Back to Journals » Drug Design, Development and Therapy » Volume 13
siRNA-based breast cancer therapy by suppressing 17β-hydroxysteroid dehydrogenase type 1 in an optimized xenograft cell and molecular biology model in vivo
Authors Li F, Zhu ZH ![]() , Xue M
, Xue M ![]() , He WH
, He WH ![]() , Zhang T, Feng LL, Lin SX
, Zhang T, Feng LL, Lin SX
Received 19 July 2018
Accepted for publication 6 November 2018
Published 22 February 2019 Volume 2019:13 Pages 757—766
DOI https://doi.org/10.2147/DDDT.S180836
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Fang Li,1,* ZhiHan Zhu,1,* Man Xue,1,* WanHong He,1 Ting Zhang,1 LingLin Feng,1 ShengXiang Lin2
1NHC Key Laboratory of Reproduction Regulation (Shanghai Institute of Planned Parenthood Research), Fudan University, and Shanghai Engineer and Technology Research Center of Reproductive Health Drug and Devices, Shanghai 200032, China; 2Axe Molecular Endocrinology and Nephrology, CHU Research Center and Department of Molecular Medicine, Laval University, Québec, G1V 4G2, QC, Canada
*These authors contributed equally to this work
Purpose: Hormone-dependent breast cancer is the most common form of breast cancer, and inhibiting 17β-HSD1 can play an attractive role in decreasing estrogen and cancer cell proliferation. However, the majority of existing inhibitors have been developed from estrogens and inevitably possess residual estrogenicity. siRNA knockdown provides a highly specific way to block a targeted enzyme, being especially useful to avoid estrogenicity. Application of 17β-HSD1-siRNA in vivo is limited by the establishment of an animal model, as well as the potential nuclease activity in vivo. We tried to reveal the in vivo potential of 17β-HSD1-siRNA-based breast cancer therapy.
Materials and methods: To establish a competent animal model, daily subcutaneous injection of an estrone micellar aqueous solution was adopted to provide the substrate for estradiol biosynthesis. The effects of three different doses of estrone (0.1, 0.5, and 2.5 µg/kg/day) on tumor growth in T47D-17β-HSD1-inoculated group were investigated and compared with the animals inoculated with wild type T47D cells. To solve in vivo delivery problem of siRNA, “17β-HSD1-siRNA/LPD”, a PEGylated and modified liposome–polycation–DNA nanoparticle containing 17β-HSD1-siRNA was prepared by the thin film hydration method and postinsertion technology. Finally, “17β-HSD1-siRNA/LPD” was tested in the optimized model. Tumor growth and 17β-HSD1 expression were assessed.
Results: Comparison with the untreated group revealed significant suppression of tumor growth in “17β-HSD1-siRNA/LPD”-treated group when HSD17B1 gene expression was knocked down.
Conclusion: These findings showed promising in vivo assessments of 17β-HSD1-siRNA candidates. This is the first report of an in vivo application of siRNA for steroid-converting enzymes in a nude mouse model.
Keywords: animal model, HSD17B1, breast cancer, estrogen, gene silencing
Introduction
Breast cancer (BC) is the most common cancer to affect women and is a major cause of death. In 2016, 249,000 women were diagnosed with BC, which resulted in 40,000 deaths.1 Most BC cases are found in women over the age of 50 and are initially hormone dependent. However, in recent years, ~11% of new BC cases in American women have been found in women younger than 45 years of age making BC a threat to all ages.2 Around 60% of premenopausal and 75% of postmenopausal BC cases are hormone dependent. Epidemiological studies indicate that a high level of estradiol contributes to cell proliferation and stimulates development of the cancer.3,4
17β-hydroxysteroid dehydrogenases (17β-HSDs) play important roles in catalyzing the interconversion of steroid hormones with different potencies. To date, 15 mammalian members of 17β-HSD superfamily have been found and the nomenclature is ranked chronologically.5 The 17β-HSDs can be classified into oxidative and reductive isoforms. Reductive 17β-HSDs (type 1, 3, 5, 7, and 12) convert the less potent estrogen form, estrone (E1), to the more potent form, estradiol (E2), using nicotinamide adenine dinucleotide phosphate (NADPH) as cofactor. Oxidative 17β-HSDs (type 2, 4, 6, 8, 9, 10, 11, and 14) perform the reverse effect using nicotinamide adenine dinucleotide (NAD) as cofactor.6,7 Among reductive 17β-HSDs, studies have shown that knocking down 17β-HSD1 significantly affects the conversion of E1 to E2, but that this is not the case for other reductive 17β-HSDs.8,9 The high capacity for E2 production has been correlated with cancer cell metastases, poor disease prognosis, and efficient cell proliferation stimulation in BC.4,10,11 Therefore, expression of 17β-HSD1 is a predominant factor in the maintenance of the E2 concentration making it a promising target for hormone-dependent BC therapy.
As early as the 1970s, research related to the important activity of 17β-HSD1 described above has focused on the search, synthesis, and testing of potential inhibitors of this enzyme. However, no 17β-HSD1 inhibitors are in clinical use to date. This is somewhat surprising since other enzymes involved in the synthesis of estrogens and androgens, eg, inhibitors of aromatase, 5α-reductases, and 17α-lyases, have been indicated in the clinical treatment of BC. The major obstacle to the therapeutic use of 17β-HSD1 inhibitors is the presence of undesirable estrogenic activity. This most likely results from the fact that the 17β-HSD1 enzyme has a high affinity for its estrogen substrates. Most designs for 17β-HSD1 inhibitors were initiated from analogs of estrogens making it difficult to eliminate the residual estrogenic activity.12
We have dedicated ourselves to the study of 17β-HSDs and have succeeded in crystallizing and determining the first three-dimensional (3D) structure of any human steroid-converting enzyme, that of the 17β-HSD1 apoenzyme and estradiol complex.13–16 Based on this work, extensive structure–function studies were carried out that demonstrated the dual functions of estrogen activation and androgen inactivation by this enzyme.17 In collaboration with Dr D. Poirier, the rational design of inhibitors has yielded a new hybrid inhibitor possessing nM-level affinity,18 and a new improved efficient inhibitor 3-(3′,17′β-dihydroxyestra-1′,3′,5′(10′)-trien-16′β-methyl) benzamide.19 However, therapeutic application of 17β-HSD1 inhibitors has been delayed due to the estrogenicity of the steroid structures and the estrogen starting molecule.
