Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Skeletal Abnormalities and VDR1 Gene Polymorphisms in Mucopolysaccharidosis Patients
Authors Alkhzouz C, Cabau G, Lazea C ![]() , Asavoaie C
, Asavoaie C ![]() , Bucerzan S, Mirea AM
, Bucerzan S, Mirea AM ![]() , Farcas M
, Farcas M ![]() , Miclaus M Jnr, Popp R, Miclea D
, Miclaus M Jnr, Popp R, Miclea D
Received 6 December 2020
Accepted for publication 22 February 2021
Published 17 March 2021 Volume 2021:14 Pages 349—358
DOI https://doi.org/10.2147/PGPM.S295241
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Camelia Alkhzouz,1,2 Georgiana Cabau,2 Cecilia Lazea,2,3 Carmen Asavoaie,3 Simona Bucerzan,1,2 Andreea Manuela Mirea,2 Marius Farcas,2 Maria Miclaus Jnr,2 Radu Popp,2 Diana Miclea1,2
1Department of Medical Genetics, Clinical Emergency Hospital for Children, Cluj-Napoca, Romania; 2“Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania; 3First Pediatric Clinic, Clinical Emergency Hospital for Children, Cluj-Napoca, Romania
Correspondence: Diana Miclea
Department of Medical Genetics, Clinical Emergency Hospital for Children, “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania
Email [email protected]
Introduction: Articular and bone damage, which is so disabling in Mucopolysaccharidosis (MPS), requires attention as to the explanatory bias of the pathogenetic mechanisms identified to date. The vitamin D receptor (VDR) has been investigated in many studies in correlation with bone metabolism, osteoporosis, and the impaired bone mineral density associated with certain polymorphisms of the VDR gene.
Aim: This study aims to observe whether there is an association between clinical features, phospho-calcium metabolism parameters and the VDR gene polymorphisms in patients with MPS.
Patients and Method: We evaluated six patients with MPS type I, 20 patients with MPS type II, two patients with MPS types IIIA and IIIB and three patients with MPS type IVB. In these patients, phospho-calcium metabolism, markers of bone formation, bone radiographs and bone densitometry were evaluated, as were four polymorphisms of the VDR gene (ApaI, BsmI, FokI and TaqI).
Results: There was a deficiency in 25 hydroxy vitamin D in MPS type I patients at the final evaluation and in MPS type II patients, both at ERT initiation and at the last evaluation. The analysed polymorphisms were not associated with modified calcium-phosphor levels, but some differences were observed regarding the level of 25 OH vitamin D. Thus, in the case of AA polymorphism, all patients have a 25 OH vitamin D deficiency, and one patient with the AA genotype and three with Aa have a 25 OH vitamin D deficiency and secondary hyperparathyroidism due to this deficiency (four patients), all of them having the Bb phenotype.
Conclusion: In MPS patients, vitamin D deficiency is observed, as it is in some patients with secondary hyperparathyroidism, which indicates vitamin D supplementation to protect bone metabolism. There are no obvious correlations between VDR polymorphism and bone metabolism in MPS patients.
Keywords: mucopolysaccharidosis, vitamin D receptor, ApaI, BsmI, FokI, TaqI
Introduction
Mucopolysaccharidoses (MPS) are characterised by the malfunctioning of lysosomal enzymes involved in the degradation of mucopolysaccharides (glycosaminoglycans or GAGs), which accumulate in various tissues, affecting cell function and the extracellular matrix. Depending on the particular deficient lysosomal enzyme involved, there are eleven MPS types (MPS I, II, IIIA, B, C, IVA, IVB, V, VI, VII, VIII and IX). Characteristic clinical features are represented by musculoskeletal, cardiovascular, CNS, ophthalmic, auditory, and pulmonary involvement. The skeletal features developed progressively in these pathologies, due to GAGs accumulation in joints and connective tissues, are very limiting regarding the mobility and activity of these patients, entailing a significant decrease in the quality of life.1,2 Bone damage is classically referred to as multiplex dysostosis, which includes vertebral abnormalities, deformity and shortening of long bones, macrocephaly, hips dysplasia, coxa valgum, genu valgum, claw hands, thorax involvement, facial dysmorphism, odontoid hypoplasia, and osteopenia. Joint and skeletal damage, so disabling in MPS, requires attention regarding the explanatory bias of the pathogenetic mechanisms known at present.
