Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Simvastatin enhances Rho/actin/cell rigidity pathway contributing to mesenchymal stem cells’ osteogenic differentiation
Authors Tai I, Wang Y, Chen C ![]() , Chuang S, Chang J, Ho M
, Chuang S, Chang J, Ho M
Received 9 March 2015
Accepted for publication 28 April 2015
Published 21 September 2015 Volume 2015:10(1) Pages 5881—5894
DOI https://doi.org/10.2147/IJN.S84273
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
I-Chun Tai,1–3 Yao-Hsien Wang,3 Chung-Hwan Chen,3,4 Shu-Chun Chuang,3 Je-Ken Chang,3–5 Mei-Ling Ho1–3,6
1Graduate Institute of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan; 2Department of Physiology, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan; 3Orthopaedic Research Center, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan; 4Department of Orthopedics, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan; 5Department of Orthopedics, Kaohsiung Municipal Ta-Tung Hospital, Kaohsiung, Taiwan; 6Department of Marine Biotechnology and Resources, National Sun Yat-sen UniVersity, Kaohsiung, Taiwan
Abstract: Recent studies have indicated that statins induce osteogenic differentiation both in vitro and in vivo. The molecular mechanism of statin-stimulated osteogenesis is unknown. Activation of RhoA signaling increases cytoskeletal tension, which plays a crucial role in the osteogenic differentiation of mesenchymal stem cells. We thus hypothesized that RhoA signaling is involved in simvastatin-induced osteogenesis in bone marrow mesenchymal stem cells. We found that although treatment with simvastatin shifts localization of RhoA protein from the membrane to the cytosol, the treatment still activates RhoA dose-dependently because it reduces the association with RhoGDIα. Simvastatin also increased the expression of osteogenic proteins, density of actin filament, the number of focal adhesions, and cellular tension. Furthermore, disrupting actin cytoskeleton or decreasing cell rigidity by using chemical agents reduced simvastatin-induced osteogenic differentiation. In vivo study also confirms that density of actin filament is increased in simvastatin-induced ectopic bone formation. Our study is the first to demonstrate that maintaining intact actin cytoskeletons and enhancing cell rigidity are crucial in simvastatin-induced osteogenesis. The results suggested that simvastatin, which is an osteoinductive factor and acts by increasing actin filament organization and cell rigidity combined with osteoconductive biomaterials, may benefit stem-cell-based bone regeneration.
Keywords: statins, RhoA, cytoskeleton, osteogenic differentiation, BMSCs
Introduction
Bone fractures are common in various accident types. Most bone fractures heal spontaneously; however, 5%–10% of bone fractions have delayed union or nonunion,1 possibly necessitating bone grafts to facilitate healing. The ideal bone graft possesses osteoinductive and osteoconductive properties. Statins inhibit 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase and are used clinically to reduce circulatory cholesterol levels.2,3 In vitro studies have revealed that statins increase the expression of osteogenic marker genes in osteoblasts and bone-marrow-derived cells,4–9 and in vivo research has demonstrated that oral administration of statins enhance trabecular bone volume in ovariectomized rats10,11 and improve bone healing by local delivery.12,13 Recent studies have indicated that statins exert bone anabolic effects4–9 and can be used as osteoconductive agents in bone tissue engineering.14–16 However, the molecular mechanisms whereby statins enhance osteogenic differentiation in mesenchymal stem cells (MSCs) are not clear. This study sought to clarify the action mechanisms of statins on MSC osteogenic differentiation for enhancing the application of statins in stem-cell-based bone tissue engineering.
The mechanical properties of the environment play a critical role in the fate of stem cells by inducing cytoskeleton organization and increasing cell rigidity.17–22 For example, MSCs cultured on the hardest substrates revealed increased formation of stress fiber and caused osteogenic differentiation.23,24 In addition, disrupting cytoskeleton or decreasing cell rigidity reduces the expression of osteoblastic gene, including alkaline phosphatase (ALP) and osteopontin,25,26 and further promotes adipogenic differentiation by increasing peroxisome proliferator-activated receptor (PPAR) gamma.18,20,27 The cytoskeletal organization and myosin-mediated cytoskeleton tension are regulated by RhoA and its effector, Rho-associated protein kinase (ROCK). Activation of RhoA causes human MSCs to undergo osteogenic differentiation by upregulating the expression of Runx2 gene; however, inactivation of RhoA causes cells to undergo adipogenic differentiation by increasing expression of the adipogenic marker gene PPAR gamma.18,20 Furthermore, inhibiting RhoA signaling promotes chondrogenic differentiation by increasing the expression of SOX-9 gene.21,22 Accordingly, the activation of RhoA and the resultant actin organization and cell rigidity may be necessary for osteogenic differentiation.
Whether RhoA signaling is related to the effects of statins on osteogenesis is unclear. One suggested association is mediated through changes in the posttranslational prenylation of Rho.28 Statins reduce the synthesis of isoprenoid groups by inhibiting HMG-CoA reductase, causing decreased prenylation of small G proteins, including Rho.29,30 For example, mevastatin inhibits the differentiation of osteoclasts by reducing the geranylgeranylation of Rho and Rac.31 Treatment with statin not only inhibits the prenylation of RhoA but also increases the cellular levels of activated RhoA.32–34 Statins are assumed to activate RhoA by reducing the binding of Rho guanine nucleotide dissociation inhibitor (RhoGDIα), a negative regulator of Rho GTPases.32
In this study, we investigated how Rho/actin/cell rigidity signaling contributes to simvastatin-induced osteogenic differentiation in mouse bone marrow mesenchymal stem cells (mBMSCs). We first examined how simvastatin affects RhoA signaling, actin reorganization, cell rigidity, and osteogenesis. We then used the pharmacological agents, ROCKII protein inhibition, myosin II ATP hydrolysis inhibition, and actin polymerization disruption to evaluate how cytoskeletal mechanics alter simvastatin-induced osteogenesis in vitro. Finally, the in vivo effects of simvastatin were assessed using an ectopic bone formation model.
Materials and methods
Cell culture of mBMSCs
The murine bone marrow mesenchymal stem cell line D1 was purchased from ATCC (Manassas, VA, USA). The cells were maintained in a bone medium (Dulbecco’s Modified Eagles Medium containing 100 μg/mL of ascorbic acid, 100 μg/mL of nonessential amino acids, 100 μg/mL of penicillin/streptomycin, and 10% fetal bovine serum) in a 37°C incubator containing 5% CO2. During drug treatment, cells were cultured in a 2% FBS osteoinduction medium containing 10 mM glycerophosphate, 0.2 mM l-ascorbic acid-2-phosphate, and 100 nM dexamethasone in the bone medium.
Drug treatment
The simvastatin solution was prepared as described in our previous study.13 The following pharmacological agents were used: 10 μM Y-27632 dihydrochloride (Tocris Bioscience, Bristol, UK), an inhibitor of p160 ROCKII activity; 10 μM blebbistatin (Tocris Bioscience), a selective inhibitor of the ATPase function of nonmuscle myosin II; and 0.5 μM cytochalasin D (Sigma-Aldrich Co., St Louis, MO, USA), which inhibits actin polymerization by capping the barbed end of F-actin polymers. Cells were pretreated with inhibitors (Y-27632 dihydrochloride, blebbistatin, and cytochalasin D) in the bone medium for 1 hour and cotreated with simvastatin and blockers in the 2% FBS osteoinduction medium. The extent of mineralization, ALP activity, expression of osteogenic gene, and osteogenic protein levels were analyzed at various time points.
