Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 13
Single-Nucleotide Polymorphisms in Genes Predisposing to Leprosy in Leprosy Household Contacts in Zhejiang Province, China
Authors Shen YL, Long SY, Kong WM, Wu LM, Fei LJ, Yao Q, Wang HS ![]()
Received 11 October 2020
Accepted for publication 26 November 2020
Published 21 December 2020 Volume 2020:13 Pages 767—773
DOI https://doi.org/10.2147/PGPM.S286270
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin Bluth
Yun-Liang Shen,1,* Si-Yu Long,2,* Wen-Ming Kong,1 Li-Mei Wu,1 Li-Juan Fei,1 Qiang Yao,1 Hong-Sheng Wang2
1Department of Leprosy Control, Zhejiang Provincial Institute of Dermatology, Huzhou, People’s Republic of China; 2Laboratory of Leprosy and Other Mycobacterial Infections, Institute of Dermatology, Chinese Academy of Medical Sciences and Peking Union Medical College, Nanjing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hong-Sheng Wang
Laboratory of Leprosy and Other Mycobacterial Infections, Institute of Dermatology, Chinese Academy of Medical Sciences and Peking Union Medical College, St 12 Jiangwangmiao, Nanjing, Jiangsu 210042, People’s Republic of China
Email [email protected]
Qiang Yao
Department of Leprosy Control, Zhejiang Provincial Institute of Dermatology, St 61, Wuyuan, Huzhou, Zhejiang 313200, People’s Republic of China
Email [email protected]
Purpose: Genome-wide association studies (GWAS) have identified multiple genetic variants associated with leprosy. To investigate the single and combined associations between single-nucleotide polymorphisms (SNPs) and the development of leprosy, we therefore performed generalized multi-analytical (GMDR) analysis in Chinese leprosy household contacts and constructed a risk prediction model.
Patients and Methods: This case–control study included 229 leprosy cases and 233 healthy household contacts in Zhejiang province, China. Participants were genotyped for 17 polymorphisms selected from GWAS. The Pearson χ2 test, logistic regression and GMDR analysis were performed to investigate gene–gene interactions and construct a risk prediction model for leprosy.
Results: The genotype and the allele distributions of rs142179458, rs2275606, rs663743 and rs73058713 were significantly different between patients and controls. rs2275606, rs6478108, rs663743 and rs73058713 showed an association after adjusting for sex and age in the logistic regression. A five-way interaction model consisting of rs2058660, rs2275606, rs4720118, rs6478108 and rs780668 was chosen as the optimal model for determining leprosy susceptibility. The model classified 237 (51.3%) into the low-risk group and 225 (48.7%) individuals into the high-risk group. The area under the curve (AUC) of this model was 0.757 (95% CI: 0.712– 0.803), and the odds ratio for leprosy between the high- and low-risk groups was 9.733 (95% CI: 6.384– 14.960; P< 0.001). The sensitivity and specificity of the model were observed to be 74.7% and 76.8%, respectively.
Conclusion: Our results suggest that rs2058660, rs2275606, rs4720118, rs6478108 and rs780668, five SNPs with a significant sole effect on leprosy, interact to confer a higher risk for the disease in leprosy household contacts (HHCs).
Keywords: leprosy, gene–gene interactions, generalized multi-analytical, risk stratification
Introduction
Leprosy, which is caused mainly by Mycobacterium leprae, remains a significant health problem, especially in low- and middle-income countries. China is still one of the countries where the disease remains endemic, mainly in southwestern provinces, including Sichuan, Hunan, Yunnan and Guizhou.1 A total of 521 new cases were detected in China in 2018, with a case detection rate of 0.036 per 100,000 people. The registered cases were 970 by the end of 2018, accounting for a prevalence rate of 0.068 per 100,000 population.2
As an infection disease, an individual’s exposure to M. leprae serves as essential in leprosy developing, so a gradually higher proportion of new cases is found amongst the contacts of known cases.3 However, only a small group of contacts will develop leprosy.4 Genetic factors play important roles in the host’s immunologic status, which can determine the clinical manifestations of leprosy; a prospective cohort study demonstrated that genetic relationship was a relevant and independent risk factor.5 At the population level, GWAS have been performed for better understanding of the human genetic background involved in the eventual outcomes of M. leprae infection, and identified a number of SNPs associated with leprosy genetic predisposition.6–12 Among the studies, SNPs located in RAB32, HIF1A, BATF3, LACC1, CTSB, TNFSF15, CDH18, SLC29A3, DEC1, FLG, NOD2, IL18RAP/IL18R1, NCKIPSD and CARD9 were significantly associated with leprosy. Several association studies have replicated these genes in other independent populations.13–17 Despite GWAS, some other risk genes have been reported or validated in Han Chinese with leprosy.18–22
Generalized multifactor dimensionality reduction (GMDR) was applied to explore the best prediction model of higher order gene–gene interactions. GMDR reduces high-dimensional genetic data to a single dimension, explores an interaction model through cross-validation and calculates score-based statistics of each subject using maximum likelihood estimates to classify subjects into two different groups (either high-risk or low-risk group), and was regarded as an objective analytical tool for evaluating multifactorial impact on an association, allowing adjustment for confounding factors.23,24
In this study, we utilized published genetic risk variants in GWAS with a case–control study and tried to evaluate the role of higher order gene–gene interaction to predict leprosy risk in leprosy HHCs in Chinese Zhejiang province.