Going beyond the classical conception of MPS pathogenesis as being due to accumulation of undegraded GAGs, the influences exerted by these molecules, as biological response modifiers, can be much more diverse, including influences on different signalling pathways (STAT, BMP4, FGFR3), on cytokines (IL2, IL6), on oxidative stress and mitochondrial dysfunction, on cathepsin activity, on autophagy, on abnormal intracellular transport and on other substrate pathways.3–7 On the other hand, MPS bone remodelling is influenced by cathepsin K activity, a lysosomal enzyme highly expressed in osteoclasts and leading to osteopenia and osteoporosis.5,8–11 Osteoporosis and osteopenia observed in MPS patients, as modifications in the extracellular matrix, should have consequences in bone phospho-calcium metabolism.
Bone mineral density and vitamin D levels were assessed and proportionally correlated in MPS patients, but there is no clear pathogenetic mechanism known so far for vitamin D metabolism in correlation with MPS, other than insufficient oral intake.12–15 It is known that the MPS therapies employed to date do not have significant efficacy in reversing the conjunctive tissue changes.16,17 Conversely, the peculiarities of phosphocalcic metabolism due to genetic changes concomitant with those responsible for MPS may influence bone metabolism in MPS patients, but there have been no clear data until now.3
A genetic change often correlated with bone metabolism involves the vitamin D receptor (VDR), through which vitamin D mediates its biological effects. VDR is a member of the nuclear hormone receptor superfamily, has a size of 75 kb, comprises 11 exons, gives rise to a protein with 427 amniotic acids, and regulates gene transcriptions with an effect on calcium and skeletal metabolism.18–20 Vitamin D (1.25 dihydroxy vitamin D) binds to the receptor (VDR) and thus influences intestinal calcium absorption as well as osteoblastic differentiation, thus leading to proper bone mineralisation and remodelling. VDR abnormalities could alter mineral metabolism, as well as other pathways such as cancer or immune response.21,22
The VDR gene has been investigated in many studies in correlation with bone metabolism, initially in relation to Type 2A-dependent vitamin D rickets, in which there is a clear cause-effect correlation, and subsequently in relation to growth, osteoporosis and impaired bone mineral density, in the case of certain polymorphisms of the VDR gene.23 There are no data about the involvement of VDR changes as aggravating factors for bone disease in MPS patients.
The aim of the study is to observe whether there is an association between clinical features, phospho-calcium metabolism parameters, and genetic polymorphisms of the VDR gene in patients with MPS.
Patients and Method
We evaluated six patients with MPS type I, 20 patients with MPS type II, two patients with MPS types IIIA and IIIB, and three patients with MPS type IVB, who presented at the Emergency Clinical Hospital for Children, Cluj-Napoca, between 1st January 2009 and 31st December 2019.
These patients were clinically evaluated, noting the clinical picture (auxological parameters, goniometry, and clinical signs). There were also investigations by different specialities to evaluate the other features of this disease: neurological and psychological assessments, ENT examinations (Ear, Nose and Throat), audiograms, ophthalmological examinations, and cardiological evaluations. The specific diagnosis was carried out by performing enzymatic and molecular tests.
The following evaluations were conducted at diagnosis (before enzyme replacement therapy, ERT), one year after ERT (for MPS I and MPS II) and at the final evaluation (between 3 and 8 years of treatment): phospho-calcium metabolism (phosphorus, calcium, PTH, 25 OH vitamin D, alkaline phosphatases), markers of bone formation (osteocalcin), bone radiographs and bone densitometry. Four polymorphisms of the VDR gene were evaluated (rs7975232-ApaI, rs1544410-BsmI, rs2228570-FokI and rs731236-TaqI).
