Back to Journals » International Journal of Nanomedicine » Volume 13
siRNA-loaded selenium nanoparticle modified with hyaluronic acid for enhanced hepatocellular carcinoma therapy
Authors Xia Y, Guo M, Xu TT, Li YH, Wang CB, Lin ZF, Zhao MQ, Zhu B ![]()
Received 19 November 2017
Accepted for publication 24 January 2018
Published 15 March 2018 Volume 2018:13 Pages 1539—1552
DOI https://doi.org/10.2147/IJN.S157519
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Yu Xia, Min Guo, Tiantian Xu, Yinghua Li, Changbing Wang, Zhengfang Lin, Mingqi Zhao, Bing Zhu
Virus Laboratory, Guangzhou Institute of Pediatrics, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangzhou, Guangdong, People’s Republic of China
Background: Small interfering RNA (siRNA) as a new therapeutic modality holds promise for cancer treatment. However, the traditional viral carriers are prone to immunogenicity and risk of insertional mutagenesis.
Methods: In order to provide a tumor-targeted delivery carrier of siRNA in cancer therapy, the hyaluronic acid (HA)-selenium (Se)-polyethylenimine (PEI) nanoparticle (NP) was fabricated by decorating SeNP with HA as a tumor-targeting moiety and by linking the polycationic polymers polyethylenimine PEI onto the surface of SeNP. The siRNA was loaded to the surface of SeNP HA-Se-PEI via the electrostatic interaction between siRNA and PEI to prepare the functionalized SeNP HA-Se-PEI@siRNA.
Results: The HA-Se-PEI@siRNA was internalized into the HepG2 cell mainly in a clathrin-mediated endocytosis manner. Owing to the active tumor-targeted effect mediated by HA, HA-Se-PEI@siRNA achieved the obvious higher transfection efficiency, greater gene silencing ability, and stronger cytotoxicity in the HepG2 cell compared with the passive tumor-targeted NP Se-PEI@siRNA. The knockdown of hairy and enhancer of split 5 by HA-Se-PEI@siRNA induced the HepG2 cell cycle arrest at the G0/G1 phase and apoptosis. Furthermore, the treatment with HA-Se-PEI@siRNA resulted in greater antitumor efficacy compared with the Se-PEI@siRNA in vitro and in vivo. In addition, the HA-Se-PEI@siRNA was almost no toxic to the key organs of mice.
Conclusion: These findings provided an alternative therapeutic route for targeted cancer treatments.
Keywords: hepatocellular carcinoma, functionalized nanoparticle, siRNA delivery, gene therapy, tumor targeting
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer and the second main cause of cancer-related deaths worldwide.1,2 Owing to the low percentage of HCC patients who are eligible for surgery and the high tumor recurrence rates, chemotherapy is still a major clinical method for HCC treatments.3 Many traditional chemotherapy drugs have been utilized for the HCC therapy, but most of them have been ineffective.4 Recently, the gene therapy using small interfering RNA (siRNA) technology has attracted increasing attention in the treatments of cancers because of the enhanced anticancer efficiency and low cytotoxicity.5,6 Virus is usually utilized for traditional gene carriers.7 However, the viral carriers are prone to immunogenicity and the risk of insertional mutagenesis.8 Therefore, nonviral carriers have great potential in the application of gene therapy due to their better safety profile.
A variety of nanosized drug carriers, including liposomes,9 gold NP,10 and mesoporous silica,11 have been developed to prepare antitumor nanomedicines with passive tumor-targeting properties due to the “enhanced permeability and retention” effect.12 Among these nanomaterials, selenium nanoparticles (SeNPs) have attracted increasing attention in the field of drug delivery vehicle in cancer therapy.13,14 Selenium (Se) is a mineral trace element of fundamental importance to humans and animals.15 The role of Se as a potential cancer chemotherapeutic and chemopreventive agent has been supported by many epidemiological, preclinical, and clinical studies.16 Previous studies have shown that Se was degradable in vivo, and the degraded Se could be used as a nutrient for many kinds of normal cells or as an antiproliferative agent for many kinds of cancer cells.17 SeNPs also showed many merits in the gene delivery systems, such as the desired size, high gene loading capacity, enhanced anticancer effect, and the low cytotoxicity.18 The previous reported literature indicated that SeNPs were capable of conjugating with siRNA to achieve enhanced anticancer activity in vitro.19 Nevertheless, there is still some deficiency in such delivery vehicle for effectively carrying the genes, such as the lack of active tumor-targeting activity.20 In the past decade, many tumor-targeting molecules have been introduced to endow NPs with active targeting ability.21 Recently, hyaluronic acid (HA) as an active tumor-targeting moiety has achieved more attention in the field of targeting delivery carriers. HA is a kind of natural acidic polysaccharide macromolecule; thus, it is generally regarded as biodegradable and nontoxic.22 The overexpression of HA-binding receptor, such as CD44, has been found on the cell surface of some malignant tumors, which brings about the broad applications of HA-based NP in active tumor targeting for anticancer drugs.23 In the present study, to prepare the actively tumor-targeted delivery carrier, the HA was conjugated onto the surfaces of SeNPs to fabricate the HA-Se NP. Then, the positively charged polycationic polymer polyethylenimine (PEI) was conjugated onto the surfaces of SeNPs for binding with siRNA.
Hairy and enhancer of split 5 (HES5) is a DNA-binding transcription factor, and it can repress the transcription of Hash1 and subsequently inhibit the cell differentiation.24 Previous studies have shown that HES5 plays an important role in the initiation or/and development of cancer and gradually developed as a crucial therapeutic target in tumor treatments.25 Therefore, in the present study, the HES5-siRNA was loaded onto the surface of SeNPs modified with tumor-targeted moiety HA to fabricate functionalized SeNP HA-Se-PEI@siRNA aiming at silencing HES5 gene for inhibiting the tumor growth. As expected, the HA-Se-PEI@siRNA could selectively accumulate in HepG2 tumors and exhibited improving gene silencing effectiveness and antitumor efficacy without obviously observable side effects. Therefore, such HA-conjugated SeNP for the delivery of HES5-siRNA to the HepG2 tumor holds great potential applications in HCC therapy.
Materials and methods
Materials
Sodium selenite (Na2SeO3), ascorbic acid (vitamin C), and HA (molecular weight [MW] =210 kDa) were purchased from Guangzhou Chemical Reagent Factory (Guangzhou, People’s Republic of China); 6-diamidino-2-pheylindole and Annexin V–fluorescein isothiocyanate (FITC) Apoptosis Kit was purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, People’s Republic of China). Cy5.5 was purchased from Beyotime (Shanghai, People’s Republic of China). All the antibodies were purchased from Cell Signaling Technology (MA, USA). The siRNA was obtained from RiboBio (Guangzhou, People’s Republic of China), and HES5-siRNA sequence was as follows: 5′-AAGGCTACTCGTGGTGCCT-3′.
Synthesis of HA-Se-PEI@siRNA
The HA-conjugated SeNPs were synthesized as previously described.26 Briefly, 5 mL of ascorbic acid (4 mM) was added to 5 mL of Na2SeO3 (1 mM), and mixed solutions were stirred for 4 hours to prepare the SeNPs; then, 0.5 mg HA dissolved with deionized water was added to the SeNP solutions, and the solutions were stirred for another 4 hours to prepare the SeNPs conjugated with HA. The 0.001% PEI (MW =25 kDa) solution was added to SeNP solution and kept for 2 hours in order to prepare HA-Se-PEI NP solution. Finally, the mixed solutions were dialyzed (3.5 kDa) to remove the redundant HA, ascorbic acid, Na2SeO3, and PEI. The HA-Se-PEI@siRNA complex was obtained as previously reported. Briefly, the HA-Se-PEI NP was dispersed in DNase-/RNase-free water and then vortexed with an siRNA solution for 1 hour to form HA-Se-PEI@siRNA complex. The NP was characterized by transmission electron microscopy (TEM) and Fourier transform infrared (FTIR) spectroscopy. Energy-dispersive X-ray (EDX) spectroscopy was conducted to examine the elemental composition of NP. The average sizes and zeta potentials of NPs were tested via Zetasizer™ nano analyzer (Malvern Instruments Ltd., Malvern, UK).
In order to examine the siRNA loading capability, we established a calibration curve against the FAM-siRNA and obtained the relative fluorescence intensity of FAM-siRNA in the NP. The test was performed in an EnSpire® Multimode Plate Reader (PerkinElmer Inc., Waltham, MA, USA; for FAM-siRNA: Ex, 465 nm; Em, 520 nm). The loading efficiency and loading contents of siRNA were calculated by using the following formulas: loading efficiency (%) = (weight of loaded siRNA/weight of siRNA in feed) × 100%; loading contents (%) = (weight of loaded siRNA/weight of HA-Se-PEI@siRNA) × 100%.
