")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 15
Sjögren’s Syndrome with Lichen Sclerosus: A Case Report
Authors Zhang J, Qi F , Zhang X, Dong J , Tong C, Zhang X, Liu F
Received 20 September 2022
Accepted for publication 17 November 2022
Published 23 November 2022 Volume 2022:15 Pages 2535—2539
DOI https://doi.org/10.2147/CCID.S389809
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Jingya Zhang, Fei Qi, Xuan Zhang, Jie Dong, Chunguang Tong, Xiuying Zhang, Fang Liu
Department of Dermatology, Beijing Chaoyang Hospital, Capital Medical University, Beijing, People’s Republic of China
Correspondence: Fang Liu, Department of Dermatology, Beijing Chaoyang Hospital, Capital Medical University, 8 Gongti South Road, Chaoyang District, Beijing, 100020, People’s Republic of China, Tel +86-10-85231688, Fax +86-10-85231217, Email [email protected]
Abstract: Sjögren’s syndrome (SJS) is a systemic disease in which the accumulated skin can include xeroderma, eyelid dermatitis, and annular erythema (AE). To the best of our knowledge, there are few reports on cases of SJS with concomitant lichen sclerosus (LS). Herein, we report the case of a 69-year-old woman with these two diseases. The patient’s skin showed atrophic leukoplakia and AE. Based on the comprehensive evaluation of general condition, the patient was diagnosed and actively received treatment. After systematic treatment, her symptoms were controlled. However, the patient’s condition requires long-term management.
Keywords: annular erythema, skin atrophy, depigmentation, skin lesions
Introduction
Sjögren’s syndrome (SJS) is a chronic autoimmune disorder of the exocrine glands associated with lymphocytic infiltration. Dry mouth and eyes result from the involvement of the salivary and lacrimal glands.1 SJS can also affect the joints, lungs, liver, nervous system, and blood system.2 In addition, skin involvement is common in SJS, and various manifestations may occur such as xeroderma, eyelid dermatitis, annular erythema (AE), and cutaneous vasculitis. Xeroderma, or dry skin, is the most common skin symptom of SJS and occurs in 67% of patients. AEs are rare cutaneous manifestations of SJS and occur in 9% of patients. The clinical presentation of AEs is an erythematous, photosensitized rash characterized by wide margins and central pallor (annular multicycle lesions), such as subacute cutaneous lupus erythematosus.3 Skin manifestations may appear earlier than the appearance of dry mouth and dry eyes, and their occurrence is related to positive Sjogren’s syndrome-related antigen A (SSA) and Sjogren’s syndrome-related antigen B (SSB) antibodies.4
Lichen sclerosus (LS) is a rare autoimmune disease that is characterized by skin atrophy and hypopigmentation. Women are more affected than men. More than 80% of the patients present with atrophic leukoplakia in the anogenital region, and those appearing in the extragenital region are rare. The typical extragenital sites include the upper torso, armpits, buttocks, and outer thighs, which are most common in adult women. The less common sites include the mouth, face, scalp, hands, feet, and nails.5 If not treated, LS can lead to scarring and an increased risk of squamous cell carcinoma.6 Herein, we report the first case of a patient with SJS and LS.
Case Report
A 69-year-old woman was simultaneously presented with two completely different types of rashes. The patient reported dry mouth for six years and dry eyes for more than three years. More than 20 months ago, the patient developed erythema of approximately 1 cm in size with a small scale on the inner surface of the right thigh without obvious incentives. The patient did not experience any itching. Subsequently, the rash developed darkened annular enlargement that subsided spontaneously within a week. However, similar skin lesions reappeared rapidly around the original rash. More than four months ago, the patient experienced hardening of the back skin that shrunk to touch, and the scope of the posterior rash moved upward, involving the back neck. More than three months ago, the patient found scattered dot-like white rashes on both upper arms, and the area gradually increased and blended into flakes. She received daily topical sodium hyaluronate eye drops.
On physical examination, the patient had depigmentation spots with mild skin atrophy around the hairline, both forearms, and on the face. Parchment-like patches, mild redness, atrophy, and some desquamations were observed on the back of the occiput and upper back. On the front neck, chest, and back, there were multiple white plaques of different sizes, with an atrophied surface, and a slight feeling of infiltration. On the right posterior scapula, inner right thigh, and right buttock, there were several circular red annular patches with diameters ranging from 2–10 cm, with a few white scales on the surface, and a depressed center. Distal separation of the two finger decks was noted. Pale red plaques were visible around the labia majora and clitoris, the surface was keratinized and desquamated, and local erosion and flaky depigmented plaques with atrophy were visible (Figure 1).