Patients and Methods
Study Participants
Two independent sets were enrolled in this study. A total of 245 leprosy patients and 245 healthy leprosy HHCs were enrolled in 2015. People affected by leprosy including those in the course of treatment and cured patients in Zhejiang province, China, were enrolled as the case group. Subjects with any kind of autoimmune-related disease were excluded. The diagnosis of leprosy was confirmed by initial clinical evaluation based on clinical manifestations, slit skin smears and histopathological examinations. Healthy leprosy HHCs from the same geographic region were included in the control group. HHCs were defined as people living under the same roof and sharing food with the patient for at least six months among the past six years and excluded those who refused to provide informed consent, and any person who received treatment for tuberculosis or leprosy within one year. HHCs in our study included relatives (first-, second- and third-degree family members) and genetically unrelated contact individuals (spouses and neighbors). The follow-up set consisted of 939 health contact individuals recruited in 2015 and followed up from 2015 to 2019. Demographic characteristics of the study individuals are listed in Table 1.
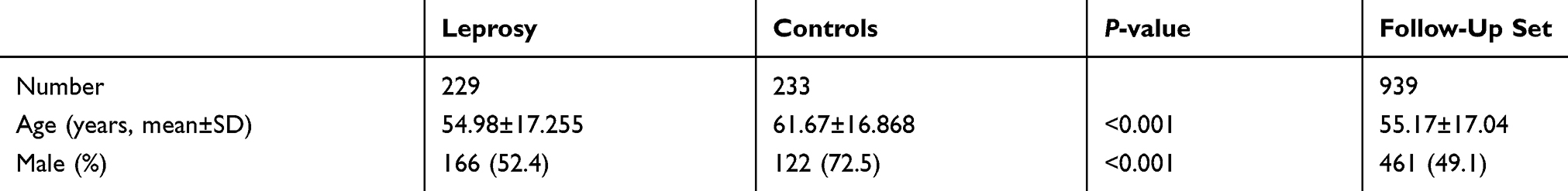 |
Table 1 General Characteristics of the Study Groups |
SNP Selection and Genotyping
We selected 17 SNPs from previously published GWAS and one study combined whole-exome sequencing and targeted next-generation sequencing within the GWAS loci,6–12 wherein was a genome-wide significant association (p<5×10−8) between the SNPs in the genes RAB32, HIF1A, BATF3, LACC1, CTSB, TNFSF15, CDH18, SLC29A3, DEC1, FLG, NOD2, IL18RAP/IL18R1, NCKIPSD and CARD9 and leprosy. This was based on multiplex polymerase chain reaction (PCR) to precisely genotype SNPs with next generation sequencing. After the PCR amplification, the products were genotyped according to the manufacturer's protocol using the Illumina Hiseq X-10 platform. Genotype call rates >95% for both SNPs and individuals, and minor allele frequency (MAF) >1% were found. Ultimately, two variants with MAF <1% were eliminated (rs149308743 and rs145562243). A total of 15 variants and 462 subjects (229 participants with leprosy, 233 controls free of leprosy) were included in the case–control analysis.
Statistical Analysis
The Pearson χ2 test was used to analyze the differences in gender between the patient group and the control group, and the two-sample t-test was used to compare age by SPSS 23.0 software. The P value was considered to be statistically significant at the 0.05 level.
