Back to Journals » Drug Design, Development and Therapy » Volume 10
Simultaneous targeting of MyD88 and Nur77 as an effective approach for the treatment of inflammatory diseases
Received 30 December 2015
Accepted for publication 5 February 2016
Published 4 May 2016 Volume 2016:10 Pages 1557—1572
DOI https://doi.org/10.2147/DDDT.S103393
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Saqib Uzma,1 Mirza S Baig2
1Divsion of Chemistry, School of Basic Sciences, 2Centre for Biosciences and Biomedical Engineering (BSBE), Indian Institute of Technology (IIT), Indore, MP, India
Abstract: Myeloid differentiation primary response protein 88 (MyD88) has long been considered a central player in the inflammatory pathway. Recent studies clearly suggest that it is an important therapeutic target in inflammation. On the other hand, a recent study on the interaction between the orphan nuclear receptor (Nur77) and p38α, leading to increased lipopolysaccharide-induced hyperinflammatory response, suggests this binary complex as a therapeutic target. In this study, we have designed inhibitors that can inhibit both MyD88 and Nur77 at the same time. Since both MyD88 and Nur77 are an integral part of the pathways involving lipopolysaccharide-induced activation of NF-κB-mediated inflammation, we tried to target both proteins with the same library in order to retrieve compounds having dual inhibitory properties. To perform this, we developed a homodimeric model of MyD88 and, along with the crystal structure of Nur77, screened a virtual library of compounds from the traditional Chinese medicine database containing ~61,000 compounds. We analyzed the resulting hits for their efficacy for dual binding and probed them for developing a common pharmacophore model that could be used as a prototype to screen compound libraries as well as to guide combinatorial library design to search for ideal dual-target inhibitors. Thus, our study explores the identification of novel leads having dual inhibiting effects due to binding to both MyD88 and Nur77 targets.
Keywords: inflammation, Nur77, MyD88, dual-target inhibitors
Introduction
Partial inhibition of a small number of targets is sometimes more efficient than the complete inhibition of a single target.1,2 This, as well as the success stories of many dual-target drugs and combinatorial therapies, led us to suggest that systematic drug-design strategies should be directed against more than one target.3–5 These days, combinations of drugs, a form of dual- or multitargeting, combining different inhibitors that target a specific single target, or a single inhibitor targeting more than one target, are the standard treatment for diseases, including cancer, type 2 diabetes mellitus, and viral and bacterial infections.6–8
Myeloid differentiation primary response protein 88 (MyD88) is a canonical adaptor protein that functions to recruit signaling proteins in the inflammatory pathways downstream of members of the Toll-like receptor and interleukin-1 (IL-1) receptor families and is associated with the induction of innate immune response.9–11 Recent studies have shown the result of MyD88 gene silencing in primary human cells in preventing lipopolysaccharide (LPS)-induced sepsis suggesting its role in systemic inflammation and the inflammatory response.12 MyD88 consists of two major domains having functional relevance: a N-terminal death domain (90 aa residues) and a C-terminal Toll/IL-1 receptor (TIR) domain (150 aa residues).13 Based on the crystal structures and mutational data, several structural models have been proposed for heteromeric TIR–TIR interactions, which commonly suggest the importance of a small BB loop in these interactions.14,15 A synthetic mimetic of the BB loop in the TIR domain of MyD88 attenuated staphylococcal enterotoxin B (SEB)-induced pro-inflammatory cytokine production.16 It is known that the BB-loop region acts as the mediator of the homo- (adaptor–adaptor) and hetero- (receptor–adaptor) dimerization, which is necessary for the functioning of TIR domains to induce MyD88-mediated signaling.9,10 Recruitment of the MyD88 dimer to the receptor–membrane complex is a requirement for MyD88-mediated signaling via the activation of the downstream kinases IL-1-associated kinase (IRAK) 1 and IRAK4.17 While working on the structure–interaction studies on MyD88 and its inhibitors so far published, a recent study by Olson et al18 has caught our attention. Their study is quite interesting in the special context of the published studies on MyD88 inhibitors so far, especially the peptide, peptidomimetic, and the recent small-molecule inhibitors. The study reveals that these molecules bind at the interface of MyD88 molecule, inhibiting its dimerization and hence the inflammatory downstream signaling mediated by MyD88. With a clear-cut role in inflammation and a recently addressed site for inhibition, there is an enormous potential of MyD88 inhibition to prevent inflammation.