Study approval was obtained from the hospital ethics committee, with informed consent being obtained for each patient from the parents or legal guardians.
Genetic Testing
Genomic DNA was extracted for all subjects from 300 µL of blood, using a commercial kit (Wizard Genomic DNA Purification kit, Promega, Cambridge, MA, USA).
The polymorphisms rs7975232-ApaI, rs1544410-BsmI, rs2228570-FokI and rs731236-TaqI were evaluated, using the adapted protocol of Kaya et al.24 The VDR gene region carrying the polymorphic restriction sites ApaI, BsmI, TaqI was amplified by PCR, using the upstream primer in exon 8 (5′-CAACCAAGACTACAAGTACCGCGTCAGTGA-3′) and the downstream primer in exon 9 (5′-CACTTCGAGCACAAGGGGCGTTAGC-3′). For the FokI polymorphic restriction site, a primer pair spanning exon 2 of the VDR gene was used for amplification (5′-AGCTGGCCCTGGCACTGACTCTGCTCT-3′ and 5′-ATGGAAACACCTTGCTTCTTCTCCCTC-3′). The PCR reaction was performed using Thermo Scientific PCR Master Mix containing Taq DNA polymerase 0.05U/µL, reaction buffer, 4 mM MgCl2, 0.4 mM of each dNTP (dATP, dCTP, dGTP and dTTP), UltraPure BSA (Invitrogen™), forward primer (0.1–1.0 µM), reverse primer (0.1–1.0 µM) and template DNA. The PCR conditions were 3 minutes at 94.5°C for initial denaturation, followed by 35 cycles of denaturation of 1 minute at 94.5°C, annealing 1 minute at 61°C and 2 minutes at 72°C for extension, with a final extension at 72°C for 7 minutes.
The amplified PCR products were digested by FastDigest FokI (10U/uL) (Thermo Scientific) restriction endonuclease for 15 minutes at 37°C, followed by enzyme inactivation at 65°C for 5 minutes, FastDigest TaqI (10U/uL) (Thermo Scientific) 15 minutes at 65°C, ApaI (10U/uL) and BsmI (10U/uL) for 16 hours at 37°C, followed by inactivation for 20 minutes at 65°C. With the enzymes ApaI, BsmI, TaqI and FokI, the respective genotypes were defined as A, B, T, and F (indicating the absence of the restriction site) or a, b, t, and f (indicating the presence of the restriction site). The digestion products were visualised under UV light using a gel electrophoresis visualising system.
Results
Six patients with MPS type I, 20 patients with MPS type II, two patients with MPS types IIIA and IIIB and three patients with MPS type IVB were analysed. Among the patients with MPS type I were three females and three males. All patients with MPS type II were males, considering the X-linked recessive transmission of this disease. The mean age of the patients at the time of ERT initiation was 9.08 years for those with MPS type I and 7 years for those with MPS type II.
The auxological characterisation of these patients can be observed in Table 1. A higher height deficit is observed at the ERT initiation in patients with MPS type I (mean −2.25SD), compared to MPS type II (mean 0.33SD) (p = 0.03). At the final evaluation, under ERT, there is a similar height growth and a higher height deficit for MPS type I (−4.80SD), compared to MPS type II patients (−1.52SD) (p = 0.02). A body mass index (BMI) within normal limits is observed for all patients with MPS.
 |
Table 1 Auxological Characterisation in MPS Patients |
Concerning the biological analysis, in Table 2 the main findings are described. For the group with MPS types I and II, data were available from the initiation of therapy to the present (between 3 and 8 years after ERT initiation). There was a deficiency in 25-Hydroxy vitamin D in MPS type I patients at the last evaluation and, in MPS type II, both at ERT initiation and at the final evaluation. Concerning 25-Hydroxy vitamin D levels, a difference was observed between MPS type I and MPS type II patients at the ERT initiation (p = 0.03) and a slight difference was also noticeable at the last evaluation (without reaching a statistically significant threshold).
 |
Table 2 Biological Assessment in MPS Patients |
There was a slightly higher level of osteocalcin in patients with MPS II than in those with MPS I, indicating a slightly more intense osteogenic process in patients with the former type, without reaching a threshold of statistical significance. The DEXA examination did not reveal osteoporosis or osteopenia in the examined patients. All patients with MPS types IIIA, IIIB and IVB also presented low levels of vitamin D (<30 µg/L), one patient with MPS IIIB and two patients with MPS type IV presenting a deficiency of 25-Hydroxy vitamin D (<20µg/L).