The Pearson χ2 test or Fisher test using PLINK v 1.07 were used to analyze the differences in distributions of genotypes and alleles between the leprosy cases and the control group. We also tested the associations between phenotypes and SNPs based on a logistic regression model adjusted for age and gender, and we constructed a weighted genetic risk score (wGRS) for each individual by summing the number of risk alleles (0/1/2) for each of the four SNPs with P <0.05 weighted by the β coefficient of each allele obtained from the logistic regression. Two-tailed P values of 0.0033 or less after Bonferroni multiple testing correction were considered statistically significant.
GMDR analysis with 10-fold cross-validation was done for assessing gene–gene interactions as an extension of the multifactor dimensionality reduction method.25 It is applicable to both dichotomous and quantitative phenotypes that allow adjustment for covariates and is more precise and accurate than multifactor dimensionality reduction. The GMDR models are evaluated on the testing balanced accuracy (TBA), the cross-validation consistency (CVC) and the statistical significance. The results of GMDR analysis were confirmed by χ2 tests. The AUC and the 95% confidence intervals (CI) were calculated for the risk model with SPSS (version 23; IBM Corporation).
Results
Baseline Characteristics of Participations
There were no subsequent cases in the period of 2015 to 2019 in the follow-up set. 229 participants with leprosy and 233 controls free of leprosy who met the inclusion criteria were included in the study. The case group consisted of 229 leprosy patients, with a mean age of 61.67±16.868 years,while 233 healthy leprosy contacts consisted the control group, with a mean age of 54.98±17.255 years. 939 healthy leprosy HHCs consisted the follow-up set, with a mean age of 55.17±17.044 years. The baseline characteristics of participants are listed in Table 1. Significant difference was found in mean age and sex ratio between leprosy patients and controls. The individuals in the case group were more likely to be older and male.
Association Between Leprosy and SNPs
Genotype distributions between leprosy cases and controls were analyzed using the χ2 test or Fisher test. Supplementary Table 1 shows the distributions of genotypes and alleles for the SNPs. The genotype and the allele distributions of rs142179458, rs2275606, rs663743 and rs73058713 were significantly different between patients and controls. Significant difference between leprosy and control groups was found in rs663743 (P=0.00299, after Bonferroni multiple testing correction).
