Back to Journals » OncoTargets and Therapy » Volume 11
Siva-1 emerges as a tissue-specific oncogene beyond its classic role of a proapoptotic gene
Authors Vachtenheim J Jr ![]() , Lischke R, Vachtenheim J
, Lischke R, Vachtenheim J ![]()
Received 4 May 2018
Accepted for publication 25 July 2018
Published 1 October 2018 Volume 2018:11 Pages 6361—6367
DOI https://doi.org/10.2147/OTT.S173001
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Takuya Aoki
Jiri Vachtenheim, Jr,1 Robert Lischke,1 Jiri Vachtenheim2
1Third Department of Surgery, First Faculty of Medicine, Charles University Prague and University Hospital Motol, Prague, Czech Republic; 2Department of Transcription and Cell Signaling, Institute of Medical Biochemistry and Laboratory Diagnostics, Charles University Prague, Czech Republic
Abstract: Siva-1 is a typical apoptotic protein commonly activated by the p53 tumor suppressor protein and should therefore participate in a barrier against the development of cancer. It has proapoptotic activities in various cell systems. Recent findings suggest that Siva-1 possesses several other apoptosis-independent functions and interacts with many other proteins not directly involved in apoptosis. It harbors the ARF E3 ubiquitin protein ligase activity, a property that is clearly prooncogenic and leads to p53 degradation through the upregulation of the Hdm2 protein level. Surprisingly, recent evidence shows that Siva-1 absence prevents the development of non-small cell lung carcinomas in a mouse model and reveals the oncogenic roles in the same types of human cells, indicating its unique function as an oncogene in the cell context-dependent manner. Herein, we review reported activities of Siva-1 in various experimental settings and comment on its ambiguous function in tumor biology.
Keywords: Siva-1, apoptosis, CD27, p53, NSCLC
Introduction
Siva-1 was originally identified as a CD27-interacting protein.1 Siva-1 was shown to associate with the cytoplasmic tail of CD27 and was cloned by the use of the yeast two-hybrid system through interaction with the CD27 cytoplasmic tail. This region is highly conserved between human and mouse2 and was used as a bait to screen the HeLa cell cDNA library. On further screening of HeLa cell and human thymocyte cDNA libraries, full-length Siva-1 cDNA containing an open reading frame with a predicted translation product comprising 189 amino acids, was identified. It has an amphipathic helical region (SAH) at the N-terminus, a death domain homology region (DDHR) in the central part, and box-B-like ring finger and two zinc finger-like cysteine-rich domains at the C-terminus.1,3 Siva-2 is an alternative splicing form, lacking exon 2 (out of total four exons) that contains the DDHR. Siva-2 also binds to the CD27 receptor protein but its apoptotic activity is greatly diminished,4 as observed in mouse cells. Intriguingly, it is able to induce apoptosis similarly as Siva-1 in T-lymphocytes.5 Siva-1 is also capable of entering the nucleus, for which the first 33 N-terminal amino acids are required (probably amino acids 1–18 are the most important).6 Siva-1 is typically transcribed through the p53 tumor suppressor, similarly as other apoptotic genes. This occurs typically during the neuronal cell death in neurodegeneration or injury.7 Siva-1 gene contains two E2F1 elements in the proximal promoter, which participate in inducing Siva-1 expression,8,9 whereas two p53 consensus sites are located in the first intron and their deletion abrogates the activity of the promoter. They bind strongly to the p53 consensus motif in gel-shift assays.8 In p53-lacking Caco2 cells, Nutlin-3-mediated degradation of Mdm2 caused the accumulation of p73, which contributed to the transcription of Siva-1, a process accompanied with increased expression of the p53-upregulated modulator of apoptosis.9 Of note, in a specific cell context (ventricular cardiac fibroblasts), Siva-1 was also transcriptionally activated by the transcription factor Egr-1, thus