The radiological skeletal findings in these MPS patients are illustrated in Figure 1. All I, II and IV MPS patients in our study group presented musculoskeletal abnormalities on imaging studies with varying degrees of severity. Dysostosis multiplex is a term that includes several radiological features typically encountered in MPS patients. Some of the most common findings associated with dysostosis multiplex in our patients are presented in Table 3.
 |
Figure 1 Radiological abnormalities seen in our patients with MPS type II. (A) Macrocephaly in 6 years old MPS II patient; (B) i,ii. Dorsolumbar kyphosis and a hypoplastic transitional vertebra with anterior beaking; (C) Short, thick clavicles, paddle-shaped ribs, scoliosis; (D) Round and flared iliac wings, an underdeveloped acetabulum and signs of hip dysplasia; (E) i,ii. Thick and hypoplastic distal radius and ulna creating a V-shaped deformity (Madelung deformity); small/underdeveloped carpal bones; bullet-shaped phalanges; (F) i,ii,iii. Small and hypoplastic tarsal bones and calcaneovalgus deformity. |
 |
Table 3 Radiological Findings Associated with Dysostosis Multiplex in MPS Patients |
The genotypes observed in our study are described in Table 4. Compared to the total GnomAD frequency, similar frequencies of these genotypes are reported. Correlations between phospho-calcium metabolism and ApaI polymorphisms are shown in Table 5.
 |
Table 4 Genotypes and Alleles Frequencies for the Variants Studied |
 |
Table 5 Correlations Between Phospho-Calcium Metabolism and ApaI Polymorphisms in Patients with MPS Type II at Last Evaluation |
In patients with MPS type II, a lower level of 25-Hydroxy vitamin D is observed in patients with homozygous phenotype AA, compared to those with heterozygous phenotype Aa (p = 0.04). An MPS type II patient with genotype AA has calcium at the upper limit of normal (10.24mg/dl), low phosphoremia (4.24 mg/dl) and 25-Hydroxy vitamin D deficiency (15.1ug/l). Also, in MPS type II patients with AA genotype, a deficiency level for vitamin D is observed in four of them (14–18ug/l), one showing secondary hyperparathyroidism due to vitamin D deficiency. Vitamin D deficiency is observed in four patients with the Aa genotype, three of them presenting secondary hyperparathyroidism from vitamin D deficiency. The four patients with vitamin D deficiency and secondary hyperparathyroidism have the heterozygous Bb (Bsm1) genotype. In patients with MPS type I, no significant changes were observed between the various ApaI genotypes and changes in phospho-calcium homeostasis. There is also a higher level of osteocalcin in MPS type II patients with the Aa genotype than in those with AA, indicating a lower level of bone synthesis in the latter.
All patients with MPS type II and the BB phenotype have a vitamin D deficiency. There is a statistically significant difference between the 25OH vitamin D level and the BB and Bb polymorphisms for Bsm1 in patients with MPS type 2, patients with BB having a lower level. All patients with the BB genotype are deficient in vitamin D 25OH (<20). The four patients with MPS type II with secondary hyperparathyroidism have a heterozygous Bb genotype. It is also observed that the PTH level is lower in MPS type II patients with the BB phenotype than in those with the bb phenotype (see Table 6).
 |
Table 6 Correlations Between Phospho-Calcium Metabolism and BsmI Polymorphisms in Patients with MPS Type II at Last Evaluation |
No statistically significant changes were observed between FokI polymorphisms and phospho-calcium homeostasis in patients with MPS types II (see Table 7).