Compared with small molecule chemical inhibitors, siRNAs can inhibit a specific target with high efficiency and are not limited to ion channels, enzymes, or nuclear hormone receptors.20,21 Most importantly, they do not possess any estrogenicity. siRNAs have been applied to the treatment of many human diseases caused by specific genes.22 Therefore, siRNA-mediated knockdown of HSD17B1 appeared to be a logical step. We have designed several siRNAs for HSD17B1 knockdown and demonstrated their remarkable in vitro efficacy.4,23,24 However, continued assessment in vivo revealed two troublesome problems:
I. Establishment of an appropriate preclinical animal model suitable for evaluating human 17β-HSD1-siRNA: due to species differences between rodents and humans, ie, differences in the amino acid sequences and tissue distributions of the 17β-HSD1 enzymes, the inhibitors designed for human 17β-HSD1 could not inhibit the rodent enzyme.25,26 Thus, the commonly used BC models have poor predictability for evaluating 17β-HSD1 inhibitors. Development of a model suitable for testing human 17β-HSD1 inhibitors appears to have more challenges: 1) how to choose an appropriate cell line for the xenograft tumor model – transfected cells or wild type cells; 2) how to simply and cost-effectively provide exogenous E1 as the substrate for activation into E2; and 3) how to determine an appropriate dose of E1 for the chosen cell line. These considerations complicate the model development and are compounded by the fact that there are a limited number of relevant studies for reference.
II. Establishment of a nanocarrier for siRNA in vivo delivery: naked siRNA is a negatively charged macromolecule (∼13 kDa). It is subject to degradation by endogenous enzymes and unable to penetrate cellular membranes efficiently.21,27 In addition, although the in vitro transfection reagent is commercially available, it is too toxic and expensive to be applied in vivo.28 Therefore, self-preparation of a low-toxicity nanocarrier for sustained and targeted delivery of 17β-HSD1-siRNA is necessary.
In the present study, we first tried to establish xenograft animal models by using not only T47D-WT but also T47D-17β-HSD1 cells. Tumor growth in response to three different doses of E1 was investigated and the comparative data for these two models were reported. Rather than using expensive osmotic mini-pumps or long-acting pellets as reported in the literature,10,26,29 subcutaneous injection of an E1 micellar aqueous solution was adopted to provide the substrate for E2 synthesis. Furthermore, “17β-HSD1-siRNA/LPD”, a new self-prepared potential suppressor for gene nanomedicine, was developed and tested in this model. This is the first report trying to reveal the in vivo efficacy of siRNA against steroid-converting enzymes.
Materials and methods
Establishing T47D-17β-HSD1 cells
T47D-17β-HSD1 cells were established according to a previously reported method with slight modifications.23 In brief, T47D-WT cells were stably transfected with a pcDNA3.1 (+) plasmid (Invitrogen Life Technologies, Carlsbad, CA, USA) expressing the human 17β-HSD1 gene (gene HSD17B1; GenBank accession no NM_000413). The transfectants were selected by using G-418 (Shanghai YeSheng Biochem Ltd., Shanghai, China).
Cell culture
T47D-WT cells were obtained from the Shanghai Institute of Biological Science (Shanghai, China). T47D-WT and T47D-17β-HSD1 cells were propagated in DMEM high glucose (HyClone Laboratories, Logan, UT, USA) supplemented with 7.5 mg/L human insulin (Sigma-Aldrich, St. Louis, MI, USA) and 10% FBS (Gibco, Grand Island, NY, USA) at 37°C under a humidified 5% CO2 atmosphere.
Western blot analysis
Total protein was extracted from cells lysed with RIPA buffer and supplemented with 1 mM phenylmethanesulfonyl fluoride and a protein inhibitor cocktail (Biotool, Houston, TX, USA). Proteins were quantified using Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientifc, San Jose, CA, USA). Equal amounts of total protein were loaded onto a 12% SDS-PAGE and transferred onto a nitrocellulose membrane (Millipore Ltd., Watford, UK). The membrane was blocked with 5% nonfat milk in PBS-Tween buffer for 1 hour at room temperature. Membranes were then incubated for 2 hours at room temperature in 5% nonfat milk in PBS Tween buffer containing a 1:10,000 dilution of the primary anti-17β-HSD1 rabbit polyclonal antibody (clone EP1682Y) (Abcam, Cambridge, UK). A 1:4,000 dilution of monoclonal anti-β-actin antibody produced in the mouse (CWBIO, Beijing, China) was used as a loading control. The respective horseradish peroxidase-conjugated antibody (Epizyme, Shanghai, China) was chosen as the secondary antibody and was diluted by a factor of 6,000. Protein signals were visualized with chemiluminescence reagent (Thermo Fisher Scientific).
Animals and inoculation
Athymic female BALB/c-nu/nu mice, 8–10 weeks of age at the study start were obtained from LingChang Biotech (Shanghai, China). The animals were maintained under specific pathogen-free conditions. The study protocol was approved by the ethics committee of Shanghai Institute of Planned Parenthood Research (No 2016–11) and followed the National Institutes of Health guide for the care and use of laboratory animals. Intact mice were used for inoculation. T47D-WT or T47D-17-HSD1 cells were harvested and resuspended in Matrigel™ Basement Membrane Matrix, Phenol-Red-Free (BD Biosciences, Bedford, MA, USA) to prepare a cell suspension of 1×108 cells/mL. An amount of 0.1 mL of the cell suspension was injected subcutaneously in the right upper flank of each mouse.
Estrogen treatment
To prepare the estrogen solution for daily subcutaneous injection, E2 (Hubei Gedian Humanwell Pharmaceutical Co., Ltd., E-zhou, Hubei, China) or E1 (National Institute for Food and Drug Control, Beijing, China) was dissolved in dimethyl sulfoxide (cell culture reagent) (Sigma) and then diluted in 10% hydroxypropyl-β-cyclodextrin (Xi’an Deli Biochemical Co., Ltd., Xi’an, Shanxi, China). From the day of inoculation, the mice received a subcutaneous injection of E2 solution at 27.5 μg/kg daily for 12 days. After tumor formation, the animals in each group (T47D-WT-inoculated or T47D-17β-HSD1-inoculated group) were then randomly assigned into four subgroups injected subcutaneously with different doses (0, 0.1, 0.5, and 2.5 μg/kg) of E1 daily throughout the duration of the study. Tumor size in the presence of E2 or E1 was determined by measuring the largest and smallest tumor diameter (d1, d2) with calipers twice weekly. Tumor volumes were calculated according to the formula V=1/2 × d1 × d22.