Four variants showed an association at P<0.05 after adjusting for sex and age in the logistic regression. These included rs2275606 at the RAB32 locus (P=0.03), rs6478108 at the TNFSF15 locus (P=0.03), rs663743 at the CCDC88B locus (P=0.0002) and rs73058713 at the CDH18 locus (P=0.005). The characteristics and association results of the 15 variants are displayed in Table 2.
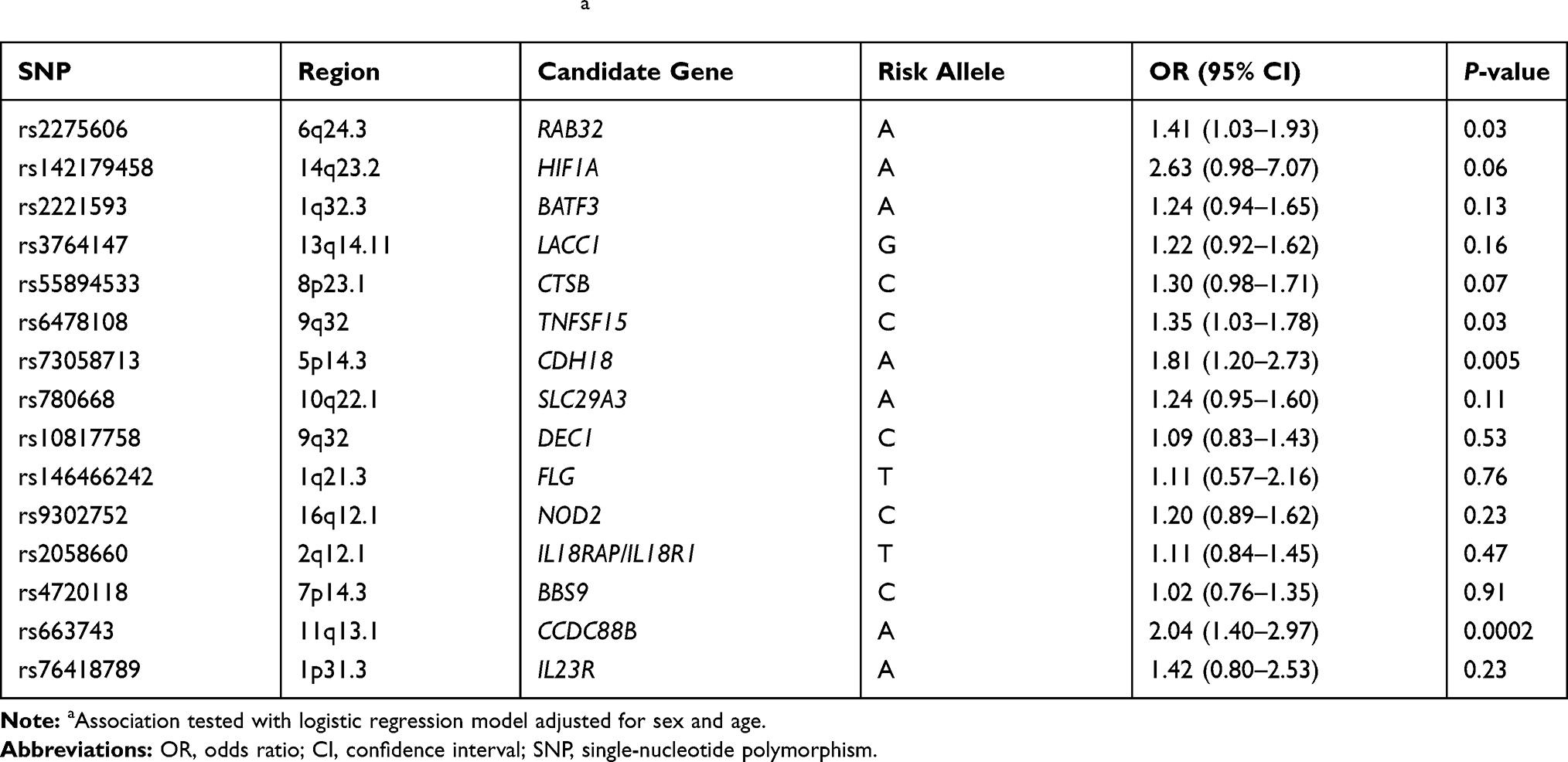 |
Table 2 Association Between SNPs and Leprosya |
Interaction Models by GMDR Analysis
The higher order gene–gene interactions were analyzed using the GMDR software with the adjustment for sex and age. The GMDR analysis yielded the best models for all possible one- to eight-locus models (Table 3). Based on the TBA, CVC and significant P values, the five-way interaction model (rs2058660, rs2275606, rs4720118, rs6478108 and rs780668) was regarded as the optimal model. The testing balanced accuracy was 0.5889, the testing accuracy was 0.5905, the testing sensitivity was 0.6036, the testing specificity was 0.5743, the testing odds ratio was 2.4807 (95% CI: 0.3472–17.7234). For the whole dataset statistics, the training accuracy was 0.7904, the training sensitivity was 0.7879, the training specificity was 0.7930, the training odds ratio was 14.2280 (95% CI: 7.3547–27.5247). There were 76 genotype combinations of the five-way interaction model having high risk of leprosy susceptibility, and the genotype combination (TC, AG, CC, TC, AG) especially showed highest risk for leprosy (Figure 1).
 |
Table 3 Gene–Gene Interaction Models for Leprosy by GMDR Analysis |
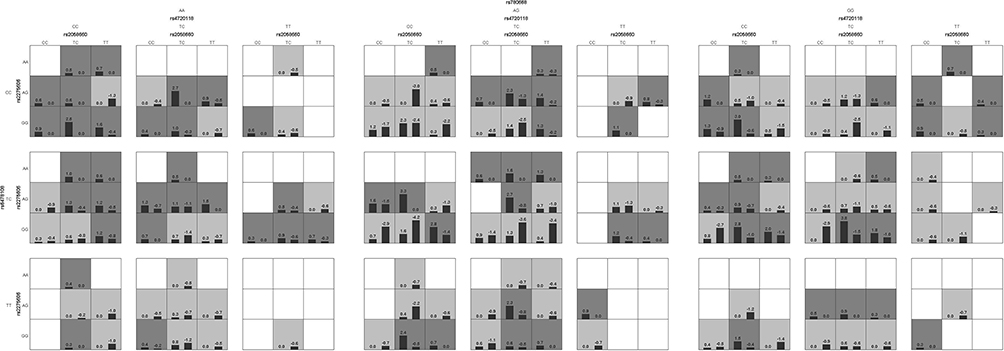 |
Figure 1 Results of adjusted GMDR analyses for the five-factor model. Dark gray and light gray boxes correspond to the high- and low-risk genotype combinations, respectively. The top number above each bar is the sum of scores for the corresponding group of individuals (cases or controls with particular five-locus genotype). The heights of the bars are proportional to the sum of scores in each group. |
Subsequently, the five-locus genotypes were classified into high-risk or low-risk due to the GMDR results and we combined 77 low-risk genotypes and 76 high-risk genotypes to perform the χ2 test to evaluate the five-locus interaction model. The model classified 237 (51.3%) into the low-risk group and 225 (48.7%) individuals into the high-risk group. 