Agarose gel retardation assay
HA-Se-PEI@siRNA NP complexes with various nitrogen/phosphorus (N/P) ratios were fabricated, and the concentrations of siRNA were fixed at 1 μM. Then, the HA-Se-PEI@siRNA NP complexes were subjected to gel electrophoresis in 1% agarose gel with 0.5 mg/mL ethidium bromide at 120 mV for 15 minutes, and the gel imaging was visualized by using an ultraviolet gel image system. The Se-PEI@siRNA NP complex was fabricated and electrophoresed according to the similar process above.
Cell culture
The HepG2 cell and Lo2 cell were purchased from the American Type Culture Collection (Manassas, VA, USA) and incubated in DMEM containing 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C in 5% CO2 atmosphere.
Cellular uptake of NP
HepG2 cells (5×104 cells/well) were seeded in 24-well plate and incubated overnight. The cells were exposed with the different formulations of FAM-siRNA at siRNA dose of 100 nM and incubated for 4 hours. Then, the cells were washed with phosphate-buffered saline (PBS) and measured by using fluorescence microscope (Leica DMi8; Leica Microsystems, Wetzlar, Germany) or flow cytometer (BD FACSCalibur System, San Jose, CA, USA).
Uptake pathways of HA-Se-PEI@siRNA
The cellular uptake of NP in the presence of different uptake inhibitors was investigated in HepG2 cells (3×105 cells), which were incubated at 4°C for 0.5 hours without inhibitors or with 3 mg/mL NaN3 +50 mM 2-deoxy-D-glucose (DOG), nystatin (5 μg/mL), chlorpromazine (5 μg/mL), and amiloride (10 μg/mL) in serum-free media at 37°C for 30 minutes. Then, HA-Se-PEI@FAM-siRNA was added to the media and incubated for another 2 hours. Then, the cells were washed with cold PBS and measured by flow cytometry.
Cell transfection
HepG2 cells (2.0×105 cells/well) were seeded in six-well plates and incubated overnight at 37°C under 5% CO2. Before transfection, the medium was discarded, and the cells were rinsed with PBS solution. Then, the cells were treated with HA-Se-PEI@siNC, Lipofectamine® 2000@siRNA, Se-PEI@siRNA, and HA-Se-PEI@siRNA, respectively (at the equivalent siRNA concentration of 100 nM). The untreated cells were used as control. After 12 hours of incubation, the medium in each well was replaced with complete medium, and the cells were cultured for another 48 hours.
Quantitative real-time polymerase chain reaction (qRT-PCR)
In this study, qRT-PCR was used to evaluate the expression of HES5-mRNA. HepG2 cells (2.0×105 cells/well) seeded in six-well plates were transfected by HA-Se-PEI@siNC, Lipofectamine 2000@siRNA, Se-PEI@siRNA, and HA-Se-PEI@siRNA, respectively (at the equivalent siRNA concentration of 100 nM) for 48 hours. Then, TRIzol® reagent was used to extract the total cell RNA, and all operations were carried out on ice. The concentration of the RNA was measured by NanoDrop™ 1000 Spectrophotometer (Thermo Scientific, South Logan, USA). The StepOne™ PCR System (Thermo Fisher Scientific, Waltham, MA, USA) was utilized to analyze the data using the 2−(ΔΔCT) method. The sequences of primer are as follows: forward 5′-GGAATTCCAATGGCCCCCAGCACTGTG-3′ and reverse 5′-GGGTACCCCACGGCCAC AGTGCTGG-3′ for HES5; and forward 5′-ATCCCATCACCATCTTCCAG-3′ and reverse 5′-ATGAGTCCTTCCACGATACC-3′ for glyceraldehyde 3-phosphate dehydrogenase.
MTT assay
The cytotoxicity was determined by MTT assay.27 Briefly, 5×104 cells were incubated in a 96-well cell culture plate for 24 hours. The cells were treated with individual agents in various siRNA equivalent concentrations for another 48 hours; 25 μL MTT solutions with the concentration of 5 mg/mL were added into each well for 4 hours of incubation at 37°C. The medium was removed carefully, and DMSO was added to dissolve the formazan dye. The OD value was tested at 570 nm.
Flow cytometer analysis
The cell cycle distributions and cell apoptosis in HepG2 cell were analyzed by flow cytometry as follows: briefly, HepG2 cells (1.0×105 cells/well) were incubated in 12-well plates, allowed to reach 50% confluence, and then treated with different formulations of siRNA (at an equivalent concentration to 100 nM of siRNA). The cells were collected after 24 hours of treatments for the cell cycle distribution analysis or after 48 hours of treatments for the cell apoptosis detection, and then, the collected cells were stained with propidium iodine (PI) or Annexin V–FITC/PI in the dark for 30 minutes, respectively. The stained cells were immediately tested by FACS flow cytometer (BD Biosciences, San Jose, CA, USA), and the results were analyzed by a FlowJo software (Tree Star Inc., Ashland, OR, USA).
Western blotting analysis
The Western blotting experiment was carried out to determine the protein expression levels. Briefly, HepG2 cells (5.0×105 cells/well) were cultured in the six-well plate for 12 hours to reach 70% confluence and treated with different formulations of siRNA (at an equivalent siRNA concentration of 100 nM) for another 24 hours; then, the cells were collected and lysed with 50 μL RIRP containing 1% phenylmethylsulfonyl fluoride. Lysates were collected by centrifugation after 15 minutes. The protein concentration was estimated by the BCA Protein Assay Kit (Thermo Fisher Scientific).
In vivo imaging of NP
First, 1×107 HepG2 cells were subcutaneously injected in the abdomen of female BALB/c nude mice (about 5 weeks of age). After the tumors reached ~200 mm3, the tumor-bearing nude mice were administrated by tail vein injection with the [email protected] and [email protected] NPs at the siRNA equivalent doses of 0.5 mg/kg, respectively. After 4 hours of intravenous injection with NPs, the fluorescence imaging of the mice was recorded at the IVIS® imaging system (Xenogen Co., Alameda, CA, USA).
Xenograft mouse model
BALB/c nude mice at ~5–6 weeks of age were used for investigating the in vivo antitumor efficacy. Approximately 1×107 HepG2 cell was resuspended in 150 μL saline and subcutaneously injected in the abdomen of the mice. When tumors grew to about 100 mm3, all mice were divided into four groups randomly; then, saline (as control group) and different formulations of siRNA (at the siRNA equivalent dose of 0.5 mg/kg) were injected to mice intravenously once every other day during the experimental period. The volumes of tumors were calculated by the following formula:
The tumor volume (mm3) = 1/2 × Length × Width2. |
Histology and immunohistochemistry
The tumors stripped from the mice and main organ (liver, heart, kidney, spleen, and lung) tissues were fixed with 3.7% paraformaldehyde and sectioned into 6-μm slices. The histological sections of tissues were used for H&E staining. The expression level of Ki67 protein associated with tumor cell growth was examined by immunohistochemical methods. The angiogenesis-related protein CD31 was tested by immunohistochemistry according to the manufacturer’s protocols. The imaging of sections was acquired by a digital microscope (Leica DMi8). All animal experiments were approved by the Ethics Committee of Guangzhou Medical University and performed according to the protocols and guidelines of the Experimental Animal Center of Guangzhou Medical University.
Statistical analysis
All data are represented as the mean ± standard deviation (SD). The statistical significance of differences was found by using a Student’s t-test. Student’s t-test was performed for comparison between the two groups; p<0.05 and p<0.01 were considered statistically significant and highly significant differences, respectively.
Results and discussion
Preparation and characterization of HA-Se-PEI NP
In this study, a novel tumor-targeted delivery carrier HA-Se-PEI was designed and synthesized. In this gene delivery system, the selenite was reduced to become Se atoms by the redox reaction after adding ascorbic acid, and the accumulating Se atoms led to a formation of SeNPs.28 The SeNPs were loaded with the HA and then linked with polycationic polymer PEI to prepare the active tumor-targeting gene carrier HA-Se-PEI NP. The HES5-siRNA was linked with the PEI on the surface of SeNP via electrostatic interaction. HA was used as the tumor-targeting molecule, and PEI was functionalized as siRNA-binding moiety. The HA-Se-PEI NP was characterized by Nano ZS particle analyzer, TEM, EDX spectroscopy, and FTIR spectroscopy. Nano ZS particle analyzer showed that the average size of HA-Se-PEI is about 136 nm (Figure 1A). TEM images indicated that HA-Se-PEI exhibited monodisperse spherical particles with the size range of 70–180 nm (Figure 1B). As shown in Figure 1C, the strong signals from the Se atoms together with the signal of C and O atoms that from HA were observed, indicating that HA was successfully loaded onto the SeNPs. Then, the FTIR spectrums were further utilized to confirm the conjugation between the SeNPs and HA. As shown in Figure 1D, the characteristic peaks of SeNPs also appeared in the spectrum of HA-Se NP, and the HA displayed the characteristic peak of amide at 1,657 cm−1 and the peak of the telescopic vibration of C−H bond in −CH2− moieties at 2,922 cm−1. After loading SeNPs with HA, the obvious characteristic peaks at 1,657 cm−1 and 2,918 cm−1 were also detected, suggesting that the HA was successfully conjugated with SeNPs.