The anti-inflammatory property of Nur77 has already been addressed previously in various cell models, where the elevation of Nur77 expression was shown to lead to the reduction of expression of several cytokines and chemokines in macrophages in response to LPS or tumor necrosis factor stimulation.19 However, a recent study explaining the mechanism of Nur77 involvement in inflammation shows a novel mechanism to target it.20 Recent investigations by Li et al20 suggested that the interaction and phosphorylation of Nur77 by p38 leads to the attenuation of its anti-inflammatory response. Nur77 interacts with p65 and blocks its binding to the κB element, leading to the downregulation of NF-κB activity. However, this anti-inflammatory effect of Nur77 is countered by its phosphorylation after binding to LPS-activated p38a, leading to the attenuation of its anti-inflammatory properties. Hence, the interference of the associated p38α–Nur77 interaction would favor Nur77’s attenuation of the LPS-induced hyperinflammatory response. The ligand binding domain (LBD) of Nur77 responsible for the direct interaction with p38α has been proposed to be the targeting point for abolishing this Nur77–p38 interaction. Disrupting this interaction may result in hypophosphorylation of Nur77 to suppress the LPS-induced inflammatory response. This would thereby let Nur77 to perform its role of restraining inflammation via binding to p65. Investigators also discovered a novel compound PDNPA (n-pentyl 2-[3,5-dihydroxy-2-(1-nonanoyl)-phenyl]acetate) from an in-house library, which targets the LBD of Nur77. The binding site for PDNPA locates among helices 4, 5, 11, and 12 with specific interactions with the conserved LSD motif and other LBD residues. By impeding the interaction between Nur77 and p38α, PDNPA prevents the p38α phosphorylation of Nur77 so that Nur77 can effectively inhibit NF-κB activity and, as a result, dampen LPS-induced inflammation. This pioneering study paved the way for the discovery of anti-inflammatory inhibitors against Nur77.
A computer-assisted strategy using a combination of molecular modeling and virtual screening comprising docking and pharmacophore design was successfully applied to screen for dual-target inhibitors against MyD88 and Nur77 as inflammatory targets. As discussed earlier, these two targets have a very defined and critical role in the signaling cascade triggering the LPS-induced inflammatory response via NF-κB. While MyD88 activation leads to the inflammatory signaling after LPS response in an indirect way via IKB-mediated NF-κB activation, Nur77 has a direct role by binding to the P65 unit of NF-κB, thus inhibiting the inflammatory response. In this work, we set out to develop compounds that could simultaneously block the homodimerization of MyD88 as well as the Nur77–p38α interaction as an anti-inflammatory strategy (Figure 1). In our protocol, we first constructed a homodimeric model of MyD88 through docking studies. Further, we used this model as well as the crystal structure of Nur77 to individually screen the traditional Chinese medicine (TCM) database of 61,000 compounds. Mechanistic analysis of the binding modes of these identified leads within the active site of both targets was performed using docking studies. Out of the many high-affinity binders retrieved, we selected only those compound hits that were binding to both the targets (dual-targeting hits). This led us to retrieve four crucial compounds that were binding efficiently to both of the targets. Further, we made an in-depth analysis of the ligand-binding patterns in both the complexes and eventually retained Compound 1 (CP1) and Compound 4 (CP4) as high-affinity, dual-target inhibitors of MyD88 and Nur77. Next, we went on to probe the critical pharmacophore features of these two pairs of complexes, which suggested important and highly critical pharmacophore points encompassing both the targets. To our surprise, we not only retrieved hits binding to both targets effectively, but after an in-depth analysis of the ligand receptor complexes, we also observed that the binding sites of both MyD88 and Nur77 have some common ligand-binding patterns that could be exploited to develop a common pharmacophore model. The developed pharmacophore model represents the binding pattern of inhibitors for both MyD88 and Nur77. This model can be used for screening large compound libraries, and the potential hits can be retrieved with high accuracy. Our study is a good starting point toward our efforts in the inhibition of inflammation by targeting key players of the inflammatory pathway, namely MyD88 homodimerization and Nur77–p38 interaction.