Preparation of “17β-HSD1-siRNA/LPD”
“17β-HSD1-siRNA/LPD” is a PEGylated liposome–polycation–DNA (LPD) nanoparticle containing 17β-HSD1-siRNA modified with Arg-Gly-Asp peptide (RGD), targeting αvβ3 integrin expressed in tumor cells. The RGD peptides were synthesized by GL Biochem Ltd. (Shanghai, China), and the duplex 17β-HSD1-siRNA with sequence 5′-CCACAGCAAGCAAGUCUUU[dT][dT] was synthesized by GenePharma (Shanghai, China). The preparation followed the method reported by Huang30,31 with some modifications. In brief, cationic liposomes composed of 1,2-dioleoyl-3-trimethylammonium-propane (Shanghai A.V.T. Pharmaceutical Co., Ltd., Shanghai, China) and cholesterol (Shanghai A.V.T. Pharmaceutical Co., Ltd.) (1:1 molar ratio) were prepared by thin film hydration followed by membrane extrusion to reduce the particle size. One hundred forty-eight microliters of protamine (2 mg/mL), 1,030 μL of deionized water, and 200 μL of a mixture of siRNA and calf thymus DNA (2 mg/mL) were mixed and maintained at room temperature for 10 minutes before addition of 1 mL of cationic liposome (10 mmol/L). After 10 minutes at room temperature, 500 μL of RGD modified distearoylphosphatidylethanolamine-poly(ethylene glycol) (DSPE-PEG-RGD) (10 mg/mL) was added and incubated at 65°C for 15 minutes. Particle size and zeta potential of “17β-HSD1-siRNA/LPD” were measured by a Zetasizer Nano-ZS instrument (Malvern Instruments Ltd., Worcestershire, UK). Free siRNA were separated by electrophoresis and visualized using a Tanon Gel System (Tanon Science & Technology Co., Ltd., Shanghai, China). The free and total amount of siRNA were signed C1 and C0, respectively. Encapsulation efficiency (EE) was calculated according to the formula: EE = (1 − C1/C0)×100%.
Administration of “17β-HSD1-siRNA/LPD”
After investigation of E1-stimulated tumor growth, the T47D-17β-HSD1-inoculated animal model treated with the intermediate dose of E1 (0.5 μg/kg/day) was chosen for drug administration. The efficacy of the inhibitor “17β-HSD1-siRNA/LPD” was tested in vivo. Injections of a 1.2 mg/kg dose of siRNA were administered via the tail vein for three consecutive days per week. The efficiency of inhibition was determined by monitoring tumor growth in the presence of E1 (0.5 μg/kg/day) with and without (vehicle only or noncoding siRNA) the inhibitor treatment. The mice were sacrificed at the end of the experiment. The wet weights of uteri were determined; the tumors were removed and fixed immediately in 4% buffered formaldehyde for examination by immunohistochemistry.
Immunohistochemistry
Formaldehyde-fixed tumors were embedded in paraffin. Five-micron-thick sections were deparaffinized, hydrated, and stained. Primary antibodies were diluted 1:100 for anti-17β-HSD1 rabbit polyclonal antibody (clone EP1682Y) (Abcam). Diaminobenzidine was used as the substrate for visualization of the immune reaction. Stained slides were scanned by Pannoramic MIDI, 3D HISTECH. For determination of labeling indices for 17β-HSD1 and apoptosis, the entire slide was screened by automatic observer and analyzed by Pannoramic viewer and Quant center software. The staining score for tumor cells was subsequently calculated. The percentage of immunostain and staining intensity were analyzed and a histochemistry score (H-score) was calculated using the following formula:
|
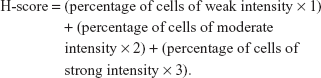
Statistical analysis
Differences in tumor growth between samples were determined by two-way ANOVA test, while differences in 17β-HSD1 expression within cells or xenograft tumor tissue were determined by Student’s t-test using GraphPad Prism 6.0 (GraphPad Software, Inc., San Diego, CA, USA).
Data and availability
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Results
Establishing T47D-17β-HSD1 cells
T47D cells were genetically engineered for inoculation. The expression of 17β-HSD1 was subsequently determined by Western bolt. Results (Figure 1) showed that the T47D-WT cells exhibited markedly lower 17β-HSD1 expression, and that the expression of 17β-HSD1 was highly elevated in T47D-17β-HSD1 cells (14.0-fold vs T47D-WT cells), demonstrating successful establishment of this transfected cell line.
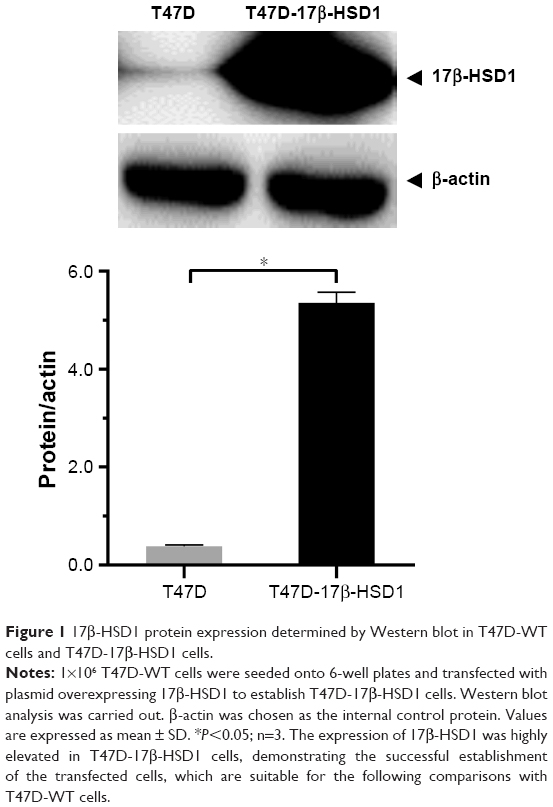 | Figure 1 17β-HSD1 protein expression determined by Western blot in T47D-WT cells and T47D-17β-HSD1 cells. |
Effects of estrogen on tumor growth in vivo
To investigate the respective differences between the two xenograft models established with T47D-WT cells or T47D-17β-HSD1 cells, the effects of estrogen on tumor growth were evaluated. Estradiol was initially used to support tumor formation. The results show that the effects of E2 on tumor growth rate are similar in both the T47D-WT-inoculated group and the T47D-17β-HSD1-inoculated group (Figure 2). After 12 days of E2 treatment, cancer formation was deemed successful with a tumor volume of 99.6±28.6 mm3 for the T47D-WT-inoculated group and 72.9±29.3 mm3 for the T47D-17β-HSD1-inoculated group. In the following experiment, E2 was replaced by E1. Three different doses of E1 were evaluated in both the T47D-WT-inoculated group and T47D-17β-HSD1-inoculated group. Tumor sizes were reduced by nearly half after 19 days in the T47-WT-inoculated group in the absence of E1 supplementation (Figure 3A, green line), implying that endogenous estrogens are not sufficient to maintain tumor growth. An elevated tumor growth rate was observed when the mice were treated with E1. The growth rate increased as the E1 dose increased (Figure 3A, blue and black lines). It is worth noting that the highest dose of E1 (up to 2.5 μg/kg/day) showed no significant facilitation of tumor growth when compared with the intermediate dose (P>0.05, Figure 3A, red line vs black line), indicating that an equilibration state was reached. The effects of the different doses on the T47-17β-HSD1-inoculated group showed a similar tendency (Figure 3B).
