Back to Journals » Drug Design, Development and Therapy » Volume 10
Sitagliptin attenuates cardiomyopathy by modulating the JAK/STAT signaling pathway in experimental diabetic rats
Authors Al-Rasheed N, Al-Rasheed N, Hasan I, Al-Amin M, Al-Ajmi H, Mahmoud A ![]()
Received 26 March 2016
Accepted for publication 8 May 2016
Published 28 June 2016 Volume 2016:10 Pages 2095—2107
DOI https://doi.org/10.2147/DDDT.S109287
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Nouf M Al-Rasheed,1 Nawal M Al-Rasheed,1,2 Iman H Hasan,1 Maha A Al-Amin,1 Hanaa N Al-Ajmi,1 Ayman M Mahmoud3
1Department of Pharmacology and Toxicology, College of Pharmacy, King Saud University, 2Department of Pharmaceutical Sciences, College of Pharmacy, Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia; 3Physiology Division, Department of Zoology, Faculty of Science, Beni-Suef University, Beni-Suef, Egypt
Abstract: Sitagliptin, a dipeptidyl peptidase-4 inhibitor, has been reported to promote cardioprotection in diabetic hearts by limiting hyperglycemia and hyperlipidemia. However, little is known about the involvement of the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway modulation in the cardioprotective effects of sitagliptin. The current study aimed to investigate the protective effects of sitagliptin against diabetic cardiomyopathy (DCM), focusing on the modulation of the JAK/STAT pathway. Diabetes was induced by streptozotocin injection, and rats received sitagliptin orally and daily for 90 days. Diabetic rats exhibited hyperglycemia, hyperlipidemia, and a significant increase in heart-to-body weight (HW/BW) ratio. Serum troponin I and creatine kinase MB, cardiac interleukin-6 (IL-6), lipid peroxidation, and nitric oxide levels showed significant increase in diabetic rats. In contrast, both enzymatic and nonenzymatic antioxidant defenses were significantly declined in the heart of diabetic rats. Histopathological study revealed degenerations, increased collagen deposition in the heart of diabetic rats. Sitagliptin alleviated hyperglycemia, hyperlipidemia, HW/BW ratio, histological architecture, oxidative stress, and inflammation, and rejuvenated the antioxidant defenses. In addition, cardiac levels of pJAK2 and pSTAT3 were increased in diabetic rats, an effect which was remarkably decreased after sitagliptin treatment. In conclusion, these results confer an evidence that sitagliptin has great therapeutic potential on DCM through down-regulation of the JAK/STAT signaling pathway.
Keywords: diabetic cardiomyopathy, DPP-4 inhibitors, JAK/STAT, oxidative stress, inflammation
Introduction
Cardiovascular complications represent the main cause of hospitalization and death in diabetic patients.1 Diabetic cardiomyopathy (DCM) is an independent complication of diabetes characterized by myocardial dysfunction in the absence of other heart diseases.2 Diabetic patients have a worse prognosis and are more susceptible to heart failure than hypertension or ischemic heart disease patients.3 Previous studies have demonstrated that hyperglycemia, hyperlipidemia, inflammation, apoptosis, and oxidative stress are probably involved in DCM pathophysiology.4–6 Hyperglycemia-derived reactive oxygen and nitrogen species (ROS and RNS) play a central role in DCM.7 In addition, chronic low-grade inflammation is a feature of DCM in human diabetics.8 As an early response to myocardial injury, inflammatory signaling in cardiomyocytes entails an activation of nuclear factor-κB and increased expression of proinflammatory cytokines such as interleukin-6 (IL-6) and tumor necrosis factor-alpha.9
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway is an intracellular signaling pathway closely related to cardiac hypertrophy.10 This pathway plays a key role in cell growth, survival, and differentiation and in regulating gene expression.11 Upon phosphorylation by JAK, STAT proteins translocate into the nucleus, bind to the promoter region of target genes, and regulate their transcription.12 In the heart, STATs regulate the expression of gene encoding proteins involved in angiogenesis, extracellular matrix composition, inflammation, apoptosis, and cellular signaling.13,14 IL-6 and its family-related proteins transduce their signals via glycoprotein 130 (gp130) mostly to STAT3 and play a key role in the development of heart failure and cardiac hypertrophy.15 It has been indicated that high glucose activates the JAK/STAT signaling of vascular endothelial cells in vitro through phosphorylation of JAK2 and subsequently STAT3, leading to the proliferation of endothelial cells.16 Thus, modulation of the IL-6-gp 130-JAK/STAT signaling pathway represents an important strategy for the treatment of DCM.
Dipeptidyl peptidase-4 (DPP-4) inhibitors, as sitagliptin, are a class of antidiabetic drugs that inhibit the degradation of insulinotropic incretins, mainly glucagon-like peptide-1 (GLP-1).17 GLP-1 is released from intestinal L-cells and regulates circulating glucose levels by enhancing insulin secretion. Studies showed that GLP-1 confers cardioprotection after congestive heart failure, ischemia, and myocardial infarction.18,19 Sitagliptin has been reported to promote cardioprotection in type 2 diabetic hearts via GLP-1 stabilization and by limiting hyperglycemia and hyperlipidemia.20 However, the issues of an involvement of the JAK/STAT pathway modulation in the protective effects of sitagliptin against DCM have not been previously reported. Therefore, we have investigated the cardioprotective role of sitagliptin in streptozotocin (STZ)-induced diabetic rats, focusing on its modulatory effect on the JAK/STAT pathway.
Materials and methods
STZ and sitagliptin (Januvia) were purchased from Sigma-Aldrich (St Louis, MO, USA) and Merck & Co., Inc. (Whitehouse, NJ, USA), respectively. Rabbit polyclonal anti-phospho-JAK2 and goat anti-phospho-STAT3 (pSTAT3) were supplied by Santa Cruz Biotechnology Inc. (Dallas, TX, USA). All other chemicals were of analytical grade and were obtained from standard commercial supplies.
Experimental animals
Male Wistar rats (10 weeks old) weighing between 140 and 160 g, obtained from the Experimental Animal Center, College of Pharmacy, King Saud University (Saudi Arabia), were used in this investigation. Rats were housed in special cages at controlled temperature of 20°C–22°C and humidity of 60%, fed a standard rat pellet chow with free access to tap water ad libitum and kept for 1 week before the experiment for acclimatization. All animal procedures were performed in accordance with the guidelines provided by the Experimental Animal Laboratory and approved by the Animal Care and Use Committee of the College of Pharmacy, King Saud University (Saudi Arabia).
Induction of type 1 diabetes mellitus and experimental design
Diabetes was induced in overnight fasted rats by a single intraperitoneal injection of 55 mg/kg body weight (BW) freshly prepared STZ, dissolved in 0.1 M citrate buffer (pH 4.5). Seventy-two hours later, blood glucose levels were determined using MEDISAFE MINI blood glucose reader (TERUMO Corporation, Hatagaya, Tokyo, Japan), and rats having blood glucose levels ≥200 mg/dL were selected for the experiment.
Thirty-two (16 normal +16 diabetic) rats were divided into four equal groups, each consisting of eight (N=8) animals as follows:
Group I: Control
Group II: Control +100 mg/kg/day sitagliptin
Group III: Diabetic
Group IV: Diabetic +100 mg/kg/day sitagliptin.
Sitagliptin was dissolved in water and supplied for 90 days via oral gavage; the dosage was balanced as indicated by any change in the BW over the entire period of study. At the end of the experiment, blood samples collected from each animal were allowed to coagulate at room temperature and then centrifuged at 3,000 rpm for 15 minutes to separate serum. Immediately after sacrifice, hearts were quickly excised, cleaned, and perfused with ice-cold saline and homogenized in phosphate-buffered saline or fixed in 10% formalin.
Determination of troponin I and creatine kinase MB
Serum levels of troponin I and activity of creatine kinase MB (CK-MB) were measured using specific rat enzyme-linked immunosorbent assay kits purchased from EIAab (Wuhan, People’s Republic of China) according to the manufacturer’s instructions. Standard curves were prepared and troponin I and CK-MB in the samples were determined from the prepared standard curves.