ALP activity staining
The Sigma-Aldrich alkaline phosphatase kit (No 85; Sigma-Aldrich Co.) was used to detect ALP activity after treatment with simvastatin. The alkaline-dye solution was prepared according to manufacturer instructions. Cells were fixed with 10% formalin-saline at room temperature for 10 minutes. After being washed once with distilled water, the alkaline-dye mixture was added to each well of a 48-well plate and incubated for 5 minutes. The staining solution was removed, and the wells were washed with distilled water. The samples were then air-dried at room temperature and dissolved in ALP staining solution with 0.1N NaOH, and the absorbance were measured at 405 nm.
Alizarin Red S staining
The mineralization of D1 cells was assayed using Alizarin Red S staining. The cells were stained in Alizarin Red solution on Day 5 of culture in the osteoinduction medium. The cells were fixed with 10% formalin in phosphate buffered saline (PBS) for 5 minutes in 48-well plates, washed twice with distilled water, and stained with Alizarin Red solution for 5 minutes. The cells were then washed with distilled water, air-dried at room temperature, resuspended in 10% acetic acid, and quantified spectrophotometrically at 450 nm.
RNA extraction and real-time PCR
Real-time polymerase chain reaction (PCR) was used to analyze the expression of osteogenic genes. Total RNA of the D1 cells was extracted using ZR RNA MiniPrep (Zymo Research, Irvine, CA, USA) after 12, 24, and 48 hours of incubation in the osteoinduction medium. Total RNA (1 μg) was reverse-transcribed into cDNA by using oligo-dT primers and a High Capacity RNA-to-cDNA Kit (Thermo Fisher Scientific, Waltham, MA, USA). Real-PCR was performed using the Bio-Rad iQ5 real-time PCR detection system (Bio-Rad Laboratories Inc., Hercules, CA, USA) and SYBR green (Applied Biosystems). The cDNA was amplified with BMP, Runx2, OC, and β-actin primers by using real-time PCR. The primers were as follows: BMP2 forward, 5′-AGCTGCAAGAGACACCCTTTG-3′; BMP2 reverse, 5′-AGCATGCCTTAGGGATTTTGGA-3′; Runx2 forward, 5′-CCCAGCCACCTTTACCTACA-3′; Runx2 reverse, 5′-TATGGAGTGCTGCTGGTCTG-3′; OC forward, 5′-GAGGGCAATAAGGTAGTGAACA-3′; OC reverse, 5′-AAGCCATACTGGTCTGATAGCTCG-3′; β-actin forward, 5′-CCAACCGTGAAAAGATGACC-3′; and β-actin reverse 5′-ACCAGAGGCATACAGGGACA-3′. The PCR conditions were as follows: 95°C for 30 seconds and 95°C for 4 minutes, followed by 40 cycles of 95°C for 10 seconds, 59°C for 15 seconds, and 72°C for 15 seconds. Melting curves were generated to confirm the specificity of the PCRs. The relative mRNA expression level was calculated from the threshold cycle (Ct) and normalized with β-actin. All real-time PCR experiments were performed in triplicate independent experiments.
Immunofluorescence staining
The D1 cells were seeded on glass coverslips. At the end of the experiment, cells were fixed for 15 minutes in 4% paraformaldehyde containing 0.5% Triton-X-100 and preincubated in 100 μL of blocking solution (2.5% bovine serum albumin in phosphate buffered saline) for 40 minutes. The cytoskeleton was detected using 0.5 μM Alexa Fluor 488-conjugated phalloidin (Molecular Probes, Invitrogen, Basel, Switzerland) and DAPI for 1 hour. Osteogenic proteins were labeled by incubation with goat anti-Runx2 (Santa Cruz Biotechnology Inc., Dallas, TX, USA), goat anti-BMP-2 (Santa Cruz Biotechnology Inc.), and rabbit antiosteocalcin (Santa Cruz Biotechnology Inc.) overnight. Cells were rinsed five times with PBST and stained with antigoat IgG conjugated with Alexa 638 (Molecular Probes) or antirabbit IgG conjugated with Alexa 638 (Molecular Probes) combined with 0.5 μM Alexa Fluor 488-conjugated phalloidin (Molecular Probes), and DAPI for 1 hour. Cells were rinsed five times in phosphate buffered saline with 0.01% Triton X-100 (PBST). Glass coverslips were fixed to the slides by using a mounting medium (35 μL) and sealed with nail polish. Images were observed using a confocal microscope (FluoView 1000; Olympus Corporation, Tokyo, Japan) at 60×.
RhoA pull-down assay
RhoA activity was measured as a function of RhoA-GTP binding to corresponding effector-binding domains fused with glutathione S-transferase (GST) in pull-down assays following the manufacturer protocol (EMD Millipore, Billerica, MA, USA). Briefly, cells were lysed in 300 μL of 50 mM Tris, pH 7.4, 10 mM MgCl2, 500 mM NaCl, 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS), 0.5% deoxycholate, and protease inhibitors. Lysates (500–750 μg) were cleared at 1,000× g for 5 minutes, and the supernatants were rotated for 45 minutes with 30 μg of GST-RBD (GST fusion protein containing the Rho-binding domain of rhotekin) bound to glutathione–Sepharose beads. Samples were washed with 50 mM Tris, pH 7.4, 10 mM MgCl2, 150 mM NaCl, 1% Triton X-100, and protease inhibitors. GST-RBD pull-downs (active-form RhoA) and lysates were detected using immunoblotting with a Rho-specific antibody (Cell Signaling Technology, Beverly, MA, USA).
Immunoprecipitations
For the immunoprecipitation experiment, D1 cells were lysed in buffer containing 200 mM Tris (pH 7.4), 1% Nonidet P-40, 0.2% SDS, protease inhibitor mix (Hoffman-La Roche Ltd., Basel, Switzerland), and phosphatase inhibitor mix (Hoffman-La Roche Ltd.). Equal amounts of total protein (500 μg) were subjected to immunoprecipitation using Catch and Release (EMD Millipore) with 2 μg of rabbit polyclonal RhoGDIα (Santa Cruz Biotechnology Inc.) according to manufacturer instructions. Immune complexes precipitated with RhoGDIα were subjected to SDS–polyacrylamide gel electrophoresis. After transferring proteins to polyvinylidene difluoride membranes, RhoA was detected using rabbit anti-RhoA (Santa Cruz Biotechnology Inc.). Immunoprecipitates were also immunoblotted, with RhoGDIα used as a loading control.
