Back to Journals » Drug Design, Development and Therapy » Volume 13
Single- and multiple-dose tolerability, safety, pharmacokinetics, and pharmacodynamics of the dual endothelin receptor antagonist aprocitentan in healthy adult and elderly subjects
Authors Sidharta PN, Melchior M, Kankam MK, Dingemanse J ![]()
Received 20 December 2018
Accepted for publication 7 February 2019
Published 22 March 2019 Volume 2019:13 Pages 949—964
DOI https://doi.org/10.2147/DDDT.S199051
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Georgios Panos
Patricia N Sidharta,1 Meggane Melchior,1 Martin K Kankam,2 Jasper Dingemanse1
1Department of Clinical Pharmacology, Idorsia Pharmaceuticals Ltd, Allschwil CH-4123, Switzerland; 2Vince and Associates Clinical Research, Overland Park, KS 66211, USA
Background: Aprocitentan is an orally active, dual endothelin (ET) receptor antagonist developed for the treatment of hypertension in which, despite available treatments, a medical need exists for drugs with a new mechanism of action.
Subjects and methods: In this study, the single- and multiple-dose tolerability, safety, pharmacokinetics (PK), and pharmacodynamics of up to 600 mg (single doses) and 100 mg once a day (qd; multiple doses) of aprocitentan were investigated in healthy male and female subjects. The effect of age on the tolerability and PK parameters was investigated at a dose of 100 mg qd.
Results: Aprocitentan was well tolerated across all doses. No serious adverse events (AEs) occurred. The most frequently reported AE was headache. Small increases in body weight were recorded in subjects receiving 100 mg qd. Plasma concentration–time profiles of aprocitentan were similar after single- and multiple-dose administration, and support a qd dosing regimen based on a half-life of 44 hours. After multiple doses, PK was dose proportional. Accumulation at steady state, reached by Day 8, was 3-fold. Only minor differences in exposure between healthy females and males, healthy elderly and adult subjects, and fed and fasted conditions were observed. Plasma ET-1 concentrations, reflecting ETB receptor antagonism, significantly increased with doses ≥25 mg. Time-matched analysis of electrocardiogram (ECG) parameters did not suggest drug-induced ECG effects. Exposure–response analysis indicated no QTc prolongations at plasma levels up to 10 µg/mL.
Conclusion: Aprocitentan was well tolerated in healthy subjects with a PK profile favorable for qd dosing.
Keywords: first-in-human study, endothelin, endothelin receptor antagonist, aprocitentan, pharmacokinetics, pharmacodynamics
Introduction
Hypertension is an important public health challenge, being the most frequent risk factor of cardiovascular disease and estimated to have a prevalence of 30%–40% of the general population.1 Despite the availability of different antihypertensive drugs, BP remains inadequately controlled in a significant number of patients due to various reasons including poor medication dosage and/or low compliance, and also secondary hypertension and resistance to actual pharmacological therapies.2 Resistant hypertension (RHT) is defined as uncontrolled BP (ie, failure to lower BP to a predefined threshold) despite concurrent administration of three antihypertensive therapies of different pharmacological classes at maximal or optimal doses, including a diuretic.3 Usually, a renin–angiotensin system (RAS) blocker is the first-line therapy, followed by calcium channel blockers and diuretics. The use of a fourth-line therapy is recommended, but the available choices often raise safety and tolerability issues. For example, the use of RAS blockers and mineralocorticoid receptor antagonists in patients with reduced renal function has been associated with an increased risk of adverse events (AEs), including hyperkalemia and heart failure.4–6 Further, hypertensive patients often display numerous comorbidities such as chronic kidney disease and diabetes mellitus, requiring additional pharmacological treatment.7,8 Therefore, there is a substantial need to develop antihypertensive drugs with new mechanisms of action that show efficacy when added to the current available therapies.9
One of the possible new targets is the endothelin (ET) system, which has an increased activity under pathological conditions, including hypertension.7,10,11 Endothelin-1 (ET-1) is a small (21 amino acids) peptide synthesized predominantly by the vascular endothelium.12,13 It is a potent vasoconstrictor that acts through two receptors, ETA and ETB, predominantly located on the vascular smooth muscle cells and the endothelial cells, respectively.14–16 Overall, in physiological conditions, ETA receptor activation leads to vasoconstriction, whereas ETB receptor activation mediates vasodilation through nitric oxide release.15 Further, ETB receptors are responsible for the clearance of ET-117,18 and are also expressed in the medullary part of the kidney, where they are responsible for sodium and water regulation.19 As RHT is frequently associated with volume expansion, which is a feature of the salt-sensitive, low-renin form of hypertension, antagonism of both ETA and ETB receptors may demonstrate greater efficacy in RHT compared to forms of hypertension with high/normal renin.20
Aprocitentan (ACT-132577) is a novel dual endothelin receptor antagonist (ERA) that potently inhibits the binding of ET-1 to ETA and ETB receptors. After administration of single oral doses of ≥10 mg/kg of aprocitentan in normotensive rats, an increase in plasma ET-1 concentrations was observed, suggesting an efficient blockade of ETB receptors.21 The potential of single- and multiple-dose administration of aprocitentan to decrease mean arterial pressure has been demonstrated in two rat models of hypertension, that is, in spontaneously hypertensive rats and in the deoxycorticosterone acetate (DOCA)-salt model of salt-dependent hypertension.21 The effect was stronger in DOCA-salt animals, a model characterized by a lower plasma renin activity. Importantly, the effect of aprocitentan was shown to be synergistic with that of RAS blockers, allowing further decrease in BP without increasing the risk of renal impairment. This suggests that aprocitentan has the potential to be safely used in combination with RAS blockers.
Aprocitentan is the active metabolite of macitentan, which is a dual ERA approved for the treatment of pulmonary arterial hypertension.22 As part of the registration package of macitentan, the tolerability, safety, pharmacokinetics (PK), and pharmacodynamics (PD) were extensively characterized.23,24 In this context, humans have been exposed to aprocitentan, albeit in a different exposure range. The PK of macitentan after multiple-dose administration showed that maximum plasma concentrations (Cmax) were reached at ~8 hours with a terminal elimination half-life (t½) varying between 14 and 19 hours. Accumulation at a steady state was limited, as indicated by an accumulation index (AI) between 1.4 and 1.7 for the different doses investigated. After single- and multiple-dose administration of macitentan, aprocitentan reached Cmax at around 8 hours with a substantial longer t½ of 48 hours. Due to the longer t½, aprocitentan had a significantly higher AI of 7–10 over the dose range tested.25
The direct administration of aprocitentan should allow reaching higher exposure at a steady state compared to that observed after administration of the currently available approved dose of 10 mg macitentan. In this first-in-human study, we investigated the tolerability, safety, PK, and PD of single- and multiple-ascending doses of aprocitentan in healthy adult male and female subjects over a wide range of doses, as well as in elderly healthy subjects and in both the presence and absence of food.
Subjects and methods
The study followed the principles of the Declaration of Helsinki and Good Clinical Practice, and the protocol was approved by an independent institutional review board (MidLands Independent Review Board, Overland Park, KS, USA). All subjects provided written informed consent prior to screening. The study was conducted at the Vince & Associate Clinical Research Unit (Overland Park, KS, USA).
Study design
This study was a three-part (pilot study, single-ascending dose [SAD] part, and multiple-ascending dose [MAD] part) single-center study, testing doses of aprocitentan ranging from 5 to 600 mg in healthy male and female subjects. The doses were chosen using safety margins based on human equivalent dose, and aimed to help identify the maximum tolerated dose. Part A (the pilot study) had an open-label design and aimed to explore the PK of aprocitentan in healthy subjects. Parts B and C (SAD and MAD) had randomized, double-blind, placebo-controlled designs and aimed to provide tolerability, safety, and PK data following different doses in healthy subjects. Additionally, in Part B, the effect of food was investigated at a dose level of 100 mg. In Part C, PD was assessed, as well as a more detailed cardiovascular analysis was performed using Holter recording. The effect of age was assessed in one group of elderly subjects at a dose of 100 mg. Screening occurred from Day −21 to Day −11. In Part A, subjects were admitted to the clinic in the evening before study treatment administration and stayed until 240 hours thereafter. For Part B, they stayed in the clinic for 96 hours after treatment administration and assessments thereafter were performed on an ambulatory basis. For Part C, subjects were admitted two evenings before the first study treatment administration and were confined to the clinic until 48 hours after the first dose administration. They were asked to come back to the clinic 24 hours before and until 48 hours after the last treatment administration, and other assessments were performed on an ambulatory basis. For all parts, end-of-study (EOS) took place within 216–240 hours after the last study treatment administration and a safety follow-up was performed 30–33 days after the last study treatment administration (by phone call for male subjects and by an outpatient visit for female subjects).