participating in apoptosis after Egr-1 activation in these cells.10 In quite different cells, the variant type of small cell lung cancer (SCLC), the TIP protein TP30 (also called CC3) acts as tumor suppressor and is capable of binding the HIV Tat protein. It is a serine/threonine kinase that phosphorylates, besides other substrates, the heptapeptide repeats of the C-terminal domain (CTD) of the largest RNA II polymerase subunit in cells, thus activating transcription, in a Tat-dependent manner. This activation resulted in an increased expression of proapoptotic genes Siva-1 and Bad and caused apoptosis in SCLC cells.11
Siva-1 is a proapoptotic protein with anticancer activity
Under normal physiological conditions, Siva-1 functions as a typical proapoptotic gene regulated by the p53 protein. Many cellular situations have been shown to lead to Siva-1-dependent apoptosis. Siva-1 interacts with several proteins that are involved in apoptosis (see above and Figure 1). These include some members of the TNFR (tumor necrosis factor receptor) super family of receptors and small proteins involved in apoptosis in mitochondria. It can therefore serve in both extrinsic and intrinsic apoptosis signaling pathways. Siva-1 interacts with the TNFR superfamily of receptors, including CD27,1 GITR (glucocorticoid-induced), OX40 (CD134), 4-1BB (CD137) and CD40.12 A common motif in the cytoplasmic domain of these receptors required for Siva-1 binding shares the sequence P(I/E)(Q/E)E (PEEE sequence in GITR and PIQE sequence in CD27 and OX40).12 Further, cotransfection of both tagged GITR and Siva-1 increased apoptosis in Cos7 cells, supporting the ability of Siva-1 to initiate apoptosis through the extrinsic, receptor pathway.12 Siva-1 has been shown to induce apoptosis in a number of different experimental settings. Apoptotic signaling can be initiated either through extrinsic (via death receptors, see above) or intrinsic pathways,13 accompanied with Bax and caspase three upregulation and BCL2 decrease. Importantly, Siva-1 synergizes with cisplatin in inducing apoptosis in breast cancer cells and the SAH in Siva-1 is necessary and sufficient for the augmentation of apoptosis.14 Cisplatin also induced Siva-1 expression in HCT116 (p53+/+) colorectal carcinoma cells. Siva-1 knockdown conferred significant resistance to cisplatin-induced apoptosis. Siva-1 also augments the cisplatin-mediated apoptosis via the thromboxane A2 receptor TP, which is accompanied with upregulation of Siva-1 protein levels, as well as levels of the antiapoptotic protein XIAP and translocation of Siva-1 from the nucleus to the cytoplasm.15 In the model whereby normal HCT 116(p53+/+) colorectal carcinoma cells and p53 genetically-null HCT 116(p53−/−) cells were treated with topotecan, a known topoisomerase I inhibitor, the profile of genes induced by topotecan mediated DNA damage was assessed by microarray was found induced (together with a set of TGFβ-related genes), thus showing the p53 activation dependency.16 This further indicates that Siva-1 can be a typical p53-dependent apoptotic protein. Intriguingly, although Siva-1 levels were found positively regulated by p53, Siva-1-induced apoptosis did not require p53. In this system, Siva-1 oligomerized with Bax followed by cytochrome c release from mitochondria.13 In retinal pigment cells, where high expression levels of antiapoptotic proteins BCL2 and Bcl-xL were observed, chemical treatment (with camptothecin or TNF-α/cycloheximide, CHX) failed to induce apoptosis. However, Nutlin-3, which maintains higher p53 levels by antagonizing Mdm2, sensitized cells to apoptosis by increasing the p53-dependent proteins Noxa, PUMA and Siva-1.17 Other studies have demonstrated phosphorylation of Siva-1 and its binding to protein kinase ARG, which is closely related to the c-Abl kinase. ARG phoshorylates Siva-1 on Y48, an event which may be important for apoptosis induced by oxidative stress.18 The proapoptotic effects of Siva-1 are abrogated by mutation of the Tyr48 site.