Determination of lipid profile and cardiovascular risk indices
Serum levels of total cholesterol, triglycerides, and high-density lipoprotein (HDL) cholesterol were assayed according to the methods of Allain et al,21 Fossati and Prencipe,22 and Burstein et al23 using commercial diagnostic kits purchased from United Diagnostics Industry (Riyadh, Saudi Arabia). Serum low-density lipoprotein (LDL) cholesterol level was calculated from the formula:
|
|
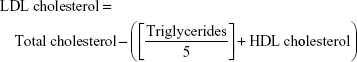
Very low-density lipoprotein (vLDL) cholesterol concentration was calculated according to the following formula:
|
|
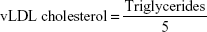
Cardiovascular risk indices were calculated according to Ross24 as follows:
|
|

|
|
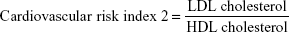
Determination of oxidative stress and antioxidant defenses
Lipid peroxidation (assayed as malondialdehyde), reduced glutathione (GSH), and nitric oxide (NO) levels were determined in heart homogenate according to the methods of Preuss et al,25 Beutler et al,26 and Montgomery and Dymock,27 respectively. Activities of the antioxidant enzymes, superoxide dismutase and catalase, were measured according to the methods of Marklund and Marklund28 and Cohen et al,29 respectively. Protein content was determined using Bradford reagent.
Determination of IL-6
Levels of IL-6 were determined in the cardiac homogenates using specific enzyme-linked immunosorbent assay kits (R&D Systems, Inc., Minneapolis, MN, USA) following the manufacturer’s instructions. Concentration of IL-6 in the samples was determined from standard curve constructed by using standard cytokine.
Histopathological study
Histological processing was prepared as described previously.30 The excised hearts were cleaned and fixed in 10% buffered formalin for 24 hours. The fixed samples were processed to prepare 5-μm thick paraffin sections, which were then stained with hematoxylin and eosin (H&E) and Masson’s trichrome stains. High-resolution digital scans of all stained slides were created with a ScanScope scanner (Aperio Technologies, Vista, CA, USA) and were viewed and analyzed using Aperio’s viewing and image analysis tools.
Immunohistochemical examination of pJAK2 and pSTAT3
Immunostaining of the heart sections for the detection of pJAK2 and pSTAT3 was performed using streptavidin-biotinylated horseradish peroxidase (Novalink Max Polymer detection system; Novocastra Laboratories, Newcastle, UK). In brief, sections were incubated in 3% hydrogen peroxide in distilled water for 5 minutes to block endogenous peroxidase activity and then washed in Tris-buffered saline (pH 7.6) for 10 minutes. Nonspecific binding of antibodies was blocked by incubation with protein block (Novocastra) for 5 minutes. Sections were incubated with rabbit polyclonal anti-pJAK2 and goat polyclonal anti-pSTAT3 primary antibodies (Santa Cruz Biotechnology Inc.) diluted 1:100 at room temperature for 1 hour. The sections were washed in Tris-buffered saline three times and incubated with biotinylated IgG (Novocastra) for 30 minutes. After washing in Tris-buffered saline and incubation with Novolink polymer (Novocastra) for 30 minutes, peroxidase was detected with diaminobenzedine substrate (Novocastra). Sections were then washed in distilled water, stained with Mayer’s hematoxylin, and mounted in DPX. The same procedure, with the omission of incubation in primary antibodies, was followed for negative control sections.
Statistical analysis
Data were analyzed using GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA), and all statistical comparisons were made by means of the one-way analysis of variance test followed by Tukey’s test post hoc analysis. Results were expressed as mean ± standard error of the mean and a P-value <0.05 was considered significant.
Results
Sitagliptin represses hyperglycemia and BW loss and prevents cardiac hypertrophy in diabetic rats
BW of control and diabetic rats showed nonsignificant (P>0.05) differences at the start of the treatment period (Table 1). After 90 days, diabetic rats exhibited a significant (P<0.001) decrease in BW when compared with the normal control rats. Treatment with sitagliptin significantly (P<0.05) protected against diabetes-induced BW loss. On the other hand, heart-to-body weight (HW/BW) ratio showed a significant (P<0.01) increase in diabetic rats when compared with the control group. Sitagliptin administration produced a significant (P<0.01) decrease in HW/BW ratio in the diabetic rats.
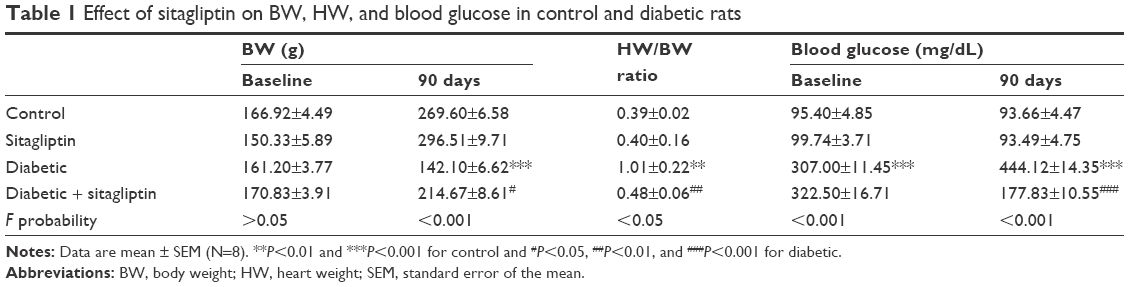 | Table 1 Effect of sitagliptin on BW, HW, and blood glucose in control and diabetic rats |
Diabetic rats showed a significant (P<0.001) increase in blood glucose levels both before and after the treatment period. Treatment of the diabetic rats with sitagliptin for 90 days significantly (P<0.001) alleviated blood glucose levels.
Of note, sitagliptin produced a nonsignificant (P>0.05) effect on BW, HW, and blood glucose parameters when supplemented to control rats.
Sitagliptin reduces circulating troponin I and CK-MB in diabetic rats
Data represented in Figure 1 show the effects of sitagliptin on troponin I levels and CK-MB activity in serum of control and diabetic rats. STZ-induced diabetic rats exhibited significant (P<0.01) increase in serum troponin I levels when compared with the control rats. Treatment of diabetic rats with sitagliptin for 90 days produced a marked (P<0.01) decrease in serum troponin I levels (Figure 1A).
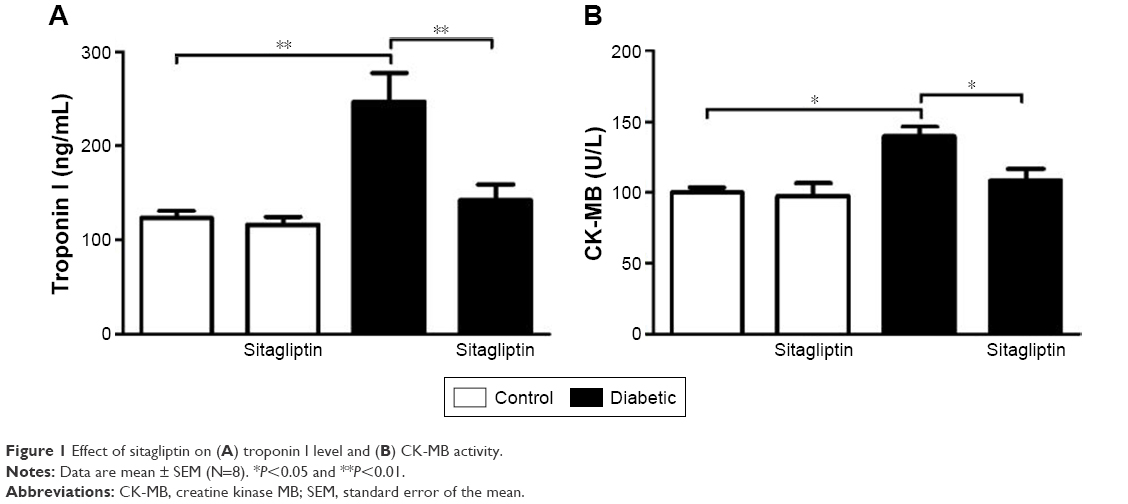 | Figure 1 Effect of sitagliptin on (A) troponin I level and (B) CK-MB activity. |
Similarly, STZ administration produced a significant (P<0.05) increase in serum CK-MB activity. Oral treatment of the STZ-induced diabetic rats with sitagliptin significantly (P<0.05) ameliorated the altered CK-MB activity, as depicted in Figure 1B. Oral supplementation of sitagliptin to the normal rats produced a nonsignificant (P<0.05) effect on either serum troponin I levels or CK-MB activity.