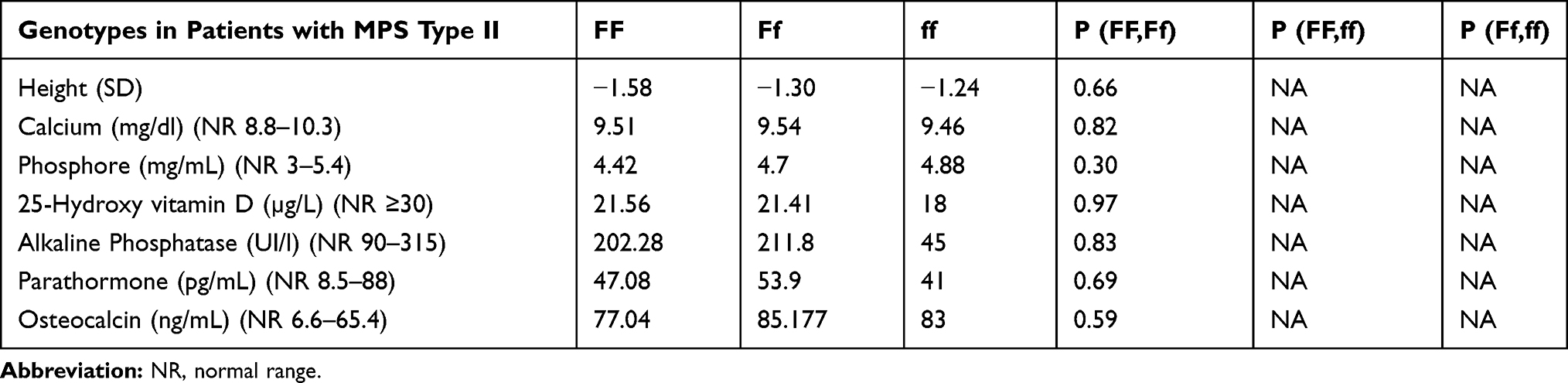 |
Table 7 Correlations Between Phospho-Calcium Metabolism and FokI Polymorphisms in Patients with MPS Type II at Last Evaluation |
Except for an association of the TT genotype with a lower level of serum phosphorus in MPS type II, no significant changes of TaqI polymorphisms with phosphocalcic homeostasis in MPS types II are observed (refer to Table 8).
 |
Table 8 Correlations Between Phospho-Calcium Metabolism and TaqI Polymorphisms in Patients with MPS Type II at Last Evaluation |
Discussion
In this study, characteristics of growth and phospho-calcium metabolism in patients with MPS have been described, as well as associations of this metabolism with genetic polymorphisms of the vitamin D receptor gene. In both MPS type I and II, a deficit of 25-hydroxy vitamin D was observed. The analysed polymorphisms were not associated with modified calcium-phosphor levels, but some differences regarding the level of 25-Hydroxy vitamin D were noted. Thus, in the case of AA polymorphism, all patients have a 25-Hydroxy vitamin D deficiency, one of them presenting secondary hyperparathyroidism. Also, three patients with the Aa have a 25-Hydroxy vitamin D deficiency and secondary hyperparathyroidism from this deficiency. All patients with secondary paratiroidism presented Bb phenotype.
The frequency of these VDR polymorphisms among patients with MPS is quite similar to that of the general population, as observed in the GnomAD database. Until now, there have been no data on these VDR polymorphisms in patients with MPS.
It is known that pathogenic variants of the VDR gene lead to vitamin D-dependent rickets type 2A. There are more subtle variations of the VDR sequence, as these polymorphisms, which are more common in the population, and while their effect is often not well known, they can influence many biological processes. These include decreased bone mineral density and osteoporosis, tall stature, hyperparathyroidism, osteoarthritis, autoimmune diseases (psoriasis, Graves’ disease, type 1 diabetes, multiple sclerosis), or infectious diseases such as tuberculosis.