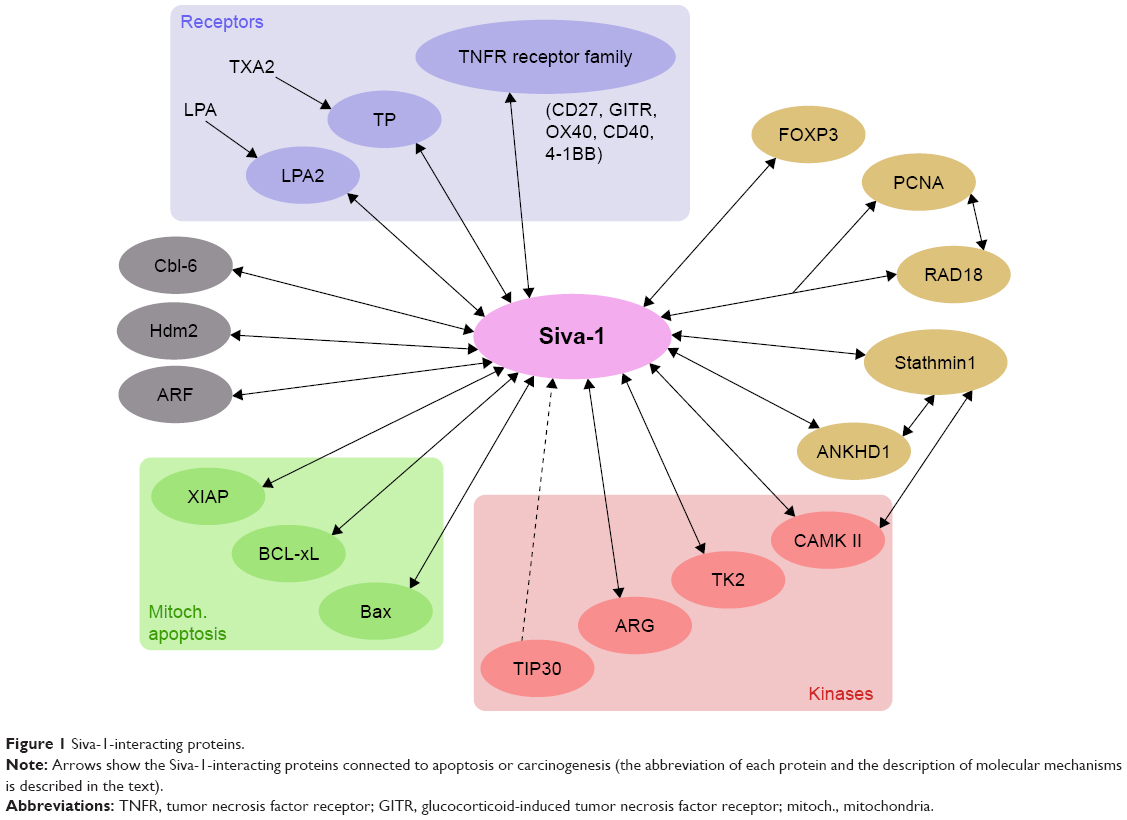 | Figure 1 Siva-1-interacting proteins. |
His-Siva-1 recombinant protein inhibited invasion and migration of HCT116 cells.19 The C-terminal domain (aa residues 84-175) expressed in bacteria as a GST-fusion protein caused apoptosis in Jurkat leukemia cells and its biophysical properties were studied.20 The authors found monodispersion of the protein in solution with a predominant unfolded and elongated shape. Recombinant Siva-1 protein was further demonstrated to induce apoptosis in cancer cells. Bacterially expressed Siva-1 displayed anticancer activities: inhibited invasion, migration and proliferation and induced apoptosis. In vivo studies on tumors xenografted into nude mice with human nasopharyngeal carcinoma cell line CNE-2 showed that recombinant Siva-1 inhibited tumor growth.21
Additionally, when Siva-1 was ectopically overexpressed in human ovarian cancer cells, it suppressed migration and invasion and induced apoptosis through facilitating both phosphorylation of stathmin and polymerization of α-tubulin.22 Siva-1 was found to interact with ANKHD1 (ankyrin repeat and KH domain containing 1 protein), which is abundantly expressed in human acute leukemia cells. Further knockdown studies implicated contrasting roles of ANKHD1 and Siva-1 in these cells. Silencing of ANKHD1 inhibited cell migration and stathmin-1 activity, whereas Siva-1 knockdown resulted in stathmin-1 activation, accompanied with the decrease in phosphorylation on S16. Thus, these findings corroborate the prooncogenic function of stathmin and situate Siva-1 as its inhibitor, while ANKHD1 is an oncogene in acute leukemias.23 In addition, in U937 cells, downregulation of Siva-1 resulted in decrease of JNK phosphorylation, Bax expression and upregulation of Bcl-xL expression, whereas the BCL2 levels remained unchanged. Taken together, the three cancer cell systems revealed a similar role of stathmin-1 and Siva-1 in promoting or restricting malignancy, respectively.