Sitagliptin alleviates lipid profile and cardiovascular risk indices in diabetic rats
Data summarized in Figure 2 represent the effect of sitagliptin on serum lipid profile and cardiovascular risk indices in both control and STZ-induced diabetic rats. Diabetic rats exhibited significant (P<0.001) increase in serum cholesterol (Figure 2A), triglycerides (Figure 2B), LDL cholesterol (Figure 2C), and vLDL cholesterol levels (Figure 2D) when compared with the corresponding control group. HDL cholesterol showed a significant (P<0.001) decrease in serum of STZ-induced diabetic rats (Figure 2E). On the other hand, diabetic rats treated with sitagliptin for 90 days showed significant (P<0.001) alleviation in all parameters of the lipid profile. Control rats treated with sitagliptin showed a nonsignificant (P>0.05) change in their lipid profile when compared with the control group.
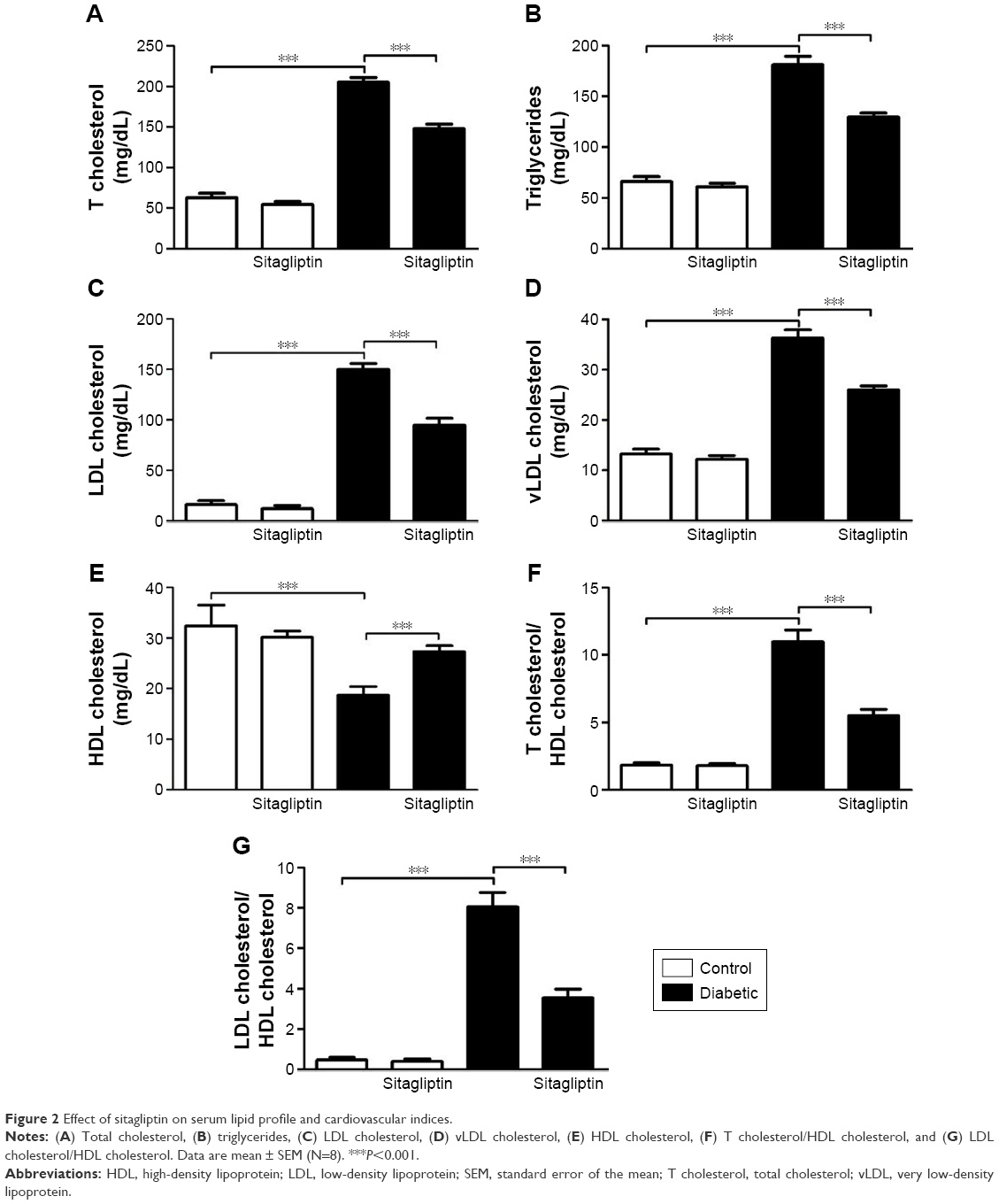 | Figure 2 Effect of sitagliptin on serum lipid profile and cardiovascular indices. |
To explore the impact of diabetes-induced hyperlipidemia and protective effect of sitagliptin on the heart, the cardiovascular risk indices, total cholesterol/HDL cholesterol (Figure 2F), and LDL cholesterol/HDL cholesterol (Figure 2G) were calculated. Diabetic rats showed significant (P<0.001) increase in both total cholesterol/HDL cholesterol and LDL cholesterol/HDL cholesterol ratios when compared with the control rats. Conversely, diabetic rats supplemented with sitagliptin for 90 days exhibited marked (P<0.001) improvement in their recorded cardiovascular risk indices. As expected, sitagliptin produced a nonsignificant (P>0.05) effect on total cholesterol/HDL cholesterol and LDL cholesterol/HDL cholesterol ratios when supplemented to normal rats.
Sitagliptin attenuates oxidative stress in heart of diabetic rats
To investigate the effect of sitagliptin on the cardiac redox status, lipid peroxidation, NO, and activity of antioxidant enzymes under homeostatic and diabetic conditions were determined.
STZ-induced diabetic rats exhibited significantly (P<0.001) increased malondialdehyde and NO levels in the heart as compared with their respective normal controls. Treatment of the diabetic rats with sitagliptin for 90 days markedly (P<0.001) decreased cardiac malondialdehyde (Figure 3A) and NO levels (Figure 3B).
