Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 9 » Issue 1
Sleep hypoventilation and daytime hypercapnia in stable chronic obstructive pulmonary disease
Authors Holmedahl N, Øverland B, Fondenes O, Ellingsen I, Hardie JA
Received 14 November 2013
Accepted for publication 16 December 2013
Published 27 February 2014 Volume 2014:9(1) Pages 265—275
DOI https://doi.org/10.2147/COPD.S57576
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Nils Henrik Holmedahl,1 Britt Øverland,2 Ove Fondenes,3 Ivar Ellingsen,1 Jon Andrew Hardie4
1Glittreklinikken LHL Helse as, Hakadal, Norway; 2Lovisenberg Diakonale Hospital, ENT Department, Oslo, Norway; 3Norwegian National Centre of Excellence in Home Mechanical Ventilation, Haukeland University Hospital, Bergen, Norway; 4Department of Clinical Science, University of Bergen, Norway
Purpose: To explore the associations between sleep hypoventilation (SH) and daytime arterial pressures of carbon dioxide (PaCO2), sleep stages, and sleep apneas/hypopneas (AHI) in subjects with chronic obstructive pulmonary disease (COPD). SH has previously been found in COPD-subjects with chronic hypercapnic respiratory failure (CHRF) using supplementary oxygen (LTOT), and has been proposed as a possible predictor for CHRF.
Patients and methods: A prospectively designed observational study in a pulmonary rehabilitation hospital of 100 (39 male) stable COPD inpatients with a mean forced expiratory volume in 1 second (FEV1) of 1.1 L (42% of predicted) and a mean age of 64 years, using polysomnography with transcutaneous measurement of carbon dioxide pressure increase (ΔPtcCO2).
Results: SH as defined by the American Academy of Sleep Medicine (AASM) was found in 15 of the subjects, seven of whom used LTOT. However, six had SH despite being normocapnic during the daytime (only one on LTOT). Subjects with SH had a greater ΔPtcCO2 increase from nonrapid eye movement (NREM) to rapid eye movement (REM) sleep stages compared to non-SH subjects (mean [standard deviation] between-groups difference =0.23(0.20) kPa, P<0.0005). Subjects with apnea/hypopnea index ≥15 (overlap, N=27) did not differ from those with COPD alone (AHI <5, N=25) in sleep ΔPtcCO2 or daytime PaCO2. A regression model with the variables FEV1, LTOT, and sleep maximum ΔPtcCO2 explained 56% of the variance in daytime PaCO2 (F(3, 94) =40.37, P<0.001).
Conclusion: In stable COPD, SH as defined by the AASM was found both in normocapnic, non-LTOT subjects and in hypercapnic, LTOT-using subjects. Between-sleep-stage increase in ΔPtcCO2 was higher in subjects with SH. Overlap subjects did not differ from simple COPD subjects in sleep ΔPtcCO2 or daytime PaCO2.
Keywords: blood gas analysis, etiology, physiopathology, carbon dioxide, polysomnography
Introduction
Chronic obstructive pulmonary disease (COPD) has a high and increasing morbidity and mortality worldwide, and is estimated to be the third leading cause of death by 2020.1 Chronic hypercapnic respiratory failure (CHRF) is associated with poor prognosis in COPD. The pathophysiological mechanisms leading to CHRF are not fully established, but sleep is of particular interest as a state of unstable respiration. Blood gas sampling during sleep is difficult, as it can interfere with sleep architecture, and only a few studies of small populations have been published. In COPD, end-tidal measurement of carbon dioxide pressure is too inaccurate to substitute for arterial pressure of carbon dioxide (PaCO2);3 however, transcutaneous measurement of carbon dioxide pressure (PtcCO2) minimally disturbs sleep, and PtcCO2 is now regarded by the American Academy of Sleep Medicine (AASM) as a surrogate for PaCO2.4
In normal sleep, changes in central respiratory control, muscle contractility, and lung mechanics lead to hypoventilation,5–7 with arterial pressure of carbon dioxide increasing to as much as 0.9 kPa above supine, awake values.8–10 In COPD this hypoventilation is more pronounced, especially during REM sleep, as a consequence of obstructed airflow, hyperinflation, respiratory muscle dysfunction, blunted ventilatory responses to hypercapnia and hypoxemia, ventilation/perfusion mismatch, and medications.11 Poor sleep quality is often reported, and polysomnography has shown a reduced total sleep time, disturbances in sleep architecture, and highly frequent arousals.12–15 Sleep hypoventilation was a common finding in two studies of stable, hypercapnic COPD subjects on long-term oxygen treatment (LTOT).16,17 It is unclear whether this hypoventilation was mainly the result of supplementary oxygen. In COPD, daytime PaCO2 correlates inversely with the forced expiratory volume in 1 second (FEV1),18 and one study reported a more severe daytime hypercapnia in subjects with coexisting COPD and obstructive sleep apnea (overlap syndrome), compared to subjects with COPD or obstructive sleep apnea alone.19 Sleep hypoventilation (SH) has recently been defined by the American Academy of Sleep Medicine as an increase of 1.3 kPa or more in PaCO2 during sleep, to a value exceeding 6.7 kPa for at least 10 minutes.4
Finding SH in normocapnic COPD subjects may be predictive of imminent hypercapnic respiratory failure, but to our knowledge, no studies have assessed SH in COPD subjects with normocapnia or in those with CHRF without LTOT.
The primary aim of this study was to determine whether SH is associated with daytime hypercapnia, with secondary aims to explore the impact of SH on the PtcCO2 increase between sleep stages, and whether SH or daytime PaCO2 is associated with the frequency of sleep apneas/hypopneas.