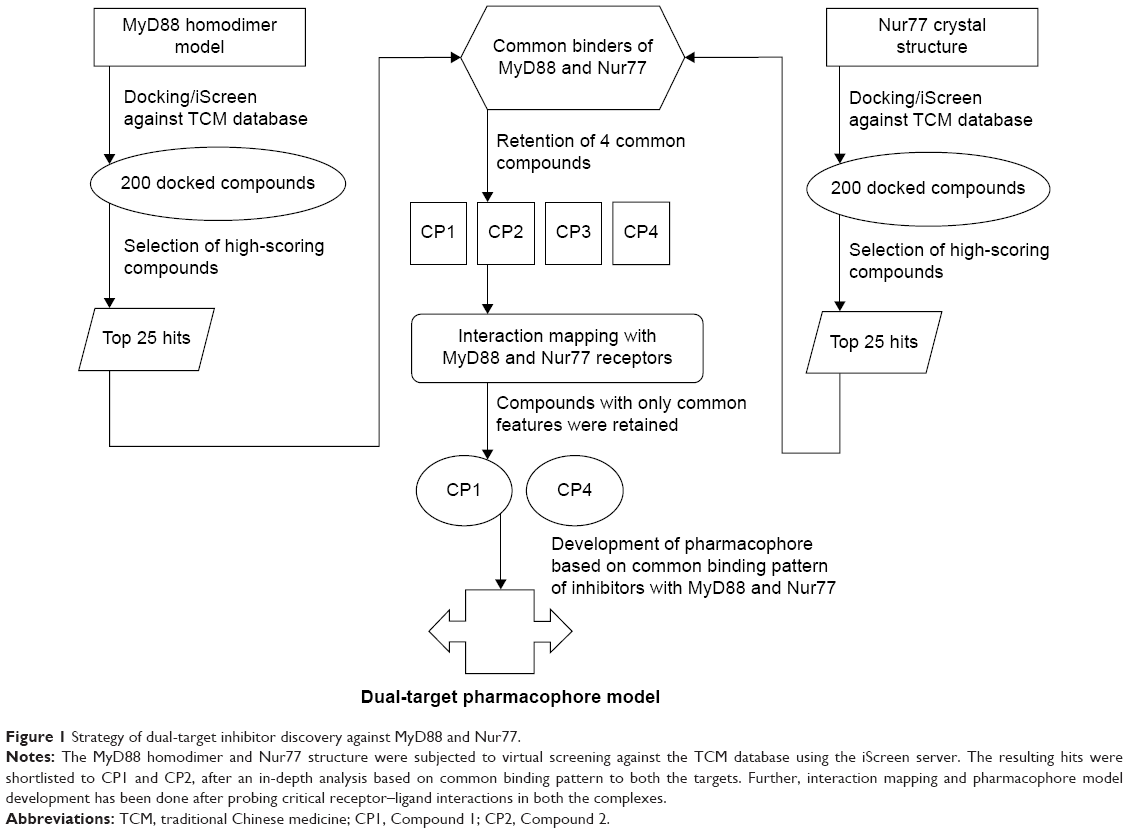 | Figure 1 Strategy of dual-target inhibitor discovery against MyD88 and Nur77. |
Results
Structural modeling via docking of the MyD88 homodimeric complex
The MyD88 monomeric protein structure is already available in the Protein Data Bank (PDB).21 However, the dimeric structure is unavailable. Recently, Olson et al18 published a homology modeled structure of the dimeric protein. We similarly modeled the dimer structure utilizing the crystal structures of monomeric MyD88 structures using two different programs: Global Range Molecular Matching (GRAMM-X) and Clustpro. When we compared the top docked poses of both the servers, we observed that they were almost superimposable (Figure 2A). Our models were very similar to that of Olson et al, specifically in terms of the highly conserved BB-loop position. As in their model, the BB-loop residues form an integral part of the dimeric interface of the structure; similarly, our top-docked conformations from both the servers also had the BB loop at the dimer interface (Figure 2B). The BB loop has residues ranging from 196 to 202 comprising the fairly conserved (25%–75%) sequence motif VLPG, with nearly invariant proline and glycine residues.22 The BB-loop region is a long coil region with flexible conformations, which could be attributed to its positioning at the interface where much action has been going on in terms of activation by dimerization and inactivation by monomerization due to various signaling events.