Siva-1 was shown to interact with the known antiapoptotic protein Bcl-xL.24 Both proteins directly bound in vitro (in pull-down assays) and in vivo (as demonstrated by immunoprecipitation). Through this association, Siva-1 strongly inhibits the antiapoptotic potency of Bcl-xL to protect cells (T-lymphocytes and breast cancer cells) against UV-radiation induced apoptosis, for which a unique SAH present in Siva-1 is essential for its binding to BCL-xL.24,25 Interestingly, increase of the proapoptotic factor Siva-1 and loss of its binding partner Bcl-xL have been also observed in neutrophils lacking the prolyl hydroxylase 3 (PHD3), one of the oxygen-sensitive PHD1-3 enzymes that participate with the hypoxia inducing factor in apoptosis under hypoxic conditions in these cells.26
Another important Siva-1-interacting protein is a potent apoptosis inhibitor X-chromosome-linked inhibitor of apoptosis (XIAP). The RING domain of XIAP and the N-terminal SAH and DDHR domains of Siva-1 are required for their binding.27 XIAP, Siva-1 and the protein TAK1 (TGF-β-associated kinase 1) form a ternary complex in Jurkat T leukemia cells. It was further shown that Siva-1 inhibited the XIAP-TAK1-mediated NFκB pathway activation, but favored the JNK signaling and caspase-3-dependent apoptosis.27 The negative effect of Siva-1 on the NFκB signaling pathway was described also by other authors.28–30 The mechanism of inhibition of the prooncogenic NFκB pathway by Siva-1 has been described in more detail. Siva-1 inhibited the activity of the prosurvival transcription factor NF-κB as well as AP-1. The mediator of these routes is the protein TRAF2, containing the ring finger domain which is necessary for its E3 ubiquitin ligase activity and its ability to activate NF-κB.29 Siva-1 decreased XIAP-induced NFκB signaling, whereas AP1 and JNK activations were enhanced. Siva-1 increased the caspase 3-dependent apoptosis, which was compromised by lysine 48-ubiquitination by XIAP.27 Siva-1 was also demonstrated to interact with the transcription factor FOXP3 in T-cells where it inhibited, as did the FOXP3, the NFκB pathway.30
Siva-1-interacting proteins not directly involved in apoptosis
The carboxy-terminal tail of the lysophosphatidic acid two receptor (LPA2), but not LPA1 or LPA3 receptors, specifically associates with the carboxyl cysteine-rich region of Siva-1. The LPA stimulation promotes the association of Siva-1 with the LPA2 receptor and targets both proteins for ubiquitination and degradation.31 Notably, tyrosine kinase 2 (TK2), a member of the Jak family of protein kinases, also associates with Siva-1 and promotes its apoptotic function, surprisingly independently of two TK2-mediated phosphorylations at tyrosines 53 and 162 of Siva-1 in a B-lymphocyte cell line Ba/F3. Expression of TK2 augmented Siva-1-induced apoptosis but this was sustained regardless of the phosphorylation of Siva-1, but was almost completely prevented by the abrogation of the TK2-Siva-1 association, indicating that the direct TK2-Siva-1 binding is essential for apoptosis.32
A paper published by Li et al33 uncovers a role of Siva-1 that is not directly linked to its apoptotic function. Siva-1 has been discovered (by using the yeast two-hybrid system) to interact with stathmin (see above), which is a microtubule destabilizer and depolymerizes microtubules through the sequestration of α/β-tubulin heterodimers. Epithelial malignancies progress through the epithelial-mesenchymal transition (EMT), a process enabling epithelial cells to acquire a more malignant, mesenchymal phenotype34 that confers cells higher invasivity and propensity to metastasize. Siva-1 inhibits stathmin’s activity and thus stabilizes microtubules and hinders EMT. Moreover, it is able to interact with calmodulin-dependent protein kinase II (CaMK II, which also interacts with stathmin) and thus inhibits stathmin indirectly by augmenting stathmin’s phosphorylation at S16 by CaMK II. Siva-1 C-terminal domain and the stathmin N-terminal region mediate the association between these two proteins. Not surprisingly, C-terminus of Siva-1 was necessary to inhibit the stathmin´s activity and prevent microtubule destabilization. Thus, a ternary complex is formed comprising Siva-1, stathmin, and CaMK II. As a result, Siva-1 suppresses EMT and cell migration and inhibits breast cancer metastasis.33
Siva-1 is an ARF E3 ubiquitin protein ligase