Material and methods
Subjects
Study participants were all Caucasians and inpatients at the Glittreklinikken Pulmonary Rehabilitation Hospital from January 2010 through June 2011. Initially, 60 subjects with PaCO2 <6.3 kPa and 60 with PaCO2 ≥6.3 kPa, all with Global initiative for Chronic Obstructive Lung Disease (GOLD)20-defined COPD, were to be included. Due to fewer hypercapnic subjects, these were oversampled by asking all to participate, whereas subjects with PaCO2 <6.3 kPa were selected randomly, stratifying for sex prior to participation request. In total, 166 subjects were screened for participation.
The following exclusion criteria were used: diagnosed obstructive sleep apnea (OSA), COPD exacerbation within 3 weeks prior, other serious lung comorbidity (ie, cancer, sarcoidosis, restrictive lung disease) or diseases affecting thoracic or abdominal movement, unstable angina pectoris, hypertension, diabetes mellitus, myocardial infarction within last 3 months, cerebral infarction, and addiction to drugs, alcohol, or narcotics.
All subjects used prescribed medication, but no respiratory depressant drugs were taken from 48 hours prior to first polysomnography (PSG) recording until end of study. Sixty-six of the 166 subjects declined, withdrew, or were excluded. The main reasons for decline/exclusion included fear of being unable to sleep with electrodes on head and body and technical artifacts in PtcCO2-signal (Figure 1, further details in Supplementary material). The remaining 100 PSGs of spontaneous sleep were analyzed.
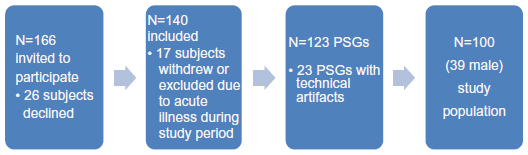 | Figure 1 Study population. |
The protocol was approved by the Regional Ethics Committee in southeastern Norway.
Measurements
Arterial blood gas samples (PICO50, Radiometer, Copenhagen, Denmark) were collected after 10 minutes seated rest at approximately 2 pm prior to PSG recording, and analyzed within 10 minutes on a Radiometer ABL720Flex (Radiometer). At sampling, all subjects were breathing room air, with the exception of those on LTOT who used their prescribed dose of supplementary oxygen. Height, weight, and lung function tests including measurement of postbronchodilator spirometry, diffusing capacity of the lung (DLCO), and body plethysmography of total lung volumes (MasterScreen Pneumo, Jaeger-Toennies, Hoechberg, Germany) were recorded during first week at the hospital. Reference values were based on equations from the European Community for Coal and Steel.21 PSG data was recorded with Embla A10 (Medcare Flaga, Reykjavik, Iceland) and PtcCO2 data with Tosca 500 (Radiometer, Basel, Switzerland) for two nights, the first night for acquaintance with the equipment, and the second night recording sleep data online to a bedside computer with Somnologica Studio Version 3.3 software (Medcare Flaga). PSGs were only recorded on weekdays, with channel setup according to the 2007 recommendations from the AASM.22 When the subject went to bed, a nurse started the recording with the subject watching TV or reading to stay awake while the signal from the Tosca probe stabilized. After approximately 30 minutes, the nurse turned off the light, this time being recorded as analysis started for the PSG. Rise time in the morning was recorded by the nurse. Subjects on LTOT used their prescribed dose of oxygen during PSG.
The PtcCO2 data were sampled with a frequency of 10 Hertz, with mean and maximum values calculated for each epoch of 30 seconds. Unpublished data collected at Glittreklinikken prior to the study showed equivalence between changes in PtcCO2 and PaCO2, but with a delay time of 54–57 seconds (see Supplementary material). Thus, PtcCO2 data were left-shifted two epochs. For each sleep stage in the PSG, the mean of each epoch’s mean PtcCO2 values were calculated, while the epoch with the highest value was reported as the maximum PtcCO2.
Sleep scoring was done independently by two experienced polysomnographists. Ten PSGs were scored by another two polysomnographists to select the scorer with the best concordance (see Supplementary material). According to recommendations from the AASM,22 a hypopnea was scored when nasal pressure dropped ≥30% for ≥10 seconds with ≥4% desaturation drop from baseline, with ≥90% of the event’s duration meeting the amplitude reduction criteria for hypopnea (criterion A).
Statistical analysis
Data were assessed for normality of distribution and homogeneity of variance. Differences between groups were analyzed using Pearson chi-square test, Fisher’s exact test, Mann– Whitney U test, and Student’s t-test, differences within groups by Student’s t-test. Relationships between groups were explored by chi-square tests for independence, within groups by Spearman’s rho and multiple regressions. No violation of the assumptions of normality, linearity, multicollinearity, and homoscedasticity was found preliminary to regression analysis. Concordance was measured by Cohen’s kappa. Two-sided P-values of ≤0.05 were considered significant, except in subgroup analyses of the SH subjects (N=15), in which two-sided P≤ 0.01 was applied. All analyses were performed using IBM SPSS Statistics version 19 (IBM Corporation, Armonk, NY, USA).
Results
As shown in Table 1, the group of 24 CHRF subjects had a greater increase in carbon dioxide pressure (ΔPtcCO2) during sleep compared to the normocapnic subjects, and SH by the AASM definition (an increase of 1.3 kPa in the PtcCO2 during sleep compared with awake supine level to a value exceeding 6.7 kPa for at least 10 minutes) was more frequent.4 Likewise, they had a higher median daytime base excess (BE), a lower PaO2, more frequent LTOT, and they were older and over-represented by females. However, no differences were found in total sleep time (TST), sleep stages as percent of TST, awakenings, arousals, apneas, or hypopneas (a table of sleep parameters according to age and compared to normal is provided in the Supplementary material). Oddly, no difference was found in body mass index (BMI); the mean BMI was 25 kg/m2, and only 13 were obese (BMI ≥30 kg/m2), whereas 26 were underweight (BMI <21 kg/m2). As would be expected, the CHRF group had more severely obstructed airways with hyperinflation and impaired gas exchange, a shorter 6-minute walking distance (6MWD), higher dyspnea scores (modified medical research council questionnaire, COPD assessment test), and a higher BMI/obstruction/dyspnea/exercise capacity (BODE)-index.