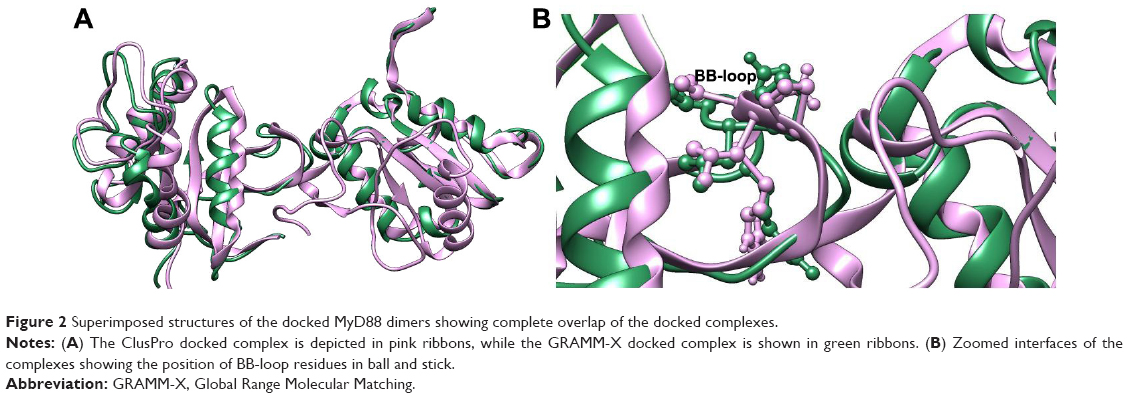 | Figure 2 Superimposed structures of the docked MyD88 dimers showing complete overlap of the docked complexes. |
Virtual screening of MyD88 homodimer and Nur77 using TCM database
The TCM database has long been used in the diagnosis and treatment of diseases. Recently, more use of this database has been observed because of the amount of data supporting its therapeutic potential. This “first-of-its-kind,” ready-to-dock database of compounds has been considered because of its enormous potential as a TCM library containing three-dimensional structural information of all its constituents. Further, this important database has been integrated into a docking and screening program, iScreen. Hence, we used iScreen for screening the TCM database against MyD88 and Nur77. The developed MyD88 model and the Nur77 crystal structure (4RZG),20 with a reasonable resolution of 2.7 Å, were selected as the input structures for the respective docking/screening runs. In the case of MyD88, the residues LPG from the B loop were specified as the probable ligand-binding sites because of their role in ligand binding, as previously reported and discussed. In Nur77, the coordinates of the bound PDNPA inhibitor were provided as the probable binding residues. From the high-scoring 200 hits, we first selected the top 25 from both the docking results (Tables 1 and 2). Further, we compared the overlapping hits from both lists. This led us to finally select four compounds common in the list of top 25 compound hits of both the targets (Table 3). We further analyzed all the four, one by one, and got many interesting facts about the similarity in ligand binding of both the targets.
 | Table 1 The top 25 virtual screening hits and their scores for MyD88 from TCM database integrated iScreen program |
 | Table 2 The top 25 virtual screening hits and their scores for Nur77 from TCM Database integrated iScreen program |
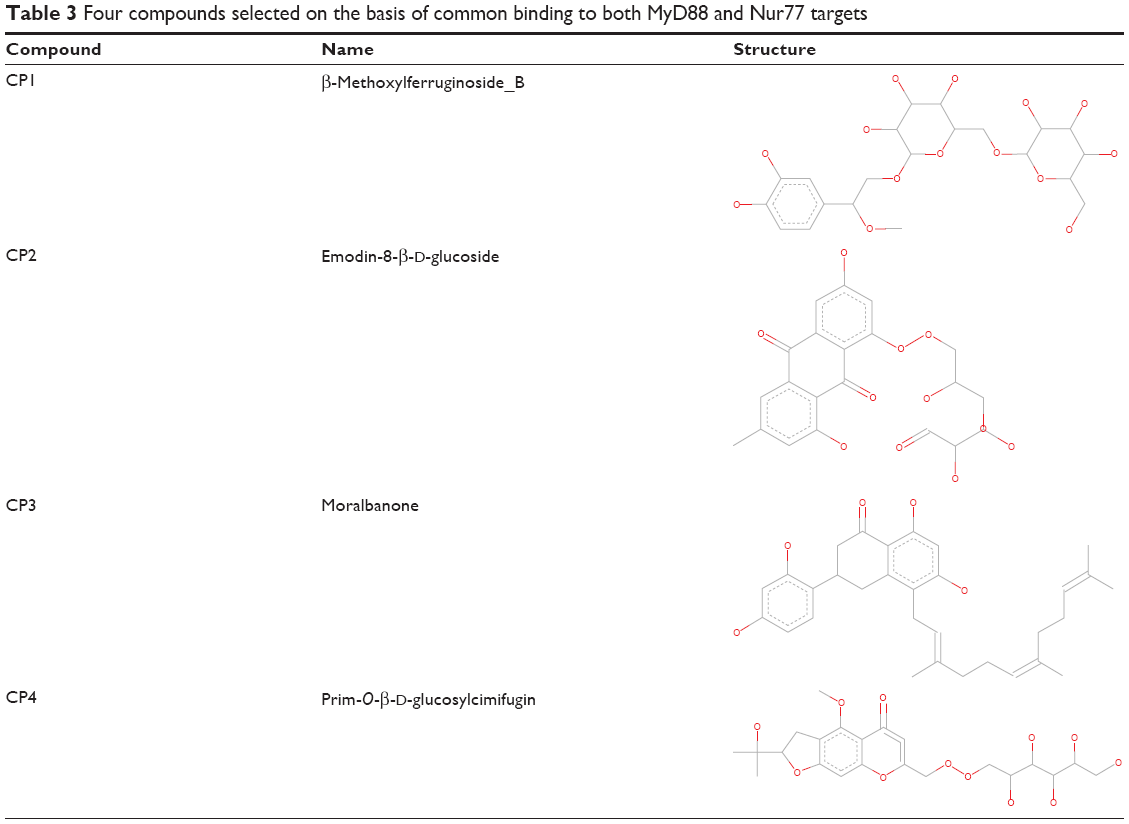 | Table 3 Four compounds selected on the basis of common binding to both MyD88 and Nur77 targets |
Interaction analysis of CP1 or β-methoxylferruginoside_B in MyD88 binding site
CP1 or β-methoxylferruginoside_B ranked 13th in the results list with a binding score of −78.3 (Table 1). Although at the 13th position, the binding energy of CP1 was different by only four units from that of the highest scoring compound rosmamrinic acid (−82.1). Since we were more interested to pursue only those hits that were common in both the targets, we selected CP1 because it was also among the top 25 of the Nur77 binders. Further, we did an in-depth analysis of the ligand–receptor interactions of CP1 in the MyD88 binding site.