Alternative reading frame protein (ARF, p14 in human and p19 in mouse) is a known tumor suppressor, acts by binding to and inhibits the oncogenic ubiquitin ligase Hdm2 (Mdm2 in mouse) which specifically attaches ubiquitin to p53 and marks it for proteasome degradation.35 The protective effect of ARF toward p53 cell levels is frequently debilitated by ARF mutations found in tumors.36 ARF is also degraded by proteasome after ubiquitination by ubiquitin ligase ULF.32 ULF interacts with ARF both in vitro and in vivo and promotes the ubiquitination and degradation of ARF. In contrast, ULF knockdown stabilizes ARF in normal human cells, allowing ARF-dependent, p53-mediated growth arrest. Surprisingly, Siva-1, which is a direct transcriptional target of p53, has been shown to augment the Mdm2-mediated p53 degradation.37 Siva-1 was shown to be an E3 ubiquitin ligase for ARF, thus representing a negative regulatory feedback loop in the regulation of p53 protein stability.38 ARF was revealed to be a Siva-1-binding protein requiring the N-terminal 20 amino acids for the interaction with Siva-1, whereas Siva-1 needed the C-terminus for binding to ARF. The ectopic Siva-1-ARF interaction proceeds in the nucleoplasm after the relocation of ARF from the nucleolus.38 Interestingly, similar negative feedback has been observed with RAP80 (receptor-associated protein 80), a protein whose expression is induced by p53 after DNA damage.39 RAP80 is capable of interacting with both Hdm2 and p53 and enhances HDM2-dependent p53 poly-ubiquitination.32 Thus, Siva-1 is an important modulator in the intricate p53 degradation process. Additionally, a protective mechanism preserving p53 levels was demonstrated in acute myeloid leukemia: E3 ubiquitin ligase Cbl-b ubiquitinates Siva-1 leading to its degradation and stabilization of p53 levels.40,41 Further, Siva-1 has a specific p53-independent function in the monoubiquitination of PCNA (proliferating cell nuclear antigen). It binds E3 ubiquitin ligase RAD18 and promotes its E3 PCNA ligase activity.41
Siva-1 as an oncogene in NSCLC cells
The interplay of apoptotic and antiapoptotic signaling plays a crucial role during the development of tumors. Apoptosis is a type of programmed cell death characterized by annexin V membrane externalization, membrane blebbing, chromatin condensation, DNA fragmentation and caspase activation in most cases. p53 tumor suppressor activates expression of many proapoptotic genes, including eg, Bax and Siva-1, thus restraining tumorigenesis. Apart from a typical proapoptotic function of Siva-1, it has been recognized as a member of a feedback loop in the regulation of p53 protein levels.38 As it is an ARF E3 ubiquitin ligase, it also plays a prosurvival role in some cancer types. Proliferation of HCT116(p53+/+) cells after Siva-1 knockdown has been found lower than in HCT116(p53−/−) cells, suggesting that Siva-1 still enhances cell proliferation in a p53-dependent manner. Possibly through the suppression of the p53 protein level, downregulation of Siva-1 by using the shRNA inhibited tumor formation in mice xenografted with the human osteosarcoma U2-OS cells (p16-null, Rb-wt and p53-wt).37
Importantly, Siva-1 has been clearly recognized as an oncogenic factor in a mutant KRAS-dependent mouse model of non-small-cell lung cancer (NSCLC) and in human NSCLC cells in culture.42,43 Siva-1 knockout mice were generated, in which the whole gene was deleted, flanked by a LoxP and a LoxP-Puro-LoxP cassette. Homozygous Siva-1-null animals revealed embryonic lethality after expressing Cre recombinase (Ad-Cre). The mice were crossed with a known KRASG12D model strain with activated KRASG12D that drives the development of lung adenocarcinomas. In these mice, an upstream floxed transcriptional stop cassette (Lox-Stop-Lox) silences KRASG12D expression until Cre recombinase is introduced. By analyzing various progeny genotypes after intratracheal Ad-Cre application, Siva-1 loss was found to impede NSCLC development, suggesting that Siva-1 is necessary to facilitate tumorigenesis in vivo. By using the cells derived from KRASG12DTrp53+/+ mice which express Siva-1, shRNA-mediated knockdown of Siva-1 resulted in reduced proliferative capacity. Depletion of Siva-1 by infecting fl/-mouse embryonic fibroblasts with Ad-Cre or knocking down Siva-1 in pancreatic cancer cell lines did not affect proliferation. This indicates cell context specific role of Siva-1.42 Further analysis showed that Siva-1 expression was not dependent on p53 in NSCLC cells. Low number of soft agar colonies after Siva-1 knockout was another result corroborating the essential function in NSCLC development. These results were recapitulated in human A549 NSCLC cells expressing oncogenic KRASG12S, wild-type p53 and high level of Siva-1. As the NFκB signaling pathway is strongly prooncogenic and is inhibited by Siva-1 (see above), its pharmacologic inhibition did not change the Siva-1 effects, indicating that NF-κB signaling does not account for the protumorigenic effects of Siva-1.