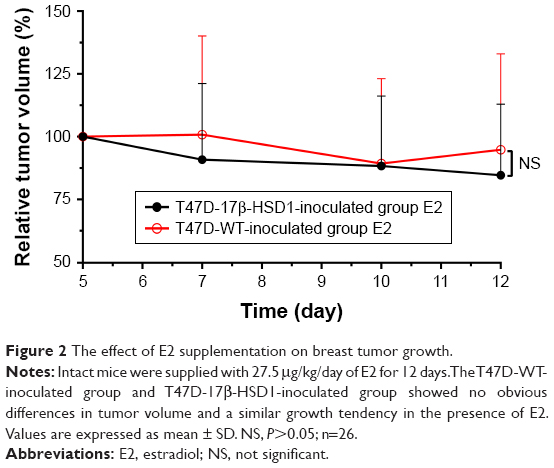 | Figure 2 The effect of E2 supplementation on breast tumor growth. |
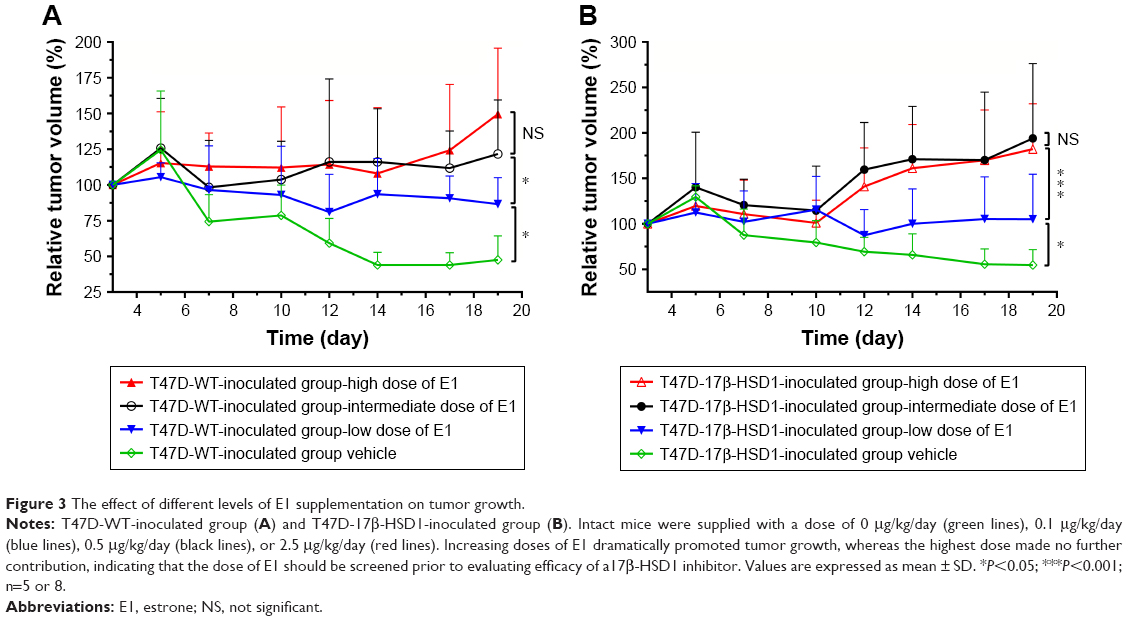 | Figure 3 The effect of different levels of E1 supplementation on tumor growth. |
Different tumor growth in vivo between T47D-WT-inoculated and T47D-17β-HSD1-inoculated groups
Although the effects of different E1 doses on T47D-17β-HSD1-inoculated group showed similar tendencies, there were still some differences between the T47D-WT-inoculated and T47D-17β-HSD1-inoculated group. Therefore, the data were subjected to additional comparisons.