171 of 229 leprosy cases were categorized into the high-risk group while 179 of 233 healthy HHCs were categorized into the low-risk group by the model. The estimated AUC of every single risk SNP with P<0.05 showed poor predictive ability. wGRS did improve the predictive ability of the model. However, the GMDR model leads to a much greater AUC increase (Table 4). The AUC of the GMDR model was 0.757 (95% CI: 0.712–0.803), and the odds ratio for leprosy between the high- and low-risk groups was 9.733 (95% CI: 6.384–14.960; P<0.001). The sensitivity and specificity of the model were observed to be 74.7% and 76.8%, respectively. We further evaluated the effectiveness of the prediction by calculating the number of high-risk subjects with this model in the follow-up set. Among the 939 healthy leprosy HHCs in the follow-up group, 375 (39.9%) contact individuals were categorized into the high-risk group and 481 (51.2%) subjects were categorized into the low-risk group by the model, which implies we should pay more attention to the higher risk contacts with longer follow-up.
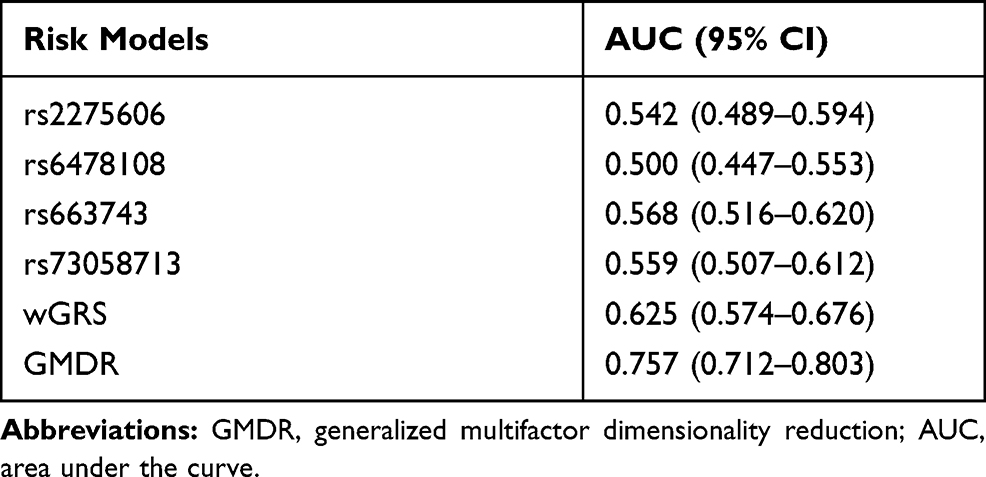 |
Table 4 Performance of Risk Models for Leprosy |
Discussion
As a millenary disease, tracing and strengthening active case finding targeted at higher-risk contact individuals of leprosy serves as a key point of interruption in the leprosy transmission. With the advances in molecular biology, numerous studies have begun to assess the contribution of specific genes and SNPs to the development of the leprosy phenotype and supported that human genetics plays an important role in leprosy susceptibility, and different sets of genes modified host predisposition to leprosy per se, its clinical forms and reactional states.26 In view of the diversity of these genes, we chose 17 independent SNP loci associated with leprosy based on GWAS and one candidate-gene study to determine the role of these genes in the development of leprosy in the Chinese population of Zhejiang province.
Our study highlights SNPs of genes encoding proteins involved in the immune response to the M. leprae infection as risk factors for developing leprosy. The genotype and the allele distributions of rs142179458, rs2275606, rs663743 and rs73058713 were significantly different between leprosy cases and HHCs. Bonferroni adjustment method was performed to correct multiple comparisons, and the allele distribution of rs663743 was found to be significant. Meanwhile, we performed logistic regression analysis to determine the role of genetic factors after adjusting the confounding factor. Four variants showed significant association, including rs2275606, rs6478108, rs663743 and rs73058713.