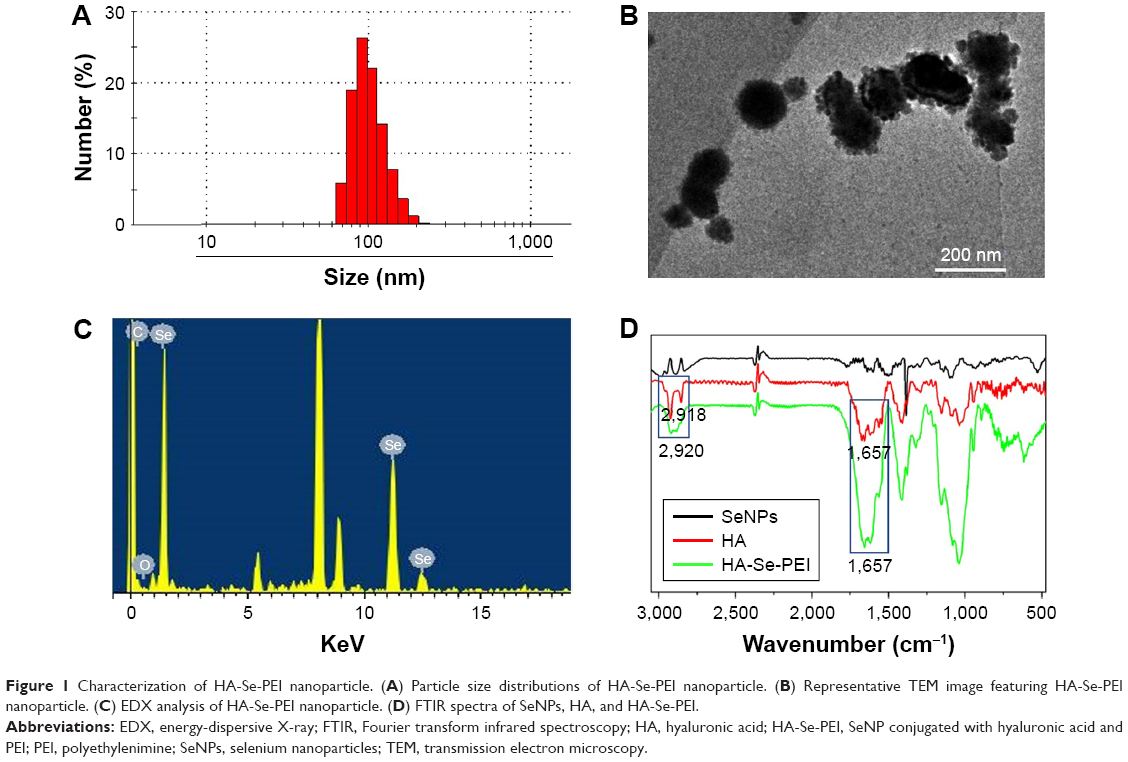 | Figure 1 Characterization of HA-Se-PEI nanoparticle. (A) Particle size distributions of HA-Se-PEI nanoparticle. (B) Representative TEM image featuring HA-Se-PEI nanoparticle. (C) EDX analysis of HA-Se-PEI nanoparticle. (D) FTIR spectra of SeNPs, HA, and HA-Se-PEI. |
The size of HA-Se-PEI@siRNA was also characterized by Nano ZS particle analyzer and TEM. Figure 2A and B shows that the size of HA-Se-PEI@siRNA was about 150 nm. The zeta potentials of NP and siRNA were shown in Figure 2C; the potential of SeNP loaded with hyaluronic acid (HA-Se) was about −22 mV, and after loading the positively charged PEI onto the surface of HA-Se NP, the potential changed from about −22 mV to +25 mV, which was helpful for the NP to load siRNA. Furthermore, the size distribution of HA-Se-PEI remained stable (<220 nm) for 15 days (Figure 2D). The favorable stability of HA-Se-PEI supports its potential application in medical area.
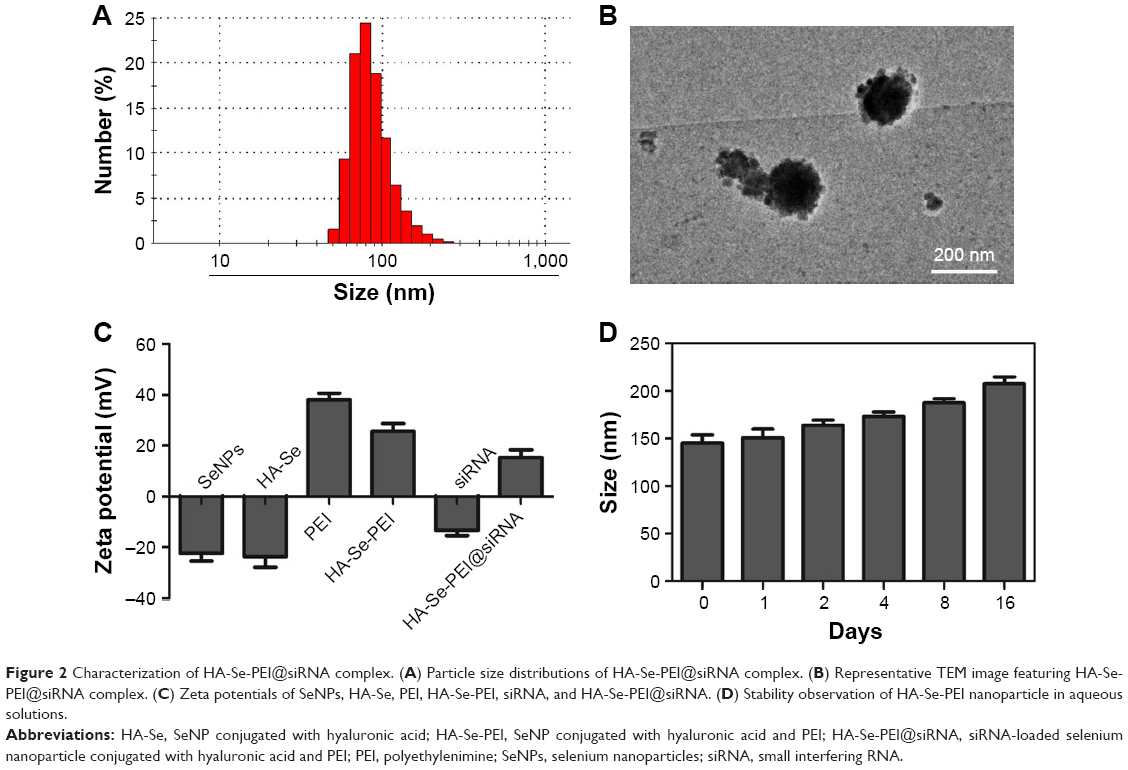 | Figure 2 Characterization of HA-Se-PEI@siRNA complex. (A) Particle size distributions of HA-Se-PEI@siRNA complex. (B) Representative TEM image featuring HA-Se-PEI@siRNA complex. (C) Zeta potentials of SeNPs, HA-Se, PEI, HA-Se-PEI, siRNA, and HA-Se-PEI@siRNA. (D) Stability observation of HA-Se-PEI nanoparticle in aqueous solutions. |
Study on siRNA loading capability
The agarose gel experiment was performed to investigate the siRNA loading ability of Se-PEI and HA-Se-PEI NPs. Complete retardations of siRNA migration were achieved at N/P rate of 8:1 in the Se-PEI@siRNA or HA-Se-PEI@siRNA groups (Figure 3), indicating that both Se-PEI and HA-Se-PEI NPs could bind the siRNA to impede degradation in the process of electrophoresis. These results showed that SeNPs could effectively protect the siRNA from degradation by serum nucleases and have potential to prolong the circulation of siRNA in the blood.