Immunoblotting
The following antibodies were used to assess protein levels: anti-RhoA (Cell Signaling Technology), antiphosphate MYPT (EMD Millipore), antiphospho-myosin light chain (MLC; Cell Signaling Technology), anti-MLC (Cell Signaling Technology), and anti-β-actin (Sigma-Aldrich Co.). The secondary antibodies used were horseradish peroxidase-conjugated goat antirabbit and antimouse IgG (Santa Cruz Biotechnology Inc.). Protein concentrations were assayed using the Bio-Rad Protein Assay Dye Reagent (Bio-Rad Laboratories, Hercules, CA, USA). Immunoprecipitates were separated on SDS gels. Samples of total protein (50 μg) were separated using 10% SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and incubated with primary antibody. Immunocomplexes were detected using the appropriate horseradish peroxidase-conjugated secondary antibody and observed using a UVP AutoChemi Image and Analysis System (UVP, Upland, CA, USA) with enhanced chemiluminescence (Amersham-Pharmacia International, Piscataway, NJ, USA).
Atomic force microscopy analysis
D1 cells (1×10−5) were seeded on 6 cm dishes, incubated for 24 hours, and treated with RhoA stimulator or inhibitor for 30 minutes. Cells were detected using atomic force microscopy (Nano-wizard II; JPK Instruments, Berlin, Germany), and cell rigidity was detected using a cantilever (ArrowTL1; Oxford Instruments, Scotts Valley, CA, USA) with a 5 μm polystyrene sphere. The indentation area targeted cell nuclei. JPK data-processing software was used to analyze cell rigidity according to the Herz model.
Mouse model of ectopic bone formation
Type I collagen gel was prepared as described in a previous study.35 D1 cells (1×106 per injection) were harvested and mixed with the collagen gel. Experimental groups included cells treated with 1 μM simvastatin (SIM), and 1 μM SIM mixed with Y27632 (10 μM), blebbistatin (Blebb) (10 μM), or cytochalasin D (CD) (0.5 μM), respectively, and untreated controls. The solution (150 μL), including type I collagen gel, D1 cells, and pharmaceuticals drugs, was subcutaneously injected into the dorsal side of 5-week-old male imprinting control region mice. All animal experiments were conducted following the Kaohsiung Medical University guidelines for animal experiments. Two weeks after injection, the animals were euthanized, and the masses at the injection sites were retrieved for detection of osteogenic gene and histological evaluation. All masses were frozen in liquid nitrogen after harvesting. Masses from each group of mice were ground into a fine powder, and RNA was extracted using a TRIzol reagent. For histological analysis, samples were embedded in paraffin and 8 μm sections were prepared. The sections were stained using Alizarin Red S and quantified using Image-Pro Plus 5.0 software (Media Cybernetics, Rockville, MD, USA).
Statistical analysis
Data are presented as the mean ± standard error of the mean (SEM) of three replicated, independent experiments. Significant differences between the groups were tested using one-way or two-way analysis of variance, and multiple comparisons were conducted using the Scheffe method. The thresholds for significance and high significance were P<0.05 and P<0.01, respectively.
Results
The osteogenic effects of mBMSCs with simvastatin treatment
We first examined the optimal dose of simvastatin stimulates osteogenesis in mesenchymal stem cells. The cell toxicity data showed that 5 μM SIM significantly enhanced cytotoxicity (Figure S1A). The osteogenic gene expression showed that simvastatin 1 uM significantly up-regulated Runx2, BMP-2 and OC gene expression within 12 hours and gradually increased until 48 hours (Figure S2B-D). The quantitative results from the ALP stain and Alizarin red staining showed that the simvastatin 0.5 and 1uM enhanced ALP activity at day 3 (Figure S1D), and mineralization at day 5 (Figure S1E) in D1 cells. We used simvastatin 1uM to complete the following experiments.
Simvastatin treatment increased active RhoA levels in mBMSCs