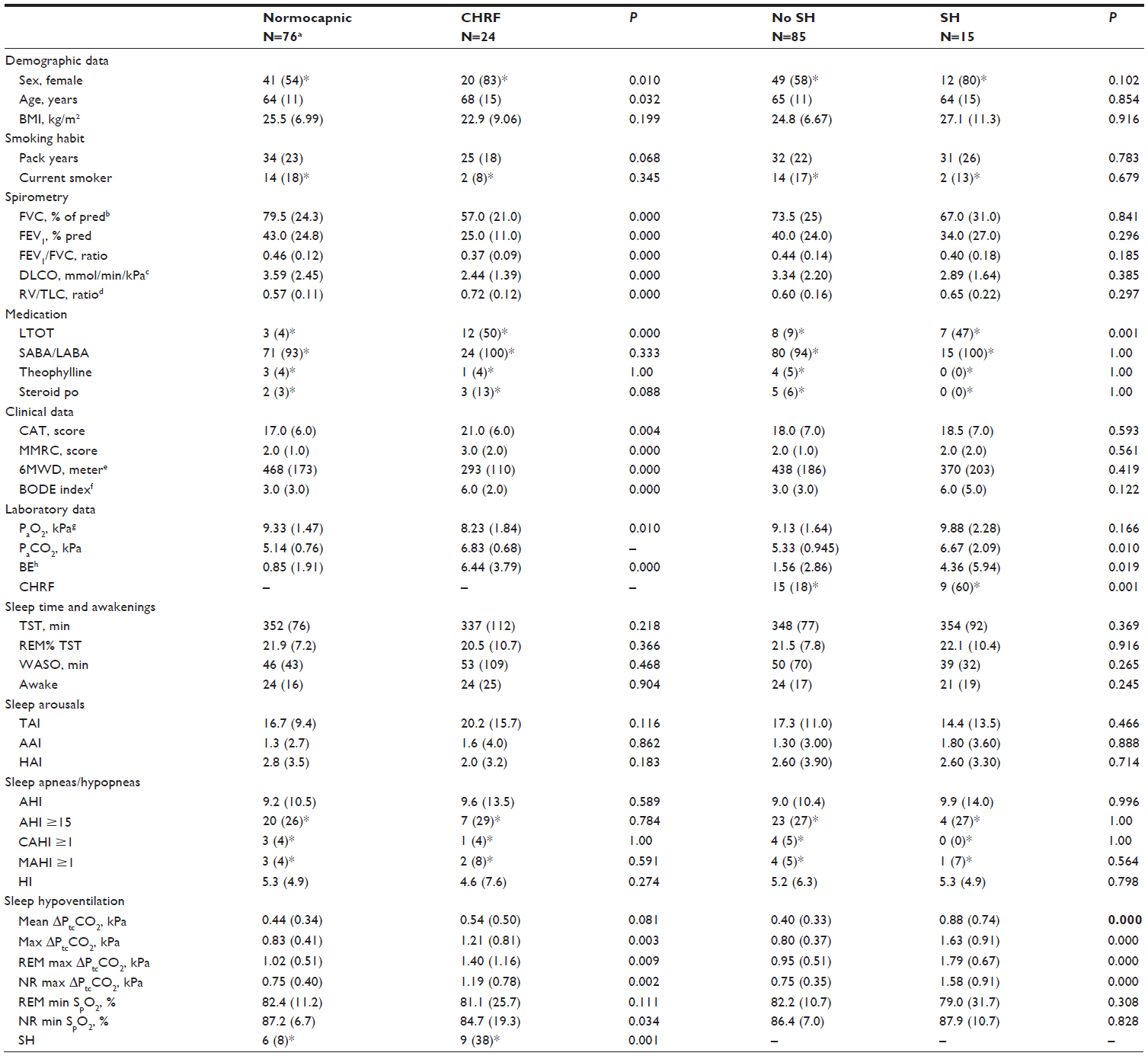 | Table 1 Differences between subjects with and without CHRF and subjects with and without SH; values as median (interquartile range), N=100 |
The group of 15 SH subjects had a higher median daytime PaCO2, and LTOT was more frequent compared to the non-SH group (Table 1); likewise, median BE was higher. No differences were found between the SH and non-SH groups in other medication, sex, age, BMI, smoking habit, spirometry, or sleep parameters mentioned above.
On average, the 15 SH subjects were severely obstructed, with a median (interquartile range [IQR]) FEV1 =0.80 (0.84) liters. Interestingly, however, six of the 15 SH-subjects were daytime normocapnic, with a median (IQR) PaCO2 of 5.00 (0.51) kPa, and only 1 used LTOT; this subgroup had a higher FEV1 compared to the group of nine hypercapnic subjects (median [IQR] 1.45 (0.81) liters versus 0.63 (0.26) liters, P=0.002). However, BMI and sleep apneas/hypopneas (AHI) did not differ between the normocapnic and hypercapnic SH-subjects.