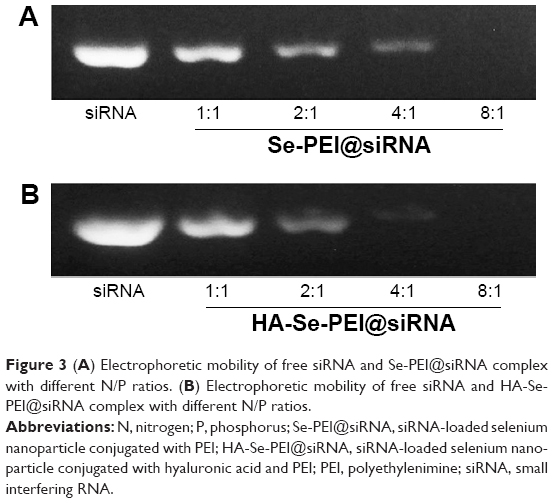 | Figure 3 (A) Electrophoretic mobility of free siRNA and Se-PEI@siRNA complex with different N/P ratios. (B) Electrophoretic mobility of free siRNA and HA-Se-PEI@siRNA complex with different N/P ratios. |
Cellular uptake of NP
Cellular uptake is closely related to siRNA delivery efficiency.29 In general, high cellular uptake of siRNA leads to high efficiency of gene silencing. In this experiment, the fluorescence microscope was used to observe the cellular uptake of passive-targeted NP Se-PEI@FAM-siRNA and active-targeted NP HA-Se-PEI@FAM-siRNA in HepG2 cells. As shown in Figure 4A and B, after 1-hour incubation, the green fluorescence from FAM-siRNA was observed in HepG2 cells exposed to Se-PEI@FAM-siRNA or HA-Se-PEI@FAM-siRNA, indicating that such NP could be internalized by HepG2 cells irrespective of the active targeting moieties. The intensity of the fluorescence in both the Se-PEI@FAM-siRNA- and HA-Se-PEI@FAM-siRNA-treatment groups increased as the incubation time prolonged to 4 hours. Interestingly, the exposure of HepG2 cells to the HA-Se-PEI@FAM-siRNA NP led to a greater cellular uptake than that in Se-PEI@FAM-siRNA group, presumably because of HA-mediated specific endocytosis.
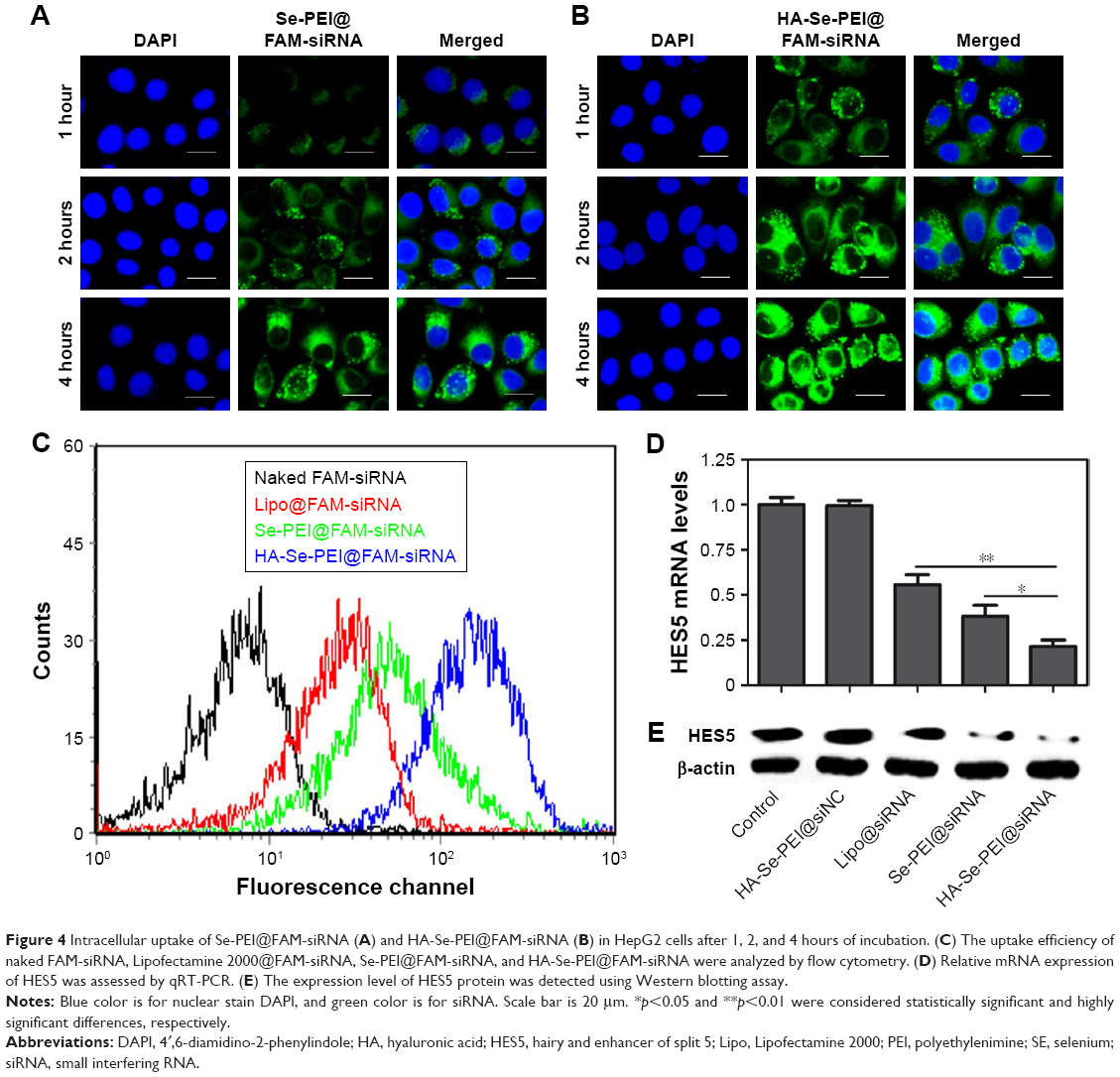 | Figure 4 Intracellular uptake of Se-PEI@FAM-siRNA (A) and HA-Se-PEI@FAM-siRNA (B) in HepG2 cells after 1, 2, and 4 hours of incubation. (C) The uptake efficiency of naked FAM-siRNA, Lipofectamine 2000@FAM-siRNA, Se-PEI@FAM-siRNA, and HA-Se-PEI@FAM-siRNA were analyzed by flow cytometry. (D) Relative mRNA expression of HES5 was assessed by qRT-PCR. (E) The expression level of HES5 protein was detected using Western blotting assay. |
The previous reports showed that the NP entered the cells through the energy-dependent endocytic routes.30 Thus, we researched whether the cellular uptake of NP was influenced by the temperature. The cellular uptake of NP in the HepG2 cells incubated at 4°C was decreased ~74.1% (Figure S1), suggesting that the HA-Se-PEI@siRNA NP was internalized into the cells in an energy-dependent manner. The pretreatment with NaN3/DOG (cell energy metabolism inhibitor) also significantly decreased the uptake of NP in HepG2 cells, which indicated that the endocytosis of the HA-Se-PEI@siRNA NP was an energy-dependent active cellular process. Moreover, various endocytic inhibitors were utilized to explore the internalization mechanism of the NP. Amiloride, nystatin, and chlorpromazine were reported to suppress micropinocytosis endocytosis, caveolae-mediated cellular uptake, and clathrin-associated cellular uptake, respectively. After pretreatment with amiloride and nystatin, the cellular uptakes of NP were decreased ~24.7% and 28.9%, respectively. However, the pretreatment with chlorpromazine resulted in 53.5% decrease in cellular uptake, which indicated that clathrin-mediated endocytosis predominantly played a key role in the cellular uptake of the HA-Se-PEI@siRNA NP in HepG2 cells.
To precisely examine the uptake efficiency of siRNA, the HepG2 cell was cocultured with naked FAM-siRNA, Lipofectamine 2000@FAM-siRNA, Se-PEI@FAM-siRNA, and HA-Se-PEI@FAM-siRNA for 24 hours, respectively, and then, the uptake efficiency of siRNA was analyzed by FACS flow cytometer. As shown in Figure 4C, the fluorescence signal of naked FAM-siRNA was set as a control group, and the fluorescence intensity in the cells cultured with HA-Se-PEI@FAM-siRNA was stronger than that in the Se-PEI@FAM-siRNA and Lipofectamine 2000@FAM-siRNA groups, suggesting that HA-Se-PEI@FAM-siRNA exhibited higher uptake efficiency. The expression levels of CD44 in normal cells and cancer cells were also examined by Western blotting assay. Figure S2 shows that the protein expression level of CD44 in HepG2 cells was remarkably higher than that in Lo2 cells, indicating that HA-mediated cellular uptake promoted the internalization of NP in HepG2 cells.
Downregulation of gene expression
The active tumor-targeted HA-Se-PEI NP was utilized to deliver siRNA into the HepG2 cell and to knockdown the gene expression of HES5 in the HepG2 cell. After the cell was transfected with different formulations of siRNA for 24 hours, the mRNA levels of HES5 were assessed by qRT-PCR. Figure 4D showed that HA-Se-PEI@siNC failed to silence HES5 expression. The Lipofectamine 2000@siRNA moderately decreased the mRNA expression of HES5 in HepG2 cells, while HA-Se-PEI@siRNA significantly downregulated the mRNA expression of HES5 in HepG2 cells. As expected, HA-Se-PEI@siRNA showed stronger ability to silence the HES5 expression compared with Se-PEI@siRNA.
The protein expression levels of HES5 in HepG2 cells were detected via Western blotting. Figure 4E shows that the delivery of HES5-siRNA by the active targeting NP HA-Se-PEI exhibited stronger activity to inhibit the protein expression of HES5 than that by the passive targeting NP Se-PEI@siRNA. However, HA-Se-PEI@siNC had no effect on the protein expression level of HES5, suggesting that no nonspecific gene silencing occurred in this kind of delivery system. The results of both qRT-PCR and Western blotting showed that the HA-Se-PEI@siRNA could significantly silence the expression of HES5.