Part A consisted of a pilot study in which the subjects received a single oral dose of 5 mg of aprocitentan after an overnight fast. Part B (SAD) consisted of four successive groups of eight healthy subjects receiving a single dose of the study treatment. In each group, after an overnight fast, six subjects received aprocitentan in ascending doses of 25, 100, 300, and 600 mg, whereas two subjects received placebo. To investigate the food effect on the tolerability and PK of aprocitentan, subjects in the 100 mg dose group returned to the clinic after a 14-day washout period and received the same study treatment after consuming a high-fat, high-calorie standardized breakfast. Part C (MAD) consisted of three successive groups of healthy subjects receiving multiple oral doses of the study treatment. In each group, six subjects received aprocitentan in ascending doses of 5, 25, and 100 mg once daily (qd) for 10 days, whereas two subjects received placebo. To investigate the effect of age on the tolerability and PK of aprocitentan, eight additional elderly subjects received the study treatment. Six elderly subjects received aprocitentan as multiple oral doses of 100 mg qd for 10 days, whereas two elderly subjects received placebo. An overview of the different elements in the study design of parts A–C is given in Table 1.
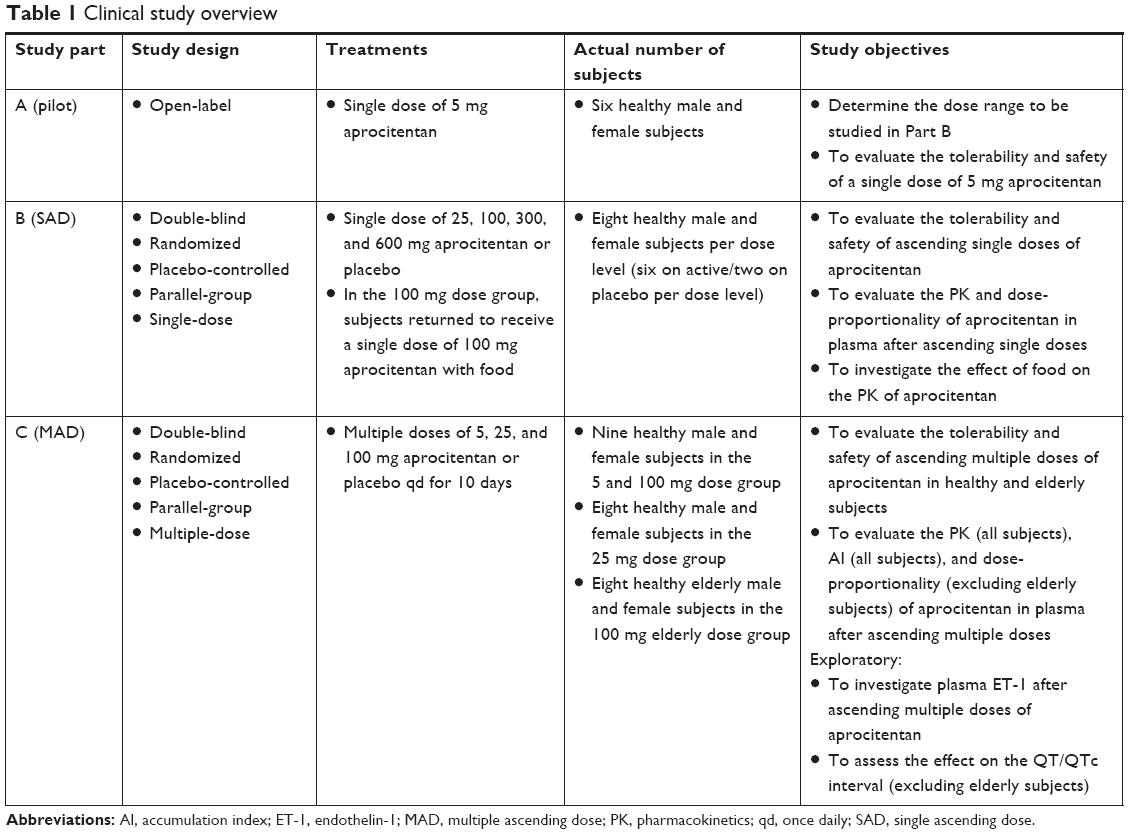 | Table 1 Clinical study overview |
Study population
In this study (AC-080–101), 6 subjects were included in Part A, 32 were included in Part B, and 24 subjects as well as 8 elderly subjects were included in Part C. The male:female ratio was 1:1 in all parts of the study. Subjects were healthy based on medical history, physical examination, electrocardiogram (ECG), vital signs, and clinical laboratory tests. Subjects had to be between 18 and 45 years of age and between 65 and 80 years of age for the elderly group. They could not participate if they smoked, had a history of drug or alcohol abuse, were allergic to any excipient of the drug formulation, were using any medication, or had participated in another clinical trial in the last 3 months. Women of childbearing potential had to have a negative pregnancy test at screening and pre-dose on Day −1 of each treatment and had to agree to use a reliable method of contraception. Male subjects had to agree to use condoms and spermicide for up to 30 days after the EOS treatment.
Tolerability assessments
All AEs that occurred after (first) study treatment administration and up to EOS were recorded.
Parts A and B: physical examination and body weight (BW) measurements were performed at screening and at EOS. Vital signs (pulse rate and standing and supine BP) and ECG were measured at screening, immediately before and at several time points after treatment administration and at EOS. Laboratory parameters (hematology, clinical chemistry, coagulation tests, and urinalysis) were performed at screening, Day 1 (before treatment administration), Day 2, and EOS.
Part C: physical examination was performed at screening and at EOS. BW, vital signs, and ECG measurements were performed at screening, immediately before treatment administration on Day 1, Day 2, Day 4, and Day 10, at various time points after study treatment administration, and at EOS. Laboratory parameters were performed at screening, immediately before treatment administration on Day 1, Day 2, Day 10, as well as on Day 11, and at EOS. In addition, for healthy adult subjects only, Holter recording was performed on Day −1 and on Day 10 for 24 hours. For the cardiodynamic analysis, ten ECG replicates were extracted from the Holter recordings from a 5-minutes window around each time point of PK blood sampling.
Before dose escalation, a thorough review of the tolerability, safety, and PK data of subjects from the previous group was performed by the safety review committee.
PK assessments
For the determination of aprocitentan in plasma, venous blood samples (4 mL each) were collected with K3-EDTA as the anticoagulant. Following centrifugation at 1,500×g for 10 minutes at 4°C, plasma was separated and frozen at −80°C±20°C pending analysis. In parts A and B, plasma samples were taken pre-dose on Day 1 and at 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 15, 24, 36, 48, 72, 96, 120, 168, and 216 hours after treatment intake. In Part C, plasma samples were taken immediately before the first dose on Day 1 and at 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, and 15 hours post-dose on Day 1, pre-dose and 12 hours post-dose on Day 2, and pre-dose on days 3, 4, 5, 8, and 10, and 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 15, 24, 36, 48, 72, 96, 120, 168, and 216 hours post-dose on Day 10.
PD assessments
For ET-1 measurements in plasma from healthy adult subjects in Part C, blood samples (2 mL each) were collected at the same time points as PK plasma samples.
Bioanalytical method
Samples were protected from light at all times. Analysis of aprocitentan was performed by a liquid chromatography coupled to tandem mass spectrometry method. To an aliquot of 30 μL of plasma, 120 μL of a 1:1 (v:v) mixture of acetonitrile/ethanol containing internal standard (penta-deuterated aprocitentan) at a concentration of 250 ng/mL was added. After protein precipitation, the samples were centrifuged for 20 minutes at 3,250×g and 4°C. Supernatants were diluted with 120 μL of water/acetonitrile (50:50, v/v), and aliquots of 2 μL were injected onto the HPLC column kept at room temperature (Kinetex C18, 50×3.0 mm ID, 2.6 μm column Phenomenex; Brechbühler, Schlieren, Switzerland). The concentrations of the analytes were determined using the internal standardization method. The response was determined by calculation of the calibration curve using a weighted linear regression (1/x2). The inter-batch precision was ≤5.8%, whereas the inter-batch accuracy was in the range from −6.7% to 0.6%. Incurred sample reanalysis was performed and met the acceptance criteria. The calibration range was 5.00–10,000 ng/mL. Study samples were 10-fold diluted when concentrations were above the upper range of the calibration range, by adding 10 μL of the study sample to 90 μL of human control plasma.
Plasma concentrations of ET-1 were determined using a validated ELISA (Quantiglo; R&D Systems GmbH, Wiesbaden-Nordenstadt, Germany) with a lower and upper limit of quantification (LOQ) of 0.3 and 15 pg/mL, respectively.
Data analysis
Tolerability parameters were analyzed descriptively. In parts B and C, all placebo-treated subjects in the different treatment groups were pooled for analysis of tolerability. Data were analyzed together for parts A and B and separately for Part C. For the cardiodynamic analysis, the relationship between aprocitentan plasma concentration and ΔQTcF (time-matched baseline-adjusted QT interval corrected with Fridericia’s formula) was quantified using a linear mixed-effects model, with ΔQTcF as the dependent variable, drug plasma concentration as the covariate, and treatment (active or placebo) and time point as the categorical factors, and a random intercept per subject.26 From the model, the slope (ie, the regression parameters for the concentration) and the treatment effect were estimated together with its two-sided 90% CI. The geometric mean of the individual Cmax values for subjects in each of the active drug groups was determined. The population average placebo-adjusted ΔQTcF (ΔΔQTcF) and its two-sided 90% CI at the geometric mean maximum plasma concentrations were derived directly from the linear mixed-effects model. The assessment of the adequacy of the linear mixed-effects model was provided by a goodness-of-fit plot (ie, the observed concentration decile–ΔΔQTcF plot). It was used to check the assumption of linearity between plasma concentration of aprocitentan and ΔΔQTcF and how well the predicted ΔΔQTcF matched the observed data in the regions of interest. The goodness-of-fit plot was generated by binning the independent variable (ie, concentrations) into deciles. The mean ΔΔQTcF with 90% CI within each decile was computed and plotted at the corresponding median concentration within the decile. The decile ranges were added in the bottom of the graphs to illustrate the span of each decile and possible skewness of the tails. The ΔΔQTcF was derived from the individual ΔQTcF for aprocitentan subtracted by the mean predicted ΔQTcF for placebo from the model. The central tendency analysis for QTcF was based on a linear mixed-effects model with ΔQTcF as the dependent variable, time (categorical), study treatment (5, 25, and 100 mg of aprocitentan, and placebo), and time by treatment interaction as factors. Subject was included as a random effect for the intercept. A categorical analysis was also performed to identify categorical outliers for QTc, PR, QRS, heart rate (HR), and T-wave morphology. Subjects dosed with placebo were analyzed as a pooled group.