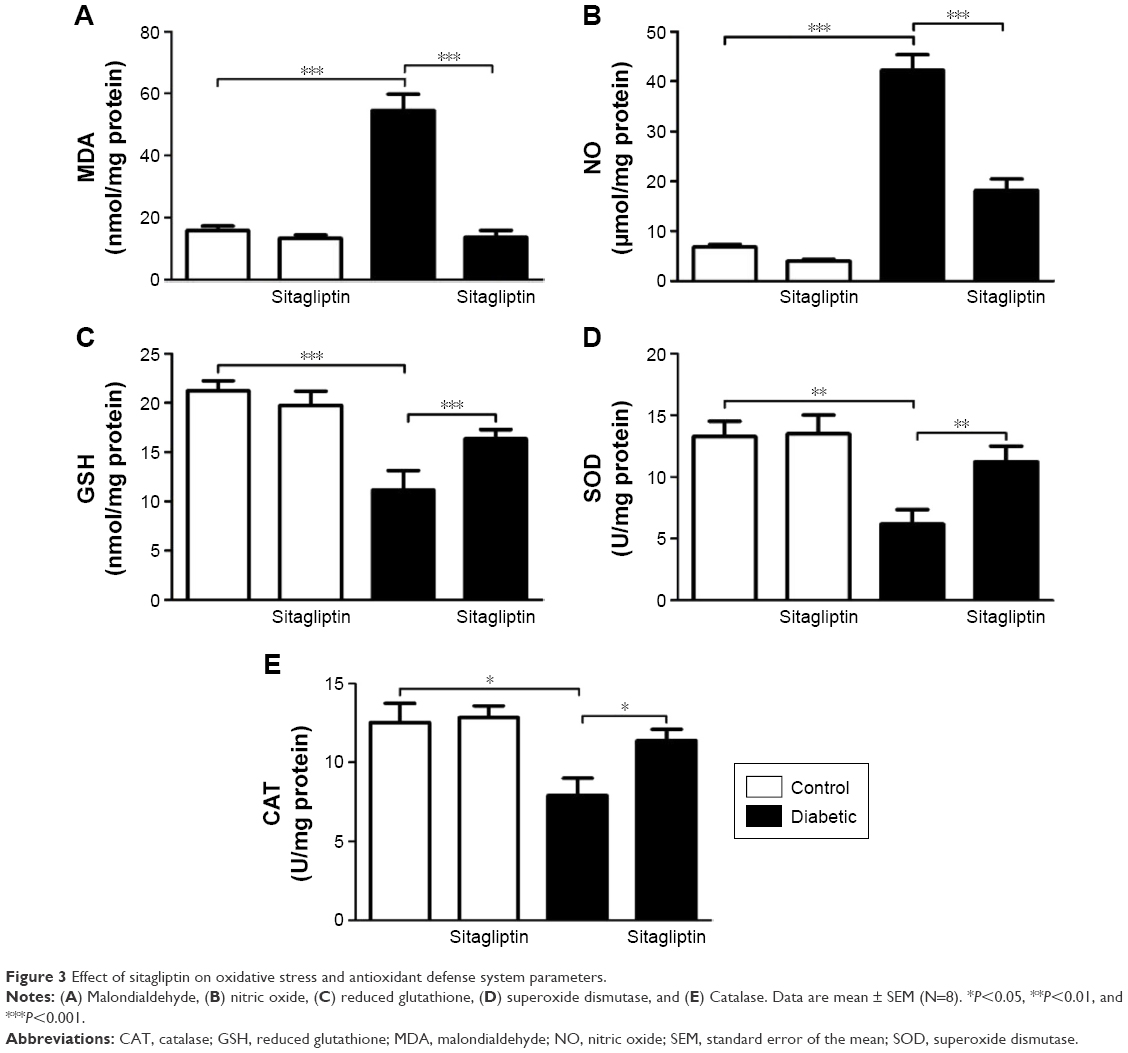 | Figure 3 Effect of sitagliptin on oxidative stress and antioxidant defense system parameters. |
In contrast, GSH content in the heart of diabetic rats was significantly (P<0.001) declined, as represented in Figure 3C. Treatment with sitagliptin produced a significant (P<0.001) alleviation of cardiac GSH content in diabetic rats. Activities of the antioxidant enzymes, superoxide dismutase (Figure 3D) and catalase (Figure 3E), were investigated in control and diabetic rats, and it was found that the activities of the enzymes exhibited a significant decrease in GSH content of the heart of diabetic rats, an effect that was significantly reduced after the administration of sitagliptin. Concerning its effects on the redox status in normal rats, sitagliptin exerted a nonsignificant effect.
Sitagliptin decreases cardiac IL-6 levels in diabetic rats
The proinflammatory cytokine IL-6 showed a significant (P<0.001) increase in the heart of diabetic rats when compared with the control group (Figure 4). Treatment of the diabetic rats with sitagliptin for 90 days significantly (P<0.001) improved cardiac levels of IL-6. In addition, sitagliptin supplementation to normal rats produced a nonsignificant (P>0.05) effect on cardiac IL-6 levels when compared with the control rats.
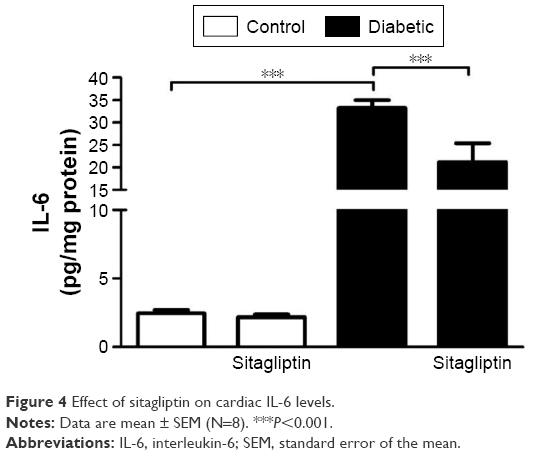 | Figure 4 Effect of sitagliptin on cardiac IL-6 levels. |
Sitagliptin improves histological architecture and decreases collagen deposition in diabetic rats
Histopathological examination of the H&E-stained heart sections of control rats showed normal heart tissue with normal features of myocardium cells, blood vessels, and endomysium (Figure 5A). Heart sections from sitagliptin-treated rats revealed apparently normal endocardium, myocardium, and endomysium (Figure 5B). On the other hand, sections in the heart of STZ-induced diabetic rats showed focal areas of myocardium degeneration and many myocardium cell degeneration (Figure 5C). Diabetic rats treated with sitagliptin (Figure 5D) showed few myocardial cell degenerations.
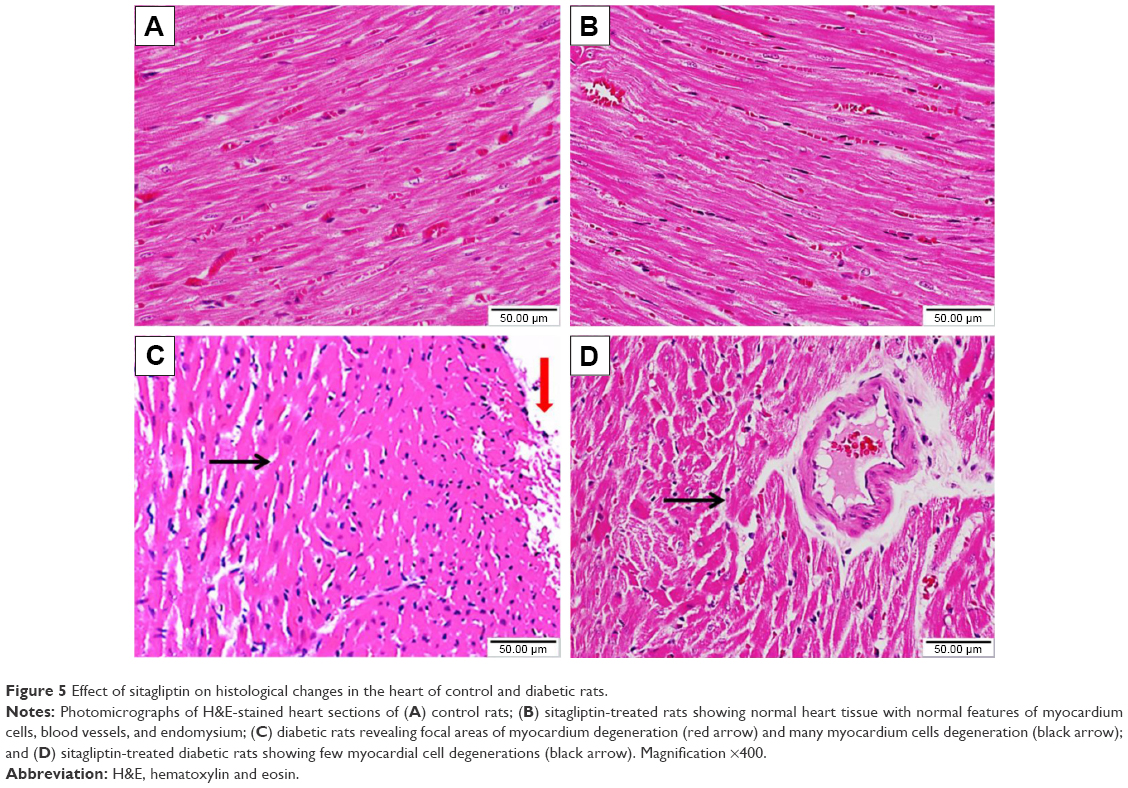 | Figure 5 Effect of sitagliptin on histological changes in the heart of control and diabetic rats. |
To demonstrate the effects of diabetes and treatment with sitagliptin on collagen deposition, heart sections were stained with Masson’s trichrome. Heart sections showed normal distribution and normal amount of cardiac fibrous tissue in both control and sitagliptin-treated rats, as represented in Figure 6A and B, respectively. Sections from the heart of STZ-induced diabetic rats (Figure 6C) showed large patches of strong fibrous tissue reactivity. STZ-induced diabetic rats treated with sitagliptin showed small patches of fibrous tissue in the Masson’s trichrome-stained heart sections (Figure 6D).