In vitro cytotoxicity study
The cytotoxicity of the NP was studied by MTT.31 Figure 5A shows that the cell viabilities in HA-Se-PEI@siNC-treatment group remained about 93.5% in comparison with the untreated cells, indicating no nonspecific cytotoxicity against HepG2 cells in this gene delivery carrier. HA-Se-PEI@siRNA showed significantly higher cytotoxicity than Se-PEI@siRNA as expected, and the Lipofectamine 2000@siRNA NP displayed moderate cell viability. The cell toxicity of Se-PEI@siRNA and HA-Se-PEI@siRNA in the normal liver cell Lo2 was also evaluated by MTT. As shown in Figure 5B, after 48 hours of treatments with Se-PEI@siRNA or HA-Se-PEI@siRNA, the Lo2 cell viabilities remained nearly 92.4% and 91.7%, respectively, indicating their low toxic and side effects.
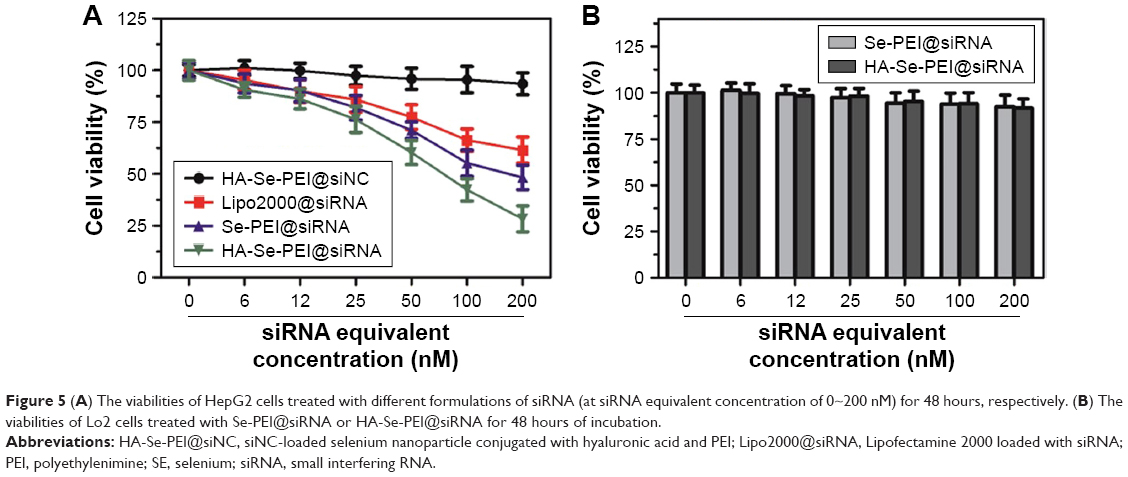 | Figure 5 (A) The viabilities of HepG2 cells treated with different formulations of siRNA (at siRNA equivalent concentration of 0~200 nM) for 48 hours, respectively. (B) The viabilities of Lo2 cells treated with Se-PEI@siRNA or HA-Se-PEI@siRNA for 48 hours of incubation. |
Cell cycle arrest and cell apoptosis
The cell cycle distribution of the HepG2 cell after treatment with different formulations of siRNA for 24 hours was analyzed by flow cytometry.32 As shown in Figure 6A, an obviously reduced populations at the S and G2/M phases were detected, and meanwhile, a prominent cell cycle arrest at G0/G1 phase occurred after the cells were incubated with the siRNA in the different formulations. Compared with Lipofectamine 2000@siRNA, the Se-PEI@siRNA and HA-Se-PEI@siRNA NP displayed slightly greater effects on the cell cycle distribution. Meanwhile, the HA-Se-PEI@siNC exhibited no obvious effect on the cell cycle distribution. This finding indicated that the knockdown of HES5 might repress the proliferation of HepG2 cell by inducing the cell cycle arrest at the G0/G1 phase.
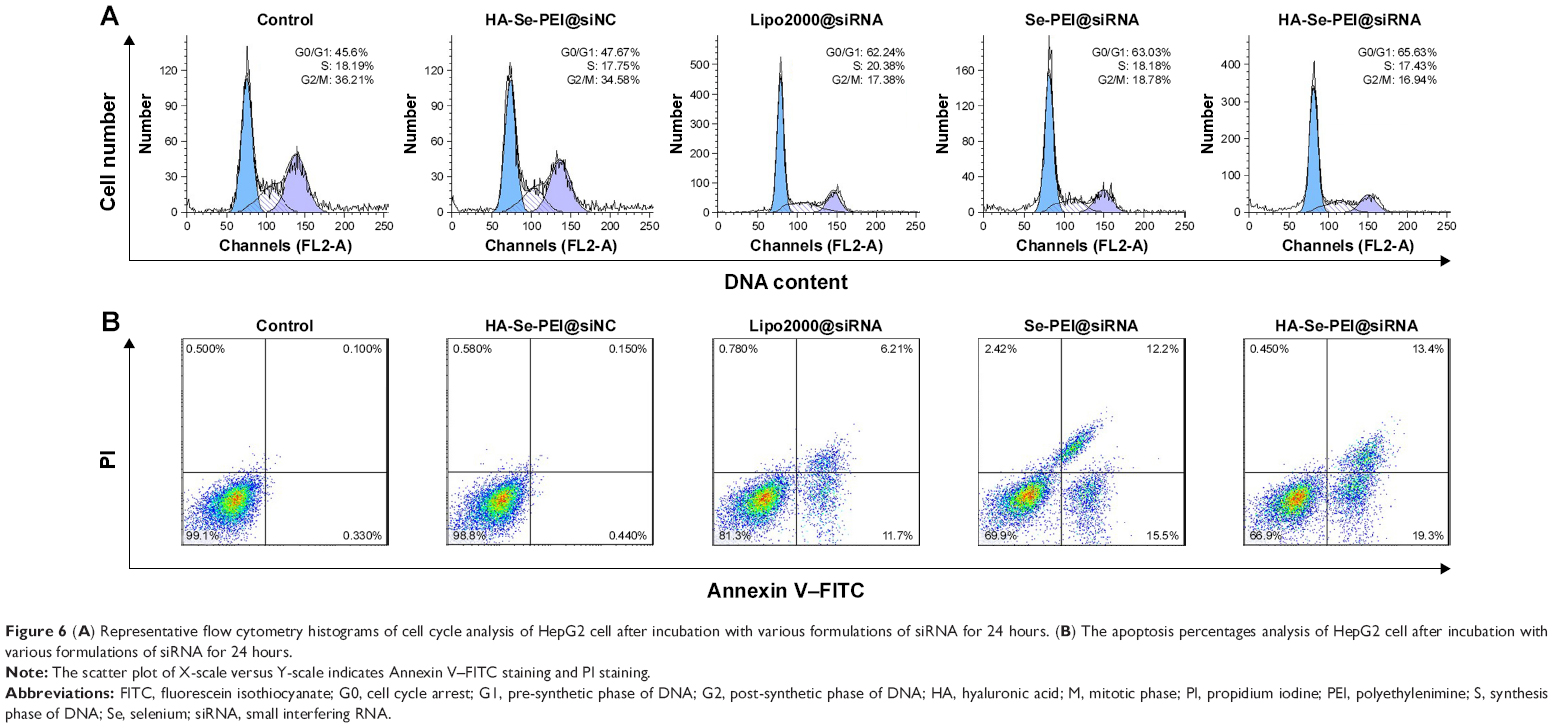 | Figure 6 (A) Representative flow cytometry histograms of cell cycle analysis of HepG2 cell after incubation with various formulations of siRNA for 24 hours. (B) The apoptosis percentages analysis of HepG2 cell after incubation with various formulations of siRNA for 24 hours. |
FITC–Annexin V/PI staining was employed to analyze the cell apoptosis by flow cytometry.33 As shown in Figure 6B, the treatment with HA-Se-PEI@siRNA markedly induced HepG2 cell apoptosis and obtained higher apoptosis rate (~32.7%) compared with Se-PEI@siRNA (~27.7%) and Lipofectamine 2000@siRNA (~17.9%). However, no obvious cell apoptosis was observed in the HA-Se-PEI@siNC-treatment group, suggesting that the silence of HES5 contributed to the apoptosis of HepG2 cells. The FACS analysis showed that HA-Se-PEI@siRNA repressed the proliferation of HepG2 cells mainly by arresting the cell cycle at G0/G1 and subsequently resulted in the cell apoptosis.