Similar to our findings, a previous study shows that rs142179458 in HIF1A contributes to leprosy risk, and the HIF1A mRNA expression was increased during M. leprae infection.7 rs2275606 located at the RAB32 locus, our research result suggesting that the Rab32-dependent pathway may act as a host defense pathway in humans. In addition, other studies showed that Rab GTPase Rab32 emerged as a key regulator of a host defense pathway that can restrict intracellular bacterial pathogens. Rab32 and its guanine nucleotide exchange factor BLOC-3 are essential to prevent the growth of the human-restricted Salmonella enterica serovar Typhi (S. Typhi) in mice.7 rs663743 at the CCDC88B locus shows significant association with leprosy in our study, and CCDC88B was reported associated with other autoimmunity and inflammatory diseases,29,30 and was required for regulating movement and migration of dendritic cells.31 rs73058713 at the CDH18 locus was found to be associated with leprosy in our study, and a previous study suspects that the CDH18 gene may be involved in neuronal development governing metabolic processes in later life.32 rs6478108 at the TNFSF15 locus was also found to be associated with leprosy in logistic regression analysis, the TNFSF15 gene is involved in mediating the switch from Th1 cells to Th2 cells, and plays an important role in the control of both pro- and anti-inflammatory cytokines.33,34
GMDR was performed as a genetic risk model to determine the influence of the combination of SNPs along with confounding factors.24,35 In our present study, GMDR was used to study the extensive gene–gene interactions of leprosy along with the adjustment of confounding factors. The best gene–gene interaction model was selected across all multi-locus models that maximized CVC, TBA and significant P values for prediction of leprosy. The five-way interaction model (rs2058660, rs2275606, rs4720118, rs6478108 and rs780668) was considered the optimal model. Furthermore, we predicted leprosy risk with this model. This model is considered useful in discriminating between high- and low-risk individuals (AUC=0.757), the AUC is higher than in a previous study,36 single SNP and wGRS models. Compared with the low-risk HHC group, the high-risk group had a 9.733 times higher risk for leprosy. This result demonstrates the considerable value of this model in risk stratification of leprosy developing. We also displayed the clinical effect of this model in the identification of an independent cohort of HHCs at higher risk of developing leprosy, and the higher-risk contact subjects should receive more frequent monitoring and educate them to increase vigilance of developing leprosy.
The strength of the study was that our study subjects were close contacts of leprosy, which leads to a more profound clinical significance compared with the general population. And we investigated 17 SNPs based on GWAS and one candidate-gene study on leprosy risk. There were a few limitations in our study. Firstly, there are some other SNPs in these genes studied in this research that showed a positive association with leprosy (Supplementary Table 2), and it might be necessary to study more polymorphisms associated with leprosy and other epidemiology factors in the future. Moreover, due to the small size of samples in this study, this five-locus model could not cover all genotypes; there were 83 (8.8%) subjects in the follow-up set who could not be categorized because their genotype combinations were not within the scope of the prediction model. And then we were unable to precisely estimate the model’s performance. Thirdly, the HHCs genetically related to index patients were included in the control group, which may have an impact on the results of association analysis, and subjects with or without genetic relationship will need to be studied separately in the future. Finally, further validation studies of the five-locus model in other prospective contact Chinese cohorts are required.
Conclusion
The results presented in our study provide evidence for a five-locus genetic interaction model in identifying higher-risk leprosy contact individuals. This model with good discrimination capacity may be translated into a tool for predicting leprosy developing and risk stratification. Medical workers should pay attention to the higher-risk contact group selected by the model when they make a decision to trace leprosy HHCs.
Ethics Approval and Informed Consent
This study was approved by the institutional ethical committee of the Institute of Dermatology, Chinese Academy of Medical Sciences and Peking Union Medical College, China (2014-KY-003) and this study was conducted in accordance with the Declaration of Helsinki. All participants provided written informed consent.
Acknowledgment
We thank all the staff who have been involved in the generation of data at each local institute.
Funding
This work was supported by the Ministry of Health of China-research special funds for public health projects [grant number: 201502008], National Science and Technology Major Project [2018ZX10101-001], Chinese Academy of Medical Sciences Innovation Fund for Medical Science [2016-I2M-1-005, 2017-I2M-B&R-14], Jiangsu Provincial Science and Technology Project [BE2018619], The Nanjing Incubation Program for National Clinical Research Center [2019060001].
Disclosure
All authors declare that they have no conflicts of interest.