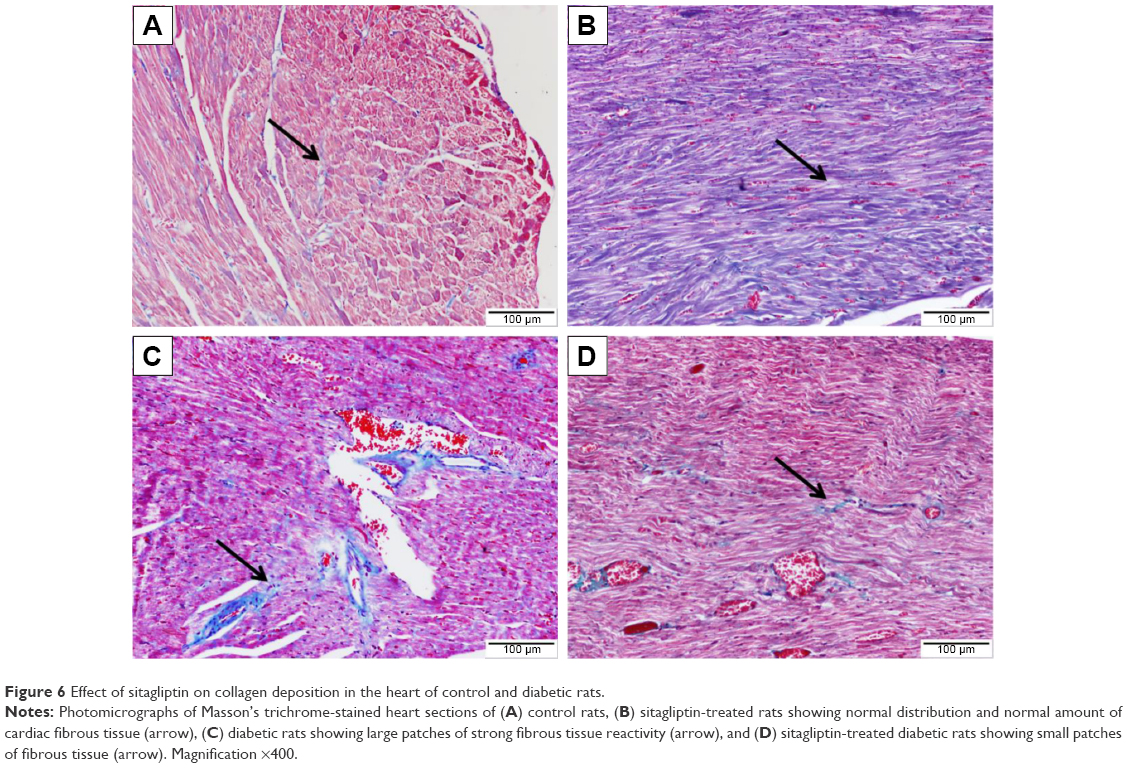 | Figure 6 Effect of sitagliptin on collagen deposition in the heart of control and diabetic rats. |
Sitagliptin reduces cardiac JAK2 and STAT3 phosphorylation in diabetic rats
The effect of sitagliptin on the expression of pJAK2 and pSTAT3 in the heart of normal and diabetic rats was determined by immunohistochemical staining. Heart sections from control and sitagliptin-treated rats, immunostained with pJAK2 primary antibody, revealed negative immune reaction of the nuclei and cytoplasm (Figure 7A and B). Heart sections of the STZ-induced diabetic rats showed strong immune reactivity of myocardium cell nuclei and moderate immune reaction of the cytoplasm (Figure 7C). In contrast, sitagliptin-treated diabetic rats showed few immunopositive nuclei (Figure 7D).
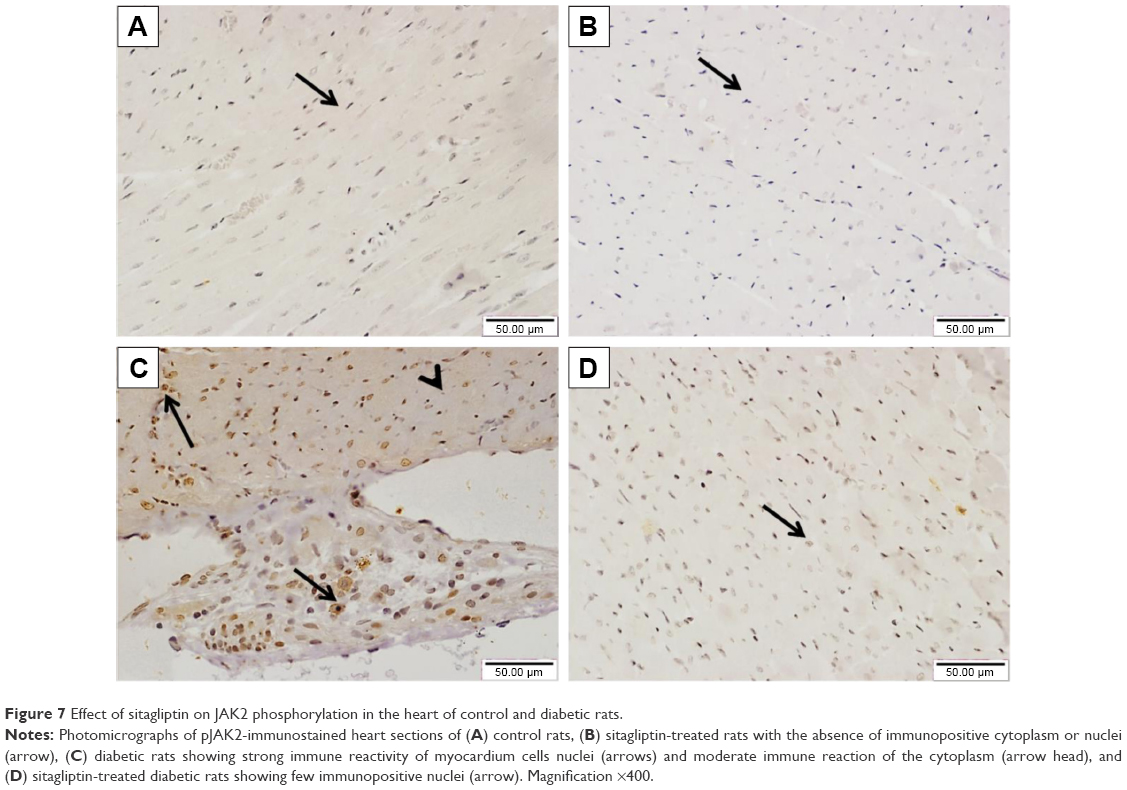 | Figure 7 Effect of sitagliptin on JAK2 phosphorylation in the heart of control and diabetic rats. |
Similarly, the immunohistochemical staining of the heart with pSTAT3 primary antibody revealed the absence of immune positivity especially in the nuclei of normal control (Figure 8A) and sitagliptin-treated rats (Figure 8B). STZ-induced diabetic rats showed strong immune reactivity of almost all of myocardium cell nuclei and the endocardium endothelium cells (Figure 8C). On the other hand, diabetic rats treated with sitagliptin for 90 days showed many immunopositive nuclei of both endocardium endothelium and myocardium cells (Figure 8D).