PK parameters were determined by non-compartmental analysis using Professional WinNonlin 6.4 (Pharsight Corp., Mountain View, CA, USA). The measured individual plasma concentrations of aprocitentan were used to directly obtain Cmax and tmax. The area under the plasma concentration–time curve from 0 to time t of the last measured concentration above the LOQ (AUC0–t) was calculated according to the linear trapezoidal rule, using the measured concentration–time values. Values below LOQ were set to zero. AUC0–∞ was calculated by combining AUC0–t and AUCextra. AUCextra represents an extrapolated value obtained by Ct/λz, where Ct is the last concentration above the LOQ and λz represents the terminal elimination rate constant determined by log-linear regression analysis of the measured plasma concentrations in the terminal elimination phase. The terminal half-life (t½) of aprocitentan was calculated as follows: t½=0.693/λz.
In addition, in Part C, trough levels of aprocitentan were used to investigate the time to attainment of steady-state conditions. This was carried out by visual inspection of the mean trough plasma concentration–time profile. The measured individual plasma concentrations at steady state were used to obtain Cmin. AUCτ was calculated according to the linear trapezoidal rule using the measured concentration–time values above the LOQ during one dosing interval. The AI was calculated by dividing AUCτ on Day 10 by AUCτ on Day 1. Cavg was calculated as AUCτ/τ, where τ represents the dosing interval. The peak-trough fluctuation was calculated using the equation: 100×(Cmax − Cmin)/Cavg. All AUC and Cmax values were assumed to be log-normally distributed.
Statistical analysis
Dose proportionality of PK of aprocitentan was explored by comparing Cmax and AUC values, corrected for dose and log transformed, using a power model described by Gough et al.27
Differences in plasma PK variables AUC0–t, AUC0–∞, and Cmax in Part B between the fed and the fasted (reference) states were investigated using a two-sided 90% CI of the ratio of the geometric means. Differences between fasted and fed tmax were explored using the median differences (non-parametric analysis) and their 90% CI using the fasted state as reference. The same analysis was used to compare plasma PK variables between healthy adult subjects (reference) and elderly subjects, and between male (reference) and female subjects.
Results
Subject disposition
Thirty-eight healthy subjects, consisting of 19 males and 19 females (age range: 18–44 years), participated in parts A and B of this study, and 34 subjects participated in Part C, consisting of 17 males (including 4 elderly) and 17 females (including 4 elderly). Age range in the healthy group was 25.1–29.3 years and in the elderly group was 67.8–70.0 years. Two subjects in two different cohorts in Part B discontinued the study prematurely (due to withdrawal of consent and for other reasons, respectively). Four subjects in Part C discontinued the study prematurely (two due to an AE and two for other reasons), among whom two were replaced after discussion between the sponsor and the investigator. All subjects were evaluable for tolerability and safety. In parts A and B, all subjects except one were evaluable for PK. One subject withdrew consent before the fed period and was excluded from the food effect analysis. In Part C, 23 subjects were evaluable for PK and 22 were evaluable for PD (the elderly subjects were not included in the PD analysis).
Tolerability of aprocitentan
No serious AEs or AEs leading to study discontinuation occurred in parts A and B. In Part C, no serious AEs occurred. Two subjects had AEs leading to discontinuation of the study treatment, the first one due to an AE of upper respiratory tract infection on Day 7, which resolved on Day 8, and the second one due to an AE of throat irritation reported after dosing on Day 1.
In parts A and B of the study, 16 of the 30 healthy subjects who were administered single doses of aprocitentan reported a total of 44 AEs (Table 2). In the placebo group, four of the eight subjects had at least one AE. Overall, most of the AEs were of mild intensity, except for moderate AEs reported by five subjects in the last three highest dose groups. The number of subjects reporting at least one AE increased with increasing dose, with ~17%, 33%, 50%, 67%, and 100% of subjects reporting at least one AE in the 5, 25, 100, 300, and 600 mg aprocitentan dose groups, respectively. The most commonly reported AEs were headache, nausea, postural orthostatic tachycardia syndrome, and nasal congestion. All AEs resolved without sequelae at EOS, with the exception of three AEs in two subjects: furuncle in the 100 mg fed cohort (the only AE reported during fed conditions) and nasal congestion and acne in one subject in the 600 mg cohort. However, acne was reported as a medical condition at baseline by this subject.
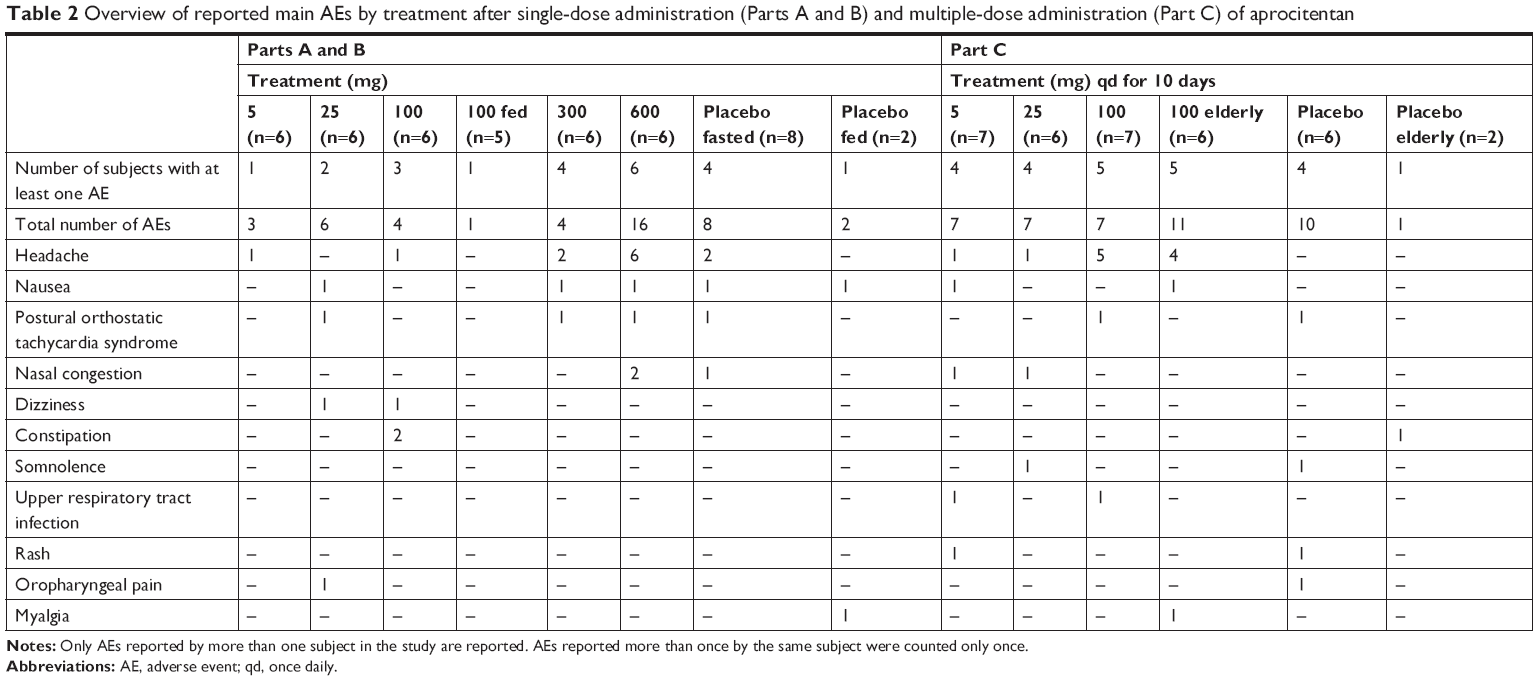 | Table 2 Overview of reported main AEs by treatment after single-dose administration (Parts A and B) and multiple-dose administration (Part C) of aprocitentan |
In Part C, 13 of the 20 healthy subjects who were administered multiple doses of aprocitentan reported a total of 21 AEs (Table 2). In the placebo group, four of the six subjects reported at least one AE, with a total of ten AEs. Most AEs were of mild intensity, except for moderate AEs reported by two subjects, one in the 100 mg cohort and one on placebo. The number of subjects reporting at least one AE increased with increasing dose, with ~57%, 67%, and 71% of subjects reporting at least one AE in the 5, 25, and 100 mg group, respectively. The most commonly reported AE was headache. All AEs resolved without sequelae at EOS, with the exception of two AEs in two subjects: leukopenia in the 5 mg cohort and vessel puncture site pain in the 25 mg group.