When treated with no or low-dose E1, the tumor growth rates of these two groups appeared similar (Figure 4A and B). This reflected that in the absence of E1 or in the presence of a low level of E1, the conversation rates to E2 in vivo were similar regardless of the expression of 17β-HSD1. There was a significant difference in tumor growth rate in response to treatment with the intermediate dose of E1, suggesting that the differential expression of 17β-HSD1 contributed to different levels of E2 in vivo. A larger inhibition interval for drug evaluation was generated when the T47D-17β-HSD1-inoculated model was used (Figure 4C). However, this difference was reduced in response to treatment with the high dose of E1 (Figure 4D). This result fits the hypothesis that excessive E1 may lead to a direct proliferative effect, thereby narrowing the proliferation difference induced by E2.26
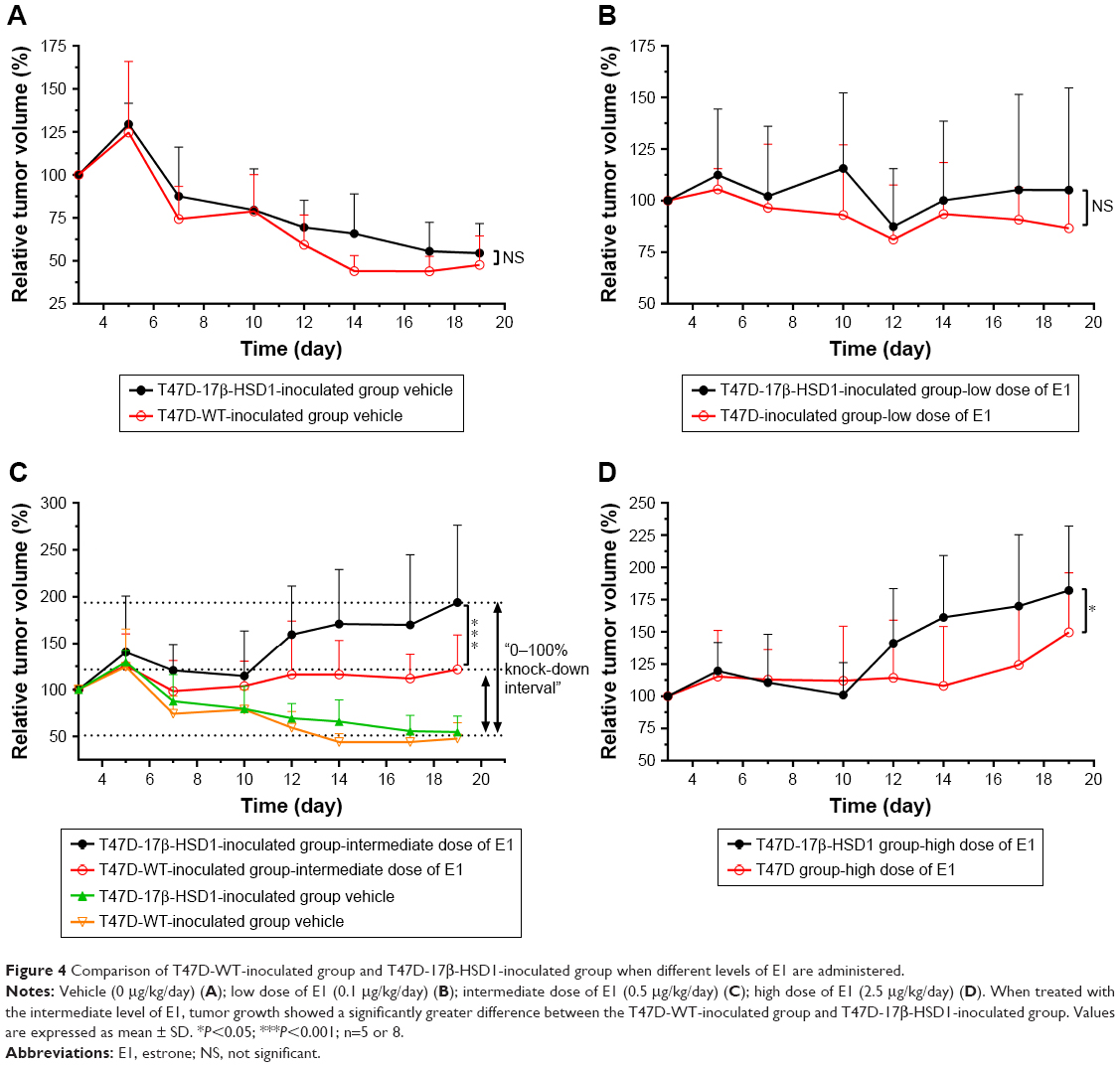 | Figure 4 Comparison of T47D-WT-inoculated group and T47D-17β-HSD1-inoculated group when different levels of E1 are administered. |
Thus, by comparative analysis, a more competent animal model was established using T47D-17β-HSD1 cells, which exhibited not only a larger inhibition interval but also an exclusively 17β-HSD1-dependent proliferation. Accordingly, the T47D-17β-HSD1-inoculated animal model with the intermediate dose of E1 (0.5 μg/kg/day) was optimized for subsequent drug administration and tumor evaluation.
Efficacy of “17β-HSD1-siRNA/LPD”
Using the optimized xenograft animal model described above, “17β-HSD1-siRNA/LPD”, a new potential suppressor for gene nanomedicine, was tested after characterization in vitro. The particle size, zeta potential, and siRNA EE of the formulation were 127.8±6.1 nm, +32.5±6.4 mV, and >90%, respectively. Tumor growth showed no significant difference between the vehicle group (n=5) and noncoding siRNA group (n=3). Thus, the vehicle group with more replicates was selected as a negative control to verify the in vivo efficiency. Tumor growth was significantly suppressed in comparison to the untreated group. In particular, on day 6 of drug administration, “17β-HSD1-siRNA/LPD” reduced the tumor volume to 56%±6% of the tumor control, and 69%±7% of the original (Figure 5). The mice weight in the medicated group rapidly returned to its previous level, although there was a slight weight loss during administration.
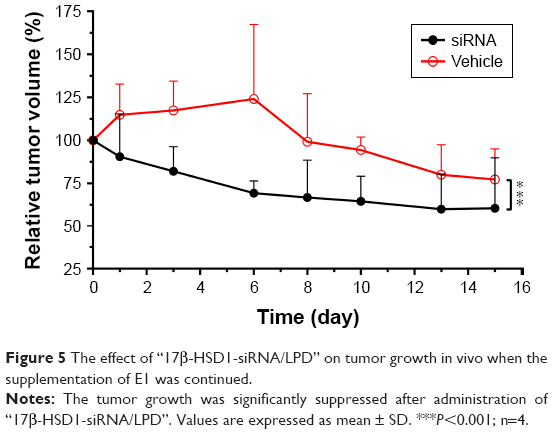 | Figure 5 The effect of “17β-HSD1-siRNA/LPD” on tumor growth in vivo when the supplementation of E1 was continued. |
Mice were sacrificed after measuring the tumor volumes. Tumors and uteri were removed for further analysis. Histochemical analysis showed lower expression of 17β-HSD1 in the “17β-HSD1-siRNA/LPD”-treated group (Figure 6B and E) than in the untreated group (Figure 6C and F) (mean H-score: 65.3 vs 145.3, Figure 6G), suggesting that deregulation of human 17β-HSD1 in BC is associated with the reduction of tumor cell proliferation.