As illustrated in Figure 2, the median of mean ΔPtcCO2 showed an increasing trend with deeper NREM sleep, and with the highest values in REM sleep, both in the non-SH group and in the SH group. A mean increase (standard deviation [SD]) from NREM to REM of 0.20 (0.16) kPa (P<0.001) was found in the non-SH group, whereas this increase was 0.46 (0.28) kPa (P<0.001) in the SH group, with the mean difference between the groups being 0.23 (0.20) kPa, (P<0.0005). The pattern of gradual increase with deeper sleep was not found for the median of maximum ΔPtcCO2 (Figure 2), especially as the values in N3 sleep were lower compared to N2. In subjects without SH the N2–N3 mean ΔPtcCO2 decrease (SD) was –0.13 (0.16) kPa (P<0.001), whereas the SH group had a mean decrease of −0.36 (0.41) kPa (P=0.005); the mean difference between the groups was 0.35 (0.31) kPa (P=0.002).
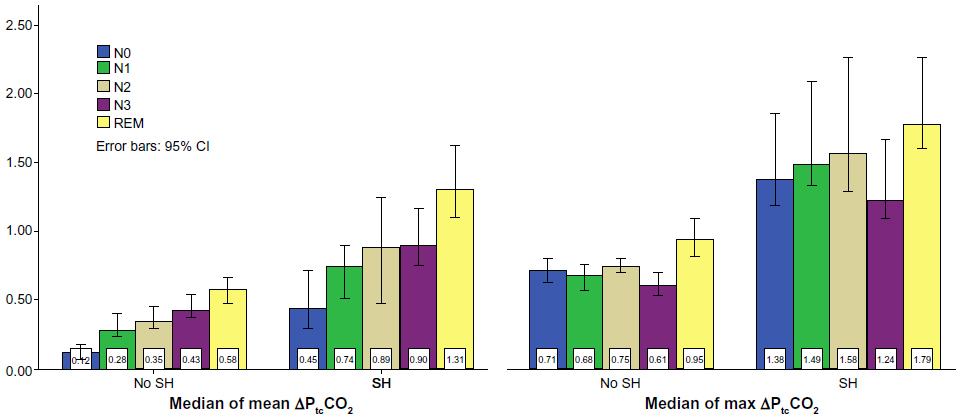 | Figure 2 ΔPtcCO2 according to sleep stages. |
With an AHI cutoff at 15/hour, concordance between the two controllers and the chosen polysomnographist was good (Kappa =1.00 between all three scorers, N=10, P=0.002). Despite exclusion of subjects with diagnosed OSA prior to study, only 25 subjects had AHI <5/hour, whereas 27 had AHI ≥15/hour; the latter chosen to define overlap syndrome as no record of daytime sleepiness was available, while COPD alone was defined by AHI <5/hour. Central and mixed apneas were found in 28 and 27 subjects, respectively, 4 having central apnea/hypopnea index ≥1/hour (highest value 4.4/hour), and five having mixed apnea/hypopnea index ≥1/hour (highest value 14.1/hour). Subjects with central and mixed apneas were included for overlap analysis (Table 2). As expected, the 27 overlap subjects had a significantly lower minimum SpO2 compared to the 25 COPD subjects, both in REM and in NREM sleep. Notably, however, the overlap subjects did not differ from those with COPD in sleep ΔPtcCO2 or daytime PaCO2 (Table 2), nor in the frequency of SH (15% in COPD versus 16% in the overlap group, P=1.00).
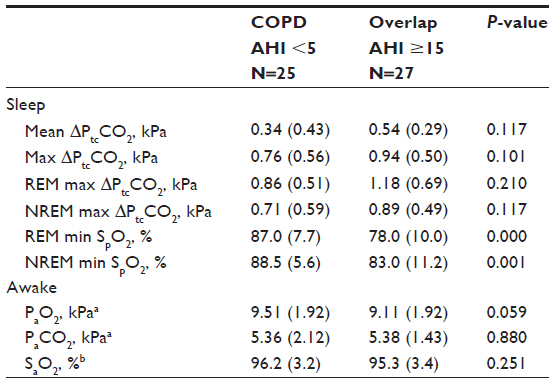 | Table 2 Blood gases in COPD and overlap subjects, as median (interquartile range), N=52 |
Finally, as sleep hypoventilation by various definitions previously has been found in severe COPD with CHRF and LTOT,16,17 a hierarchical multiple regression was performed to assess whether the maximum CO2 increase during sleep (sleep max ΔPtcCO2) independently predicts daytime PaCO2 when controlling for COPD severity (FEV1) and the use of LTOT. FEV1 (liters) and LTOT (yes/no) were entered at Step 1, explaining 52% of the variance in daytime PaCO2. After entry of sleep max ΔPtcCO2 (kPa) at Step 2, the total variance explained by the model was 56%, F(3, 94) =40.37, P<0.001. Thus, sleep max ΔPtcCO2 explained an additional 4% of the variance in daytime PaCO2 after controlling for LTOT and FEV1, R square change =0.043, F change (1, 94) =9.24, P=0.003. In the full model, FEV1 recorded the highest beta value (−0.44, P<0.001) over LTOT (0.39, P<0.001) and sleep max ΔPtcCO2 (0.22, P=0.003).
Discussion
As previously shown by others, we find SH to be common in stable COPD subjects with CHRF using LTOT. However, the novelty in this study is that SH is also found in COPD with CHRF without LTOT, and even in some normocapnic, non-LTOT subjects. We also find the mean PtcCO2 increasing with depth of sleep, the between-sleep-stage differences being greater in subjects with SH. Finally, overlap subjects in this study do not differ from COPD subjects in SH or in daytime PaCO2.