In the elderly cohort, five of the six subjects (83.3%) on 100 mg aprocitentan and one of the two subjects on placebo reported at least one AE. Headache was the most commonly reported AE, with four subjects on aprocitentan. All AEs were of mild intensity. One subject in this cohort had an AE of elevated ALT (3.8× upper limit of normal [ULN] vs 1.3× ULN at baseline) and AST (2.5× ULN vs 1.0× ULN at baseline), which was considered related to the study treatment by the investigator and resolved ~2.5 weeks after last study treatment administration.
Clinical laboratory evaluation revealed a decrease from baseline in hemoglobin following single-dose administration of 25, 100, 300, and 600 mg aprocitentan. However, this was not dependent on the dose. In Part C, the review of hemoglobin at steady state on Day 10 suggested a dose-related decrease with median decreases from baseline of 1.00, 1.30, and 1.60 g/dL for 5, 25, and 100 mg aprocitentan, respectively, whereas the median decrease from baseline was 1.10 g/dL in the placebo group. A similar observation was made in the elderly group, which displayed a median decrease from baseline of 1.5 g/dL at steady state on Day 10 compared to 0.70 g/dL in the elderly subjects treated with placebo. After multiple-dose administration of aprocitentan, similar mean and median decreases in hematocrit and red blood cells from baseline were observed in the aprocitentan and placebo groups. There were no other clinically relevant findings on laboratory variables and vital signs.
In Part C, the cardiodynamic analysis of change-from-baseline and time-matched, placebo-adjusted change-from-baseline values revealed no change in ECG variables QTcF, HR, PR, and QRS in relation to time of dosing and dose level. There were no subjects with a QTcF value >450 ms at any time point on Day 10, and only one subject receiving 25 mg aprocitentan had a QTcF value ≥30 ms at one time point. The results of the exposure–response analysis are presented in Figure 1. The adequacy and assumption of linearity of the linear mixed-effects model were confirmed by the goodness-of-fit plot. In the analysis, no concentration-dependent effect of aprocitentan on QTcF was identified, with a statistically not significant slope of the relationship of 4.17×10−4 ms/ng/mL (90% CI: −1.03, 9.36) and an intercept (ie, treatment effect: active – placebo) of −0.0040 ms. The predicted ΔΔQTcF at the geometric peak of aprocitentan plasma concentration was 0.34 (90% CI: −5.52, 6.20) for the 5 mg group, 1.48 (90% CI: −4.07, 7.03) for the 25 mg group, and 5.53 (90% CI: −1.60, 12.66) for the 100 mg aprocitentan dose group. Overall, the exposure–response analysis demonstrated that at plasma levels below ~10 μg/mL, aprocitentan did not cause clinically relevant QTc prolongation. Results of the categorical analyses for HR, PR, and QRS showed no treatment-related pattern. There were no subjects with a QTcF value >450 ms at any time point on Day 10, and one subject in the aprocitentan 25 mg group displayed a ΔQTcF value >30 ms at one time point. One subject in the aprocitentan 100 mg group exhibited a notched T-wave at one time point on Day 10.
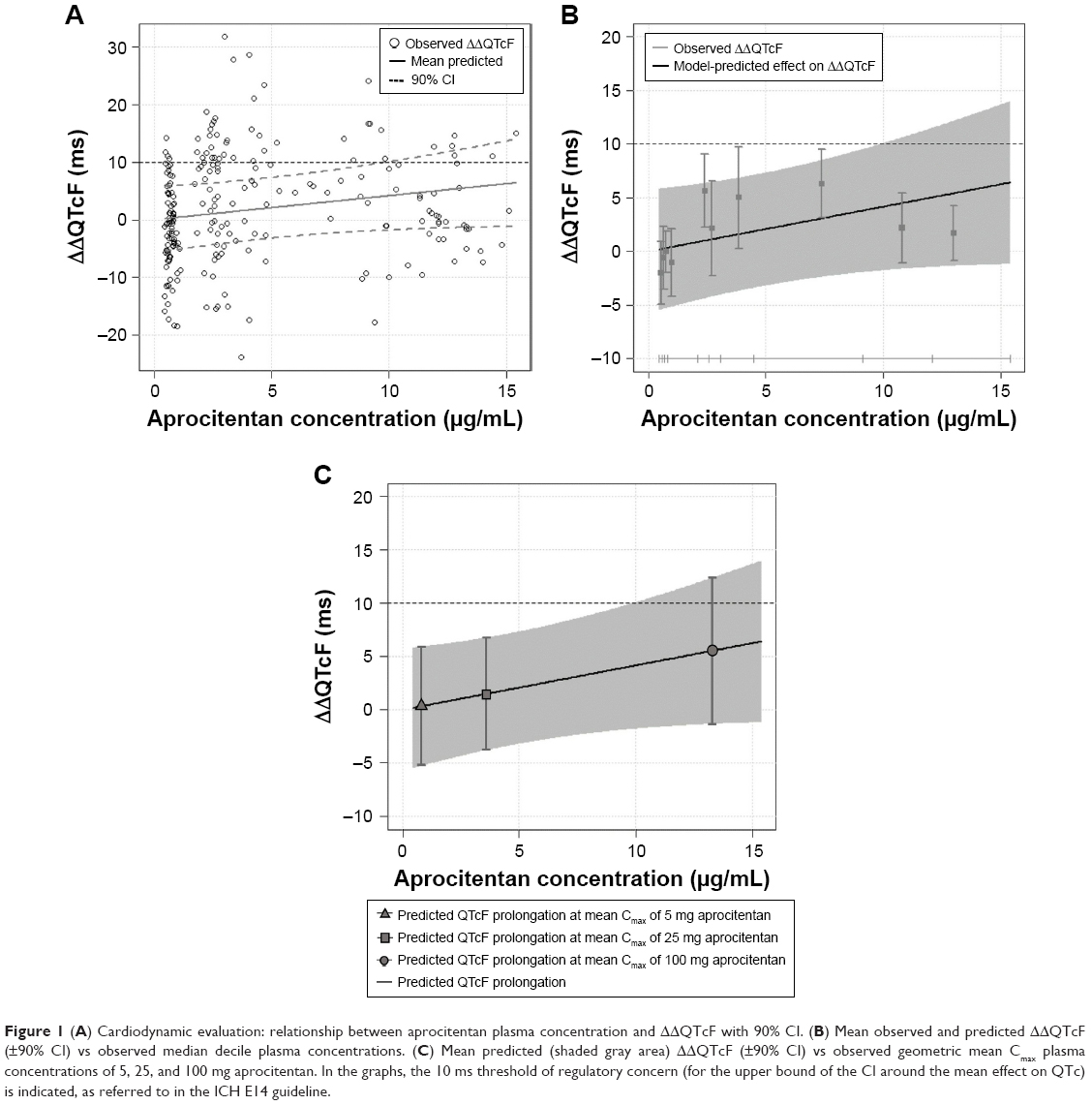 | Figure 1 (A) Cardiodynamic evaluation: relationship between aprocitentan plasma concentration and ΔΔQTcF with 90% CI. (B) Mean observed and predicted ΔΔQTcF (±90% CI) vs observed median decile plasma concentrations. (C) Mean predicted (shaded gray area) ΔΔQTcF (±90% CI) vs observed geometric mean Cmax plasma concentrations of 5, 25, and 100 mg aprocitentan. In the graphs, the 10 ms threshold of regulatory concern (for the upper bound of the CI around the mean effect on QTc) is indicated, as referred to in the ICH E14 guideline. |
In Part C, after multiple-dose administration, mean increases from baseline in BW were observed in the aprocitentan dose groups, especially in the 100 mg dose group and the elderly group. In these groups, mean changes from baseline in BW ranged from −0.5 to +2.5 kg during the study (Figure 2). In the placebo group, mean changes from baseline ranged from −1.1 to 0.9 kg.