As can be seen from Figure 3A, CP1 binds to MyD88 with strong polar and nonpolar interactions, specifically with the BB-loop residues 196–201 (49–54 in our structure). Since we modeled the structure of the MyD88 homodimer via docking two different monomer crystal structures, the residue numbers changed in the final docked model. Hence, we will use the nomenclature as in our model from now on. We have tabulated the corresponding residue numbers in both the model and the original sequence of MyD88 in Uniprot (Q99836) in Table 4 for easy reference. Among the major interactions, Asp50 and Val51 were acting as hydrogen-bond (H-bond) acceptors from the hydroxyl (–OH) substituents of the tetrahydropyran ring in CP1. Further, Gln29 acts as both the H-bond donor and acceptor for the –OH substituents of the phenyl ring. Another important interaction observed between the residues Pro53, Pro14, and the phenyl ring of the compound was a π–alkyl bond. Gln26, Leu52, and the nearby residues were found to have numerous Van der Waals (VdW) and carbon–hydrogen (C–H) bonds with the H atoms of the alkyl hub. Many residues making the VdW contacts are not shown for the purpose of clarity. Overall, the conformation of CP1 seemed to fit closely into the MyD88 active site.
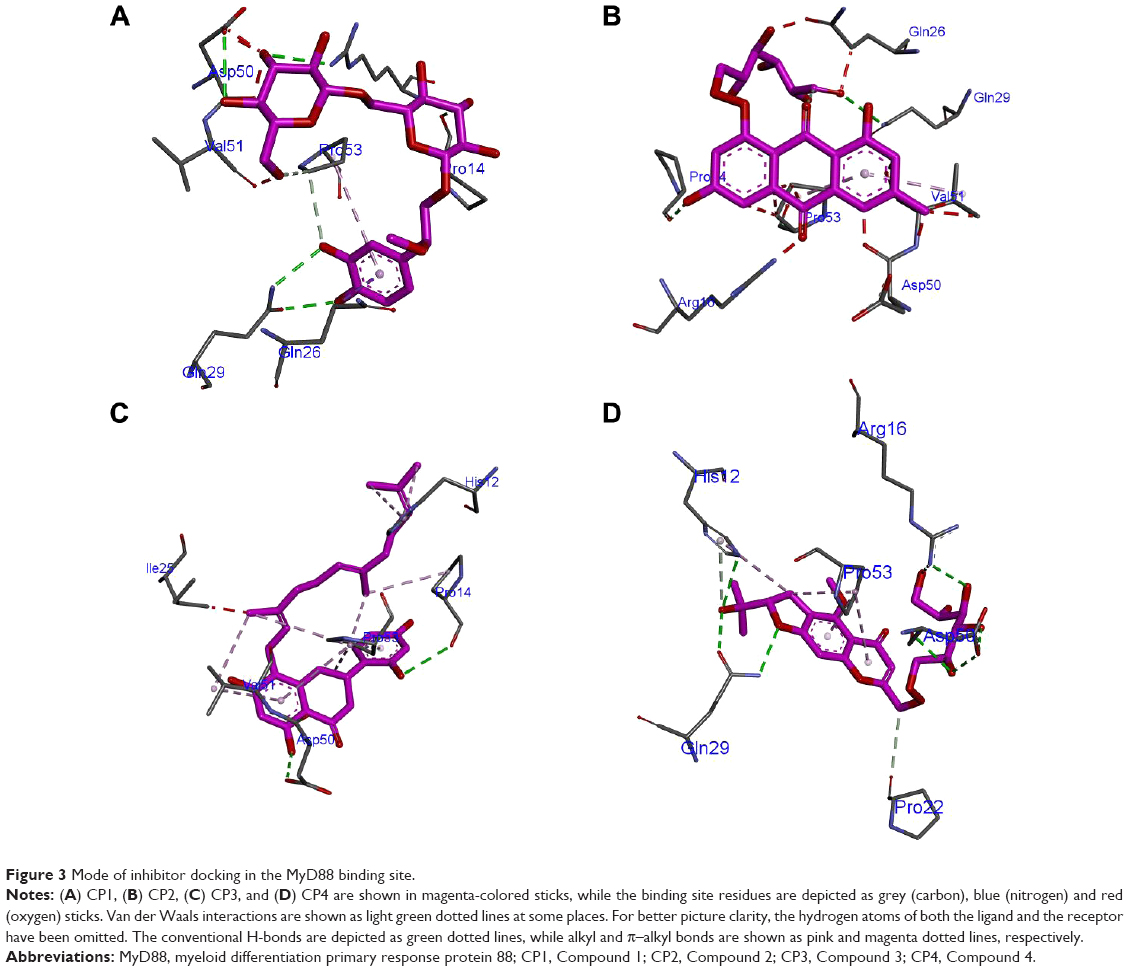 | Figure 3 Mode of inhibitor docking in the MyD88 binding site. |
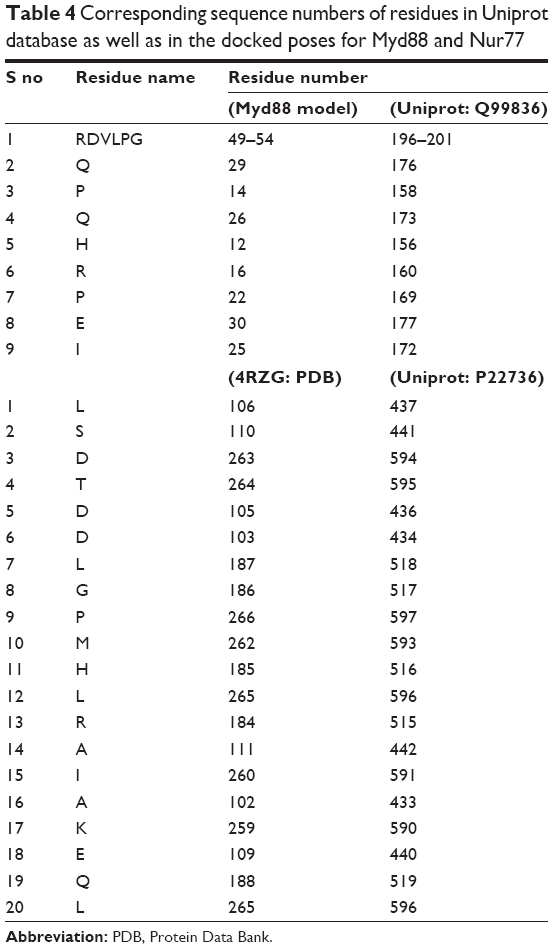 | Table 4 Corresponding sequence numbers of residues in Uniprot database as well as in the docked poses for Myd88 and Nur77 |
Interaction analysis of CP2 or emodin-8-β-d-glucoside in MyD88 binding site