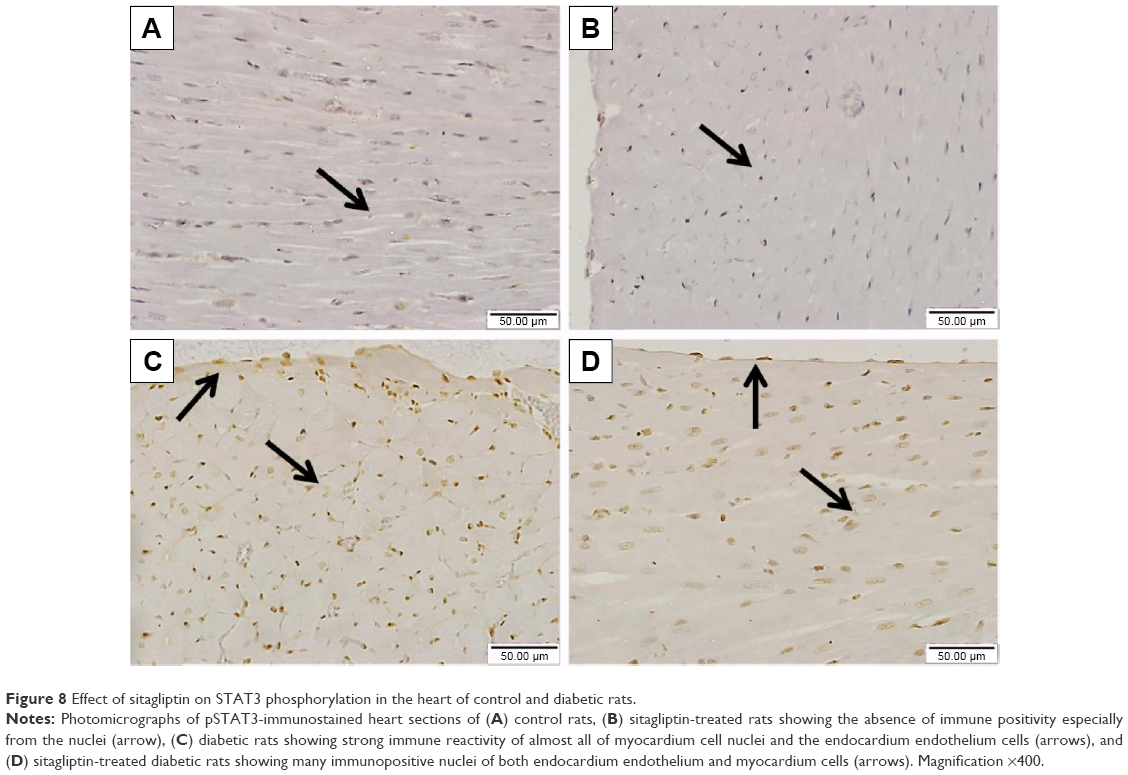 | Figure 8 Effect of sitagliptin on STAT3 phosphorylation in the heart of control and diabetic rats. |
Discussion
The JAK/STAT signaling pathway regulates different genes involved in inflammation, fibrosis, and cell growth and differentiation.11,12 The present study outlines the protective effects of sitagliptin against diabetes-induced cardiomyopathy in STZ-induced diabetic rat model. Sitagliptin attenuated cardiac oxidative stress, inflammation, and collagen deposition and reduced the JAK/STAT expression, thus suggesting the potential benefit of sitagliptin to halt the progression of DCM.
As previously reported STZ-induced diabetic rats exhibited BW loss and increased blood glucose.31,32 Treatment of the STZ-induced diabetic rats with sitagliptin was associated with reduced blood glucose that may have contributed to the greatly alleviated BW. In contrast, we observed no difference in blood glucose levels between sitagliptin-treated and normal rats. Similar findings were reported in the study of Connelly et al33 who studied the effect of DPP-4 inhibition on cardiac dysfunction in experimental diabetic rats. Although not necessarily seen in type 1 diabetic patients, these results are typical of STZ-induced diabetic animals in which high glucose provoked glucagon and insulin secretion is impaired.34
Here, the STZ-induced diabetic rats showed notable increase in the HW/BW ratio, which demonstrates cardiac hypertrophy. The diabetic heart is metabolically characterized by increased fatty acid oxidation, leading to lipid accumulation in the myocardium.35,36 In the same context, Wang et al37 reported that STZ-induced diabetic rats exhibited cardiac hypertrophy after 8 weeks of diabetes induction. Sitagliptin treatment produced a positive effect on HW/BW ratio and was thus able to prevent diabetes-associated cardiac hypertrophy. DPP-4 inhibition has been reported to reduce cardiac myocyte hypertrophy and alleviate diastolic dysfunction in diabetic rats with myocardial infarction.33
This beneficial effect of sitagliptin could be directly attributable to the lowered serum lipids and increased HDL cholesterol levels. Serum total cholesterol, triglycerides, and LDL cholesterol were significantly increased, and HDL cholesterol was significantly decreased in diabetic rats, an effect that was reversed following sitagliptin treatment. In diabetes, the elevated level of serum lipids causes the risk of coronary heart disease38 while HDL was found to be cardioprotective.39 In addition, the recorded values of atherogenic indices showed the bad impact of diabetes-induced dyslipidemia on the cardiovascular system and were further confirmed by the elevated levels of CK-MB and troponin I. Increases in circulating levels of CK-MB represent a sensitive and powerful predictor of increased cardiac complications.40 Furthermore, studies have demonstrated the relation between CK-MB and troponin I elevation and the onset of myofibrillar disintegration and increased permeability caused by inflammation.41 Recently, we have demonstrated elevations in CK-MB and troponin I in rats with cardiac hypertrophy.30 Reduction of these indices in sitagliptin-treated diabetic rats strongly supports the notion that DPP-4 inhibitors may reduce the risk of developing heart diseases.
Oxidative stress constitutes an important factor implicated in the development of DCM. Several lines of evidence indicate that hyperglycemia in diabetes is associated with increased mitochondrial production of ROS and RNS.42 In addition, elevated free fatty acids in the diabetic heart modulates the mitochondrial electron chain and activates nicotinamide adenine dinucleotide phosphate oxidases to generate superoxide.43 Superoxide can combine with NO forming highly reactive and damaging peroxynitrite species.44 The current findings showed increased lipid peroxidation and NO levels in the heart of STZ-induced diabetic rats, demonstrating oxidative stress condition. Lipid peroxidation, produced from the reaction of ROS with membrane lipids, has been associated with altered membrane structure and enzyme inactivation.45 This enhancement of lipid peroxidation might be due to hyperglycemia-induced free radical production. The elevated NO levels could be directly connected to activated inducible NO synthase. A previous study has demonstrated that inducible NO synthase is able to generate superoxide anions independent of NO production.46
By comparison, STZ-induced diabetic rats exhibited significant decrease in cardiac GSH content as well as activity of antioxidant defense enzymes. GSH depletion has been linked to declined cellular defense against ROS-induced injury, leading to necrotic cell death.47 Superoxide dismutase catalyzes the dismutation of superoxide radicals to oxygen and hydrogen peroxide. The latter is converted to molecular oxygen and water. Glutathione peroxidase serves a similar antioxidant role by degrading hydrogen peroxide and lipid peroxides to water and lipid alcohols.48 Therefore, these antioxidants represent a vital line of defense against diabetes-induced oxidative stress.
Interestingly, diabetic rats treated with sitagliptin showed significant decrease in cardiac lipid peroxidation and NO levels, with concomitant enhancement of the antioxidant defenses. In agreement with our findings, treatment with sitagliptin for 6 weeks decreased lipid peroxidation in the heart of Zucker diabetic fatty rats.49 In addition, Kelleni et al50 demonstrated a protective effect of sitagliptin against doxorubicin-induced cardiotoxicity and oxidative stress in rats. Another supporting study recently conducted by Alam et al51 showed that sitagliptin prevents oxidative stress of heart and kidney in two kidney and one clip rats. These data support the premise that sitagliptin can protect against DCM via suppressing oxidative stress.
Cardiac inflammation, fibrosis, and apoptosis have been recognized in DCM52,53 as a consequence of oxidative stress.54 Therefore, diabetes induces both functional and structural disturbances in the myocardium. In the present investigation, the proinflammatory cytokine IL-6 showed a significant increase in the heart of diabetic rats. Studies demonstrated that concentrations of inflammatory cytokines including IL-6 increased in the serum of experimental animal models55 and diabetic patients,56 leading to diastolic dysfunction. The beneficial effect of sitagliptin on cardiac oxidative stress was accompanied by an improvement of IL-6 in the heart of diabetic rats. These findings point to an anti-inflammatory effect exerted by sitagliptin. In this context, Ferreira et al49 and Kelleni et al50 reported the anti-inflammatory effect of sitagliptin in Zucker diabetic fatty rats and doxorubicin-induced cardiotoxicity animal model, respectively.