Active RhoA was detected using pull-down assays with or without the treatment of simvastatin. Simvastatin increased the levels of active RhoA within 30 minutes of treatment, with peak levels reaching within 3 hours of treatment followed by a slow decrease for 24 hours (Figure 1A). The effect of simvastatin was dose-dependent (Figure 1B). We used a membrane protein extraction assay to determine whether simvastatin alters the membrane anchoring of RhoA. Simvastatin dose-dependently increased the amount of cytosolic RhoA and reduced membrane-bound RhoA (Figure 1C). To further investigate the mechanism of RhoA activation by simvastatin, we assessed the association of RhoA with RhoGDIα by using immunoprecipitation. The amount of RhoA bound to RhoGDIα was lower in simvastatin-treated cells than in untreated cells (Figure 1D). The treatment with simvastatin increased the level of phospho-MYPT, a ROCK substrate molecule, suggesting increased ROCK activity (Figure 1E). Simvastatin increased the phosphorylation of MLC (Figure 1F).
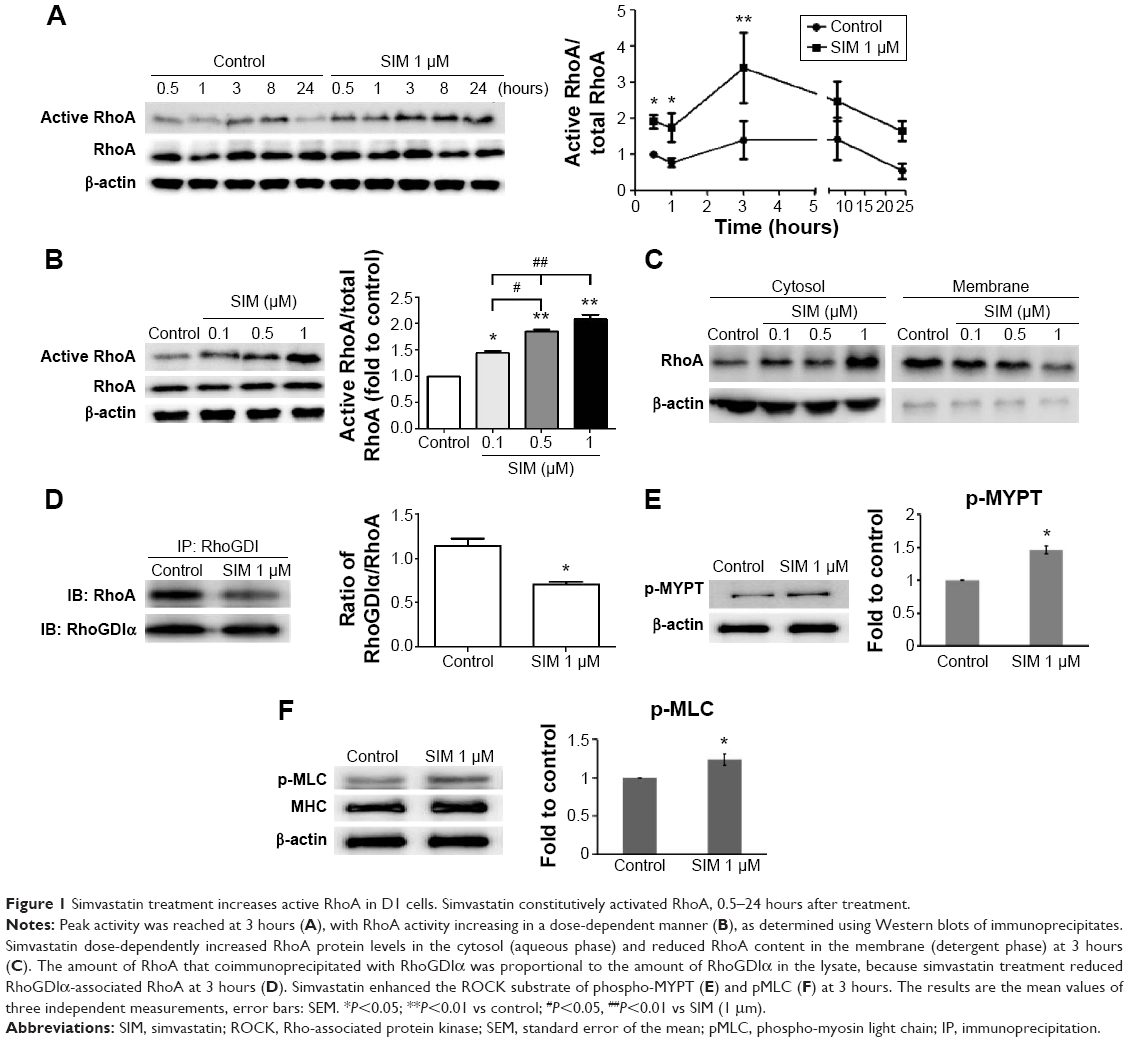 | Figure 1 Simvastatin treatment increases active RhoA in D1 cells. Simvastatin constitutively activated RhoA, 0.5–24 hours after treatment. |
Simvastatin-induced osteogenic differentiation and effects of cytoskeletal alterations
Biochemical agents, including Y-27632, blebbistatin, and cytochalasin D, were used to determine whether RhoA/actin/cell rigidity signaling was involved in simvastatin-induced osteogenesis. Cells treated with Y-27632 or blebbistatin became stellate-shaped and lacked actin fibril organization. Cells treated with cytochalasin D exhibited disrupted actin fibril organization and were clustered into irregular aggregates, even in the cultures treated with simvastatin (Figure 2A). Confocal images revealed clear increases in focal adhesion after treatment with simvastatin and decreases after adding Y-27632, blebbistatin, and cytochalasin D (Figure 2B). These biochemical agents not only disrupted fibril organization but also reduced simvastatin-enhanced cell rigidity (Figure 2C). Next, we measured the mRNA and protein levels of osteogenic genes (Runx2, BMP-2, and OC) in D1 cells treated with simvastatin alone or in combination with Y-27632, blebbistatin, or cytochalasin D. We found that incubation with Y-27632 or blebbistatin or disrupting actin polymerization with cytochalasin D significantly reduced the simvastatin-induced gene expression of Runx2, BMP-2, and OC at 12 and 24 hours (Figure 3A and B). Confocal images showed that simvastatin-induced increases in Runx2 protein in nuclei were lower at 12 and 24 hours (Figure 3C) and that simvastatin-induced increases in BMP-2 and OC protein levels were lower at 12 hours of culture in Y-27632, blebbistatin, or cytochalasin D (Figure 3D and E). These blockers also inhibited simvastatin-induced ALP activity on Day 3 and mineralization effects on Day 5 after treatment with simvastatin (Figure 4A and B). These results demonstrated that simvastatin-induced osteogenesis involves RhoA signaling, actin rearrangement, and increased cell rigidity.
 | Figure 2 Simvastatin increased actin filament formation, focal adhesions, and cellular tension. |
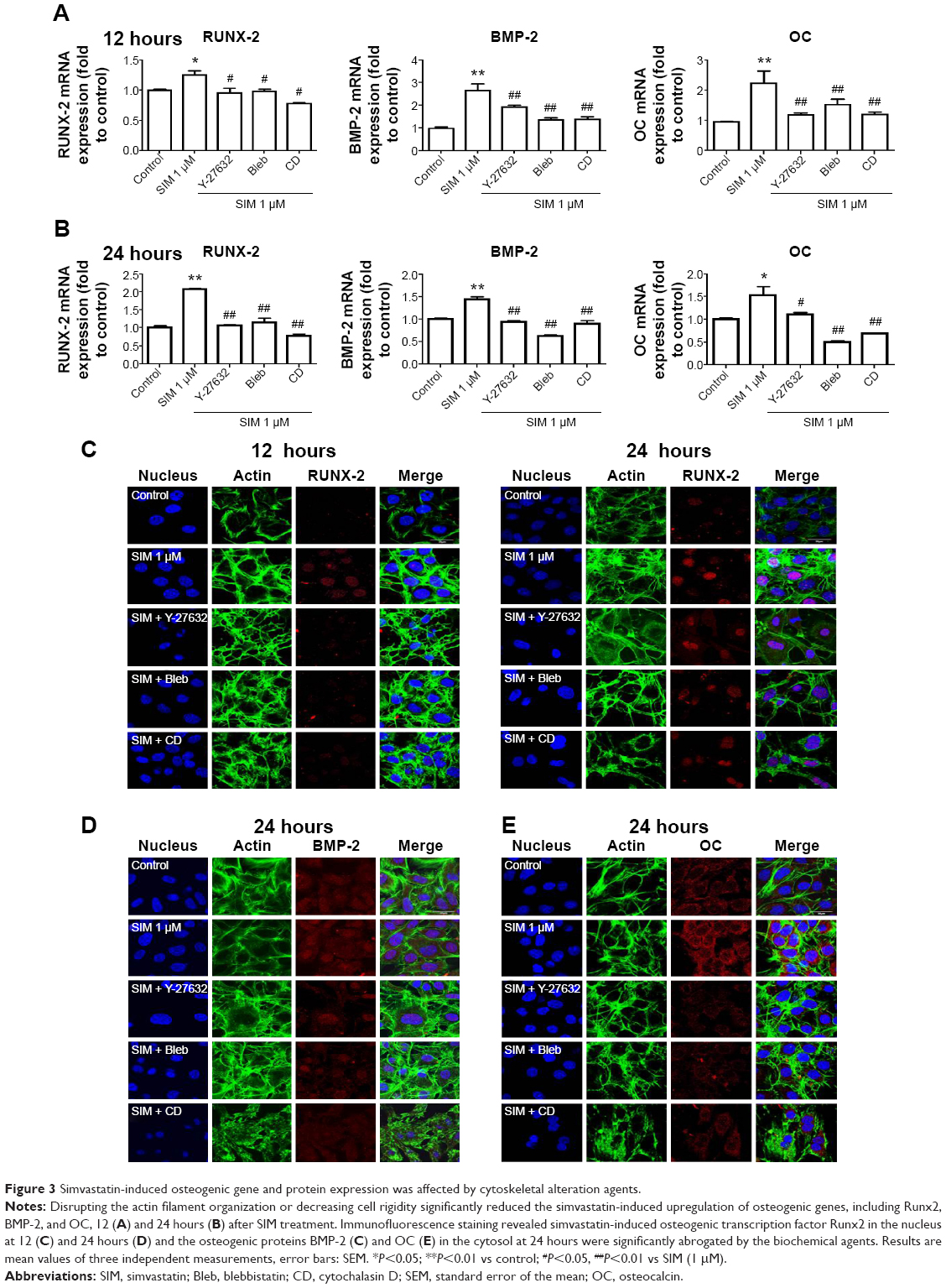 | Figure 3 Simvastatin-induced osteogenic gene and protein expression was affected by cytoskeletal alteration agents. |
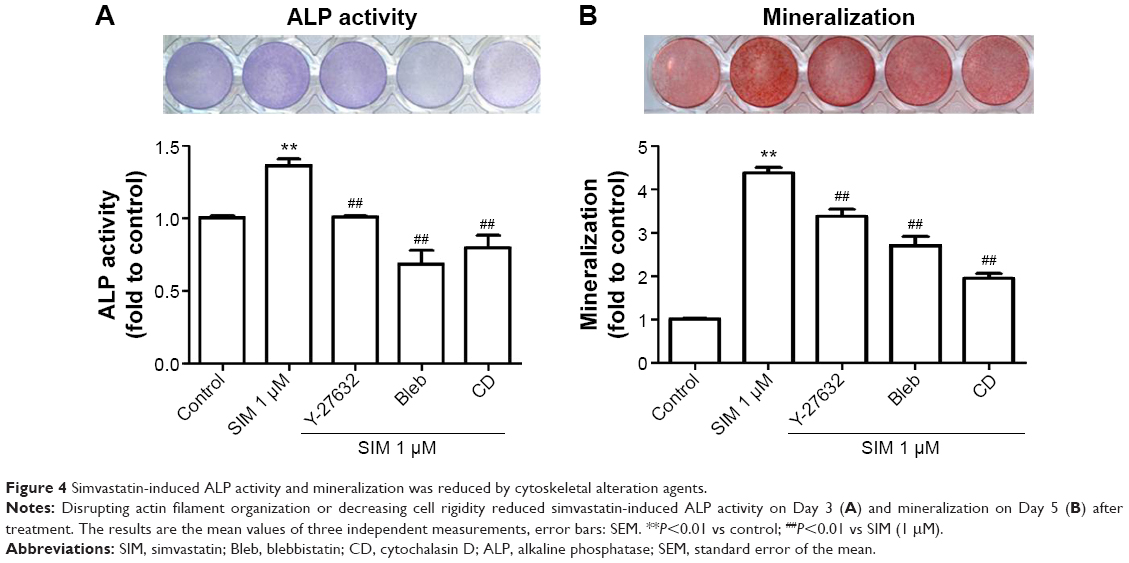 | Figure 4 Simvastatin-induced ALP activity and mineralization was reduced by cytoskeletal alteration agents. |
Simvastatin-induced bone formation and effects of cytoskeletal alterations
We used an ectopic bone formation model in imprinting control region mice to confirm that Rho/actin/cell rigidity signaling is involved in simvastatin-induced osteogenesis. Confocal images revealed clear increases in density of actin fibril after treatment with simvastatin, and actin fibril was attenuated by Y-27632, blebbistatin, or cytochalasin D, 1 week after injection (Figure 5A). In addition, simvastatin-induced increases in expression of Runx2, BMP-2, and OC mRNA were also significantly reduced by the inhibitors, 1 week after injection (Figure 5B). Alizarin Red S images and quantification results demonstrated that simvastatin induced the mineralization of ectopic bone mass; this effect was decreased by treatment with Y-27632, blebbistatin, or cytochalasin D, 2 weeks after injection (Figure 5C).
 | Figure 5 Simvastatin-induced osteogenic differentiation in an animal model of ectopic bone formation was reduced by cytoskeletal alteration agents. |
Discussion