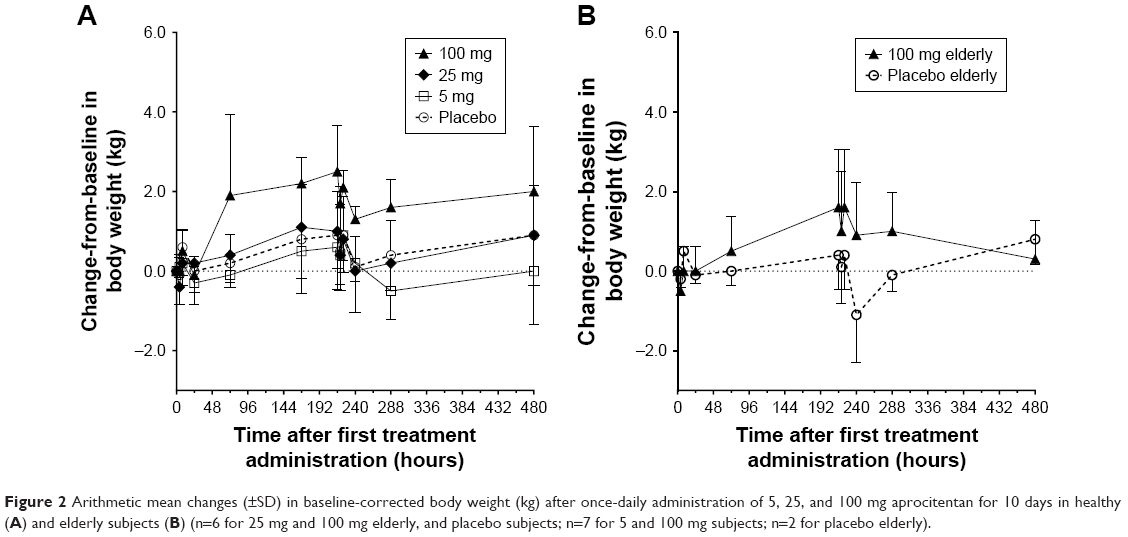 | Figure 2 Arithmetic mean changes (±SD) in baseline-corrected body weight (kg) after once-daily administration of 5, 25, and 100 mg aprocitentan for 10 days in healthy (A) and elderly subjects (B) (n=6 for 25 mg and 100 mg elderly, and placebo subjects; n=7 for 5 and 100 mg subjects; n=2 for placebo elderly). |
Single- and multiple-dose administration of aprocitentan up to 600 and 100 mg qd, respectively, was well tolerated in male, female, adult, and elderly subjects. The tolerability and safety profile were not affected by food.
PK results
In parts A and B, aprocitentan plasma concentrations were measurable for all subjects administered aprocitentan. The plasma concentration–time profiles of aprocitentan showed that Cmax was reached 3–4 hours after administration of low (5 and 25 mg) doses, whereas absorption was slower for higher doses with a Cmax reached 8.5–9 hours after administration (Figure 3A). Thereafter, aprocitentan concentrations decreased slowly, with an apparent t1/2 ranging from 41.3 to 46.8 hours across the dose groups. A summary of aprocitentan PK parameters after single-dose administration is presented in Table 3.
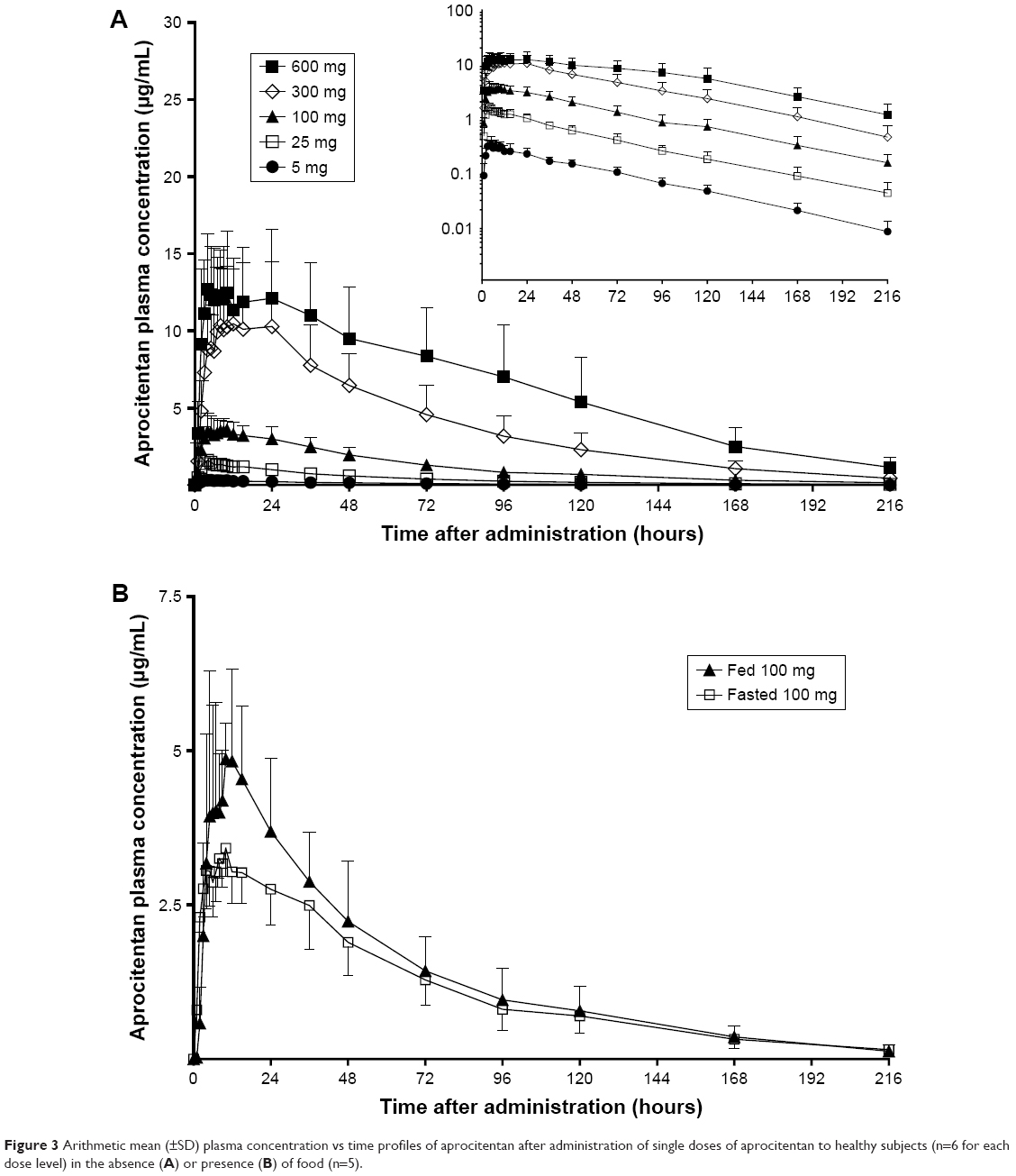 | Figure 3 Arithmetic mean (±SD) plasma concentration vs time profiles of aprocitentan after administration of single doses of aprocitentan to healthy subjects (n=6 for each dose level) in the absence (A) or presence (B) of food (n=5). |
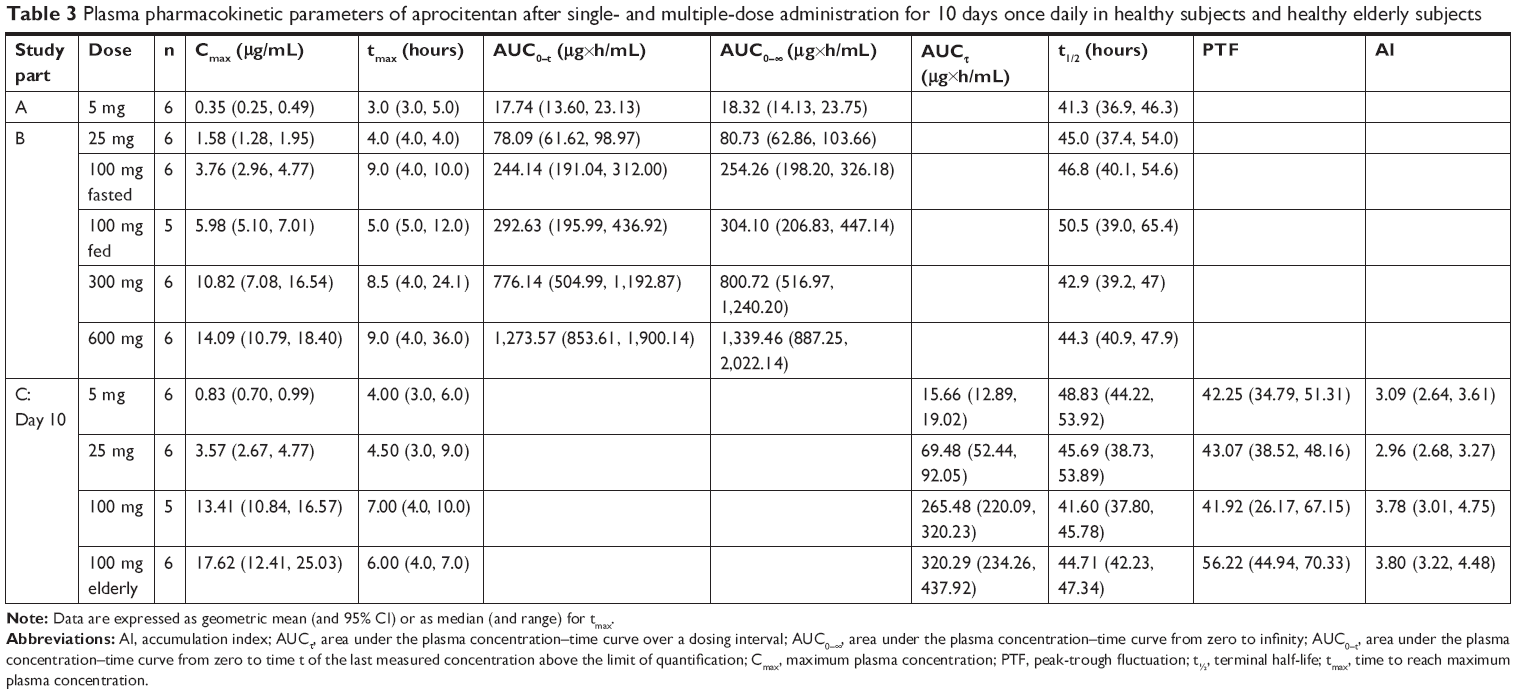 | Table 3 Plasma pharmacokinetic parameters of aprocitentan after single- and multiple-dose administration for 10 days once daily in healthy subjects and healthy elderly subjects |
Results of the Gough test indicate that after single-dose administration of doses up to 600 mg of aprocitentan, exposure to aprocitentan increased less than dose proportionally (Table 4). Indeed, the slope estimate and 90% CI of the estimate for Cmax (0.78 and 0.73, 0.84, respectively) were outside of the critical interval for slope (0.86, 1.14). For AUC0–∞, the lower 90% CI (0.85) was just outside the critical limit of 0.86.
 | Table 4 Dose proportionality and influence of food, sex, and age |
After a high-fat, high-calorie standardized diet, the rate of absorption was increased, as indicated by a median tmax reached 1 hour earlier in the fed condition (Table 4). When compared with the fasted state, exposure as based on AUC0–t and AUC0–∞ was not substantially changed, with respective geometric ratios (fed vs fasted) of 1.24 and 1.23. The elimination rate was unchanged, based on a geometric mean ratio of t1/2 of 1.05 (Figure 3B; Tables 3 and 4).