O’Donoghue et al found SH in 43% of 54 stable COPD subjects with CHRF, all using LTOT, when defining CHRF as daytime PaCO2 >6.12 kPa and SH as a ΔPtcCO2 increase of ≥1.33 kPa for ≥20% of total sleep time.17 Likewise, Tarrega et al studied stable, hypercapnic COPD subjects on LTOT and found nocturnal hypoventilation (NHV) in 21% of their 80 subjects, defining NHV as an increase in PaCO2 >1.33 kPa in any nocturnal blood gas sample as compared to the awake levels.16 Using the AASM definition, we find SH in 15% of our 100 subjects, and in seven of the 15 (47%) subjects on LTOT. Hence, this study supports O’Donoghue et al and Tarrega et al in the notion of SH being commonly found in COPD with CHRF using LTOT. Finding SH in three subjects with CHRF not using LTOT indicates that SH is associated with daytime PaCO2 independent of LTOT. However, CHRF is not necessary either, as SH was also found in five normocapnic, non-LTOT subjects.
Figure 2 illustrates the mean PtcCO2 increase with sleep stages, and the greater between-stages increase in subjects with SH versus without SH, supporting the notion that SH in COPD primarily is the consequence of a failing respiratory pump. As arterial CO2 increases due to hypoventilation, the brains’ extracellular fluid bicarbonate builds up, blunting the central ventilatory drive, which in turn will lead to further elevation of CO2.23 In patients without COPD this vicious circle is broken by an arousal when PaCO2 reaches 7.3–8.7 kPa.24 As changes in PtcCO2 are too sluggish in comparison to PaCO2, and in lack of a control group, this study did not provide data comparable to an arousal threshold. However, considering normal sleep maximum ΔPCO2 is in the range of 0.3–0.9 kPa,8 our findings of maximum ΔPtcCO2 values of 1.2–1.8 kPa (Figure 2) suggests that COPD subjects with SH have elevated CO2 thresholds. With the increased levels of ΔPtcCO2 and BE in subjects with SH, an elevated arousal threshold would be expected, hence unchanged frequencies of arousals and awakenings. Indeed, we find no significant differences in arousals, number of awakenings, or minutes of REM sleep percentage of TST between subjects with or without SH (Table 1). The lowest NREM ΔPtcCO2 values recorded in stage N3 both in SH and non-SH subjects (Figure 2) is in line with the normal hypercapnic ventilatory response described by Douglas et al.25
The overlap subjects do not differ from those with COPD alone in the frequency of SH, in sleep CO2-increase or in daytime PaCO2 (Table 2), neither are there significant differences in AHI between subjects with SH versus without SH (Table 1). This can be explained by the heterogeneity of COPD, as both the lungs (gas exchange system) and the respiratory pump (ventilatory system) are affected in various degrees of severity. Frequent apneas/hypopneas are not likely to result in sleep hypercapnia in patients with well-functioning respiratory pumps, as the retained CO2 following an apnea/hypopnea will be exhaled during the next few breaths. When scoring PSG in severely hyperinflated COPD patients, however, desaturation events resulting from a failing diaphragm can be classified as hypopneas or apneas, thus contributing to the AHI, even though there is no obstruction of the upper airways (as in OSA). In this case, one would expect to find AHI correlated to sleep CO2 increase. Hence, severe COPD with frequent apneas/hypopneas will be classified as overlap syndrome regardless of the etiology of the desaturation events.
Contrary to the findings of Resta et al,19 we did not find a significant difference in daytime PaCO2 in the overlap group as compared to the COPD group (Table 2). A main anthropometric difference between the study populations is body weight, as Resta et al reported a mean BMI of 36 kg/m2 in the overlap versus 31 kg/m2 in the COPD group, while our subjects had a median BMI of 25 kg/m2 with no significant differences between overlap and COPD subjects. In a multiple regression model, Resta et al found the best predictors of PaCO2 in overlap subjects to be PaO2, FEV1, and weight, O’Donoghue et al found the severity of SH in COPD subjects best predicted by a combination of baseline PaCO2, BMI, and percent REM sleep,17 and Tarrega et al found NHV related to BMI.16 Obesity might therefore explain the discrepancy between the findings of Resta et al and our findings.19
The variables FEV1, use of LTOT, and sleep maximum ΔPtcCO2 explain 56% of the variance in daytime PaCO2. A low FEV1 can indicate poor function of the respiratory pump (chronic obstruction leading to hyperinflation); LTOT is known to affect the central respiratory drive to breathe,26 and increased sleep ΔPtcCO2 can indicate a blunted respiratory drive as a consequence of the failing respiratory pump. However, the intriguing subgroup of five normocapnic, non-LTOT SH subjects does not fit in the picture, as they were not obese, nor severely obstructive or hyperinflated, and they had a median AHI of only 12.8/hour. Thus, their respiratory pump should have sufficient reserves to expel CO2 even during REM sleep.
To our knowledge, there are no previously published studies with PSG and PtcCO2 data from COPD subjects including as many as 100 subjects. This study has its limitations, however. First, as previous studies indicate the prevalence of OSA being the same in COPD as in the normal population,27 the study protocol did not include collection of daytime sleepiness data. Hence, the diagnosis of overlap syndrome based exclusively on the AHI is admittedly uncertain, and prevalence could not be analyzed. However, as OSA is defined either by AHI ≥5 and daytime sleepiness symptoms or AHI ≥15 in asymptomatic subjects,28 we chose the latter to define overlap; the most conservative criterion would include only certain OSA subjects in this group, whereas AHI <5 was chosen to define subjects with COPD alone, thus reducing the risk of masking any differences between overlap and COPD groups.
Second, we do not regard the demographic profile in this study as representative for the entire COPD population in Norway, as a selection bias can be present for several reasons, including the referral/selection for lung rehabilitation, the oversampling of subjects with PaCO2 ≥6.3 kPa, and subjects declining to participate. As the study population had a somewhat high BODE index of 4, indicating a 4-year mortality of about 32%,29 the results presented does not reflect the status of the entire COPD population, and the prevalence of SH is undoubtedly lower in patients with mild COPD.