It was further demonstrated that Siva-1 knockdown increases autophagy and decreases metabolic activity. Importantly, it has been demonstrated that it enhances the mTOR pathway activity. The phospho-S1462-TSC2 levels were diminished upon Siva-1 knockdown. Although no change in RAPTOR phosphorylation was noted in Siva-1 downregulated cells, however phosphorylated pS6K, p4EBP1 and pS6 proteins were decreased.42 Despite the fact that the changes in mTOR pathway members phosphorylation were not dramatic, this mechanism may at least in part contribute to the oncogenic function of Siva-1. No relevant clinical data that would clarify the aggressiveness, the course of the disease (relapse, death of cancer) if Siva-1 protein level is increased in NSCLC is not yet available. Together, these findings reveal a role of Siva-1 in promoting mTOR and metabolic activity which may participate in enhancing proliferation of NSCLC cells.
Conclusion
Contradictory evidence suggests pro- and antiapoptotic roles for Siva-1. Apoptotic Siva-1 signaling can be accomplished through both extrinsic (through death receptors) or intrinsic (mitochondria-dependent) pathways, resulting in the activation of caspases. This apoptotic activity in various cell systems is dependent on the classical p53 pathway. Siva-1 interacts with several proteins (Figure 1), and its various interactions have been revealed through the two-hybrid yeast system, though some of which are not directly associated with apoptosis. Siva-1 plays a role in restricting EMT by inhibiting the activity of stathmin-1 in tumor cells. Intriguingly, Siva-1 has an E3 ARF ubiquitin ligase activity, through which it activates the Hdm2 activity, thus downmodulating the p53 levels as a negative feedback loop. Interestingly, Siva-1 is also a context-dependent oncogenic protein. This activity has been observed so far in osteosarcoma U2-OS cells (dependent on p53 pathway regulation) when xenografted into nude mice and in NSCLC in vitro and in vivo (p53-independent). Especially its oncogenic function demonstrated in a mouse NSCLC KRAS model and in NSCLC human cells in culture is challenging and requires further research. Conclusively, Siva-1 serves different roles in various cells, and the mechanism of its action relies on the specific cell type.
Acknowledgment
This review was supported by the institutional funding program PROGRESQ25 from the Charles University, Prague, Czech Republic.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Prasad KV, Ao Z, Yoon Y, et al. CD27, a member of the tumor necrosis factor receptor family, induces apoptosis and binds to Siva, a proapoptotic protein. Proc Natl Acad Sci U S A. 1997;94(12):6346–6351. | ||
Gravestein LA, Blom B, Nolten LA, et al. Cloning and expression of murine CD27: comparison with 4-1BB, another lymphocyte-specific member of the nerve growth factor receptor family. Eur J Immunol. 1993;23(4):943–950. | ||
Mei Y, Wu M. Multifaceted functions of Siva-1: more than an Indian God of Destruction. Protein Cell. 2012;3(2):117–122. | ||
Yoon Y, Ao Z, Cheng Y, Schlossman SF, Prasad KV. Murine Siva-1 and Siva-2, alternate splice forms of the mouse Siva gene, both bind to CD27 but differentially transduce apoptosis. Oncogene. 1999;18(50):7174–7179. | ||
Py B, Slomianny C, Auberger P, Petit PX, Benichou S. Siva-1 and an alternative splice form lacking the death domain, Siva-2, similarly induce apoptosis in T lymphocytes via a caspase-dependent mitochondrial pathway. J Immunol. 2004;172(7):4008–4017. | ||