Compared to male subjects, female subjects had longer tmax (median tmax 3–11 vs 3–8 hours) and a slightly higher exposure to aprocitentan in terms of both Cmax and AUC. The ratios (females vs males) of geometric mean Cmax and AUC0–∞ were 1.25 (90% CI: 1.04, 1.49) and 1.27 (90% CI: 1.07, 1.52), respectively.
For Part C, the plasma concentration–time profiles are presented in Figure 4A. Visual inspection of trough concentrations indicated that steady-state conditions were reached by Day 8 of dosing (Figure 4B). After 10 days of multiple-dose oral administration of aprocitentan, the median tmax ranged from 4 to 7 hours. Both Cmax and AUCτ increased with dose (Table 3). At a steady state, t1/2 ranged from 41.6 to 48.8 hours and was not dependent on the dose. Accumulation of aprocitentan was moderate, with accumulation factors of 3.0–3.8 for the different doses when comparing the AUCτ on days 1 and 10.
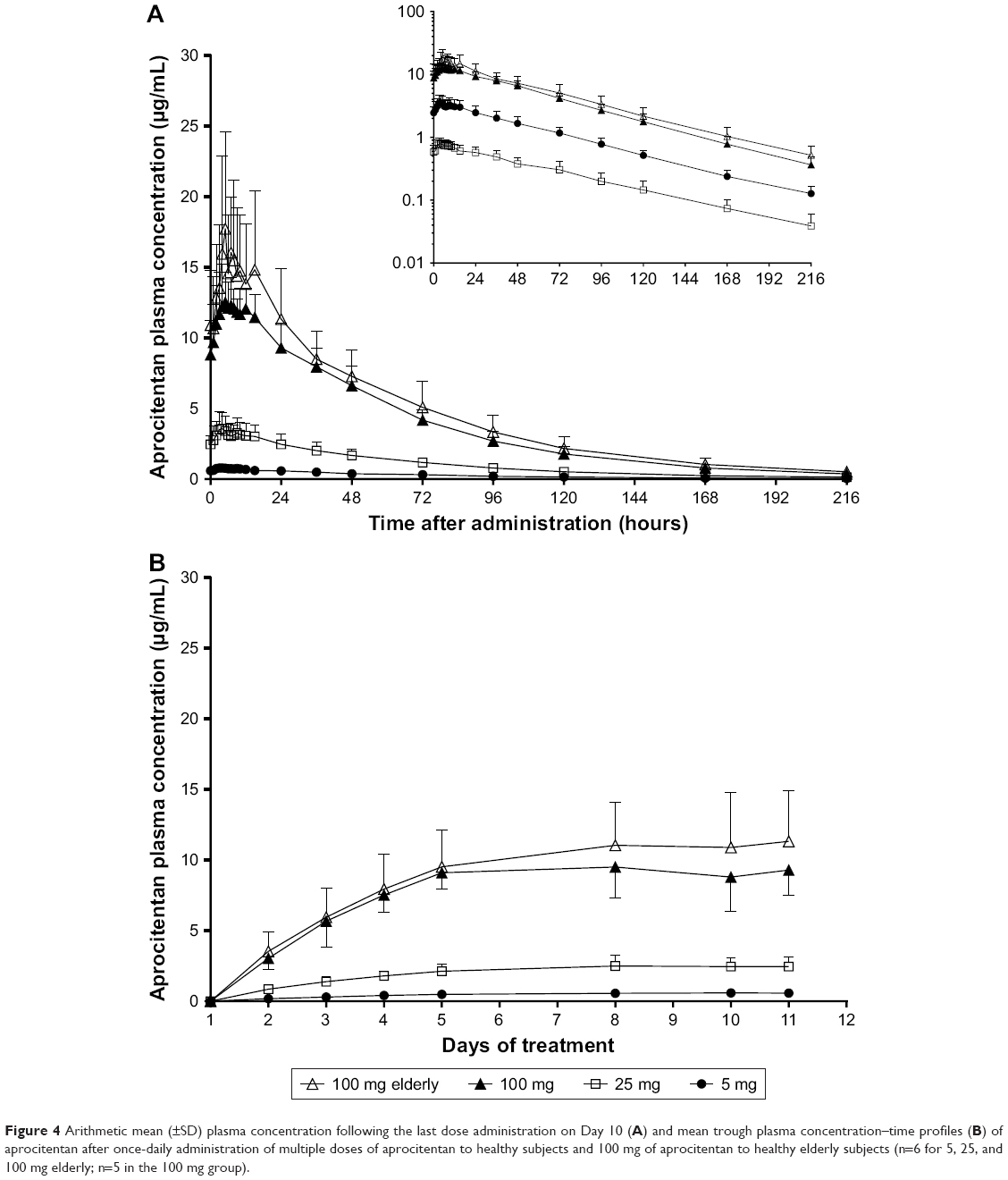 | Figure 4 Arithmetic mean (±SD) plasma concentration following the last dose administration on Day 10 (A) and mean trough plasma concentration–time profiles (B) of aprocitentan after once-daily administration of multiple doses of aprocitentan to healthy subjects and 100 mg of aprocitentan to healthy elderly subjects (n=6 for 5, 25, and 100 mg elderly; n=5 in the 100 mg group). |
After multiple-dose administration, at a steady state, results of the Gough test showed that Cmax and AUCτ increased dose proportionally for doses up to 100 mg aprocitentan, with a slope estimate of 0.93 (90% CI: 0.85, 1.00) for Cmax and of 0.94 (90% CI: 0.87, 1.02) for AUCτ (Table 4).
Compared to male subjects, female subjects had slightly higher exposure to aprocitentan in terms of both Cmax and AUCτ. The ratios (females vs males) of geometric mean Cmax and AUCτ for the last dosing interval were 1.38 (90% CI: 1.18, 1.60) and 1.37 (90% CI: 1.02, 1.56), respectively (Table 4).
In the elderly population receiving 100 mg aprocitentan qd, the rate of absorption was increased, as indicated by a median tmax reached 1.5 hours earlier (90% CI: −4.0, 2.0) for the last dosing interval (Tables 3 and 4). Compared to healthy adult subjects, elderly subjects had a slightly higher exposure to aprocitentan in terms of both Cmax and AUCτ both on Day 1 and Day 10. The ratios (elderly vs adults) of geometric mean Cmax and AUCτ for the last dosing interval were 1.31 (90% CI: 0.97, 1.78) and 1.21 (90% CI: 0.92, 1.58), respectively. No effect of age on the t1/2 of aprocitentan was observed.
PD results
ET-1 plasma concentrations were similar between the placebo and 5 mg aprocitentan dose group. With doses of 25 and 100 mg aprocitentan, ET-1 plasma concentrations increased dose dependently. The increase was more pronounced on Day 10, when steady-state conditions had been reached. After the last dose of treatment on Day 10, plasma ET-1 concentrations slowly declined and returned to baseline by the end of the observation period at 216 hours after the dose.
After multiple-dose oral administration of 25 or 100 mg aprocitentan, ET-1 AUCτ increased with dose both on Day 1 and Day 10 (Figure 5), with the increase being more pronounced on Day 10 when steady-state conditions had been reached. No relevant differences between males and females were observed for any dose. The ratios (females vs males) of geometric mean AUCτ for the first and last dosing interval were 0.93 (90% CI: 0.79, 1.10) and 1.02 (90% CI: 0.82, 1.26), respectively.