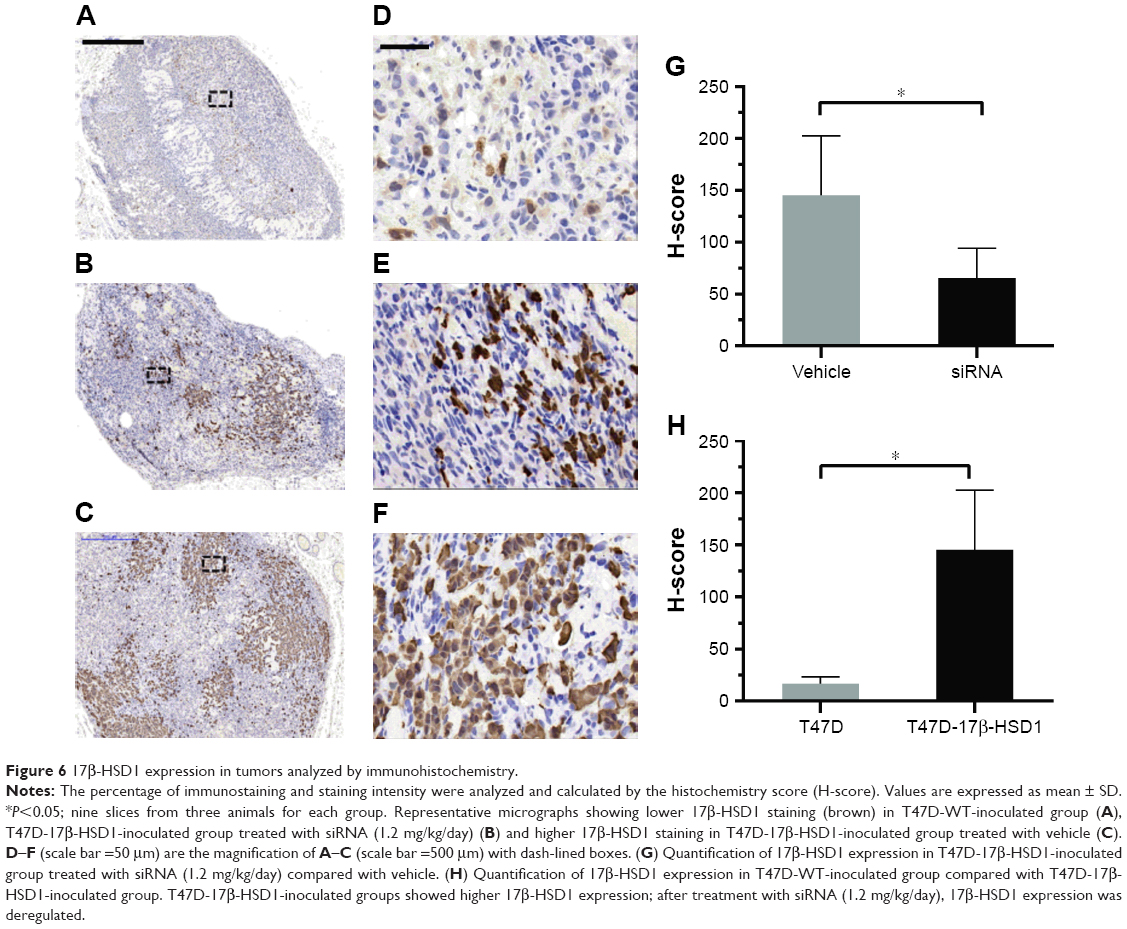 | Figure 6 17β-HSD1 expression in tumors analyzed by immunohistochemistry. |
Moreover, as for the immunohistochemical results, the tumors from the T47D-WT-inoculated group (Figure 6A and D) exhibited an extremely low level of 17β-HSD1 (mean H-score: 16.4) (8.8-fold, T47D-17β-HSD1-inoculated group vs T47D-WT-inoculated group, Figure 6H). The in vivo results are in agreement with the in vitro Western blot results, providing additional support that T47D-17β-HSD1-inoculated cells are well suited for evaluation of the 17β-HSD1 inhibitor.
Furthermore, the uterine weights showed no significant difference between the “17β-HSD1-siRNA/LPD”-treated group and the untreated group (data not shown), indicating that this siRNA had no estrogenic activity and was shown to be a promising 17β-HSD1 suppressor.
Discussion
The expression of 17β-HSD1 has been shown to be upregulated in many hormone-dependent BC cases in the clinic.32,33 The choice of a suitable cell line appeared to be of particular importance in order to establish an appropriate xenograft animal model that accurately reflects this aggressive phenotype. T47D and MCF7 cells are the two most extensively used estrogen-responsive BC epithelial cell lines. Although the expression of 17β-HSD1 is higher in T47D than in MCF7 cells, the level of 17β-HSD1 is still low, accounting for 3% of all 17β-HSD mRNAs in T47D cells.33,34 Therefore, we deemed it necessary to genetically engineer these estrogen-responsive cell lines in order to develop a preclinical animal model with which to screen potential 17β-HSD1 inhibitors.
It was anticipated that established MCF7-17β-HSD1 cells could be used for this model. To date, studies with T47D cell lines have used T47D-WT cells rather than T47D-17β-HSD1. In order to provide more information to help other researchers with the choice of an appropriate cell line for this model, both T47D-WT and T47D-17β-HSD1 cells were used and compared in the present study.
Estrogen supplementation was the second troublesome problem encountered during the establishment of this model. Since the requirement for estrogen is well known for estrogen-dependent tumor growth,35 we used E2 to support tumor formation at first to increase the success rate of engraftment. After successful tumor formation in both T47D-WT-inoculated and T47D-17β-HSD1-inoculated groups, E2 was replaced by 17β-HSD1-substrate E1. Husen et al used osmotic mini-pumps in order to provide a continuous in vivo level of E1 in mice.10,26 Day et al recommended expensive long-acting E1 pellets because they stimulated tumor growth more effectively than equivalent daily subcutaneous injections of an E1 propylene glycol solution.29 However, these two long-acting preparations are too expensive for wide application especially when multidrug candidates and their effective dose need to be screened. A cost-effective method for E1 supplementation was adopted in this study: in order to avoid skin irritation in nude mice by propylene glycol solution, a daily subcutaneous injection of E1 as a micellar aqueous solution containing HP-β-cyclodextrin was used.
After choosing a method of estrogen supplementation, caution was still required with regard to the dose of E1 in order to establish an exclusively 17β-HSD1-induced BC model. An excess of E1 could activate the estrogen receptor directly, instead of via 17β-HSD1 activity.26 Husen et al also screened different doses of E1, but the effects of these different doses on tumor growth were not reported. In this study, a total of eight tumor growth curves are shown and compared to determine the ideal dose (Figure 3). The intermediate dose of E1 was proven to be appropriate to establish this exclusively 17β-HSD1-induced cancer model (Figure 3, black lines). A slow tumor growth rate in which the original tumor size was essentially maintained was observed when the T47D-WT-inoculated group was treated with the intermediate dose of E1 (Figure 4C, red line). Therefore, we speculated that the success rate of engraftment would be very low if only E1 rather than E2 was used to support T47D-WT-inoculated tumor formation during the early days of the study. As a result, we believe that in addition to the lower inhibition interval, the markedly slower tumor growth rate, which may lead to a low tumor formation rate, is another factor to be considered for the T47D-WT cell line.
