–20. doi:10.1016/j.ejphar.2010.05.063
141. Kumar J, Solaiman A, Mahakkanukrauh P, et al. Sleep Related Epilepsy and Pharmacotherapy: an Insight. Front Pharmacol. 2018;9:1088. doi:10.3389/fphar.2018.01088
142. Gambardella A, Annesi G, De Fusco M, et al. A new locus for autosomal dominant nocturnal frontal lobe epilepsy maps to chromosome 1. Neurology. 2000;55(10):1467–1471. doi:10.1212/WNL.55.10.1467
143. Oldani A, Manconi M, Zucconi M, et al. Topiramate treatment for nocturnal frontal lobe epilepsy. Seizure. 2006;15(8):649–652. doi:10.1016/j.seizure.2006.07.002
144. Romigi A, Marciani MG, Placidi F, et al. Oxcarbazepine in nocturnal frontal-lobe epilepsy: a further interesting report. Pediatr Neurol. 2008;39(4):298. doi:10.1016/j.pediatrneurol.2008.07.015
145. Raju GP, Sarco DP, Poduri A, et al. Oxcarbazepine in children with nocturnal frontal-lobe epilepsy. Pediatr Neurol. 2007;37(5):345–349. doi:10.1016/j.pediatrneurol.2007.06.013
146. Mullen SA, Carney PW, Roten A, et al. Precision therapy for epilepsy due to KCNT1 mutations: a randomized trial of oral quinidine. Neurology. 2018;90(1):e67–e72. doi:10.1212/WNL.0000000000004769
147. Samarasekera SR. A case series of lacosamide as adjunctive therapy in refractory sleep-related hypermotor epilepsy (previously nocturnal frontal lobe epilepsy). J Sleep Res. 2018;27(5):e12669. doi:10.1111/jsr.12669
148. Puligheddu M, Melis M, Pillolla G, et al. Rationale for an adjunctive therapy with fenofibrate in pharmacoresistant nocturnal frontal lobe epilepsy. Epilepsia. 2017;58(10):1762–1770. doi:10.1111/epi.13863
149. Derry CP, Harvey AS, Walker MC, et al. NREM arousal parasomnias and their distinction from nocturnal frontal lobe epilepsy: a video EEG analysis. Sleep. 2009;32(12):1637–1644. doi:10.1093/sleep/32.12.1637
150. Tinuper P, Provini F, Bisulli F, et al. Movement disorders in sleep: guidelines for differentiating epileptic from non-epileptic motor phenomena arising from sleep. Sleep Med Rev. 2007;11(4):255–267. doi:10.1016/j.smrv.2007.01.001
151. Tinuper P, Bisulli F. From nocturnal frontal lobe epilepsy to Sleep-Related Hypermotor Epilepsy: A 35-year diagnostic challenge. Seizure. 2017;44:87–92. doi:10.1016/j.seizure.2016.11.023
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.