The histological findings showed focal areas of myocardium degeneration and large patches of strong fibrous tissue reactivity in the heart sections of STZ-induced diabetic rats. These alterations could be directly connected to the hyperglycemia-induced oxidative stress and inflammation. Increased ROS are known to cause cardiac dysfunction by direct damage to DNA and proteins and by inducing apoptosis. Cardiac fibrosis occurs as a consequence of inflammation and cell injury57 as confirmed by various studies4,58 where suppression of inflammation has been shown to improve DCM, enhance cardiac function, and reduce cardiac fibrosis. Furthermore, studies have demonstrated the role of oxidative stress as an important regulator of pro-fibrotic processes in the heart.59 The study by Suzuki et al53 showed ameliorated hyperglycemia-induced cardiac fibrosis by treatment with antioxidant. In addition, Alam et al51 revealed the antifibrotic activity of sitagliptin in kidneys and heart of two kidney and one clip rats. Here, sitagliptin was able to protect the heart against diabetes-induced cellular injury and fibrosis probably through its dual ability to reduce chronic inflammation and improve the redox balance.
We hypothesized that the beneficial effects of sitagliptin in DCM might be associated with the modulation of the JAK/STAT pathway. To verify our findings, phosphorylated JAK2 and STAT3 were determined in the heart of diabetic and sitagliptin-treated rats. Diabetic rats exhibited marked increase in phosphorylation levels of JAK2 and STAT3 as seen in the immunohistochemically stained heart sections. Therefore, it is clearly apparent that diabetes induces upregulation of the JAK/STAT pathway in the heart of rats as recently reported in type 2 diabetic rats.60 JAK/STAT pathway is closely related to cardiac hypertrophy caused by heart failure, pressure overload,10 and isoproterenol administration.30 The upregulated JAK2/STAT3 signaling in the present investigation was positively correlated with the cardiac IL-6 levels. IL-6 transduces hypertrophic signals via gp130 predominantly to STAT315 in cardiac myocytes and satellite cells.61 Therefore, the IL-6-gp130-JAK/STAT signaling pathway has a key role in cardiac hypertrophy.30 Compared with DCM rats, the phosphorylation levels of JAK2 and STAT3 in myocardium of rats treated with sitagliptin were declined. Because JAK/STAT signaling is induced by IL-6, the recorded down-regulation could be attributed to the anti-inflammatory effect of sitagliptin. Modulation of this pathway mediates the antifibrotic effect of sitagliptin in diabetic rat heart. Previous studies have demonstrated that inhibiting the JAK/STAT signaling pathway can decrease the synthesis of transforming growth factor-beta 1 in mesangial cells cultured in high glucose12 and type 2 diabetic rat heart.60 Transforming growth factor-beta 1 is a polypeptide that regulates cell growth and differentiation, and promotes myocardial fibrosis.62 These findings demonstrate that sitagliptin has a protective effect on diabetes-induced myocardial injury by down-regulating JAK/STAT signaling pathway.
Conclusion
The present study demonstrates that sitagliptin exerts protective effects against DCM. Sitagliptin has shown strong modulatory effect against hyperglycemia-induced cardiac inflammation and oxidative stress via enhancement of antioxidant defenses and suppression of ROS production. In addition, sitagliptin showed strong antihyperglycemic and antihyperlipidemic effects, attenuated collagen deposition and cardiac hypertrophy, and reduced severity of histopathological lesions in heart of diabetic rats. The study provides evidence that the cardioprotective effect of sitagliptin is related to down-regulation of the JAK/STAT signaling pathway in diabetic rats. Thus, sitagliptin could be considered as a potential candidate for the prevention/management of DCM.
Acknowledgment
This research project was supported by a grant from the Research Center of the Female Scientific and Medical Colleges, Deanship of Scientific Research, King Saud University, Saudi Arabia.
Disclosure
The authors report no conflicts of interest in this work.
References
Bauters C, Lamblin N, Mc Fadden EP, Van Belle E, Millaire A, de Groote P. Influence of diabetes mellitus on heart failure risk and outcome. Cardiovasc Diabetol. 2003;2:1. | ||
Huynh K, McMullen JR, Julius TL, et al. Cardiac-specific IGF-1 receptor transgenic expression protects against cardiac fibrosis and diastolic dysfunction in a mouse model of diabetic cardiomyopathy. Diabetes. 2010;59(6):1512–1520. | ||
Solang L, Malmberg K, Ryden L. Diabetes mellitus and congestive heart failure. Further knowledge needed. Eur Heart J. 1999;20(11):789–795. | ||
Westermann D, Walther T, Savvatis K, et al. Gene deletion of the kinin receptor B1 attenuates cardiac inflammation and fibrosis during the development of experimental diabetic cardiomyopathy. Diabetes. 2009;58(6):1373–1381. | ||
Falcao-Pires I, Leite-Moreira AF. Diabetic cardiomyopathy: understanding the molecular and cellular basis to progress in diagnosis and treatment. Heart Fail Rev. 2012;17(3):325–344. | ||
Yu W, Wu J, Cai F, et al. Curcumin alleviates diabetic cardiomyopathy in experimental diabetic rats. PLoS One. 2012;7(12):e52013. | ||
Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–1625. | ||
Schilling JD, Mann DL. Diabetic cardiomyopathy: bench to bedside. Heart Fail Clin. 2012;8(4):619–631. | ||
Aoyagi T, Matsui T. The cardiomyocyte as a source of cytokines in cardiac injury. J Cell Sci Ther. 2012;(S5):003. | ||
Kiu H, Nicholson SE. Biology and significance of the JAK/STAT signalling pathways. Growth Factors. 2012;30(2):88–106. | ||
Soebiyanto RP, Sreenath SN, Qu CK, Loparo KA, Bunting KD. Complex systems biology approach to understanding coordination of JAK-STAT signaling. Biosystems. 2007;90(3):830–842. | ||
Boengler K, Buechert A, Heinen Y, et al. Cardioprotection by ischemic postconditioning is lost in aged and STAT3-deficient mice. Circ Res. 2008;102(1):131–135. | ||
Hilfiker-Kleiner D, Hilfiker A, Drexler H. Many good reasons to have STAT3 in the heart. Pharmacol Ther. 2005;107(1):131–137. | ||
Snyder M, Huang XY, Zhang JJ. Identification of novel direct Stat3 target genes for control of growth and differentiation. J Biol Chem. 2008;283(7):3791–3798. | ||
Fischer P, Hilfiker-Kleiner D. Survival pathways in hypertrophy and heart failure: the gp130-STAT3 axis. Basic Res Cardiol. 2007;102(4):279–297. | ||
Marrero MB, Schieffer B, Paxton WG, et al. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature. 1995;375(6528):247–250. | ||
Drucker DJ. Dipeptidyl peptidase-4 inhibition and the treatment of type 2 diabetes: preclinical biology and mechanisms of action. Diabetes Care. 2007;30(6):1335–1343. | ||
Nikolaidis LA, Mankad S, Sokos GG, et al. Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation. 2004;109(8):962–965. | ||
Sokos GG, Nikolaidis LA, Mankad S, Elahi D, Shannon RP. Glucagon-like peptide-1 infusion improves left ventricular ejection fraction and functional status in patients with chronic heart failure. J Card Fail. 2006;12(9):694–699. | ||
Picatoste B, Ramirez E, Caro-Vadillo A, et al. Sitagliptin reduces cardiac apoptosis, hypertrophy and fibrosis primarily by insulin-dependent mechanisms in experimental type-II diabetes. Potential roles of GLP-1 isoforms. PLoS One. 2013;8(10):e78330. | ||