After the optimization of the abovementioned three factors, “17β-HSD1-siRNA/LPD”, a nanocarrier, was prepared to encapsulate and protect siRNA. Due to the positive expressions of integrin αvβ1, αvβ5, and α5β1 in T47D cells,36–38 this nanocarrier was further modified with RGD, one of the most studied integrin ligands, to improve the efficiency of drug delivery.
Finally, “17β-HSD1-siRNA/LPD” was tested in the optimized model. It was noted that tumor proliferation in the untreated group was not observed after 8 days although the supplementation of E1 was continued (Figure 5, red line). One explanation is degradation of E1 in the micellar aqueous solution leading to a reduced daily injection dosage. However, stability tests showed that the E1 micellar aqueous solution was stable under ambient temperature for at least 2 weeks (data not shown). Another major reason was the age of the nude mice. It spent around 8 weeks to establish the animal model suitable for evaluating human 17β-HSD1-siRNA, longer than most of other tumor bearing animal model. Thus, the nude mice were considered old for administration (around 16 weeks), for the life span of nude mice is normally 6 months to a year.39 It is reported that the “malignant” characteristics of tumors (ie, rapid growth and metastases) appear to be less prominent in the elderly. Host features such as the fibrotic, angiogenic, or immune response may be altered by the aging process and may render the host “soil” less fertile for “malignant” tumor growth.40,41 Therefore, to create such a special tumor model able to proliferate throughout the duration of the experiment, younger mice of around 3 weeks of age for inoculation may be more suitable.42
Conclusion
In summary, in this study we have successfully optimized and established a xenograft nude mouse model suitable for evaluating 17β-HSD1-siRNA by using T47D-17β-HSD1 cells. Furthermore, this model was used to test 17β-HSD1-siRNA. To our knowledge, this is the first report involving a T47D-17β-HSD1-innoculated nude mouse model and the first report of an in vivo application of siRNA for steroid-converting enzymes.
Acknowledgments
This work was supported by the National Key Technologies R & D Program of China during the 13th Five-Year Plan Period (grant No. 2016YFC1000902), the Major Key Basic Research Fundamental Program of Shanghai Science and Technology Committee (grant No. 14JC1492300), the Scientific Research Project of Shanghai Municipal Health and Family Planning Commission (grant number: 20144Y0049) and Shanghai Biomedical Science and Technology Support Projects (grant number: 14431902000). The authors thank Dr Qianxi Zhu from Shanghai Institute of Planned Parenthood Research for the great help in the statistical analysis and Dr Muriel Kelly for the help in English language improvement of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. | ||
Center for disease control and prevention. What Are The Risk Factors For Breast Cancer?. Available from: https://wwwcdcgov/cancer/breast/basic_info/risk_factorshtm. Accessed 9 Mar 2017. | ||
Aka JA, Calvo EL, Lin SX. Estradiol-independent modulation of breast cancer transcript profile by 17 beta-hydroxysteroid dehydrogenase type 1. Mol Cell Endocrinol. 2017;439:175–186. | ||
Aka JA, Zerradi M, Houle F, Huot J, Lin SX. 17beta-hydroxysteroid dehydrogenase type 1 modulates breast cancer protein profile and impacts cell migration. Breast Cancer Res. 2012;14(3):R92. | ||
He W, Gauri M, Li T, Wang R, Lin SX. Current knowledge of the multifunctional 17β-hydroxysteroid dehydrogenase type 1 (HSD17B1). Gene. 2016;588(1):54–61. | ||
Brozic P, Lanisnik Risner T, Gobec S. Inhibitors of 17beta-hydroxysteroid dehydrogenase type 1. Curr Med Chem. 2008;15(2):137–150. | ||
Lukacik P, Kavanagh KL, Oppermann U. Structure and function of human 17beta-hydroxysteroid dehydrogenases. Mol Cell Endocrinol. 2006;248(1–2):61–71. | ||
Zhang CY, Chen J, Yin DC, Lin SX. The contribution of 17beta-hydroxysteroid dehydrogenase type 1 to the estradiol-estrone ratio in estrogen-sensitive breast cancer cells. PLoS One. 2012;7(1):e29835. | ||
Kocbek P, Teskac K, Brozic P, Rizner T, Gobec S, Kristl J. Effect of free and in poly(η-caprolactone) nanoparticles incorporated new type 1 17β-hydroxysteroid dehydrogenase inhibitors on cancer cells. Current Nanoscience. 2010;6(1):69–76. | ||
Husen B, Huhtinen K, Saloniemi T, Messinger J, Thole HH, Poutanen M. Human hydroxysteroid (17-beta) dehydrogenase 1 expression enhances estrogen sensitivity of MCF-7 breast cancer cell xenografts. Endocrinology. 2006;147(11):5333–5339. | ||
Hilborn E, Stål O, Jansson A. Estrogen and androgen-converting enzymes 17β-hydroxysteroid dehydrogenase and their involvement in cancer: with a special focus on 17β-hydroxysteroid dehydrogenase type 1, 2, and breast cancer. Oncotarget. 2017;8(18):30552–30562. | ||
Lin SX, Poirier D, Adamski J. A challenge for medicinal chemistry by the 17β-hydroxysteroid dehydrogenase superfamily: an integrated biological function and inhibition study. Curr Top Med Chem. 2013;13(10):1164–1171. | ||
Zhu DW, Lee X, Breton R, et al. Crystallization and preliminary X-ray diffraction analysis of the complex of human placental 17 beta-hydroxysteroid dehydrogenase with NADP+. J Mol Biol. 1993;234(1):242–244. | ||
Ghosh D, Pletnev VZ, Zhu DW, et al. Structure of human estrogenic 17 beta-hydroxysteroid dehydrogenase at 2.20 A resolution. Structure. 1995;3(5):503–513. | ||