In vivo imaging of NP
As efficacious active tumor-targeted delivery vehicles, the vehicles should be capable of selectively delivering the genes to tumors for obtaining the high-efficiency treatment of cancer.34 The in vivo distribution of Se-PEI@siRNA and HA-Se-PEI@siRNA loaded with cy5.5 was assessed via the ex vivo fluorescence imaging after 4 hours of intravenous injection. The mice without injection with NP were set as control group. As shown in Figure 7, no fluorescence signal was captured in control group, suggesting that there was no autofluorescence in the tumor-bearing mice. Nevertheless, obvious fluorescence signals were observed in tumors after 4 hours of intravenous injection with [email protected] or [email protected], and the significant, higher fluorescence intensity was observed in the [email protected] group. These results indicated that the HA-Se-PEI@siRNA NP achieved more efficient tumor accumulation following systemic administration, presumably due to the enhanced avidity of HA ligands to its receptor CD44 molecule overexpressed on HepG2 cells.
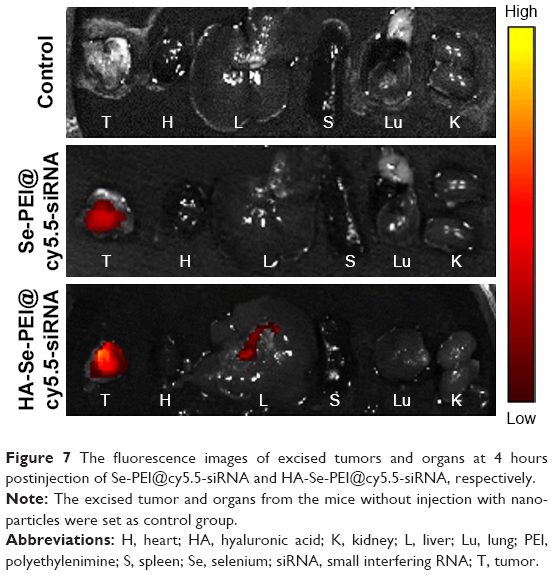 | Figure 7 The fluorescence images of excised tumors and organs at 4 hours postinjection of [email protected] and [email protected], respectively. |
In vivo anticancer activity
The antitumor activity of Se-PEI@siRNA and HA-Se-PEI@siRNA NP was evaluated using HepG2 tumor xenograft model. The mice were intravenously injected with saline, HA-Se-PEI, Se-PEI@siRNA, and HA-Se-PEI@siRNA NPs to test the antitumor activity, respectively. The rapid growth in tumor size over time in different treatment groups was observed in Figure 8A. Compared with the group treated with saline, the HA-Se-PEI did not exhibit obvious inhibition of tumor growth, indicating that the gene carrier HA-Se-PEI NP was low toxic in vivo. The Se-PEI@siRNA exhibited moderate inhibition of tumor growth. However, HA-Se-PEI@siRNA showed greater inhibition of tumor growth compared with Se-PEI@siRNA. The enhanced antitumor activity in the HA-Se-PEI@siRNA-treatment group might attribute to active tumor-targeting ability mediated by HA, resulting in higher drug accumulation in the tumor sites. The tumor images (Figure 8B) also confirmed the remarkable antitumor activity of HA-Se-PEI@siRNA. The body weight was measured every other day up to 21 days. No noteworthy body weight loss was recorded after various treatments, indicating the minimal side effect of the drugs at the tested doses (Figure 8C). In order to verify whether tumor growth inhibition was related to the downregulation of HES5, the mRNA expression level of HES5 in tumors was determined through qRT-PCR. As shown in Figure 8D, as expected, HA-Se-PEI@siRNA exhibited greater ability to downregulate the mRNA expression of HES5 in the tumor compared with Se-PEI@siRNA. However, there was no obvious downregulation of HES5 expression level after the treatment with gene carrier HA-Se-PEI NP, indicating that HA-Se-PEI@siRNA inhibited the tumor growth possibly via the downregulation of HES5 gene.
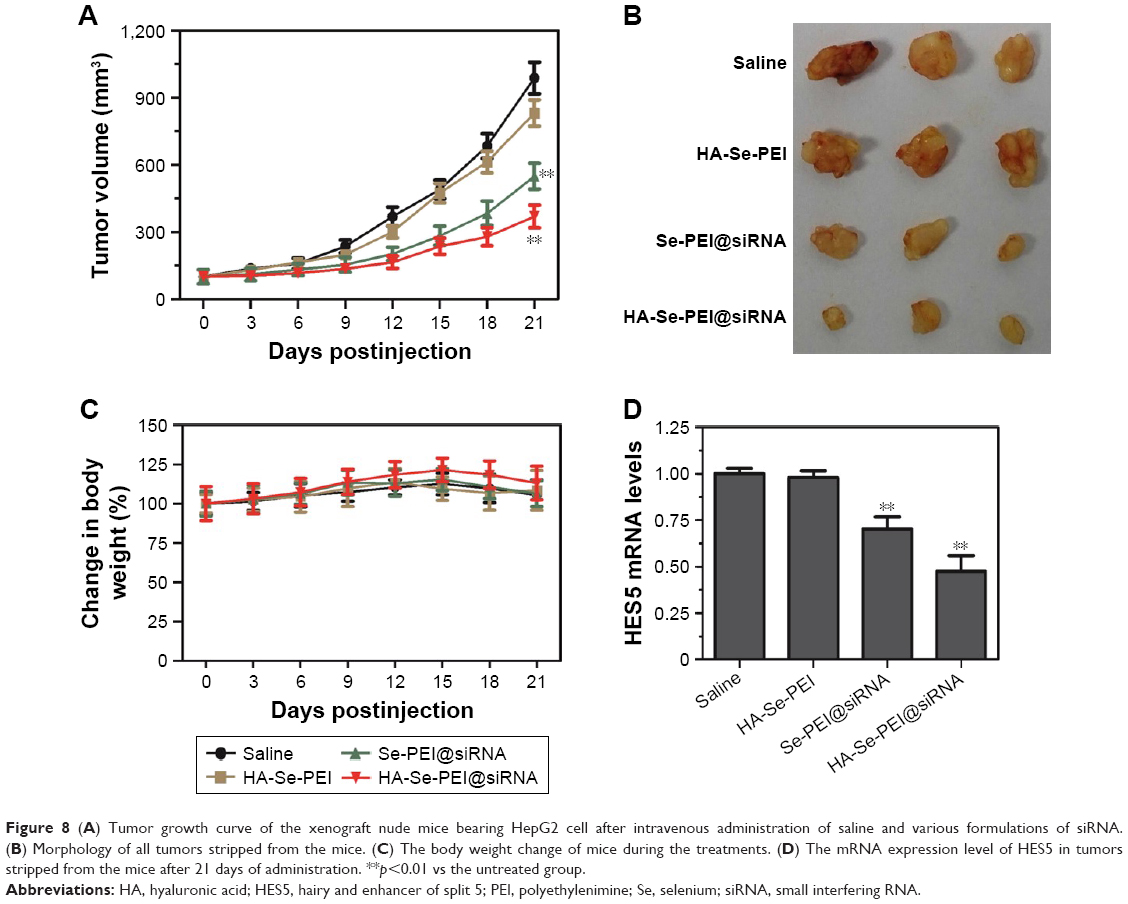 | Figure 8 (A) Tumor growth curve of the xenograft nude mice bearing HepG2 cell after intravenous administration of saline and various formulations of siRNA. (B) Morphology of all tumors stripped from the mice. (C) The body weight change of mice during the treatments. (D) The mRNA expression level of HES5 in tumors stripped from the mice after 21 days of administration. **p<0.01 vs the untreated group. |
To investigate the mechanism of in vivo antitumor of HA-Se-PEI@siRNA NP, the tumor tissues were stained by the H&E,35 and Ki6736 and the expression of CD31,37 a marker for neovasculature, were examined. The tumor necrotic fraction is usually identified by H&E staining.
As shown in Figure 9A, the H&E-stained section of tumor tissue in saline and HA-Se-PEI groups appeared to be hypercellular, and the nuclear polymorphism in tumor tissue was very evident. In contrast, most of the cancer cells in HA-Se-PEI@siRNA-treatment group were lysed and destroyed, indicating that the HA-Se-PEI@siRNA treatment had induced significant cancer cell apoptosis and necrosis in vivo. Ki67 is a nuclear protein linked with cellular proliferation, and the reduction of Ki67 positive cells in tumor sections suggests the proliferation inhibition of cancer cells. In addition, HA-Se-PEI@siRNA treatment resulted in an obvious decrease of Ki67-positive cancer cell, suggesting that the cancer cell proliferation had been significantly suppressed by HA-Se-PEI@siRNA. CD31 reflects the microvessel density of the tumor. The immunostaining with anti-CD31 antibody was used to visualize the formation of microvessels in the tumor mass.38 As shown in Figure 9A, the angiogenesis-positive cells with brown color broadly existed in the saline group. The microvessel density was greatly reduced after the treatment with HA-Se-PEI@siRNA. As expected, the antitumor effect was more pronounced in the HA-Se-PEI@siRNA-treated group as compared to the Se-PEI@siRNA-treated group. These results suggested that HA-Se-PEI@siRNA might inhibit the tumor growth in vivo via inhibiting cancer cell proliferation and CD31-induced vessel formation.