Chen JY, Yang LX, Huang ZF. The N-terminal 33 amino acid domain of Siva-1 is sufficient for nuclear localization. Braz J Med Biol Res. 2013;46(12):1021–1027. | ||
Jacobs SB, Basak S, Murray JI, Pathak N, Attardi LD. Siva is an apoptosis-selective p53 target gene important for neuronal cell death. Cell Death Differ. 2007;14(7):1374–1385. | ||
Fortin A, MacLaurin JG, Arbour N, et al. The proapoptotic gene SIVA is a direct transcriptional target for the tumor suppressors p53 and E2F1. J Biol Chem. 2004;279(27):28706–28714. | ||
Ray RM, Bhattacharya S, Johnson LR. Mdm2 inhibition induces apoptosis in p53 deficient human colon cancer cells by activating p73- and E2F1-mediated expression of PUMA and Siva-1. Apoptosis. 2011;16(1):35–44. | ||
Zins K, Pomyje J, Hofer E, Abraham D, Lucas T, Aharinejad S. Egr-1 upregulates Siva-1 expression and induces cardiac fibroblast apoptosis. Int J Mol Sci. 2014;15(1):1538–1553. | ||
Xiao H, Palhan V, Yang Y, Roeder RG. TIP30 has an intrinsic kinase activity required for up-regulation of a subset of apoptotic genes. Embo J. 2000;19(5):956–963. | ||
Spinicelli S, Nocentini G, Ronchetti S, Krausz LT, Bianchini R, Riccardi C. GITR interacts with the pro-apoptotic protein Siva and induces apoptosis. Cell Death Differ. 2002;9(12):1382–1384. | ||
Barkinge JL, Gudi R, Sarah H, et al. The p53-induced Siva-1 plays a significant role in cisplatin-mediated apoptosis. J Carcinog. 2009;8:2. | ||
Chu F, Barkinge J, Hawkins S, Gudi R, Salgia R, Kanteti PV. Expression of Siva-1 protein or its putative amphipathic helical region enhances cisplatin-induced apoptosis in breast cancer cells: effect of elevated levels of BCL-2. Cancer Res. 2005;65(12):5301–5309. | ||
Iorio-Morin C, Germain P, Roy S, Génier S, Labrecque P, Parent JL. Thromboxane A2 modulates cisplatin-induced apoptosis through a Siva1-dependent mechanism. Cell Death Differ. 2012;19(8):1347–1357. | ||
Daoud SS, Munson PJ, Reinhold W, et al. Impact of p53 knockout and topotecan treatment on gene expression profiles in human colon carcinoma cells: a pharmacogenomic study. Cancer Res. 2003;63(11):2782–2793. | ||
Bhattacharya S, Ray RM, Chaum E, Johnson DA, Johnson LR. Inhibition of Mdm2 sensitizes human retinal pigment epithelial cells to apoptosis. Invest Ophthalmol Vis Sci. 2011;52(6):3368–3380. | ||
Cao C, Ren X, Kharbanda S, et al. The ARG tyrosine kinase interacts with Siva-1 in the apoptotic response to oxidative stress. J Biol Chem. 2001;276(15):11465–11468. | ||
Zhang YH, Yu LG, Zhu WZ, et al. Preliminary research on the expression, purification and function of the apoptotic fusion protein, Siva1. Asian Pac J Cancer Prev. 2014;15(20):8685–8688. | ||
Dantas LE, Saad ST, Ramos CH, Bénichou S. Overexpression and characterization of the C-terminal domain of human Siva1, a proapoptotic factor and cytoskeleton binding protein. Protein Pept Lett. 2016;23(1):43–50. | ||
Chen GH, Xue QQ, Li J, Gao TL, Sun QS, Bai GP. Anticancer activity of recombinant Siva1 protein in human nasopharyngeal carcinoma cell line CNE-2. Cancer Biomark. 2015;15(6):833–841. | ||
Ma Y, Liu T, Song X, et al. Siva 1 inhibits proliferation, migration and invasion by phosphorylating stathmin in ovarian cancer cells. Oncol Lett. 2017;14(2):1512–1518. | ||
Machado-Neto JA, Lazarini M, Favaro P, et al. ANKHD1 silencing inhibits stathmin 1 activity, cell proliferation and migration of leukemia cells. Biochim Biophys Acta. 2015;1853(3):583–593. | ||