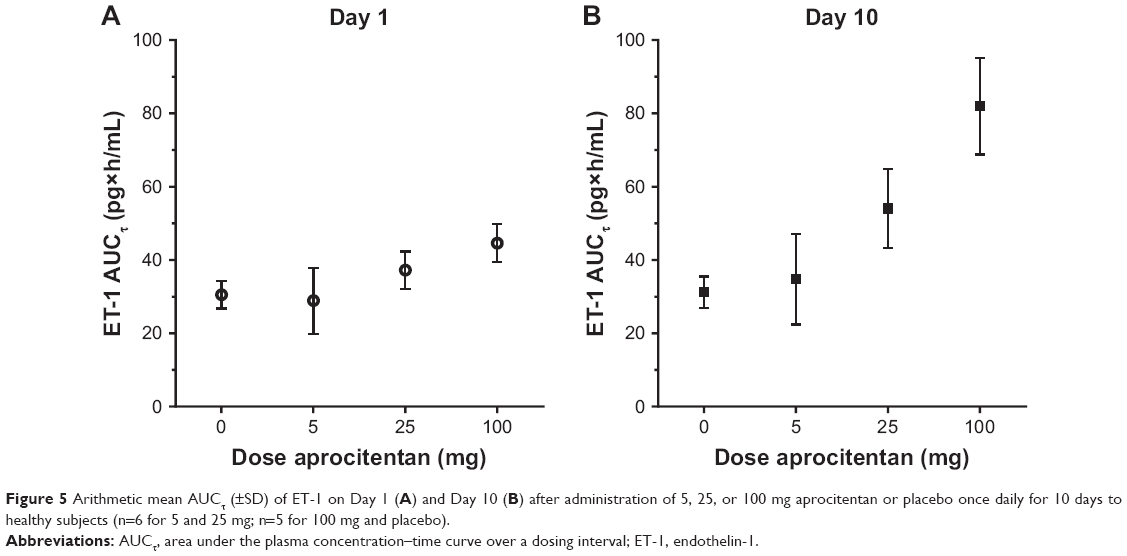 | Figure 5 Arithmetic mean AUCτ (±SD) of ET-1 on Day 1 (A) and Day 10 (B) after administration of 5, 25, or 100 mg aprocitentan or placebo once daily for 10 days to healthy subjects (n=6 for 5 and 25 mg; n=5 for 100 mg and placebo). |
Discussion
In this study, aprocitentan was administered for the first time to healthy adult and elderly male and female subjects. Single- and multiple-dose administration of aprocitentan was well tolerated up to 600 and 100 mg qd, respectively. AEs reported in this study were mostly mild and the most frequently reported one was headache. Cardiac evaluation of 24 hours Holter data after multiple-dose administration of aprocitentan did not suggest ECG effects both in the time-matched ECG interval analysis as well as exposure–response analysis. The use of concentration–response modeling of QTc data in early clinical studies, coupled with an extensive preclinical program, was proposed as an alternative to perform thorough QT studies to assess the risk of QT prolongation. In this study, the only out-of-range observation was the predicted ΔΔQTc after 10 days of treatment with 100 mg aprocitentan. However, the analysis included a low number of subjects per dose and on placebo and the variability of ECG parameters across groups was relatively large. The categorical analysis revealed no QTcF intervals higher than 450 ms. Overall, this suggests that at plasma levels below ~10 μg/mL, aprocitentan will not cause clinically relevant QTc prolongation and hence displays a low pro-arrhythmic potential.
No effect on BW was observed in parts A and B. In Part C, small increases in BW from baseline compared to placebo-treated subjects were detected only after multiple-dose administration of 100 mg aprocitentan. As BW gain could be the first indirect indicator of fluid retention, an important safety consideration with use of ERAs,28–31 this observation should be considered when selecting doses for further clinical studies. However, due to the limited number of subjects per dose level and choice of a healthy subject population, more clinical studies are needed to confirm the results obtained. In particular, more data in elderly patients are needed as they are likely to have impaired kidney function and other comorbidities which can make them more prone to fluid retention.32 At a steady state, there was a minor treatment- and dose-related decrease in hemoglobin and hematocrit, as reported with other ERAs.33 No treatment-related pattern was observed in other clinical laboratory parameters or in vital signs. The presence of food did not influence aprocitentan tolerability.
After single- and multiple-dose administration, the plasma concentration–time profiles of aprocitentan were characterized by a Cmax attained between 3 and 9 hours after administration and a long t½ of ~44 hours. The tmax increased with the dose of aprocitentan, suggesting that the absorption is slower at doses higher than 100 mg, probably due to the poor water solubility of the compound. Steady-state conditions of aprocitentan were reached after 8 days and accumulation was 3-fold. At a steady state, AUC and Cmax were dose proportional over the tested dose range. These data support a qd dosing approach. The long t½ of the compound could also be convenient for patients treated chronically in case of missing doses, as has been suggested with amlodipine, an antagonist of calcium channels with a half-life of around 40–60 hours also used in the treatment of hypertension.34 In that study, lower BP fluctuations were observed after 2 days without treatment compared to diltiazem, which has a shorter t½.
Food increased the exposure to aprocitentan in terms of Cmax and AUC, with geometric mean ratios (fed/fasted) of 1.73 and 1.23, respectively. The increase of Cmax coupled with the fact that tmax was reached about 1 hour earlier indicates a higher rate of absorption of aprocitentan after food intake. It is possible that changes in gastrointestinal pH after food administration might lead to an increased solubility and facilitate the absorption of the drug. However, the change in AUC is smaller than the change in Cmax, meaning that the overall extent of drug absorption is not significantly changed after a high-fat diet.
As the prevalence of hypertension, including RHT, is higher in the elderly population,35,36 we compared the PK parameters of multiple-dose administration of aprocitentan in elderly and healthy adult subjects. Elderly had an increased exposure in terms of Cmax and AUC, with geometric mean ratios (elderly/adult) of 1.31 and 1.21, respectively. The increased exposure might be explained, among others, by an age-related decline in renal function in the elderly population.37 However, the limited increase in AUC is not considered to be of clinical relevance. Therefore, aprocitentan can be administered with or without food and no dose adjustments for age are needed.
After multiple-dose administration of aprocitentan, plasma ET-1 increased with doses ≥25 mg aprocitentan in a dose-dependent fashion. The effect was more pronounced on Day 10 at a steady state. The increase in plasma ET-1 is attained through blockade of ETB receptors, which are responsible for the clearance of ET-1.38 Aprocitentan has a stronger inhibitory effect on ETA than on ETB receptors (inhibitory potency ratio 16:1), meaning that at doses at which an increase in plasma ET-1 can be observed, ETA receptors should be fully blocked by aprocitentan.39
In this study, no treatment-related changes in vital signs were observed. However, this does not exclude that aprocitentan has a BP-lowering effect. The current study was performed in healthy subjects in whom the ET-1 system is not disrupted. In pathological conditions, for example, patients suffering from hypertension, the balance of the ET-1 system is altered,40,41 which would suggest that treatment with aprocitentan would have a larger impact on BP in patients when compared to healthy subjects. Further clinical data are needed to corroborate this hypothesis.
Clinical trials with ERAs investigating the effect on BP are not numerous, but have shown encouraging results in terms of decrease in BP. For example, macitentan showed a dose-dependent decrease in BP within 4 weeks of treatment in a dose-finding study in patients with mild-to-moderate essential hypertension.42 Also, in a proof-of-concept study in hypertensive patients with chronic renal failure, ETA and/or ETB receptor antagonist administration (BQ-123 and BQ-788, respectively) significantly reduced BP compared to placebo.43 Further, in a Phase 3 study in patients with RHT, treatment with the selective ETA receptor antagonist darusentan on top of three or more background antihypertensive drugs induced a mean decrease in SBP and DBP of 17/10 mmHg with 50 mg darusentan, 18/10 mmHg with 100 mg, and 18/11 mmHg with 300 mg compared to 9/5 mmHg with placebo. The main AEs in this study were fluid retention and edema,30 and development of darusentan was discontinued after it failed to reach its primary endpoints in the reduction of BP in the second Phase 3 study.44,45
To ensure that aprocitentan has the potential of an antihypertensive treatment, further studies are needed to determine PK parameters in sensitive populations as well as the optimal effective dose(s). For that purpose, a Phase 2 study to assess the effect on BP of different doses of aprocitentan in patients with essential hypertension was conducted (study NCT02603809). Patients with hypertension often suffer from concomitant renal diseases that might interfere with the elimination process of the drug and thereby modify the PK of the compound.8 Moreover, since patients with hypertension often display several comorbidities requiring pharmacological treatment,7,10 the drug–drug interaction potential of aprocitentan needs to be further investigated.
In summary, single- and multiple-dose administration of aprocitentan was well tolerated up to doses of 100 mg qd for 10 days in both healthy adult and elderly subjects. The tolerability, safety, and PK/PD profile of aprocitentan confirms the potential of the drug in the treatment of hypertension and further clinical development of this compound.
Disclosure
JD is a fellow of the American College of Clinical Pharmacology. PNS and JD are current employees of Idorsia Pharmaceuticals Ltd and former employees of Actelion Pharmaceuticals Ltd. MM is a current employee of Idorsia Pharmaceuticals Ltd. MKK was the principal investigator of the study that was sponsored by Actelion Pharmaceuticals Ltd. The authors report no other conflicts of interest in this work.