Ghosh D, Wawrzak Z, Pletnev V, et al. Molecular mechanism of inhibition of steroid dehydrogenases by licorice-derived steroid analogs in modulation of steroid receptor function. Ann N Y Acad Sci. 1995;761:341–343. | ||
Azzi A, Rehse PH, Zhu DW, Campbell RL, Labrie F, Lin SX. Crystal structure of human estrogenic 17 beta-hydroxysteroid dehydrogenase complexed with 17 beta-estradiol. Nat Struct Biol. 1996;3(8):665–668. | ||
Gangloff A, Shi R, Nahoum V, Lin SX. Pseudo-symmetry of C19 steroids, alternative binding orientations, and multispecificity in human estrogenic 17beta-hydroxysteroid dehydrogenase. Faseb J. 2003;17(2):274–276. | ||
Qiu W, Campbell RL, Gangloff A, et al. A concerted, rational design of type 1 17beta-hydroxysteroid dehydrogenase inhibitors: estradiol-adenosine hybrids with high affinity. Faseb J. 2002;16(13):1829–1831. | ||
Mazumdar M, Fournier D, Zhu DW, Cadot C, Poirier D, Lin SX. Binary and ternary crystal structure analyses of a novel inhibitor with 17beta-HSD type 1: a lead compound for breast cancer therapy. Biochem J. 2009;424(3):357–366. | ||
Dim N, Perepelyuk M, Gomes O, et al. Novel targeted siRNA-loaded hybrid nanoparticles: preparation, characterization and in vitro evaluation. J Nanobiotechnology. 2015;13:61. | ||
Vaissière A, Aldrian G, Konate K, et al. A retro-inverso cell-penetrating peptide for siRNA delivery. J Nanobiotechnology. 2017;15(1):34. | ||
Goldberg MS. siRNA delivery for the treatment of ovarian cancer. Methods. 2013;63(2):95–100. | ||
Aka JA, Mazumdar M, Chen CQ, Poirier D, Lin SX. 17beta-hydroxysteroid dehydrogenase type 1 stimulates breast cancer by dihydrotestosterone inactivation in addition to estradiol production. Mol Endocrinol. 2010;24(4):832–845. | ||
Zhang CY, Wang WQ, Chen J, Lin SX. Reductive 17beta-hydroxysteroid dehydrogenases which synthesize estradiol and inactivate dihydrotestosterone constitute major and concerted players in ER+ breast cancer cells. J Steroid Biochem Mol Biol. 2015;150:24–34. | ||
Marchais-Oberwinkler S, Henn C, Möller G, et al. 17β-Hydroxysteroid dehydrogenases (17β-HSDs) as therapeutic targets: protein structures, functions, and recent progress in inhibitor development. J Steroid Biochem Mol Biol. 2011;125(1–2):66–82. | ||
Husen B, Huhtinen K, Poutanen M, Kangas L, Messinger J, Thole H. Evaluation of inhibitors for 17beta-hydroxysteroid dehydrogenase type 1 in vivo in immunodeficient mice inoculated with MCF-7 cells stably expressing the recombinant human enzyme. Mol Cell Endocrinol. 2006;248(1–2):109–113. | ||
Piktel E, Niemirowicz K, Wątek M, Wollny T, Deptuła P, Bucki R. Recent insights in nanotechnology-based drugs and formulations designed for effective anti-cancer therapy. J Nanobiotechnology. 2016;14(1):39. | ||
Peng H, Yang H, Song L, et al. Sustained delivery of siRNA/PEI complex from in situ forming hydrogels potently inhibits the proliferation of gastric cancer. J Exp Clin Cancer Res. 2016;35:57. | ||
Day JM, Foster PA, Tutill HJ, et al. 17beta-hydroxysteroid dehydrogenase Type 1, and not Type 12, is a target for endocrine therapy of hormone-dependent breast cancer. Int J Cancer. 2008;122(9):1931–1940. | ||
Li S, Rizzo MA, Bhattacharya S, Huang L. Characterization of cationic lipid-protamine-DNA (LPD) complexes for intravenous gene delivery. Gene Ther. 1998;5(7):930–937. | ||
Chen Y, Wu JJ, Huang L. Nanoparticles targeted with NGR motif deliver c-myc siRNA and doxorubicin for anticancer therapy. Mol Ther. 2010;18(4):828–834. | ||
Irahara N, Miyoshi Y, Taguchi T, Tamaki Y, Noguchi S. Quantitative analysis of aromatase, sulfatase and 17beta-HSD(1) mRNA expression in soft tissue metastases of breast cancer. Cancer Lett. 2006;243(1):23–31. | ||
Laplante Y, Rancourt C, Poirier D. Relative involvement of three 17beta-hydroxysteroid dehydrogenases (types 1, 7 and 12) in the formation of estradiol in various breast cancer cell lines using selective inhibitors. Mol Cell Endocrinol. 2009;301(1–2):146–153. | ||
Poutanen M, Moncharmont B, Vihko R. 17 beta-hydroxysteroid dehydrogenase gene expression in human breast cancer cells: regulation of expression by a progestin. Cancer Res. 1992;52(2):290–294. | ||
Yue W, Brodie A. MCF-7 human breast carcinomas in nude mice as a model for evaluating aromatase inhibitors. J Steroid Biochem Mol Biol. 1993;44(4–6):671–673. | ||
Meyer T, Marshall JF, Hart IR. Expression of alphav integrins and vitronectin receptor identity in breast cancer cells. Br J Cancer. 1998;77(4):530–536. | ||
Perks CM, Newcomb PV, Norman MR, Holly JM. Effect of insulin-like growth factor binding protein-1 on integrin signalling and the induction of apoptosis in human breast cancer cells. J Mol Endocrinol. 1999;22(2):141–150. | ||
Nieberler M, Reuning U, Reichart F, et al. Exploring the role of RGD-recognizing integrins in cancer. Cancers (Basel). 2017;9(9):E116. | ||
Wikipedia. Nude Mouse. 2014. Available from: https://en.wikipedia.org/wiki/Nude_mouse. Accessed October 17, 2018. | ||
Ershler WB. The change in aggressiveness of neoplasms with age. Geriatrics. 1987;42(1):99–103. | ||
Gravekamp C, Sypniewska R, Gauntt S, Tarango M, Price P, Reddick R. Behavior of metastatic and nonmetastatic breast tumors in old mice. Exp Biol Med (Maywood). 2004;229(7):665–675. | ||
Cariati M, Marlow R, Dontu G. Xenotransplantation of breast cancers. Methods Mol Biol. 2011;731:471–482. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.