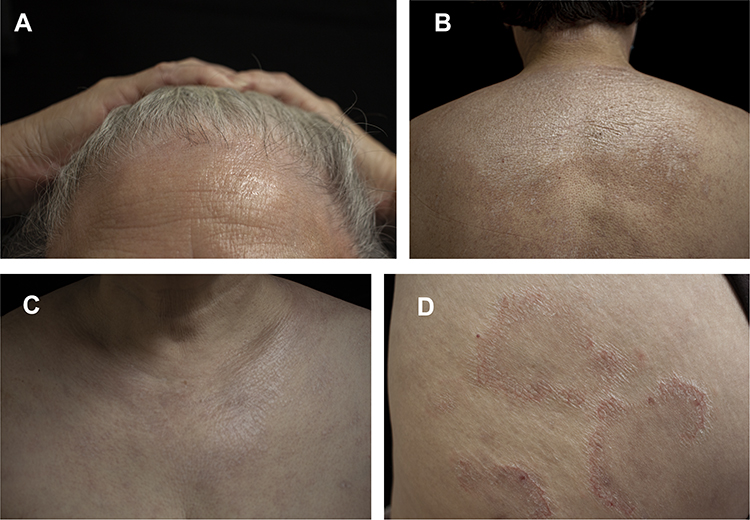 |
Figure 1 (A–C) Depigmented spots on the hairline, posterior occipital, upper back, and anterior neck, with superficial atrophy. (D) Right scapular circular red annular patch with a few white scales on the surface and central regression. |
Histopathological examination of the patient’s back revealed hyperkeratosis of the epidermis, presence of epidermal processes, basal edema, lymphocyte-predominant infiltration in the superficial dermis, and perivascular infiltration and thickening of the collagen fibers. Leg tissue biopsy revealed epidermal hyperkeratosis, focal parakeratosis, and lymphocyte-predominant infiltration around the superficial and middle dermis (Figure 2).
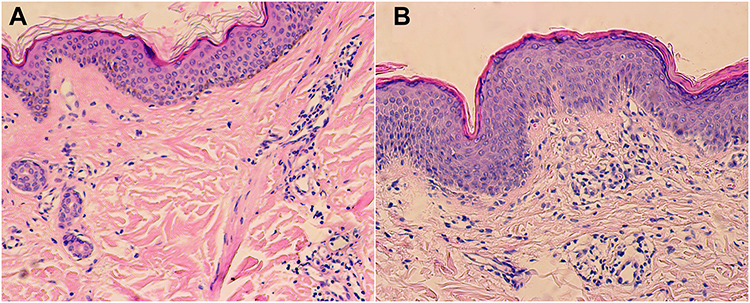 |
Figure 2 (A) Back. (B) Legs. |
Fungal microscopic examination of the patient’s skin lesions was negative. The anti-SSA antibody was positive, and the anti-nuclear antibody titer was 1:100. The Schirmer I test was positive and the salivary flow rate was positive. The white blood cell and platelet counts dropped to 2.87*10^9/L and 101*10^9/L, respectively. Albumin, aspartate aminotransferase, alanine aminotransferase, bilirubin, and coagulation levels were mildly abnormal. High-resolution computed tomography showed mild interstitial lung disease and cirrhosis.
According to the American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) recommendation (2016), the patient was diagnosed with LS combined with Sjogren’s syndrome.
She was treated with compound glycyrrhizin 40 mL/day intravenously, Compound Danshen Tablet (three tablets thrice daily) and glucosidorum tripterygll totorum (two tablets daily) orally. As the leukocyte count decreased, we stopped glucosidorum tripterygll totorum one day later. Tacrolimus ointment and mometasone furoate cream were alternately used on the face and vulva each day. Halometasone cream was used on the trunk and limbs; mucopolysaccharide polysulfate cream was used on the rashes of the whole body every day. One month after the patient was discharged from the hospital, AE of the whole body had completely subsided, and the area of atrophic leukoplakia was smaller than before. However, new skin lesions appeared. Three months later, hydroxychloroquine (100 mg BID) was administered to the patient. One month later, the skin lesions decreased in size (please see Figure 3: Timeline).