Although statins were developed as inhibitors of cholesterol synthesis, recent studies have indicated that these agents exert pleiotropic effects.36 In this study, we demonstrated that simvastatin enhances osteogenic differentiation in cultured mBMSCs by activating the RhoA/ROCK pathway. We also revealed that this effect is not related to simvastatin-inhibited prenylation. Simvastatin also increases actin cytoskeleton organization, focal adhesion formation, and cellular tension. These simvastatin-induced osteogenic effects are significantly attenuated by blockers of the RhoA-signaling pathway in mBMSCs and in a mouse ectopic bone formation model. Our findings indicated that the treatment with simvastatin enhances osteogenic differentiation in mBMSCs.
Statins are known to block cholesterol synthesis by inhibiting the HMG-CoA reductase that converts HMG-CoA to mevalonate.37 The mevalonate pathway provides the source of isoprenoid intermediates such as farnesylpyrophosphate and geranylgeranylpyrophosphate for the prenylation, cell membrane anchoring, and function of small G proteins.38,39 Therefore, statins inhibit the prenylation of small G proteins and block their related signaling pathways. However, recent studies have reported that statins increase active-form RhoA,32–34,40 including increases in Rho-GTP in cardiac myofibroblasts33 and Rho-GTP and Rac-GTP in THP-1 monocytes and endothelial cells.34,40 In human erythroleukemia cells, lovastatin was reported as reducing the association between Rho and RhoGDα, thereby increasing cytosolic Rho-GTP.32 Notably, the present study found that treatment with simvastatin altered the distribution of RhoA from the membrane to the cytosol (Figure 2E) but still activated RhoA (Figure 2A and B). These results suggested that simvastatin-induced activation of RhoA does not require its anchoring to the membrane. We further observed that the treatment with simvastatin reduces the association between Rho and RhoGDIα (Figure 2D). Accordingly, we suggest that the key mechanism whereby simvastatin activates RhoA is decreasing the Rho/RhoGα association.
Small GTPase signaling occurs in various cellular events, including proliferation, differentiation, and cell survival.41,42 The constitutive expression of active RhoA is reported to upregulate expression of Runx2 mRNA and cause osteoblastic differentiation in human MSCs.18,20 The attenuation of RhoA activity upregulates expression of PPARγ mRNA and causes adipogenic differentiation.18,20 In this study, we investigated the role of RhoA in the osteogenic differentiation of BMSCs by transfecting constitutively active RhoA (RhoV14) and dominant negative RhoA (RhoV19) (Supplementary Materials and methods). We confirmed that expression of RhoV14 increased the expression of osteogenic marker genes, including Runx2, BMP-2, and OC, and enhanced mineralization (Figure S2). These results indicated that RhoA signaling plays a crucial role in osteogenic differentiation in mBMSCs. Furthermore, our data demonstrated that the treatment with simvastatin increased the active form of RhoA, and cotreatments with RhoA blockers reduced simvastatin-induced osteogenic differentiation, indicating that simvastatin-induced osteogenic differentiation occurs, at least partly, through the activation of RhoA signaling.
Several studies have used the ROCK inhibitor Y-27632 to investigate the role of the RhoA signaling pathway in osteogenic differentiation, with conflicting results.43,44 Y-27632 has been reported as increasing mineralization in mouse calvarial osteoblasts43,44 and inducing ectopic bone formation in response to rhBMP-2.45 However, ROCK activation has also been reported as enhancing osteogenic differentiation in MSCs under mechanical stimulation18,46–49 or treatment with BMP-2.50 These opposing effects of ROCK in osteogenic differentiation may be due to different roles played by ROCK during different stages of differentiation in MSCs and/or in MSCs from different species. In this study, we observed that Y-27632 reduced simvastatin-induced expression of osteogenic gene, ALP activity, and mineralization in mBMSCs (Figure 4). Most importantly, our results are the first to demonstrate that Rho/ROCK signaling mediates simvastatin-enhanced osteogenic differentiation in mBMSCs.