Allain CC, Poon LS, Chan CS, Richmond W, Fu PC. Enzymatic determination of total serum cholesterol. Clin Chem. 1974;20(4):470–475. | ||
Fossati P, Prencipe L. Serum triglycerides determined colorimetrically with an enzyme that produces hydrogen peroxide. Clin Chem. 1982;28(10):2077–2080. | ||
Burstein M, Scholnick HR, Morfin R. Rapid method for the isolation of lipoproteins from human serum by precipitation with polyanions. J Lipid Res. 1970;11(6):583–595. | ||
Ross R. The pathogenesis of atherosclerosis. In: Braunwald E, editor. Heart Disease: A Textbook of Cardiovascular Medicine. 4th ed. Philadelphia (PA): WB Saunders; 1992:1106–1124. | ||
Preuss HG, Jarrell ST, Scheckenbach R, Lieberman S, Anderson RA. Comparative effects of chromium, vanadium and gymnema sylvestre on sugar-induced blood pressure elevations in SHR. J Am Coll Nutr. 1998;17(2):116–123. | ||
Beutler E, Duron O, Kelly BM. Improved method for the determination of blood glutathione. J Lab Clin Med. 1963;61:882–888. | ||
Montgomery HAC, Dymock JF. The determination of nitrite in water. Analyst. 1961;86:414–416. | ||
Marklund S, Marklund G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem. 1974;47(3):469–474. | ||
Cohen G, Dembiec D, Marcus J. Measurement of catalase activity in tissue extracts. Anal Biochem. 1970;34(1):30–38. | ||
Al-Rasheed NM, Al-Oteibi MM, Al-Manee RZ, et al. Simvastatin prevents isoproterenol-induced cardiac hypertrophy through modulation of the JAK/STAT pathway. Drug Des Devel Ther. 2015;9:3217–3229. | ||
Sarkhail P, Rahmanipour S, Fadyevatan S, et al. Antidiabetic effect of Phlomis anisodonta: effects on hepatic cells lipid peroxidation and antioxidant enzymes in experimental diabetes. Pharmacol Res. 2007;56(3):261–266. | ||
Guimaraes JF, Muzio BP, Rosa CM, et al. Rutin administration attenuates myocardial dysfunction in diabetic rats. Cardiovasc Diabetol. 2015;14(6):90. | ||
Connelly KA, Zhang Y, Advani A, et al. DPP-4 inhibition attenuates cardiac dysfunction and adverse remodeling following myocardial infarction in rats with experimental diabetes. Cardiovasc Ther. 2013;31(5):259–267. | ||
Meier JJ, Ueberberg S, Korbas S, Schneider S. Diminished glucagon suppression after beta-cell reduction is due to impaired alpha-cell function rather than an expansion of alpha-cell mass. Am J Physiol Endocrinol Metab. 2011;300(4):E717–E723. | ||
Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404(6779):787–790. | ||
Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes. 2003;52(1):1–8. | ||
Wang GG, Li W, Lu XH, Zhao X, Xu L. Taurine attenuates oxidative stress and alleviates cardiac failure in type I diabetic rats. Croat Med J. 2013;54(2):171–179. | ||
Leite AC, Araujo TG, Carvalho BM, Silva NH, Lima VL, Maia MB. Parkinsonia aculeata aqueous extract fraction: biochemical studies in alloxan-induced diabetic rats. J Ethnopharmacol. 2007;111(3):547–552. | ||
Keul P, Sattler K, Levkau B. HDL and its sphingosine-1-phosphate content in cardioprotection. Heart Fail Rev. 2007;12(3–4):301–306. | ||
Howard-Alpe GM, Sear JW, Foex P. Methods of detecting atherosclerosis in non-cardiac surgical patients; the role of biochemical markers. Br J Anaesth. 2006;97(6):758–769. | ||
Apple FS. Tissue specificity of cardiac troponin I, cardiac troponin T and creatine kinase-MB. Clin Chim Acta. 1999;284(2):151–159. | ||
Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112(17):2686–2695. | ||
Steinberg HO, Paradisi G, Hook G, Crowder K, Cronin J, Baron AD. Free fatty acid elevation impairs insulin-mediated vasodilation and nitric oxide production. Diabetes. 2000;49(7):1231–1238. | ||
McKim SE, Gäbele E, Isayama F, et al. Inducible nitric oxide synthase is required in alcohol-induced liver injury: studies with knockout mice. Gastroenterology. 2003;125(6):1834–1844. | ||
Silambarasan T, Raja B. Diosmin, a bioflavonoid reverses alterations in blood pressure, nitric oxide, lipid peroxides and antioxidant status in DOCA-salt induced hypertensive rats. Eur J Pharmacol. 2012;679(1–3):81–89. | ||
Amaral LM, Pinheiro LC, Guimaraes DA, et al. Antihypertensive effects of inducible nitric oxide synthase inhibition in experimental pre-eclampsia. J Cell Mol Med. 2013;17(10):1300–1307. | ||
Mahmoud AM, Al Dera HS. 18b-Glycyrrhetinic acid exerts protective effects against cyclophosphamide-induced hepatotoxicity: potential role of PPARgamma and Nrf2 upregulation. Genes Nutr. 2015;10(6):41. | ||
Arthur JR. The glutathione peroxidases. Cell Mol Life Sci. 2000;57(13–14):1825–1835. | ||
Ferreira L, Teixeira-de-Lemos E, Pinto F, et al. Effects of sitagliptin treatment on dysmetabolism, inflammation, and oxidative stress in an animal model of type 2 diabetes (ZDF rat). Mediators Inflamm. 2010;2010:592760. | ||
Kelleni MT, Amin EF, Abdelrahman AM. Effect of metformin and sitagliptin on doxorubicin-induced cardiotoxicity in rats: impact of oxidative stress, inflammation, and apoptosis. J Toxicol. 2015;2015:424813. | ||
Alam MA, Chowdhury MR, Jain P, Sagor MA, Reza HM. DPP-4 inhibitor sitagliptin prevents inflammation and oxidative stress of heart and kidney in two kidney and one clip (2K1C) rats. Diabetol Metab Syndr. 2015;7:107. | ||
Rajesh M, Mukhopadhyay P, Batkai S, et al. Cannabidiol attenuates cardiac dysfunction, oxidative stress, fibrosis, and inflammatory and cell death signaling pathways in diabetic cardiomyopathy. J Am Coll Cardiol. 2010;56(25):2115–2125. | ||
Suzuki H, Kayama Y, Sakamoto M, et al. Arachidonate 12/15-lipoxygenase-induced inflammation and oxidative stress are involved in the development of diabetic cardiomyopathy. Diabetes. 2015;64(2):618–630. | ||
Seddon M, Looi YH, Shah AM. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart. 2007;93(8):903–907. | ||
Mahmoud AM, Ashour MB, Abdel-Moneim A, Ahmed OM. Hesperidin and naringin attenuate hyperglycemia-mediated oxidative stress and proinflammatory cytokine production in high fat fed/streptozotocin-induced type 2 diabetic rats. J Diabetes Complications. 2012;26(6):483–490. | ||
Dinh W, Futh R, Nickl W, et al. Elevated plasma levels of TNF-alpha and interleukin-6 in patients with diastolic dysfunction and glucose metabolism disorders. Cardiovasc Diabetol. 2009;8:58. | ||
Duerrschmid C, Crawford JR, Reineke E, et al. TNF receptor 1 signaling is critically involved in mediating angiotensin-II-induced cardiac fibrosis. J Mol Cell Cardiol. 2013;57:59–67. | ||
Westermann D, Van Linthout S, Dhayat S, et al. Tumor necrosis factor-alpha antagonism protects from myocardial inflammation and fibrosis in experimental diabetic cardiomyopathy. Basic Res Cardiol. 2007;102(6):500–507. | ||
Zhao W, Zhao T, Chen Y, Ahokas RA, Sun Y. Oxidative stress mediates cardiac fibrosis by enhancing transforming growth factor-beta1 in hypertensive rats. Mol Cell Biochem. 2008;317(1–2):43–50. | ||
Wang L, Li J, Li D. Losartan reduces myocardial interstitial fibrosis in diabetic cardiomyopathy rats by inhibiting JAK/STAT signaling pathway. Int J Clin Exp Pathol. 2015;8(1):466–473. | ||
Serrano AL, Baeza-Raja B, Perdiguero E, Jardi M, Munoz-Canoves P. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 2008;7(1):33–44. | ||
Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.