Polymorphisms of the VDR gene have been studied in different diseases, but the most important associations were found in osteoporosis studies. Also, amongst all the genes studied in osteoporosis, the VDR gene polymorphism was the most relevant. ApaI polymorphism was associated with protection against osteoporosis;23,25 however, there are other studies that find some detrimental effects or no effect at all on osteoporosis.26–28 BsmI polymorphism was associated with an increased risk of osteoporosis,23 but other authors have not found any association.28,29
FokI and TaqI were significantly associated with the development of osteoporosis.23,25,28 Fok1 is located in exon 2 of the gene and leads to the formation of a new start codon, resulting in a shorter VDR product that interacts more efficiently with the transcription factor TFIIB.30 BsmI and ApaI are located in intron 8 of the VDR gene and influence mRNA stability by altering a splice site or altering intronic regulatory elements.30 TaqI polymorphism is located in exon 8 and represents a synonymous substitution, not influencing the VDR protein but being involved in the regulation of mRNA stability.30 Other authors have observed that, if VDR polymorphisms are not clearly associated with bone health, a haplotype of these polymorphisms, such as Bat or BAt, could be more relevant in association with bone mineral density.31,32
The variability of skeletal involvement in MPS patients could also be influenced by epistatic gene interactions or gene-environment interactions. Other factors which exacerbate bone mineralisation abnormalities in MPS could be secondary to immobilisation, poor nutritional status, vitamin D deficiencies, and little sun exposure.33
Among the findings arising from this study is the lack of data regarding associated factors that could exacerbate skeletal disease in MPS patients. VDR polymorphisms have not been evaluated until now in MPS patients. Phosphocalcic metabolism in MPS patients has not often been evaluated in the literature,33 so data regarding these factors is needed. Vitamin D deficiency was also observed by Zuber et al in MPS patients, and our study reinforces the idea that these patients need vitamin D supplementation to protect a particular bone metabolism.31,33,34
The limitations of this study are the low number of MPS patients. However, the low prevalence of this disorder in the general population makes it very difficult to recruit a higher number of patients: these patients represent all the patients treated with ERT in our country. Another limitation is the retrospective nature of the study, which has resulted in an absence of data for some patients in relation to skeletal metabolism, such as bone mineral density.
Conclusions
- Vitamin D deficiency is observed in MPS type I and type II patients and in some patients associated with secondary hyperparathyroidism, indicating supplementation with vitamin D in order to protect the bone metabolism.
- There are no obvious correlations between VDR polymorphism and bone metabolism in patients with MPS types I and II.
Details of Ethics Approval
The research study was approved by the Ethics Committee of Clinical Emergency Hospital for Children, Cluj-Napoca, Romania.
Patient Consent Statement
The written, informed consent was obtained from participants or their guardians prior to the genetic analysis. All genetic procedures were done in accordance with the ethical standards on human experimentation, of the hospital committee, and with the Helsinki Declaration of 1975, as revised in 2000. Approval from the Institutional Committee for Care and Use of Laboratory Animals. This article does not contain any studies with animal subjects performed by any of the authors.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
Dr. Camelia Alkhzouz, Dr. Carmen Asavoaie and Dr. Diana Miclea report personal fees from Speaker in conference organized by Takeda Pharmaceuticals Company, Romania, “News on integrated care and management of patients with Hunter’s syndrome”, Cluj Napoca, 08 November 2019. The authors report no other conflicts of interest in this work.
References
1. Hendriksz CJ, Berger KI, Lampe C, et al. Health-related quality of life in mucopolysaccharidosis: looking beyond biomedical issues. Orphanet J Rare Dis. 2016;11(1):119. doi:10.1186/s13023-016-0503-2