Actin cytoskeletal organization and resultant contractility are regulated by RhoA/ROCK signaling, which is crucial in regulating stem cell differentiation.18,20,51 In previous studies, it was found that stem cells that were cultured on a stiff matrix underwent osteogenic differentiation,23,24,52 whereas those on a soft matrix underwent adipogenic differentiation.53–55 These results indicated that actin filament organization and actin-related cell rigidity directly affect the fate of stem cells. In addition, disrupting actin cytoskeleton or decreased cell rigidity because of cytochalism D or blebbistatin reduces osteogenic differentiation even under a rigid substrate48,56 but promotes adipogenic differentiation18,57 in human MSCs. In this study, we found that blocking actin filament organization or losing cell rigidity significantly reduced the simvastatin-enhanced expression of osteogenic marker gene and protein but significantly increased expression of adipogenic marker gene (Figure S3). Furthermore, in our in vivo study, disrupting the actin filament or decreasing cell rigidity reduced simvastatin-enhanced actin filament density and bone formation. These results strongly indicated that maintaining actin filament intact and enhancing cell rigidity are required for the osteogenic differentiation of MSCs.
Conclusion
We found that simvastatin enhances the osteogenic differentiation of mBMSCs by activating the RhoA/actin/cell rigidity signaling pathway. Both disrupting actin cytoskeletal organization and losing cell rigidity decrease simvastatin-enhanced osteogenic differentiation in mBMSCs. Our study also confirmed that RhoA/actin/cell rigidity signaling plays a crucial role in simvastatin-induced bone formation in vivo. These results suggested that simvastatin is a potent osteoinductive factor that can increase actin filament organization and cell rigidity in MSCs and that combined use with osteoconductive materials may benefit stem-cell-based bone tissue engineering.
Acknowledgments
The authors thank Professor Tang Ming-Jer from the Physiology Department of National Cheng Kung University, Tainan, Taiwan, for providing atomic force microscopy and technical support. We thank the Center for Research Resources and Development of Kaohsiung Medical University in protein identification using Olympus Confocal. We gratefully acknowledge support for this research provided by the Ministry of Economic Affairs, Ministry of Science and Technology, Kaohsiung Medical University Hospital and Kaohsiung Medical University Aim for the Top Universities Grant under grant numbers 102-EC-17-A-19-S1-176, MST 102-2314-B-037-021-MY2, KMUH-1I-01, KMUH-2R-35, KMUH-2R-36, and KMU-TP103B01, respectively.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest in this work.
References
Yelin EH, Felts WR. A summary of the impact of musculoskeletal conditions in the United States. Arthritis Rheum. 1990;33(5):750–755. | ||
Corsini A, Bellosta S, Baetta R, Fumagalli R, Paoletti R, Bernini F. New insights into the pharmacodynamic and pharmacokinetic properties of statins. Pharmacol Ther. 1999;84(3):413–428. | ||
Schachter M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam Clin Pharmacol. 2005;19(1):117–125. | ||
Maeda T, Matsunuma A, Kawane T, Horiuchi N. Simvastatin promotes osteoblast differentiation and mineralization in MC3T3-E1 cells. Biochem Biophys Res Commun. 2001;280(3):874–877. | ||
Song C, Guo Z, Ma Q, et al. Simvastatin induces osteoblastic differentiation and inhibits adipocytic differentiation in mouse bone marrow stromal cells. Biochem Biophys Res Commun. 2003;308(3):458–462. | ||
Hwang R, Lee EJ, Kim MH, et al. Calcyclin, a Ca2+ ion-binding protein, contributes to the anabolic effects of simvastatin on bone. J Biol Chem. 2004;279(20):21239–21247. | ||
Baek KH, Lee WY, Oh KW, et al. The effect of simvastatin on the proliferation and differentiation of human bone marrow stromal cells. J Korean Med Sci. 2005;20(3):438–444. | ||
Solomon DH, Finkelstein JS, Wang PS, Avorn J. Statin lipid-lowering drugs and bone mineral density. Pharmacoepidemiol Drug Saf. 2005; 14(4):219–226. | ||
Maritz FJ, Conradie MM, Hulley PA, Gopal R, Hough S. Effect of statins on bone mineral density and bone histomorphometry in rodents. Arterioscler Thromb Vasc Biol. 2001;21(10):1636–1641. | ||
Ho ML, Chen YH, Liao HJ, et al. Simvastatin increases osteoblasts and osteogenic proteins in ovariectomized rats. Eur J Clin Invest. 2009; 39(4):296–303. | ||
Mundy G, Garrett R, Harris S, et al. Stimulation of bone formation in vitro and in rodents by statins. Science. 1999;286(5446): 1946–1949. | ||
Skoglund B, Forslund C, Aspenberg P. Simvastatin improves fracture healing in mice. J Bone Miner Res. 2002;17(11):2004–2008. | ||
Tai IC, Fu YC, Wang CK, Chang JK, Ho ML. Local delivery of controlled-release simvastatin/PLGA/HAp microspheres enhances bone repair. Int J Nanomedicine. 2013;8:3895–3905. | ||
Stein D, Lee Y, Schmid MJ, et al. Local simvastatin effects on mandibular bone growth and inflammation. J Periodontol. 2005;76(11): 1861–1870. | ||
Thylin MR, McConnell JC, Schmid MJ, et al. Effects of simvastatin gels on murine calvarial bone. J Periodontol. 2002;73(10):1141–1148. | ||
Nyan M, Sato D, Oda M, et al. Bone formation with the combination of simvastatin and calcium sulfate in critical-sized rat calvarial defect. J Pharmacol Sci. 2007;104(4):384–386. | ||
Discher DE, Mooney DJ, Zandstra PW. Growth factors, matrices, and forces combine and control stem cells. Science. 2009;324(5935): 1673–1677. | ||
McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. 2004;6(4):483–495. | ||
Sun Y, Chen CS, Fu J. Forcing stem cells to behave: a biophysical perspective of the cellular microenvironment. Annu Rev Biophys. 2012; 41:519–542. | ||
Arnsdorf EJ, Tummala P, Kwon RY, Jacobs CR. Mechanically induced osteogenic differentiation – the role of RhoA, ROCKII and cytoskeletal dynamics. J Cell Sci. 2009;122(pt 4):546–553. | ||
Woods A, Wang G, Beier F. RhoA/ROCK signaling regulates Sox9 expression and actin organization during chondrogenesis. J Biol Chem. 2005;280(12):11626–11634. | ||
Woods A, Beier F. RhoA/ROCK signaling regulates chondrogenesis in a context-dependent manner. J Biol Chem. 2006;281(19): 13134–13140. | ||
Tan S, Fang JY, Yang Z, Nimni ME, Han B. The synergetic effect of hydrogel stiffness and growth factor on osteogenic differentiation. Biomaterials. 2014;35(20):5294–5306. | ||