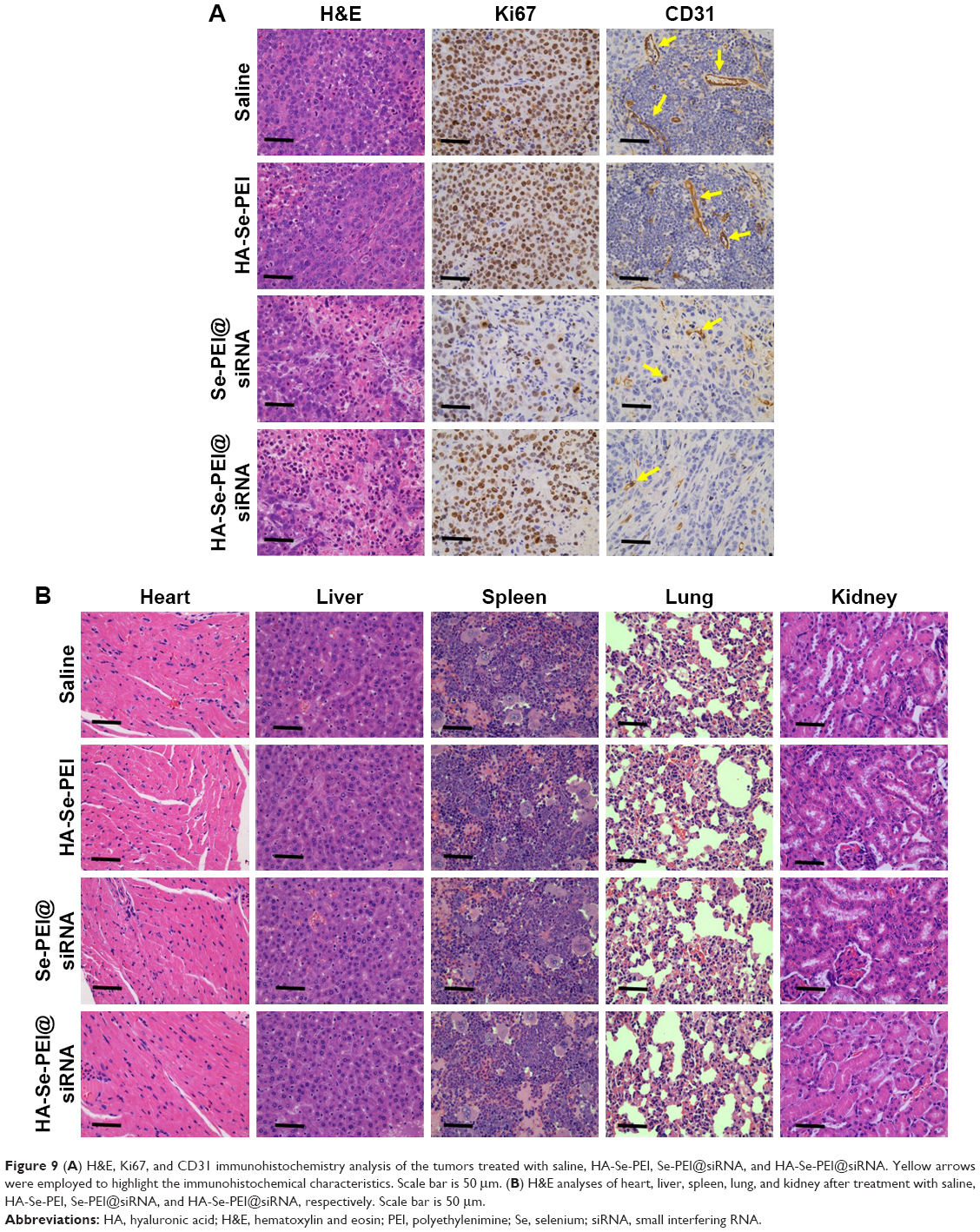 | Figure 9 (A) H&E, Ki67, and CD31 immunohistochemistry analysis of the tumors treated with saline, HA-Se-PEI, Se-PEI@siRNA, and HA-Se-PEI@siRNA. Yellow arrows were employed to highlight the immunohistochemical characteristics. Scale bar is 50 μm. (B) H&E analyses of heart, liver, spleen, lung, and kidney after treatment with saline, HA-Se-PEI, Se-PEI@siRNA, and HA-Se-PEI@siRNA, respectively. Scale bar is 50 μm. |
Side effects analysis
Apart from the antitumor efficacy, the in vivo toxicity assessment is very crucial for cancer therapy.39 Histological analysis of main organs was performed by H&E staining. As shown in Figure 9B, compared with the saline-treatment group, no obvious differences were observed in all other treatment groups. The integrated results certified that HA-Se-PEI@siRNA NPs were well tolerated in vivo at the tested dose. By analyzing these issues, HA-Se-PEI@siRNA NPs have a great potential as a tumor-targeted delivery system for effective HCC therapy with a reduced systemic toxicity.
Conclusion
In this work, we have developed functionalized SeNP conjugated with tumor-targeting moiety HA as a carrier to deliver HES5-siRNA for HCC therapy. The active tumor-targeted functionalized SeNP HA-Se-PEI@siRNA exhibited higher siRNA transfection efficiency than that of Se-PEI@siRNA and effectively knocked down the expression of HES5 in vitro and in vivo. Meanwhile, HA-Se-PEI@siRNA NPs were able to inhibit the HepG2 cell proliferation and arrest the cell at the G0/G1 phase. HA-Se-PEI@siRNA inhibited the tumor growth via the silence of HES5 gene in vivo. The enhanced antitumor activity of HA-Se-PEI@siRNA NP was consistent with its high tumor accumulation in vivo. Moreover, no obvious in vivo toxicity was observed in the main organs of NP-treated mice, suggesting its good compatibility in this gene delivery system. In a word, these studies provided a potential strategy for HCC-targeted gene therapy.
Acknowledgments
This work was supported by the China Postdoctoral Science Foundation (number 2017M612632), the Technology Planning Project of Guangdong Province (number 2014A020212697), and the Technology Planning Project of Guangzhou City (number 201607010120).
Disclosure
The authors report no conflicts of interest in this work.