References
Mills KT, Bundy JD, Kelly TN, et al. Global disparities of hypertension prevalence and control: a systematic analysis of population-based studies from 90 countries. Circulation. 2016;134(6):441–450. | ||
Moser M, Setaro JF. Clinical practice. Resistant or difficult-to-control hypertension. N Engl J Med. 2006;355(4):385–392. | ||
Mancia G, Fagard R, Narkiewicz K, et al; Task Force Members. 2013 ESH/ESC guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens. 2013;31(7):1281–1357. | ||
Makani H, Bangalore S, Desouza KA, Shah A, Messerli FH. Efficacy and safety of dual blockade of the renin–angiotensin system: meta-analysis of randomised trials. BMJ. 2013;346:f360. | ||
Yusuf S, Teo KK, Pogue J, et al. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358(15):1547–1559. | ||
Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation study. N Engl J Med. 2004;351(6):543–551. | ||
Horowitz B, Miskulin D, Zager P. Epidemiology of hypertension in CKD. Adv Chronic Kidney Dis. 2015;22(2):88–95. | ||
Gijón-Conde T, Graciani A, Banegas JR. Resistant hypertension: demography and clinical characteristics in 6,292 patients in a primary health care setting. Rev Esp Cardiol (Engl Ed). 2014;67(4):270–276. | ||
US Food and Drug Administration. Hypertension: Conducting Studies of Drugs to Treat Patients on a Background of Multiple Antihypertensive Drugs Guidance for Industry; Silver Spring, MD: US Food and Drug Administration; 2018. | ||
Donatelli M, Colletti I, Bucalo ML, Russo V, Verga S. Plasma endothelin levels in NIDDM patients with macroangiopathy. Diabetes Res. 1994;25(4):159–164. | ||
Cardillo C, Kilcoyne CM, Waclawiw M, Cannon RO, Panza JA. Role of endothelin in the increased vascular tone of patients with essential hypertension. Hypertension. 1999;33(2):753–758. | ||
Davenport AP, Hyndman KA, Dhaun N, et al. Endothelin. Pharmacol Rev. 2016;68(2):357–418. | ||
Yanagisawa M, Kurihara H, Kimura S, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332(6163):411–415. | ||
Davenport AP, O’Reilly G, Kuc RE. Endothelin ETA and ETB mRNA and receptors expressed by smooth muscle in the human vasculature: majority of the ETA sub-type. Br J Pharmacol. 1995;114(6):1110–1116. | ||
Zhang M, Luo B, Chen SJ, Abrams GA, Fallon MB. Endothelin-1 stimulation of endothelial nitric oxide synthase in the pathogenesis of hepatopulmonary syndrome. Am J Physiol. 1999;277(5):G944–G952. | ||
Winkles JA, Alberts GF, Brogi E, Libby P. Endothelin-1 and endothelin receptor mRNA expression in normal and atherosclerotic human arteries. Biochem Biophys Res Commun. 1993;191(2):1081–1088. | ||
Dupuis J, Goresky CA, Fournier A. Pulmonary clearance of circulating endothelin-1 in dogs in vivo: exclusive role of ETB receptors. J Appl Physiol (1985). 1996;81(4):1510–1515. | ||
Dupuis J, Stewart DJ, Cernacek P, Gosselin G. Human pulmonary circulation is an important site for both clearance and production of endothelin-1. Circulation. 1996;94(7):1578–1584. | ||
Ge Y, Bagnall A, Stricklett PK, et al. Collecting duct-specific knockout of the endothelin B receptor causes hypertension and sodium retention. Am J Physiol Renal Physiol. 2006;291(6):F1274–F1280. | ||
Gaddam KK, Nishizaka MK, Pratt-Ubunama MN, et al. Characterization of resistant hypertension: association between resistant hypertension, aldosterone, and persistent intravascular volume expansion. Arch Intern Med. 2008;168(11):1159–1164. | ||
Trensz F, Bortolamiol C, Kramberg M, et al. Pharmacological characterization of APROCITENTAN, a dual endothelin receptor antagonist, alone and in combination with blockers of the renin angiotensin system, in two models of experimental hypertension. J Pharmacol Exp Ther. 2019;368(3):462–473. | ||
Pulido T, Adzerikho I, Channick RN, et al; SERAPHIN Investigators. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(9):809–818. | ||
Dingemanse J, Sidharta PN, Maddrey WC, Rubin LJ, Mickail H. Efficacy, safety and clinical pharmacology of macitentan in comparison to other endothelin receptor antagonists in the treatment of pulmonary arterial hypertension. Expert Opin Drug Saf. 2014;13(3):391–405. | ||
Sidharta PN, Treiber A, Dingemanse J. Clinical pharmacokinetics and pharmacodynamics of the endothelin receptor antagonist macitentan. Clin Pharmacokinet. 2015;54(5):457–471. | ||
Sidharta PN, van Giersbergen PLM, Dingemanse J. Safety, tolerability, pharmacokinetics, and pharmacodynamics of macitentan, an endothelin receptor antagonist, in an ascending multiple-dose study in healthy subjects. J Clin Pharmacol. 2013;53(11):1131–1138. | ||
US Food and Drug Administration. U.S. Food and Drug Administration, Center for Drug Evaluation and Research (CDER): Guidance for Industry: E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-antiarrhythmic Drugs – Questions and Answers (R3). Silver Spring, MD: June 2017. October 2017:1550–7416. | ||
Gough K, Hutchison M, Keene O, et al. Assessment of dose proportionality: report from the statisticians in the pharmaceutical industry/pharmacokinetics UK joint working Party. Drug Inf J. 1995;29(3):1039–1048. | ||
Krum H, Viskoper RJ, Lacourciere Y, Budde M, Charlon V. The effect of an endothelin-receptor antagonist, bosentan, on blood pressure in patients with essential hypertension. Bosentan Hypertension Investigators. N Engl J Med. 1998;338(12):784–791. | ||
Packer M, McMurray JJV, Krum H, et al; ENABLE Investigators and Committees. Long-term effect of endothelin receptor antagonism with bosentan on the morbidity and mortality of patients with severe chronic heart failure: primary results of the enable trials. JACC Heart Fail. 2017;5(5):317–326. | ||
Weber MA, Black H, Bakris G, et al. A selective endothelin-receptor antagonist to reduce blood pressure in patients with treatment-resistant hypertension: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;374(9699):1423–1431. | ||
Black HR, Bakris GL, Weber MA, et al. Efficacy and safety of darusentan in patients with resistant hypertension: results from a randomized, double-blind, placebo-controlled dose-ranging study. J Clin Hypertens (Greenwich). 2007;9(10):760–769. | ||
Stevens LA, Viswanathan G, Weiner DE. Chronic kidney disease and end-stage renal disease in the elderly population: current prevalence, future projections, and clinical significance. Adv Chronic Kidney Dis. 2010;17(4):293–301. | ||
Epstein BJ. Efficacy and safety of darusentan: a novel endothelin receptor antagonist. Ann Pharmacother. 2008;42(7):1060–1069. | ||
Leenen FH, Fourney A, Notman G, Tanner J. Persistence of anti-hypertensive effect after “missed doses” of calcium antagonist with long (amlodipine) vs short (diltiazem) elimination half-life. Br J Clin Pharmacol. 1996;41(2):83–88. | ||
Brambilla G, Bombelli M, Seravalle G, et al. Prevalence and clinical characteristics of patients with true resistant hypertension in central and eastern Europe: data from the BP-CARE study. J Hypertens. 2013;31(10):2018–2024. | ||
Daugherty SL, Powers JD, Magid DJ, et al. Incidence and prognosis of resistant hypertension in hypertensive patients. Circulation. 2012;125(13):1635–1642. | ||
Wetzels JF, Kiemeney LA, Swinkels DW, Willems HL, den Heijer M. Age- and gender-specific reference values of estimated GFR in z: the Nijmegen Biomedical Study. Kidney Int. 2007;72(5):632–637. | ||
Löffler BM, Breu V, Clozel M. Effect of different endothelin receptor antagonists and of the novel non-peptide antagonist Ro 46-2005 on endothelin levels in rat plasma. FEBS Lett. 1993;333(1–2):108–110. | ||
Iglarz M, Binkert C, Morrison K, et al. Pharmacology of macitentan, an orally active tissue-targeting dual endothelin receptor antagonist. J Pharmacol Exp Ther. 2008;327(3):736–745. | ||
Iglarz M, Steiner P, Wanner D, Rey M, Hess P, Clozel M. Vascular effects of endothelin receptor antagonists depends on their selectivity for ETA versus ETB receptors and on the functionality of endothelial ETB receptors. J Cardiovasc Pharmacol. 2015;66(4):332–337. | ||
Kakoki M, Hirata Y, Hayakawa H, et al. Effects of hypertension, diabetes mellitus, and hypercholesterolemia on endothelin type B receptor-mediated nitric oxide release from rat kidney. Circulation. 1999;99(9):1242–1248. | ||
EMA. Assessment report. Opsumit. Procedure N° EMEA/H/C/002697/0000; 2013. Available from: https://www.ema.europa.eu/en/documents/assessment-report/opsumit-epar-public-assessment-report_en.pdf. Accessed December 6, 2018. | ||
Goddard J, Johnston NR, Hand MF, et al. Endothelin-A receptor antagonism reduces blood pressure and increases renal blood flow in hypertensive patients with chronic renal failure: a comparison of selective and combined endothelin receptor blockade. Circulation. 2004;109(9):1186–1193. | ||
Bakris GL, Lindholm LH, Black HR, et al. Divergent results using clinic and ambulatory blood pressures: report of a darusentan-resistant hypertension trial. Hypertension. 2010;56(5):824–830. | ||
Science G. Second pivotal Phase III study of Gilead’s darusentan for resistant hypertension misses primary endpoints; 2009. Available from: www.gilead.com. Accessed December 6, 2018. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.