2. Guarany NR, Vanz AP, Bressan Wilke MVM, et al. Mucopolysaccharidosis: caregiver quality of life. JIEMS. 2015;3:1–7.
3. Oussoren E, Brands MMMG, Ruijter GJG, van der Ploeg AT, Reuser AJJ. Bone, joint and tooth development in mucopolysaccharidoses: relevance to therapeutic options. Biochim Biophys Acta. 2011;1812(11):1542–1556. doi:10.1016/j.bbadis.2011.07.013
4. Parker H, Bigger BW. The role of innate immunity in mucopolysaccharide diseases. J Neurochem. 2019;148(5):639–651. doi:10.1111/jnc.14632
5. Opoka-Winiarska V, Jurecka A, Emeryk A, Tylki-Szymańska A. Osteoimmunology in mucopolysaccharidoses type I, II, VI and VII. Immunological regulation of the osteoarticular system in the course of metabolic inflammation. Osteoarthr Cartil. 2013;21(12):1813–1823. doi:10.1016/j.joca.2013.08.001
6. Lee S, Takeda Y, Kawano H, et al. Expression and regulation of a gene encoding neural recognition molecule NB-3 of the contactin/F3 subgroup in mouse brain. Gene. 2000;245(2):253–266. doi:10.1016/S0378-1119(00)00031-7
7. Fujitsuka H, Sawamoto K, Peracha H, et al. Biomarkers in patients with mucopolysaccharidosis type II and IV. Mol Genet Metab Rep. 2019;19:100455. doi:10.1016/j.ymgmr.2019.100455
8. Li Z, Yasuda Y, Li W, et al. Regulation of collagenase activities of human cathepsins by glycosaminoglycans. J Biol Chem. 2004;279(7):5470–5479. doi:10.1074/jbc.M310349200
9. Wilson S, Brömme D. Potential role of cathepsin K in the pathophysiology of mucopolysaccharidoses. J Pediatr Rehabil Med. 2010;3(2):139–146. doi:10.3233/PRM-2010-0116
10. Yasuda Y, Kaleta J, Brömme D. The role of cathepsins in osteoporosis and arthritis: rationale for the design of new therapeutics. Adv Drug Deliv Rev. 2005;57(7):973–993. doi:10.1016/j.addr.2004.12.013
11. Wilson S, Hashamiyan S, Clarke L, et al. Glycosaminoglycan-mediated loss of cathepsin K collagenolytic activity in MPS I contributes to osteoclast and growth plate abnormalities. Am J Pathol. 2009;175(5):2053–2062. doi:10.2353/ajpath.2009.090211
12. Fung EB, Johnson JA, Madden J, Kim T, Harmatz P. Bone density assessment in patients with mucopolysaccharidosis: a preliminary report from patients with MPS II and VI. J Pediatr Rehabil Med. 2010;3(1):13–23.
13. Nur BG, Nur H, Mihci E. Bone mineral density in patients with mucopolysaccharidosis type III. J Bone Miner Metab. 2017;35(3):338–343. doi:10.1007/s00774-016-0762-y
14. Monteiro VCL, de Oliveira Silva JA, Oliveira RB, et al. Evaluation of food intake in patients with mucopolysaccharidosis. Nutrire. 2018;43:9. doi:10.1186/s41110-018-0066-1
15. Chakraborty PP, Biswas SN, Ray S, Dey SK. Mucopolysaccharidosis type I disguised as rickets. BMJ Case Rep. 2016;2016:bcr2016215416. doi:10.1136/bcr-2016-215416
16. Chen HH, Sawamoto K, Mason RW, et al. Enzyme replacement therapy for mucopolysaccharidoses; past, present, and future. J Hum Genet. 2019;64(11):1153–1171. doi:10.1038/s10038-019-0662-9
17. Tomatsu S, Alméciga-Díaz CJ, Montaño AM, et al. Therapies for the bone in mucopolysaccharidoses. Mol Genet Metab. 2015;114(2):94–109. doi:10.1016/j.ymgme.2014.12.001
18. Baker AR, Mcdonnellt DP, Hughest M, et al. Cloning and expression of full-length cDNA encoding human vitamin D receptor. Proc Natl Acad Sci U S A. 1988;85(10):3294–3298. doi:10.1073/pnas.85.10.3294