References
1. Chokkakula S, Shui T, Jiang H, et al. Genotyping of Mycobacterium leprae for understanding the distribution and transmission of leprosy in endemic provinces of China. Int J Infect Dis. 2020;98:6–13. doi:10.1016/j.ijid.2020.06.032
2. Global leprosy update, 2018: moving towards a leprosy- free world. Available from: https://www.who.int/publications/i/item/who-wer9435-36. Accessed December 15, 2020.
3. Gillini L, Cooreman E, Wood T, Pemmaraju VR, Saunderson P, Phillips RO. Global practices in regard to implementation of preventive measures for leprosy. PLoS Negl Trop Dis. 2017;11(5):e5399. doi:10.1371/journal.pntd.0005399
4. Le W, Haiqin J, Danfeng H, et al. Monitoring and detection of leprosy patients in Southwest China: a retrospective study, 2010–2014. Sci Rep. 2018;8(1):11407.
5. Moet FJ, Pahan D, Schuring RP, Oskam L, Richardus JH. Physical distance, genetic relationship, age, and leprosy classification are independent risk factors for leprosy in contacts of patients with leprosy. J Infect Dis. 2006;193(3):346–353. doi:10.1086/499278
6. Zhang FR, Huang W, Chen SM, et al. Genomewide association study of leprosy. N Engl J Med. 2009;361(27):2609–2618. doi:10.1056/NEJMoa0903753
7. Wang D, Fan Y, Malhi M, et al. Missense variants in HIF1A and LACC1 contribute to leprosy risk in Han Chinese. Am J Hum Genet. 2018;102(5):794–805. doi:10.1016/j.ajhg.2018.03.006
8. Liu H, Wang Z, Li Y, et al. Genome-wide analysis of protein-coding variants in leprosy. J Invest Dermatol. 2017;137(12):2544–2551. doi:10.1016/j.jid.2017.08.004
9. Wang Z, Sun Y, Fu X, et al. A large-scale genome-wide association and meta-analysis identified four novel susceptibility loci for leprosy. Nat Commun. 2016;7:13760. doi:10.1038/ncomms13760
10. Liu H, Irwanto A, Fu X, et al. Discovery of six new susceptibility loci and analysis of pleiotropic effects in leprosy. Nat Genet. 2015;47(3):267–271. doi:10.1038/ng.3212
11. Liu H, Irwanto A, Tian H, et al. Identification of IL18RAP/IL18R1 and IL12B as leprosy risk genes demonstrates shared pathogenesis between inflammation and infectious diseases. Am J Hum Genet. 2012;91(5):935–941. doi:10.1016/j.ajhg.2012.09.010
12. Zhang F, Liu H, Chen S, et al. Identification of two new loci at IL23R and RAB32 that influence susceptibility to leprosy. Nat Genet. 2011;43(12):1247–1251. doi:10.1038/ng.973
13. Berrington WR, Macdonald M, Khadge S, et al. Common polymorphisms in the NOD2 gene region are associated with leprosy and its reactive states. J Infect Dis. 2010;201(9):1422–1435. doi:10.1086/651559
14. Wong SH, Hill AV, Vannberg FO. Genomewide association study of leprosy. N Engl J Med. 2010;362(15):
15. Cobat A, Abel L, Alcaïs A, Schurr E. A general efficient and flexible approach for genome-wide association analyses of imputed genotypes in family-based designs. Genet Epidemiol. 2014;38(6):560–571.
16. Grant AV, Alter A, Huong NT, et al. Crohn’s disease susceptibility genes are associated with leprosy in the Vietnamese population. J Infect Dis. 2012;206(11):1763–1767. doi:10.1093/infdis/jis588
17. Li GD, Wang D, Zhang DF, et al. Fine mapping of the GWAS loci identifies SLC35D1 and IL23R as potential risk genes for leprosy. J Dermatol Sci. 2016;84(3):322–329. doi:10.1016/j.jdermsci.2016.09.018
18. Wang D, Feng JQ, Li YY, et al. Genetic variants of the MRC1 gene and the IFNG gene are associated with leprosy in Han Chinese from Southwest China. Hum Genet. 2012;131(7):1251–1260. doi:10.1007/s00439-012-1153-7
19. Zhang DF, Huang XQ, Wang D, Li YY, Yao YG. Genetic variants of complement genes ficolin-2, mannose-binding lectin and complement factor H are associated with leprosy in Han Chinese from Southwest China. Hum Genet. 2013;132(6):629–640. doi:10.1007/s00439-013-1273-8
20. Wang D, Xu L, Lv L, et al. Association of the LRRK2 genetic polymorphisms with leprosy in Han Chinese from Southwest China. Genes Immun. 2015;16(2):112–119. doi:10.1038/gene.2014.72
21. Xiang YL, Zhang DF, Wang D, Li YY, Yao YG. Common variants of OPA1 conferring genetic susceptibility to leprosy in Han Chinese from Southwest China. J Dermatol Sci. 2015;80(2):133–141. doi:10.1016/j.jdermsci.2015.09.001
22. Wang D, Zhang DF, Li GD, et al. A pleiotropic effect of the APOE gene: association of APOE polymorphisms with multibacillary leprosy in Han Chinese from Southwest China. Br J Dermatol. 2018;178(4):931–939. doi:10.1111/bjd.16020
23. Liu FH, Song JY, Shang XR, Meng XR, Ma J, Wang HJ. The gene-gene interaction of INSIG-SCAP-SREBP pathway on the risk of obesity in Chinese children. Biomed Res Int. 2014;2014:538564.