CP2 ranked 19th in the result list, with a binding score of −77.1 in the MyD88 binding site (Table 1). It is the composed of a modified anthracene ring and an alkyl chain with many substituted polar oxy (=O) and –OH groups. Figure 3B shows the major H-bond interactions, of which His12, Pro14, Arg16, Pro53, and Gln29 were the prominent ones. His12, Arg16, and Gln29 acted as the H-bond donors, while Pro14, Pro53, and Gln29 were H-bond acceptors from the various polar substituents of the anthracene ring and alkyl chain. Further, Gln26, Pro53, Val51, Pro22, and Leu52 were making various π–σ and π–alkyl bonds and VdW interactions with various substituents of the anthracene ring.
Interaction analysis of CP3 or moralbanone in MyD88 binding site
The structure of CP3 was different from that of CP1 and CP2, although both too carry a three-membered ring system where a substituted napthyl ring is separated from the phenyl ring by a bond, the rest of the alkyl chain was devoid of any substituted polar groups, especially =O and –OH. This might be a reason for it to show up at the 23rd position with a score of −75.8924 (Table 1) in the MyD88 binding site. The major H-bond interactions it exhibited with Asp50, Arg16, and Pro14 were mostly through its substituted rings (Figure 3C). The MyD88 binding site residues Pro53 and Val51 were making multiple π–alkyl interactions with the rings as well as the long alkyl side chain of CP3. His12 was also making some nonpolar interactions with the alkyl side chain. Gln26, Gln29, Leu52, etc were also found to make a few VdW interactions with the long alkyl side chains.