19. Mohri T, Nakajima M, Takagi S, Komagata S, Yokoi T. MicroRNA regulates human vitamin D receptor. Int J Cancer. 2009;125(6):1328–1333. doi:10.1002/ijc.24459
20. Lee SM, Meyer MB, Benkusky NA, O’Brien CA, Pike JW. The impact of VDR expression and regulation in vivo. J Steroid Biochem Mol Biol. 2018;177:36–45. doi:10.1016/j.jsbmb.2017.06.002
21. Zheng Y, Trivedi T, Lin RC, et al. Loss of the vitamin D receptor in human breast and prostate cancers strongly induces cell apoptosis through downregulation of Wnt/β-catenin signaling. Bone Res. 2017;5:17023. doi:10.1038/boneres.2017.23
22. Lin R. Crosstalk between vitamin D metabolism, VDR signalling, and innate immunity. Biomed Res Int. 2016;2016:1375858. doi:10.1155/2016/1375858
23. Zhang L, Yin X, Wang J, et al. Associations between VDR gene polymorphisms and osteoporosis risk and bone mineral density in postmenopausal women: a systematic review and meta-analysis. Sci Rep. 2018;8(1):981. doi:10.1038/s41598-017-18670-7
24. Kaya T, Erdal EM, Tursen U, et al. Association between vitamin D receptor gene polymorphism and psoriasis among the Turkish population. Arch Dermatol Res. 2002;294(6):286–289. doi:10.1007/s00403-002-0326-y
25. Ahmad I, Jafar T, Mahdi F, et al. Association of vitamin D receptor gene polymorphism (TaqI and Apa1) with bone mineral density in North Indian postmenopausal women. Gene. 2018;659:123–127. doi:10.1016/j.gene.2018.03.052
26. Meng D, Ding X, Lan J, et al. Association of vitamin D receptor apai gene polymorphism with osteoporosis susceptibility in postmenopausal Han Chinese women in Xinjiang. Biomed Rep. 2018;9(6):483–490. doi:10.3892/br.2018.1155
27. Dundar U, Solak M, Kavuncu V, et al. Evidence of association of vitamin D receptor Apa I gene polymorphism with bone mineral density in postmenopausal women with osteoporosis. Clin Rheumatol. 2009;28(10):1187–1191. doi:10.1007/s10067-009-1220-1
28. Yadav U, Kumar P, Rai V. Vitamin D receptor (VDR) gene FokI, BsmI, ApaI, and TaqI polymorphisms and osteoporosis risk: a meta-analysis. Egypt J Med Hum Genet. 2020;21:15. doi:10.1186/s43042-020-00057-5
29. Yu M, Chen GQ, Yu F. Lack of association between vitamin D receptor polymorphisms ApaI (rs7975232) and BsmI (rs1544410) and osteoporosis among the Han Chinese population: a meta-analysis. Kaohsiung J Med Sci. 2016;32(12):599–606.
30. Vasilovici AF, Grigore LE, Ungureanu L, et al. Vitamin D receptor polymorphisms and melanoma (Review). Oncol Lett. 2019;17(5):4162–4169. doi:10.3892/ol.2018.9733
31. Fung E, Kim T, Johnson JA, Madden J, Stege E, Harmatz P. Bone health and vitamin D deficiency in MPS II and VI. Mol Genet Metab. 2009;96(2):S24. doi:10.1016/j.ymgme.2008.11.057
32. Thakkinstian A, D’Este C, Attia J. Haplotype analysis of VDR gene polymorphisms: a meta-analysis. Osteoporos Int. 2004;15(9):729–734. doi:10.1007/s00198-004-1601-x
33. Zuber Z, Jurecka A, Król-Zdechlikiewicz A, Rózdzyńska-͆wiatkowska A, Tylki-Szymańska A. Bone metabolism in patients with mucopolysaccharidosis type II. Reumatologia. 2014;52(6):354–361. doi:10.5114/reum.2014.47228
34. Gudin MA, Campistol J, Chavez B, Hernàndez S, Vilaseca MA. Hurler's syndrome, west’s syndrome, and vitamin D-dependent rickets. J Child Neurol. 2002;17(2):149–151. doi:10.1177/088307380201700214
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.