24. Agarwal G, Tulsyan S, Lal P, Mittal B. Generalized Multifactor Dimensionality Reduction (GMDR) analysis of drug-metabolizing enzyme-encoding gene polymorphisms may predict treatment outcomes in Indian breast cancer patients. World J Surg. 2016;40(7):1600–1610. doi:10.1007/s00268-015-3263-6
25. Lou XY, Chen GB, Yan L, et al. A generalized combinatorial approach for detecting gene-by-gene and gene-by-environment interactions with application to nicotine dependence. Am J Hum Genet. 2007;80(6):1125–1137. doi:10.1086/518312
26. Dallmann-Sauer M, Correa-Macedo W, Schurr E. Human genetics of mycobacterial disease. Mamm Genome. 2018;29(7–8):523–538. doi:10.1007/s00335-018-9765-4
27. Spanò S, Gao X, Hannemann S, Lara-Tejero M, Galán JE. A bacterial pathogen targets a host rab-family GTPase defense pathway with a GAP. Cell Host Microbe. 2016;19(2):216–226. doi:10.1016/j.chom.2016.01.004
28. Gerondopoulos A, Langemeyer L, Liang JR, Linford A, Barr FA. BLOC-3 mutated in Hermansky-Pudlak syndrome is a Rab32/38 guanine nucleotide exchange factor. Curr Biol. 2012;22(22):2135–2139. doi:10.1016/j.cub.2012.09.020
29. Mells GF, Floyd JA, Morley KI, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet. 2011;43(4):329–332. doi:10.1038/ng.789
30. Ryan FJ, Ahern AM, Fitzgerald RS, et al. Colonic microbiota is associated with inflammation and host epigenomic alterations in inflammatory bowel disease. Nat Commun. 2020;11(1):1512. doi:10.1038/s41467-020-15342-5
31. Olivier JF, Fodil N, Al HS, et al. CCDC88B is required for mobility and inflammatory functions of dendritic cells. J Leukoc Biol. 2020;108(6):1787–1802. doi:10.1002/JLB.3A0420-386R
32. Schlicht K, Nyczka P, Caliebe A, et al. The metabolic network coherence of human transcriptomes is associated with genetic variation at the cadherin 18 locus. Hum Genet. 2019;138(4):375–388. doi:10.1007/s00439-019-01994-x
33. Meylan F, Davidson TS, Kahle E, et al. The TNF-family receptor DR3 is essential for diverse T cell-mediated inflammatory diseases. Immunity. 2008;29(1):79–89. doi:10.1016/j.immuni.2008.04.021
34. Kadiyska T, Tourtourikov I, Popmihaylova AM, Kadian H, Chavoushian A. Role of TNFSF15 in the intestinal inflammatory response. World J Gastrointest Pathophysiol. 2018;9(4):73–78. doi:10.4291/wjgp.v9.i4.73
35. Jakobsdottir J, Conley YP, Weeks DE, Ferrell RE, Gorin MB, Weedon MN. C2 and CFB genes in age-related maculopathy and joint action with CFH and LOC387715 genes. PLoS One. 2008;3(5):e2199. doi:10.1371/journal.pone.0002199
36. Wang N, Wang Z, Wang C, et al. Prediction of leprosy in the Chinese population based on a weighted genetic risk score. PLoS Negl Trop Dis. 2018;12(9):e6789. doi:10.1371/journal.pntd.0006789
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.