Interaction analysis of CP4 or prim-O-β-d-glucosylcimifugi in MyD88 binding site
CP4 scored 12th rank among the top 25 compounds. Its score (−78.4) and rank were quite close to those of CP1 in the score list (Table 1); hence we expected to see a common scaffold between the two. However, they did not share any. The modified pyran benzofuran ring was making multiple π–alkyl interactions with Pro53 and His12 of MyD88 binding site (Figure 3D). However, the key H-bond interactions were between the Gln29, Asp50, and Arg16 residues and various polar hydroxyl and oxy groups of the molecule. Further, Pro22 and Val51 were making a few C–H bonds with the side alkyl chain of CP4. Gln26, Glu30, Leu52, Gly54, Pro14, and Ile25 were also making multiple VdW interactions with many side-chain groups of the molecule.
Interaction analysis of CP1 or β-methoxylferruginoside_B in Nur77 binding site
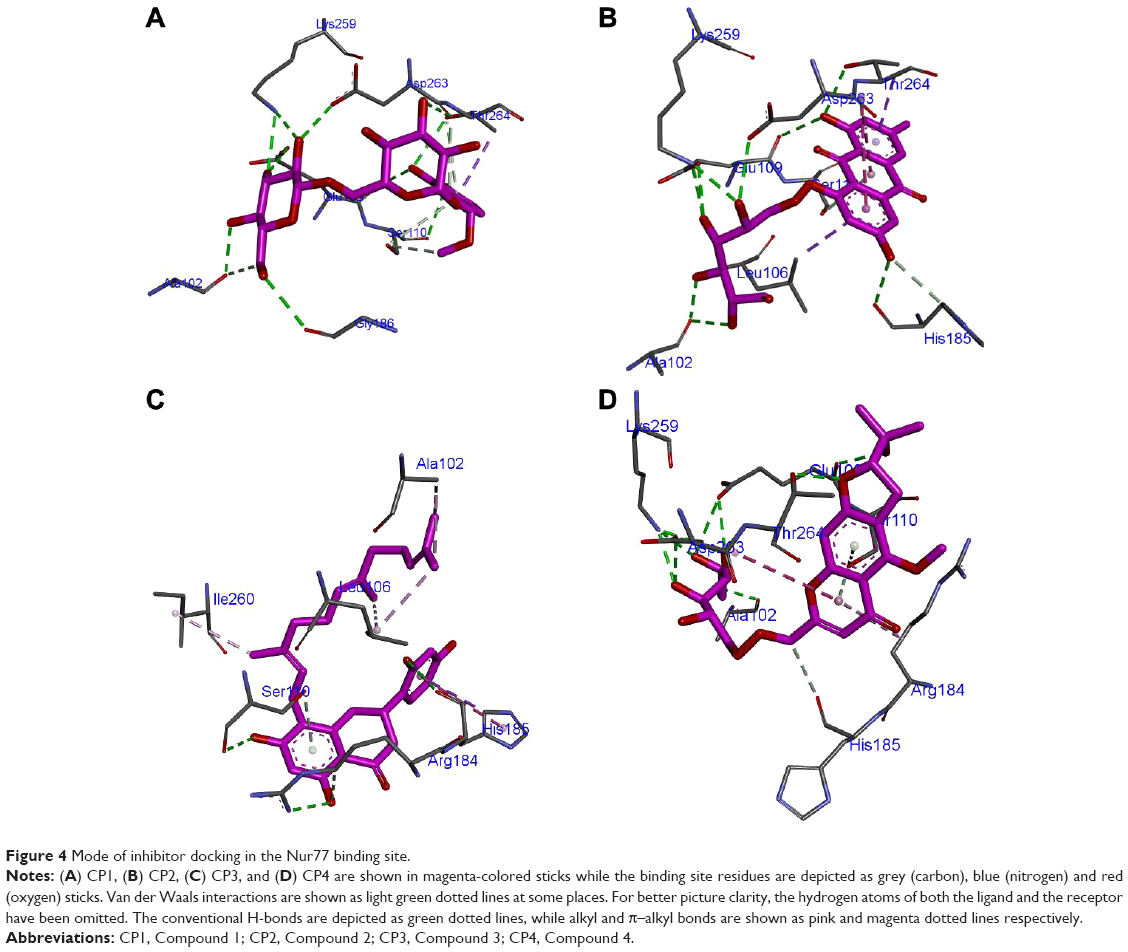
CP1 occupies 12th rank with a binding score of −95.2541 in the Nur77 binding site (Table 2). The residue numbers in the crystal structure of Nur77 (4RZG) have been altered from the original sequence in Uniprot. Hence, we will keep the residue numbers as in the crystal structure. For easy reference, we have tabulated the corresponding numbers in both the crystal structure (4RZG) and the Uniprot sequence (P22736) in Table 4. The LBD comprising the residues L437, Ser441, and Asp594 hosts the binding site for Nur77. Earlier, the compound PDNPA had been identified as a potent inhibitor for this target. As with PDNPA, CP1 interacts with the LBD residues in a similar fashion. The interactions involve an array of H-bonds between the polar groups at the two pyran rings as well as the phenyl ring of CP1 and Nur77 binding site residues Ala102, Lys259, Asp263, Thr264, Glu109, Ser110, etc (Figure 4A). There was an evident π–amide/π–alkyl bond between Asp263 and the phenyl ring of CP1. Besides, a few C–H bonds and multiple VdW interactions were present between Ser110, Thr264, Asp263, Asp105, Asp103, Leu187, Gly186, Leu106, Pro266, Met262, His185, Leu265, Arg184, Ala111 and Ile260 and the alkyl hub of CP1.
 | Figure 4 Mode of inhibitor docking in the Nur77 binding site. |
Interaction analysis of CP2 or emodin-8-β-d-glucoside in Nur77 binding site
CP2 occupies the sixth rank with a binding score of −97.7 in the Nur77 binding site (Table 2). Similar residues make H-bond interactions with the ring and side-chain substituents of the compounds as in CP1, namely Ala102, Lys259, Asp263, His185, Glu109, and Thr264 (Figure 4B). At the same time, Asp263 makes a novel amide–π-stacked bond with the rings 1 and 2 of the anthracene ring. Similarly, Leu106 makes π–alkyl bonds with ring 1 of anthracene. The VdW contacts remain similar to those of CP1.