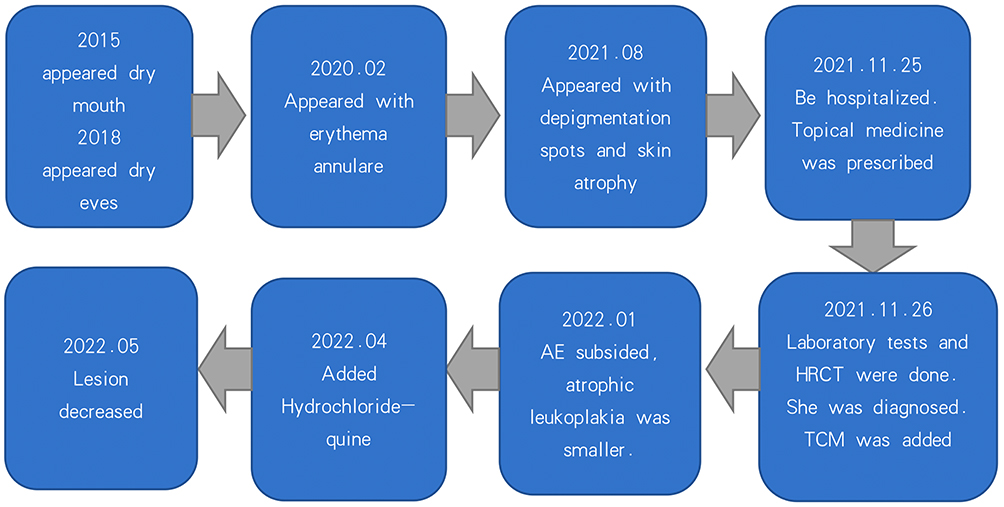 |
Figure 3 Timeline. |
Discussion
Here, we report the case of a patient with SJS and LS. Patients with SJS exhibit obvious systemic symptoms. Our patient had liver and blood abnormalities such as liver cirrhosis, leukopenia, and thrombocytopenia. SJS lesions can also occur on the skin. In this patient, AE on the right scapula, hip, and thigh was considered the skin manifestation of Sjogren’s syndrome. In addition, the patient also had LS, which has not been previously reported. It has been reported that a variety of immune system diseases often coexist, which may be related to the patient’s systemic immune disorder.7,8
The patient’s EULAR Sjogren’s syndrome disease activity index (ESSDAI) score was 10, indicating moderate systemic active disease. The recommended dose of methylprednisolone is < 0.5 mg/kg/d. When the patient is in the inactive period and the ESSDAI score is less than 1, glucocorticoids should be stopped. With the help of immunosuppressants, methylprednisolone should be reduced to ≤ 5 mg/d. There is little data to support the selection of immunosuppressants, whose main purpose is to reduce the side effects of hormones. As the patient refused to use glucocorticoids, immunosuppressive agents, and biologics, traditional Chinese medicine was prescribed. Volume replacement and lubrication using artificial tears and ocular gels are the first choice for the treatment of ocular dryness. Immunosuppressive-containing drops, serum eye drops, topical ocular non-steroidal anti-inflammatory drugs, or corticosteroids can be used under the evaluation of an experienced ophthalmologist. When the patient has severe glandular dysfunction, saliva substitution is recommended.9 Topical corticosteroids (TCS) can be used in the treatment of AE. Tumor necrosis factor, interleukin-1, and B-cell inhibitors can be used in some patients whose traditional treatment is ineffective.10 SJS is a chronic disease requiring long-term management. Doctors need to assess the patients’ ESSDAI scores to adjust the treatment plan.
All national guidelines believe that potent TCS is the first choice for the treatment of vulvar LS; once daily for 12 weeks is recommended. Topical tacrolimus is considered a second-line treatment.11–13 For our patients, we alternately used TCS and tacrolimus to reduce adverse reactions. For skin lesions other than those in the vulva, UVA1 phototherapy is recommended for 10 weeks (evidence level 1 + / recommendation level B). Potent to ultrapotent topical steroids (evidence level 3 / recommendation level D), tacrolimus ointment 0.1% (evidence level 3 / recommendation level D), and systemic steroids / methotrexate (evidence level 3 / recommendation level D) can also be used. Due to the inconvenience of phototherapy, we chose TCS and tacrolimus for treatment.14
Systemic diseases often begin with the development of skin lesions. Dermatologists should carefully ask about their medical history to avoid misdiagnosis.