Third, as arterial blood gas samples from the subjects using LTOT were collected when using their prescribed oxygen flow, the PO2 and PCO2 referred in Table 1 do not reflect the untreated values of the study population. However, as all subjects on LTOT were stable and adapted to their supplementary oxygen, blood gases measured reflects their habitual status.
Conclusion
Sleep hypoventilation as defined by the AASM is frequently found in subjects with severe COPD and CHRF using supplementary oxygen. The novelty of this study is the finding of SH also in CHRF without LTOT, and even in some normocapnic, non-LTOT subjects with only moderate COPD. The carbon dioxide pressure increases with deeper sleep, with differences between sleep stages being greater in subjects with SH than those without SH. Subjects with AHI ≥15/hour (overlap) do not differ from those with AHI <5 (COPD alone) in SH, sleep ΔPtcCO2, or in daytime PaCO2. A regression model with the predictors FEV1, LTOT, and sleep maximum ΔPtcCO2 explains half of the variance in daytime PaCO2.
SH is currently used in clinical practice at some centers as an independent indicator for the initiation of noninvasive positive pressure ventilation in COPD,30 and correction of nocturnal PtcCO2 to normocapnic values has shown to improve lung function and may improve survival.31,32 To the best of our knowledge, however, no case-control or prospective studies have been undertaken to investigate the exact mechanisms behind SH or SH’s role as a predictor for CHRF, so further research is clearly needed.
Acknowledgments
This work was supported by grants from the Norwegian ExtraFoundation for Health and Rehabilitation, LHL’s research fund, Glittreklinikken LHL-Helse as, and the Norwegian National Centre of Excellence in Home Mechanical Ventilation. Minor grants were received from the Norwegian Lung Medicine Society/Takeda Nycomed and Major Eckbo’s endowments. PSG hardware and software were provided by ResMed Norway.
Disclosure
This is not an industry-supported study. Nils Henrik Holmedahl has received lecture honoraria from Glaxo Smith Kline and a grant from the Norwegian Lung Medicine Society/Takeda Nycomed. Britt Øverland, Ove Fondenes, Ivar Ellingsen, and Jon Andrew Hardie report no conflicts of interest.
References
Lopez AD, Shibuya K, Rao C, et al. Chronic obstructive pulmonary disease: current burden and future projections. Eur Respir J. 2006;27(2):397–412. | |
Costello R, Deegan P, Fitzpatrick M, McNicholas WT. Reversible hypercapnia in chronic obstructive pulmonary disease: a distinct pattern of respiratory failure with a favorable prognosis. Am J Med. 1997;102(3):239–244. | |
Hatle L, Rokseth R. The arterial to end-expiratory carbon dioxide tension gradient in acute pulmonary embolism and other cardiopulmonary diseases. Chest. 1974;66(4):352–357. | |
Berry RB, Budhiraja R, Gottlieb DJ, et al; American Academy of Sleep Medicine. Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med. 2012;8(5):597–619. | |
Meadows GE, Dunroy HM, Morrell MJ, Corfield DR. Hypercapnic cerebral vascular reactivity is decreased, in humans, during sleep compared with wakefulness. J Appl Physiol (1985). 2003;94(6):2197–2202. | |
Meadows GE, Kotajima F, Vazir A, et al. Overnight changes in the cerebral vascular response to isocapnic hypoxia and hypercapnia in healthy humans: protection against stroke. Stroke. 2005;36(11):2367–2372. | |
Douglas NJ, White DP, Pickett CK, Weil JV, Zwillich CW. Respiration during sleep in normal man. Thorax. 1982;37(11):840–844. | |
Sleep-related breathing disorders in adults: recommendations for syndrome definition and measurement techniques in clinical research. The Report of an American Academy of Sleep Medicine Task Force. Sleep. 1999;22(5):667–689. | |
Birchfield RI, Sieker HO, Heyman A. Alterations in blood gases during natural sleep and narcolepsy; a correlation with the electroencephalographic stages of sleep. Neurology. 1958;8(2):107–112. | |
Bristow JD, Honour AJ, Pickering TG, Sleight P. Cardiovascular and respiratory changes during sleep in normal and hypertensive subjects. Cardiovasc Res. 1969;3(4):476–485. | |
McNicholas WT, Lee R. Obstructive sleep apnoea and COPD. European Respiratory Monograph. 2013(59):135–143. | |
Cormick W, Olson LG, Hensley MJ, Saunders NA. Nocturnal hypoxaemia and quality of sleep in patients with chronic obstructive lung disease. Thorax. 1986;41(11):846–854. | |
Fleetham J, West P, Mezon B, Conway W, Roth T, Kryger M. Sleep, arousals, and oxygen desaturation in chronic obstructive pulmonary disease. The effect of oxygen therapy. Am Rev Respir Dis. 1982;126(3):429–433. | |
Valipour A, Lavie P, Lothaller H, Mikulic I, Burghuber OC. Sleep profile and symptoms of sleep disorders in patients with stable mild to moderate chronic obstructive pulmonary disease. Sleep Med. 2011;12(4):367–372. | |
McSharry DG, Ryan S, Calverley P, Edwards JC, McNicholas WT. Sleep quality in chronic obstructive pulmonary disease. Respirology. 2012;17(7):1119–1124. | |
Tarrega J, Anton A, Guell R, et al. Predicting nocturnal hypoventilation in hypercapnic chronic obstructive pulmonary disease patients undergoing long-term oxygen therapy. Respiration. 2011; 82(1):4–9. | |
O’Donoghue FJ, Catcheside PG, Ellis EE, et al; Australian trial of Noninvasive Ventilation in Chronic Airflow Limitation investigators. Sleep hypoventilation in hypercapnic chronic obstructive pulmonary disease: prevalence and associated factors. Eur Respir J. 2003;21(6):977–984. | |