References
Wang G, Gao X, Gu G, et al. Polyethylene glycol-poly(epsilon-benzyloxycarbonyl-l-lysine)-conjugated VEGF siRNA for antiangiogenic gene therapy in hepatocellular carcinoma. Int J Nanomedicine. 2017;12:3591–3603. | ||
Yassin AM, El-Deeb NM, Metwaly AM, El Fawal GF, Radwan MM, Hafez EE. Induction of apoptosis in human cancer cells through extrinsic and intrinsic pathways by Balanites aegyptiaca furostanol saponins and saponin-coated silvernanoparticles. Appl Biochem Biotechnol. 2017;182(4):1675–1693. | ||
Zhang P, Cao Y, Chen H, Zhou B, Hu W, Zhang L. Preparation and evaluation of glycyrrhetinic acid-modified and honokiol-loaded acoustic nanodroplets for targeted tumor imaging and therapy with low-boiling-point phase-change perfluorocarbon. J Mater Chem B. 2017;5(29):5845–5853. | ||
Xia Y, Lin Z, Li Y, et al. Targeted delivery of siRNA using RGDfC-conjugated functionalized selenium nanoparticles for anticancer therapy. J Mater Chem B. 2017;5(33):6941–6952. | ||
Li J, Xue S, Mao Z. Nanoparticle delivery systems for siRNA-based therapeutics. J Mater Chem B. 2016;4(41):6620–6639. | ||
Zhao J, Tu K, Liu Y, et al. Photo-controlled aptamers delivery by dual surface gold-magnetic nanoparticles for targeted cancer therapy. Mater Sci Eng C Mater Biol Appl. 2017;80:88–92. | ||
Encabo-Berzosa MM, Sancho-Albero M, Sebastian V, et al. Polymer functionalized gold nanoparticles as nonviral gene delivery reagents. J Gene Med. 2017;19(6–7):e2964. | ||
Rabbani PS, Zhou A, Borab ZM, et al. Novel lipoproteoplex delivers Keap1 siRNA based gene therapy to accelerate diabetic wound healing. Biomaterials. 2017;132:1–15. | ||
Sun C, Ding Y, Zhou L, et al. Noninvasive nanoparticle strategies for brain tumor targeting. Nanomedicine. 2017;13(8):2605–2621. | ||
Kang SH, Revuri V, Lee SJ, et al. Oral siRNA delivery to treat colorectal liver metastases. ACS Nano. 2017;11(10):10417–10429. | ||
Lin JT, Du JK, Yang YQ, et al. pH and redox dual stimulate-responsive nanocarriers based on hyaluronic acid coated mesoporous silica for targeted drug delivery. Mater Sci Eng C Mater Biol Appl. 2017;81:478–484. | ||
Yamamoto S, Kato A, Sakurai Y, Hada T, Harashima H. Modality of tumor endothelial VEGFR2 silencing-mediated improvement in intratumoral distribution of lipid nanoparticles. J Control Release. 2017;251:1–10. | ||
Chen Q, Xu M, Zheng W, Xu T, Deng H, Liu J. Se/Ru-decorated porous metal-organic framework nanoparticles for the delivery of pooled siRNAs to reversing multidrug resistance in taxol-resistant breast cancer cells. ACS Appl Mater Interfaces. 2017;9(8):6712–6724. | ||
Yang Y, Xie Q, Zhao Z, et al. Functionalized selenium nanosystem as radiation sensitizer of 125I seeds for precise cancer therapy. ACS Appl Mater Interfaces. 2017;9(31):25857–25869. | ||
Wu H, Zhu H, Li X, et al. Induction of apoptosis and cell cycle arrest in A549 human lung adenocarcinoma cells by surface-capping selenium nanoparticles: an effect enhanced by polysaccharide-protein complexes from Polyporus rhinocerus. J Agric Food Chem. 2013;61(41):9859–9866. | ||
Ahmad MS, Yasser MM, Sholkamy EN, Ali AM, Mehanni MM. Anticancer activity of biostabilized selenium nanorods synthesized by Streptomyces bikiniensis strain Ess_amA-1. Int J Nanomedicine. 2015;10:3389–3401. | ||
Kumari M, Ray L, Purohit MP, et al. Curcumin loading potentiates the chemotherapeutic efficacy of selenium nanoparticles in HCT116 cells and Ehrlich’s ascites carcinoma bearing mice. Eur J Pharm Biopharm. 2017;117:346–362. | ||
Zheng W, Yin T, Chen Q, et al. Co-delivery of Se nanoparticles and pooled SiRNAs for overcoming drug resistance mediated by P-glycoprotein and class III beta-tubulin in drug-resistant breast cancers. Acta Biomater. 2016;31:197–210. | ||
Li Y, Lin Z, Zhao M, et al. Multifunctional selenium nanoparticles as carriers of HSP70 siRNA to induce apoptosis of HepG2 cells. Int J Nanomedicine. 2016;11:3065–3076. | ||
Xia Y, Xu T, Wang C, et al. Novel functionalized nanoparticles for tumor targeting co-delivery of doxorubicin and siRNA to enhance cancer therapy. Int J Nanomedicine. 2018;13:143–159. | ||
Wang M, Guo Y, Yu M, Ma PX, Mao C, Lei B. Photoluminescent and biodegradable polycitrate-polyethylene glycol-polyethyleneimine polymers as highly biocompatible and efficient vectors for bioimaging-guided siRNA and miRNA delivery. Acta Biomater. 2017;54:69–80. | ||
Zhou Z, Li H, Wang K, et al. Bioreducible cross-linked hyaluronic acid/calcium phosphate hybrid nanoparticles for specific delivery of siRNA in melanoma tumor therapy. ACS Appl Mater Interfaces. 2017;9(17):14576–14589. | ||
Hou X, Yang C, Zhang L, et al. Killing colon cancer cells through PCD pathways by a novel hyaluronic acid-modified shell-core nanoparticle loaded with RIP3 in combination with chloroquine. Biomaterials. 2017;124:195–210. | ||
Wakasaki T, Niiro H, Jabbarzadeh-Tabrizi S, et al. Musashi-1 is the candidate of the regulator of hair cell progenitors during inner ear regeneration. BMC Neurosci. 2017;18(1):64. | ||
Sun L, Ke J, He Z, et al. HES1 promotes colorectal cancer cell resistance to 5-Fu by inducing of EMT and ABC transporter proteins. J Cancer. 2017;8(14):2802–2808. | ||
Xia Y, You P, Xu F, Liu J, Xing F. Novel functionalized selenium nanoparticles for enhanced anti-hepatocarcinoma activity in vitro. Nanoscale Res Lett. 2015;10(1):1051. | ||
Xia Y, Chen Q, Qin X, Sun D, Zhang J, Liu J. Studies of ruthenium(ii)-2,2′-bisimidazole complexes on binding to G-quadruplex DNA and inducing apoptosis in HeLa cells. New J Chem. 2013;37(11):3706–3715. | ||
Huang Y, He L, Liu W, et al. Selective cellular uptake and induction of apoptosis of cancer-targeted selenium nanoparticles. Biomaterials. 2013;34(29):7106–7116. | ||
Wang Y, Malcolm DW, Benoit DSW. Controlled and sustained delivery of siRNA/NPs from hydrogels expedites bone fracture healing. Biomaterials. 2017;139:127–138. | ||
Wong NKY, Shenoi RA, Abbina S, Chafeeva I, Kizhakkedathu JN, Khan MK. Nontransformed and cancer cells can utilize different endocytic pathways to internalize dendritic nanoparticle variants: implications on nanocarrier design. Biomacromolecules. 2017;18(8):2427–2438. | ||
Li Y, Guo M, Lin Z, et al. Polyethylenimine-functionalized silver nanoparticle-based co-delivery of paclitaxel to induce HepG2 cell apoptosis. Int J Nanomedicine. 2016;11:6693–6702. | ||
Feio DCA, de Oliveira NCL, Pereira ELR, et al. Organic effects of associating paclitaxel with a lipid-based nanoparticle system on a nonhuman primate, Cebus apella. Int J Nanomedicine. 2017;12:3827–3837. | ||
Yuan YG, Gurunathan S. Combination of graphene oxide-silver nanoparticle nanocomposites and cisplatin enhances apoptosis and autophagy in human cervical cancer cells. Int J Nanomedcine. 2017;12:6537–6558. | ||
Feiner-Gracia N, Beck M, Pujals S, et al. Super-resolution microscopy unveils dynamic heterogeneities in nanoparticle protein corona. Small. 2017;13(41):1701631. | ||
Kirui DK, Khalidov I, Wang Y, Batt CA. Targeted near-IR hybrid magnetic nanoparticles for in vivo cancer therapy and imaging. Nanomedicine. 2013;9(5):702–711. | ||
Elnegaard MP, List M, Christiansen H, Schmidt S, Mollenhauer J, Block I. Protein-based nanotoxicology assessment strategy. Nanomedicine. 2017;13(3):1229–1233. | ||
Choi JY, Ramasamy T, Kim SY, et al. PEGylated lipid bilayer-supported mesoporous silica nanoparticle composite for synergistic co-delivery of axitinib and celastrol in multi-targeted cancer therapy. Acta Biomater. 2016;39:94–105. | ||
Muralidharan R, Babu A, Amreddy N, et al. Tumor-targeted nanoparticle delivery of HuR siRNA inhibits lung tumor growth in vitro and in vivo by disrupting the oncogenic activity of the RNA-binding protein HuR. Mol Cancer Ther. 2017;16(8):1470–1486. | ||
Xue H, Yu Z, Liu Y, et al. Delivery of miR-375 and doxorubicin hydrochloride by lipid-coated hollow mesoporous silica nanoparticles to overcome multiple drug resistance in hepatocellular carcinoma. Int J Nanomedicine. 2017;12:5271–5287. |
Supplementary materials
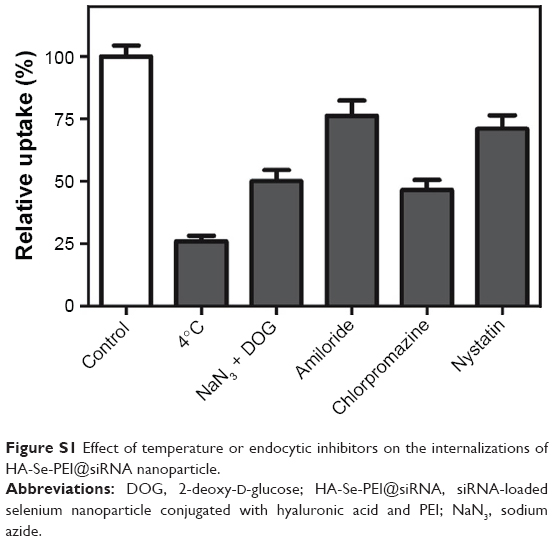 | Figure S1 Effect of temperature or endocytic inhibitors on the internalizations of HA-Se-PEI@siRNA nanoparticle. |
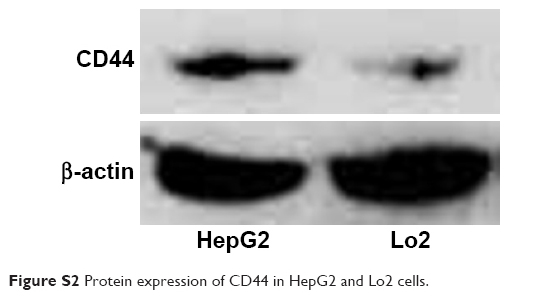 | Figure S2 Protein expression of CD44 in HepG2 and Lo2 cells. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.