Conclusion
The cases of SJS with LS are rarely reported. Potent TCS is the first choice for the treatment of vulvar LS. SJS is a systemic disease that may present as accumulated skin. Once suspected, a detailed patient medical history, systematic examinations are necessary. ESSDAI scores need to be evaluated to formulate specific treatment plans. As Sjogren’s syndrome is a chronic disease, patients need long-term follow-up to control the disease. Given the rarity of such cases, we report this case to provide a reference for dermatologists.
Ethics Approval and Informed Consent
Written informed consent was provided by the patient to have the case details and any accompanying images published. Publication of the case details was approved by Beijing Chaoyang Hospital, Capital Medical University.
Acknowledgments
We thank the patient and physicians for participating in our study. We would like to thank Editage for English language editing.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fox RI. Sjögren’s syndrome. Lancet. 2005;366(9482):321–331. doi:10.1016/s0140-6736(05)66990-5
2. Brito-Zeron P, Baldini C, Bootsma H, et al. Sjogren syndrome. Nat Rev Dis Primers. 2016;2:16047. doi:10.1038/nrdp.2016.47
3. Generali E, Costanzo A, Mainetti C, Selmi C . Cutaneous and mucosal manifestations of Sjogren’s syndrome. Clin Rev Allergy Immunol. 2017;53(3):357–370. doi:10.1007/s12016-017-8639-y
4. Brito-Zeron P, Theander E, Baldini C, et al. Early diagnosis of primary Sjogren’s syndrome: EULAR-SS task force clinical recommendations. Expert Rev Clin Immunol. 2016;12(2):137–156. doi:10.1586/1744666X.2016.1109449
5. Lewis FM, Tatnall FM, Velangi SS, et al. British association of dermatologists guidelines for the management of lichen sclerosus, 2018. Br J Dermatol. 2018;178(4):839–853. doi:10.1111/bjd.16241
6. Chamli A, Souissi A. Lichen Sclerosus. Treasure Island (FL): StatPearls; 2022.
7. Tran DA, Tan X, Macri CJ, Goldstein AT, and Fu SW . Lichen Sclerosus: an autoimmunopathogenic and genomic enigma with emerging genetic and immune targets. Int J Biol Sci. 2019;15(7):1429–1439. doi:10.7150/ijbs.34613
8. Bieber AK, Steuer AB, Melnick LE, Wong PW, and Pomeranz MK . Autoimmune and dermatologic conditions associated with lichen sclerosus. J Am Acad Dermatol. 2021;85(1):228–229. doi:10.1016/j.jaad.2020.08.011
9. Ramos-Casals M, Brito-Zeron P, Bombardieri S, et al. EULAR recommendations for the management of Sjogren’s syndrome with topical and systemic therapies. Ann Rheum Dis. 2020;79(1):3–18. doi:10.1136/annrheumdis-2019-216114
10. Seror R, Nocturne G, Mariette X. Current and future therapies for primary Sjogren syndrome. Nat Rev Rheumatol. 2021;17(8):475–486. doi:10.1038/s41584-021-00634-x
11. The American College of Obstetricians and Gynecologists. Diagnosis and management of vulvar skin disorders: ACOG practice bulletin summary, number 224. Obstet Gynecol. 2020;136(1):222–225. doi:10.1097/AOG.0000000000003945
12. van der Meijden WI, Boffa MJ, Ter Harmsel WA, et al. 2016 European guideline for the management of vulval conditions. J Eur Acad Dermatol Venereol. 2017;31(6):925–941. doi:10.1111/jdv.14096
13. Edwards SK, Bates CM, Lewis F, Sethi G, and Grover D . 2014 UK national guideline on the management of vulval conditions. Int J STD AIDS. 2015;26(9):611–624. doi:10.1177/0956462414554271
14. Kirtschig G, Becker K, Gunthert A, et al. Evidence-based (S3) Guideline on (anogenital) Lichen sclerosus. J Eur Acad Dermatol Venereol. 2015;29(10):e1–e43. doi:10.1111/jdv.13136
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.