Saure EW, Eagan TM, Jensen RL, et al. Explained variance for blood gases in a population with COPD. Clin Respir J. 2012;6(2):72–80. | |
Resta O, Foschino Barbaro MP, Brindicci C, Nocerino MC, Caratozzolo G, Carbonara M. Hypercapnia in overlap syndrome: possible determinant factors. Sleep Breath. 2002;6(1):11–18. | |
Global Initiative for Chronic Obstructive Pulmonary Disease (GOLD). Global strategy for the diagnosis, treatment, and prevention of chronic obstructive pulmonary disease. Updated 2013. Available from: http://www.goldcopd.org/uploads/users/files/GOLD_Report_2013_Feb20.pdf. Accessed December 19, 2013. | |
Quanjer PH, Tammeling GJ, Cotes JE, Pedersen OF, Peslin R, Yernault JC. Lung volumes and forced ventilatory flows. Report Working Party Standardization of Lung Function Tests, European Community for Steel and Coal. Official Statement of the European Respiratory Society. Eur Respir J Suppl. 1993;16:5–40. | |
Iber C, Ancoli-Israel S, Chesson AL Jr, Quan SF; for the American Academy of Sleep Medicine. The AASM Manual for the Scoring of Sleep and Associated Events: Rules,Terminology and Technical Specifications. Westchester, IL: American Academy of Sleep Medicine; 2007. | |
Javaheri S, Shore NS, Rose B, Kazemi H. Compensatory hypoventilation in metabolic alkalosis. Chest. 1982;81(3):296–301. | |
Berthon-Jones M, Sullivan CE. Ventilation and arousal responses to hypercapnia in normal sleeping humans. J Appl Physiol Respir Environ Exerc Physiol. 1984;57(1):59–67. | |
Douglas NJ. Control of ventilation during sleep. Clin. Chest Med. 1985;6(4):563–575. | |
Aubier M, Murciano D, Milic-Emili J, et al. Effects of the administration of O2 on ventilation and blood gases in patients with chronic obstructive pulmonary disease during acute respiratory failure. Am Rev Respir Dis. 1980;122(5):747–754. | |
Weitzenblum E, Chaouat A, Kessler R, Canuet M. Overlap syndrome: obstructive sleep apnea in patients with chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2008;5(2):237–241. | |
Hauri PJ (Ed). The International Classification of Sleep Disorders. 2nd ed. Diagnostic and Coding Manual. Westchester 2005. | |
Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(10):1005–1012. | |
Windisch W, Walterspacher S, Siemon K, Geiseler J, Sitter H; German Society for Pneumology. Guidelines for non-invasive and invasive mechanical ventilation for treatment of chronic respiratory failure. Published by the German Society for Pneumology (DGP). Pneumologie. 2010;64(10):640–652. | |
Windisch W, Haenel M, Storre JH, Dreher M. High-intensity non-invasive positive pressure ventilation for stable hypercapnic COPD. Int J Med Sci. 2009;6(2):72–76. | |
McEvoy RD, Pierce RJ, Hillman D, et al; Australian trial of non-invasive Ventilation in Chronic Airflow Limitation (AVCAL) Study Group. Nocturnal non-invasive nasal ventilation in stable hypercapnic COPD: a randomised controlled trial. Thorax. 2009;64(7):561–566. |
Supplementary material
Subjects
Reasons given from the 26 subjects not participating: Anxiety/fear of not being able to sleep with electrodes connected to head/body (ten subjects); not willing to discontinue sedentary medication (three subjects); fear that sleep studies would interfere with the rehabilitation program (three subjects); did not want sleep investigated (three subjects); fear of pain from blood gas sampling (two subjects). Two subjects declined because an interventional part of the project (not published) implied sleeping with supplementary oxygen or taking alcohol or zopiclone before sleep for a third polysomnography. One subject did not want to be seen with wires connected to the head, one needed to go outdoors for smoking at night and one did not give a reason.
Reasons for drop out of 17 subjects after initial consent given: Felt it impossible to sleep with wires on the body (seven subjects); exacerbation of COPD (six subjects); technical or protocol failure (two subjects); illness/disease of other cause (one subject); medication which excluded (one subject).
Twenty-three polysomnographies were excluded because of technical artifacts in transcutaneous measurement of PCO2; 17 because the PtcCO2 probe periodically lost contact with skin (low PtcCO2-signal); six because the probe was compressed by pillow or hand/arm (high PtcCO2-signal).
PtcCO2 versus PaCO2
The correlation between PaCO2 and PtcCO2 has been studied in stable subjects with lung diseases,1 in critically ill subjects,2–5 during cardio-pulmonary exercise testing6 and in circulatory stable subjects; both inactive and during exercise.7,8 Conclusions varied as to whether PtcCO2 can substitute PaCO2. Hence, clinical data were obtained from 18 stable, supine COPD subjects with an indwelling catheter at Glittreklinikken (mean FEV1(SD)=46(26)% of predicted, N=4 had PaCO2> 6.3 kPa). Every twentieth minute for up to six hours a pair of PaCO2 and PtcCO2 values were recorded. From 204 pairs, a mean difference (SD) PtcCO2 – PaCO2 of 0.233 (0.312) kPa was found; this SD regarded as too high a variance to let the absolute values of PtcCO2 substitute those of PaCO2. However, the within subject mean difference (SD) PtcCO2 – PaCO2 was 0.229 (0.186) kPa. We concluded that the within subject SD of <0.2 kPa indicates that the changes in PtcCO2 can substitute the changes in PaCO2. Consequently, when studying COPD subjects with PSG and PtcCO2 we recorded the changes in PtcCO2 during sleep, not the absolute values.