Zouani OF, Kalisky J, Ibarboure E, Durrieu MC. Effect of BMP-2 from matrices of different stiffnesses for the modulation of stem cell fate. Biomaterials. 2013;34(9):2157–2166. | ||
Takeuchi Y, Suzawa M, Kikuchi T, Nishida E, Fujita T, Matsumoto T. Differentiation and transforming growth factor-beta receptor down-regulation by collagen-alpha2beta1 integrin interaction is mediated by focal adhesion kinase and its downstream signals in murine osteoblastic cells. J Biol Chem. 1997;272(46):29309–29316. | ||
Toma CD, Ashkar S, Gray ML, Schaffer JL, Gerstenfeld LC. Signal transduction of mechanical stimuli is dependent on microfilament integrity: identification of osteopontin as a mechanically induced gene in osteoblasts. J Bone Miner Res. 1997;12(10):1626–1636. | ||
Feng T, Szabo E, Dziak E, Opas M. Cytoskeletal disassembly and cell rounding promotes adipogenesis from ES cells. Stem Cell Rev. 2010; 6(1):74–85. | ||
Adamson P, Marshall CJ, Hall A, Tilbrook PA. Post-translational modifications of p21rho proteins. J Biol Chem. 1992;267(28):20033–20038. | ||
Wong WW, Dimitroulakos J, Minden MD, Penn LZ. HMG-CoA reductase inhibitors and the malignant cell: the statin family of drugs as triggers of tumor-specific apoptosis. Leukemia. 2002;16(4):508–519. | ||
Laufs U, Liao JK. Targeting Rho in cardiovascular disease. Circ Res. 2000;87(7):526–528. | ||
Woo JT, Nakagawa H, Krecic AM, et al. Inhibitory effects of mevastatin and a geranylgeranyl transferase I inhibitor (GGTI-2166) on mononuclear osteoclast formation induced by receptor activator of NF kappa B ligand (RANKL) or tumor necrosis factor-alpha (TNF-alpha). Biochem Pharmacol. 2005;69(1):87–95. | ||
Turner SJ, Zhuang S, Zhang T, Boss GR, Pilz RB. Effects of lovastatin on Rho isoform expression, activity, and association with guanine nucleotide dissociation inhibitors. Biochem Pharmacol. 2008; 75(2):405–413. | ||
Porter KE, Turner NA, O’Regan DJ, Balmforth AJ, Ball SG. Simvastatin reduces human atrial myofibroblast proliferation independently of cholesterol lowering via inhibition of RhoA. Cardiovasc Res. 2004; 61(4):745–755. | ||
Vecchione C, Brandes RP. Withdrawal of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors elicits oxidative stress and induces endothelial dysfunction in mice. Circ Res. 2002;91(2):173–179. | ||
Wang YH, Chiu WT, Wang YK, et al. Deregulation of AP-1 proteins in collagen gel-induced epithelial cell apoptosis mediated by low substratum rigidity. J Biol Chem. 2007;282(1):752–763. | ||
Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005;45:89–118. | ||
Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343(6257):425–430. | ||
Xia Z, Tan MM, Wong WW, Dimitroulakos J, Minden MD, Penn LZ. Blocking protein geranylgeranylation is essential for lovastatin-induced apoptosis of human acute myeloid leukemia cells. Leukemia. 2001; 15(9):1398–1407. | ||
Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annu Rev Biochem. 1996;65:241–269. | ||
Cordle A, Koenigsknecht-Talboo J, Wilkinson B, Limpert A, Landreth G. Mechanisms of statin-mediated inhibition of small G-protein function. J Biol Chem. 2005;280(40):34202–34209. | ||
Boureux A, Vignal E, Faure S, Fort P. Evolution of the Rho family of ras-like GTPases in eukaryotes. Mol Biol Evol. 2007;24(1):203–216. | ||
Bustelo XR, Sauzeau V, Berenjeno IM. GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays. 2007;29(4):356–370. | ||
Kanazawa I, Yamaguchi T, Yano S, Yamauchi M, Sugimoto T. Activation of AMP kinase and inhibition of Rho kinase induce the mineralization of osteoblastic MC3T3-E1 cells through endothelial NOS and BMP-2 expression. Am J Physiol Endocrinol Metab. 2009; 296(1):E139–E146. | ||
Harmey D, Stenbeck G, Nobes CD, Lax AJ, Grigoriadis AE. Regulation of osteoblast differentiation by Pasteurella multocida toxin (PMT): a role for Rho GTPase in bone formation. J Bone Miner Res. 2004;19(4): 661–670. | ||
Yoshikawa H, Yoshioka K, Nakase T, Itoh K. Stimulation of ectopic bone formation in response to BMP-2 by Rho kinase inhibitor: a pilot study. Clin Orthop Relat Res. 2009;467(12):3087–3095. | ||
Wang G, Woods A, Sabari S, Pagnotta L, Stanton LA, Beier F. RhoA/ROCK signaling suppresses hypertrophic chondrocyte differentiation. J Biol Chem. 2004;279(13):13205–13214. | ||
Hamamura K, Swarnkar G, Tanjung N, et al. RhoA-mediated signaling in mechanotransduction of osteoblasts. Connect Tissue Res. 2012; 53(5):398–406. | ||
Khatiwala CB, Kim PD, Peyton SR, Putnam AJ. ECM compliance regulates osteogenesis by influencing MAPK signaling downstream of RhoA and ROCK. J Bone Miner Res. 2009;24(5):886–898. | ||
McMurray RJ, Gadegaard N, Tsimbouri PM, et al. Nanoscale surfaces for the long-term maintenance of mesenchymal stem cell phenotype and multipotency. Nat Mater. 2011;10(8):637–644. | ||
Wang YK, Yu X, Cohen DM, et al. Bone morphogenetic protein-2-induced signaling and osteogenesis is regulated by cell shape, RhoA/ROCK, and cytoskeletal tension. Stem Cells Dev. 2012;21(7): 1176–1186. | ||
Sarasa-Renedo A, Tunc-Civelek V, Chiquet M. Role of RhoA/ROCK-dependent actin contractility in the induction of tenascin-C by cyclic tensile strain. Exp Cell Res. 2006;312(8):1361–1370. | ||
Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126(4):677–689. | ||
Trappmann B, Gautrot JE, Connelly JT, et al. Extracellular-matrix tethering regulates stem-cell fate. Nat Mater. 2012;11(7):642–649. | ||
Fu J, Wang YK, Yang MT, et al. Mechanical regulation of cell function with geometrically modulated elastomeric substrates. Nat Methods. 2010;7(9):733–736. | ||
Park JS, Chu JS, Tsou AD, et al. The effect of matrix stiffness on the differentiation of mesenchymal stem cells in response to TGF-beta. Biomaterials. 2011;32(16):3921–3930. | ||
Shih YR, Tseng KF, Lai HY, Lin CH, Lee OK. Matrix stiffness regulation of integrin-mediated mechanotransduction during osteogenic differentiation of human mesenchymal stem cells. J Bone Miner Res. 2011;26(4):730–738. | ||
Arnsdorf EJ, Tummala P, Jacobs CR. Non-canonical Wnt signaling and N-cadherin related beta-catenin signaling play a role in mechanically induced osteogenic cell fate. PLoS One. 2009;4(4):e5388. |
Supplementary materials
Lactate dehydrogenase (LDH) assay
The LDH concentration was assayed utilizing a commercial kit (Cytotoxicity Detection Kit, Roche), which quantifies the LDH release from the cells into the culture medium. The D1 cells were seeded 5,000 cells per well in 96-well plates for 24 hours to promote adherence and then treated with simvastatin 0.1, 0.5, 1, and 5 μM for 24 hours. Absorbance was read at 490 nm by using a microplate reader.