Xue L, Chu F, Cheng Y, et al. Siva-1 binds to and inhibits BCL-X(L)-mediated protection against UV radiation-induced apoptosis. Proc Natl Acad Sci U S A. 2002;99(10):6925–6930. | ||
Chu F, Borthakur A, Sun X, et al. The Siva-1 putative amphipathic helical region (SAH) is sufficient to bind to BCL-XL and sensitize cells to UV radiation induced apoptosis. Apoptosis. 2004;9(1):83–95. | ||
Walmsley SR, Chilvers ER, Thompson AA, et al. Prolyl hydroxylase 3 (PHD3) is essential for hypoxic regulation of neutrophilic inflammation in humans and mice. J Clin Invest. 2011;121(3):1053–1063. | ||
Resch U, Schichl YM, Winsauer G, Gudi R, Prasad K, de Martin R. Siva1 is a XIAP-interacting protein that balances NFκB and JNK signalling to promote apoptosis. J Cell Sci. 2009;122(Pt 15):2651–2661. | ||
Gudi R, Barkinge J, Hawkins S, et al. Siva-1 negatively regulates NF-κB activity: effect on T-cell receptor-mediated activation-induced cell death (AICD). Oncogene. 2006;25(24):3458–3462. | ||
Gudi R, Barkinge J, Hawkins S, Prabhakar B, Kanteti P. Siva-1 promotes K-48 polyubiquitination of TRAF2 and inhibits TCR-mediated activation of NF-kappaB. J Environ Pathol Toxicol Oncol. 2009;28(1):25–38. | ||
Hench VK, Su L. Regulation of IL-2 gene expression by Siva and FOXP3 in human T cells. BMC Immunol. 2011;12:54. | ||
Lin FT, Lai YJ, Makarova N, Tigyi G, Lin WC. The lysophosphatidic acid 2 receptor mediates down-regulation of Siva-1 to promote cell survival. J Biol Chem. 2007;282(52):37759–37769. | ||
Shimoda HK, Shide K, Kameda T, Matsunaga T, Shimoda K. Tyrosine kinase 2 interacts with the proapoptotic protein Siva-1 and augments its apoptotic functions. Biochem Biophys Res Commun. 2010;400(2):252–257. | ||
Li N, Jiang P, Du W, et al. Siva1 suppresses epithelial-mesenchymal transition and metastasis of tumor cells by inhibiting stathmin and stabilizing microtubules. Proc Natl Acad Sci U S A. 2011;108(31):12851–12856. | ||
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. | ||
Sherr CJ, Weber JD. The ARF/p53 pathway. Curr Opin Genet Dev. 2000;10(1):94–99. | ||
Chen D, Shan J, Zhu WG, Qin J, Gu W. Transcription-independent ARF regulation in oncogenic stress-mediated p53 responses. Nature. 2010;464(7288):624–627. | ||
Du W, Jiang P, Li N, et al. Suppression of p53 activity by Siva1. Cell Death Differ. 2009;16(11):1493–1504. | ||
Wang X, Zha M, Zhao X, et al. Siva1 inhibits p53 function by acting as an ARF E3 ubiquitin ligase. Nat Commun. 2013;4:1551. | ||
Yan J, Menendez D, Yang XP, Resnick MA, Jetten AM. A regulatory loop composed of RAP80-HDM2-p53 provides RAP80-enhanced p53 degradation by HDM2 in response to DNA damage. J Biol Chem. 2009;284(29):19280–19289. | ||
Park IK, Blum W, Baker SD, Caligiuri MA. E3 ubiquitin ligase Cbl-b activates the p53 pathway by targeting Siva1, a negative regulator of ARF, in FLT3 inhibitor-resistant acute myeloid leukemia. Leukemia. 2017;31(2):502–505. | ||
Han J, Liu T, Huen MS, Hu L, Chen Z, Huang J. SIVA1 directs the E3 ubiquitin ligase RAD18 for PCNA monoubiquitination. J Cell Biol. 2014;205(6):811–827. | ||
van Nostrand JL, Brisac A, Mello SS, Jacobs SB, Luong R, Attardi LD. The p53 target gene SIVA enables non-small cell lung cancer development. Cancer Discov. 2015;5(6):622–635. | ||
Resnick-Silverman L, Manfredi JJ. Two faces of SIVA. Cancer Discov. 2015;5(6):581–583. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.