PtcCO2 delay time
To find the time delay from an alveolar change in PCO2 until the first response in PtcCO2, we studied nine COPD subjects (six male) with a mean FEV1(SD) of 41(20)% of predicted, and PaCO2< 6.3 kPa. The supine subjects wore a tight fitting face mask, inlet selecting either room air or a gas mixture of 4% CO2 in air. Data were collected in three phases, each lasting 200 sec: 1) stable phase breathing room air; 2) increasing phase breathing 4% CO2; and 3) decreasing phase after switching back to room air. PtcCO2 was recorded every 5th second for 2 minutes, then every 10th second in each phase. Arterial blood samples were taken from an indwelling catheter three times during phase 1), every 5th second the first 30 sec of phase 2) and 3), then every 30th second for a total of 150 seconds. The results showed a first response time (frt) meaning time from change in alveolar PCO2 to PtcCO2>2SD off stable phase as follows: Mean frt(SD) in increasing phase: 54(5,6) sec; Mean frt(SD) in decreasing phase: 57(15) sec. For arterial PCO2: 13,3(5,6) sec and 11,7(2,5) sec, accordingly.
Sleep scoring
All PSG’s were initially scored by two independent, registered polysomnographists (RPSG’s) blinded to subject data except sex, age, height and weight. As the scorings from these two RPSG’s differed substantially on several sleep parameters (eg, AHI, HI, sleep stages) a random selection of 10 PSG’s were scored by another two RPSG’s who were blinded regarding the results from the first two scorers. The scorings from the initial RPSG with the best match to the last two RPSG’s were then selected for data analysis.
Results, table supplement
When compared to normal subjects,9,10 the study-subjects had twice to threefold more awakenings and N1-sleep. However, they had a normal amount of REM-sleep and about twice the percentage of slow wave (N3) sleep (table 1).
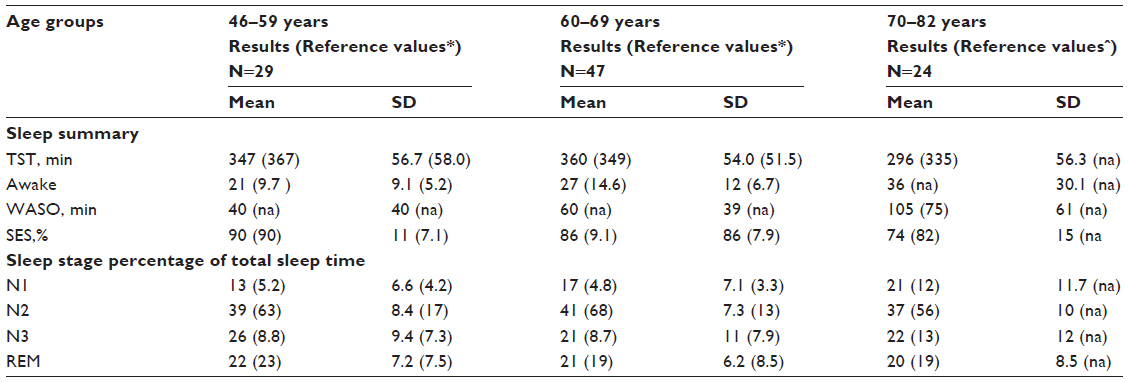 | Table S1 Sleep parameters according to age, compared to reference populations with normal lung function, N=100 |
References
Parker SM, Gibson GJ. Evaluation of a transcutaneous carbon dioxide monitor (“TOSCA”) in adult patients in routine respiratory practice. Respir. Med. Feb 2007;101(2):261–264. | |
Senn O, Clarenbach CF, Kaplan V, Maggiorini M, Bloch KE. Monitoring carbon dioxide tension and arterial oxygen saturation by a single earlobe sensor in patients with critical illness or sleep apnea. Chest. Sep 2005;128(3):1291–1296. | |
Bolliger D, Steiner LA, Kasper J, Aziz OA, Filipovic M, Seeberger MD. The accuracy of non-invasive carbon dioxide monitoring: a clinical evaluation of two transcutaneous systems. Anaesthesia. Apr 2007;62(4):394–399. | |
Rodriguez P, Lellouche F, Aboab J, Buisson CB, Brochard L. Transcutaneous arterial carbon dioxide pressure monitoring in critically ill adult patients. Intensive Care Med. Feb 2006;32(2):309–312. | |
Storre JH, Steurer B, Kabitz HJ, Dreher M, Windisch W. Transcutaneous PCO2 monitoring during initiation of noninvasive ventilation. Chest. Dec 2007;132(6):1810–1816. | |
Stege G, van den Elshout FJ, Heijdra YF, van de Ven MJ, Dekhuijzen PN, Vos PJ. Accuracy of transcutaneous carbon dioxide tension measurements during cardiopulmonary exercise testing. Respiration. 2009;78(2):147–153. | |
Janssens JP, Perrin E, Bennani I, de Muralt B, Titelion V, Picaud C. Is continuous transcutaneous monitoring of PCO2 (TcPCO2) over 8 h reliable in adults? Respir. Med. May 2001;95(5):331–335. | |
Janssens JP, Laszlo A, Uldry C, Titelion V, Picaud C, Michel JP. Non-invasive (transcutaneous) monitoring of PCO2 (TcPCO2) in older adults. Gerontology. May–Jun 2005;51(3):174–178. | |
Hirshkowitz M, Moore CA, Hamilton CR, 3rd, Rando KC, Karacan I. Polysomnography of adults and elderly: sleep architecture, respiration, and leg movement. J. Clin. Neurophysiol. Jan 1992;9(1):56–62. | |
Ohayon MM, Carskadon MA, Guilleminault C, Vitiello MV. Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: developing normative sleep values across the human lifespan. Sleep. Nov 1 2004;27(7):1255–1273. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.