Constitutively active and dominant negative RhoA transfection
RhoA-V14, RhoA-N19, and GFP recombinant plasmids (donated by Dr Tang Ming-Jer at National Cheng Kung University) were transfected into 90% confluent D1 cells by using Lipofectamine 2000 (Invitrogen, Grand Island, NY, USA) according to the manufacturer protocol. In brief, the cells were cultured in a growth medium without antibiotics before transfection. Transfection was performed using Opti-MEM (250 μL) mixed with 4 μg of RhoA-V14, RhoA-N19, or GFP recombinant plasmids and Lipofectamine 2000 (6 μL). Following incubation at room temperature for 20 minutes, the mixture was added to the cells in six-well plates. After 4–6 hours, the medium was exchanged with the growth medium overnight and cells selected after using G418 for 1 week. After selection, cells were harvested to detect RhoA activity and antiphospho-MLC. The cells were cultured in the growth medium, and gene expression was detected on Days 1, 3, and 5. The cells were cultured in the osteoinduction medium and mineralization was measured on Day 5.
PPARγ primers
The cDNA was amplified using PPARγ primers with real-time PCR. The primers were PPARγ forward, 5′-TGGGTGAAACTCTGGGAGATTC-3′; and PPARγ reverse, 5′-GGAGCAGAAATGCTGGAGAAATC-3′.
 | Figure S1 Simvastatin enhanced osteogenesis in D1 cells. |
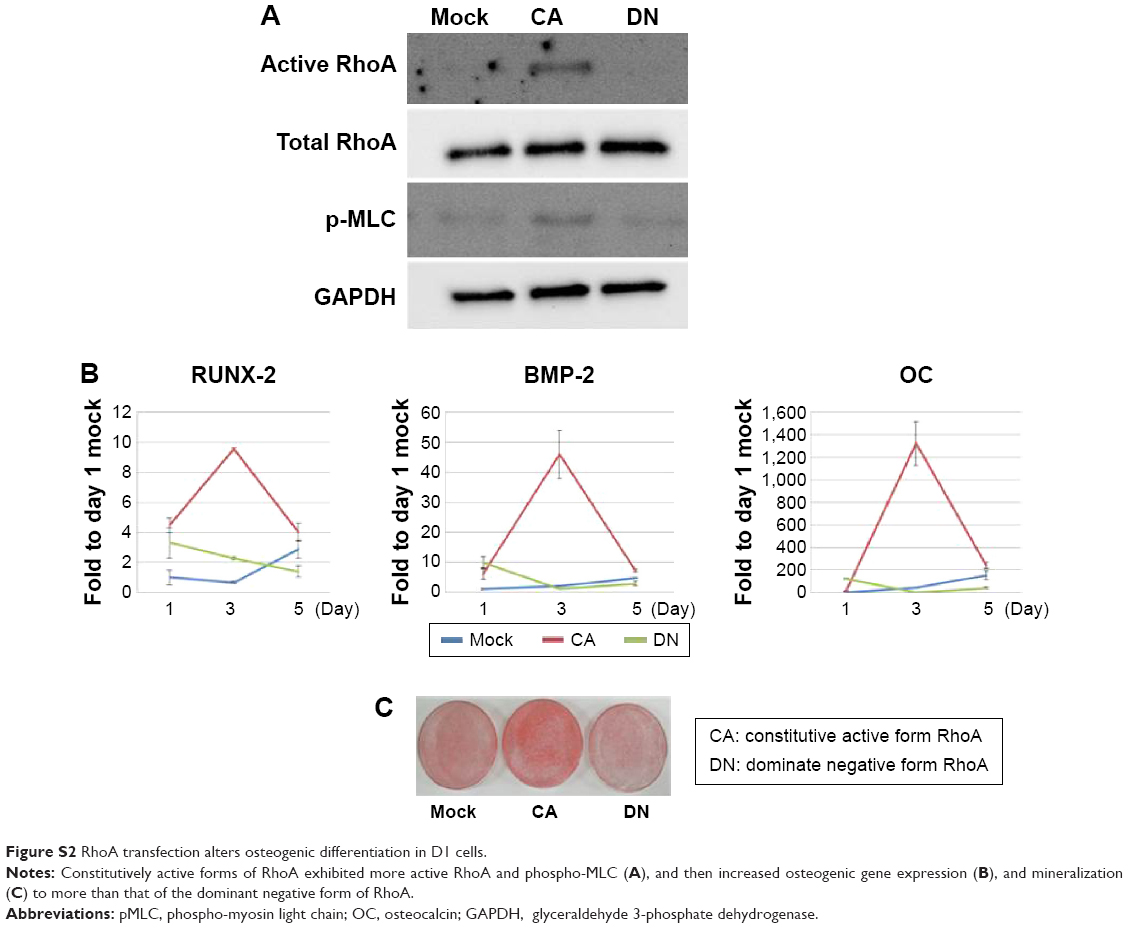 | Figure S2 RhoA transfection alters osteogenic differentiation in D1 cells. |
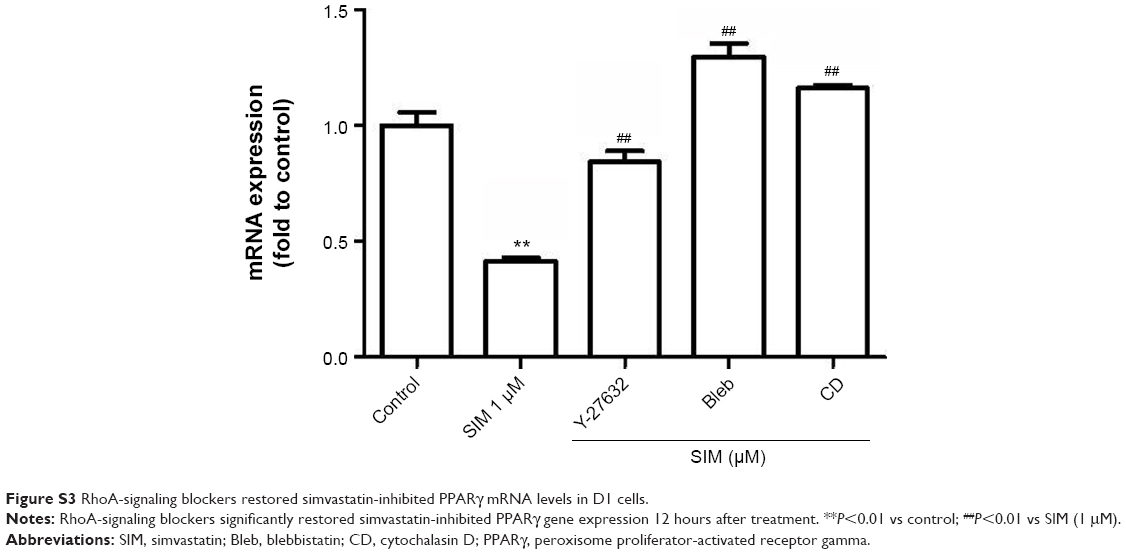 | Figure S3 RhoA-signaling blockers restored simvastatin-inhibited PPARγ mRNA levels in D1 cells. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.