Interaction analysis of CP3 or moralbanone in Nur77 binding site
CP3 ranks 16th in the top compound list with a score of −94.3 in the Nur77 binding site (Table 2). As described previously for MyD88, the lack of polar groups at the alkyl side chains of this compound prevents it from making many H-bond interactions with the polar Nur77 binding site. The only few ones are between the Ser110, Arg184, His185 residues of Nur77 binding site and the substituted napthyl and phenyl rings of CP3 (Figure 4C). However, the major nonpolar interactions include those between Ile260, Leu106, Ala102, and the side-chain alkyl substituents of CP3, which are quite prominent. His185 makes a similar π–σ bond with ring 3 as in CP2. Further, as expected, the VdW forces were prevalent in CP3 due to presence of the alkyl side chain and included those between Ala111, Glu109, Thr264, Asp263, Lys259, Leu187, Asp103, Gln188, and Gly186 with its long side alkyl chain.
Interaction analysis of CP4 or prim-O-β-d-glucosylcimifugi in Nur77 binding site
Although CP4 ranks 24th in the top scoring compound list in the Nur77 binding site, its score is not far from the rest of three compounds discussed earlier, which is −92.5 (Table 2). It makes several H-bonds with the polar residues of the Nur77 binding site through its highly polar hydroxyl and oxy substituents (Figure 4D). Among them, the major ones are Ala102, Lys259, Asp263, Thr264, Glu109, and Arg184. Here, again, Asp263 makes its signature π–amide/π–alkyl bond with the pyran ring of CP4. At the same time, Arg184 also makes a π–alkyl bond with the CP4 pyran ring. A similar set of VdW interactions is observed in CP4 as in CP1 and CP2 due to the position of its alkyl hub in the close vicinity of these residues including Asp105, Asp103, Leu187, Gly186, Leu106, Pro266, Met262, His185, Leu265, Arg184, Ala111, and Ile260.
Final selection of compounds based on “only common feature” criterion
The common features, when compiled, led us to interesting conclusions. When all four compounds were compared on the basis of their interactions in the MyD88 binding site (Figure 5A–D), it was observed that the H-bonds between Gln29 and the polar groups –OH and =O seemed to be common in CP1, CP2, and CP4, while the H-bond between the acceptor =O of Asp50 accepting H’s from the donor –OH groups of compounds was present only in CP1, CP3, and CP4. Further, Gln26 was making VdW/C–H bonds in all the four compounds. Similarly, Pro53 was also making a common π–alkyl bond in all the four compounds.
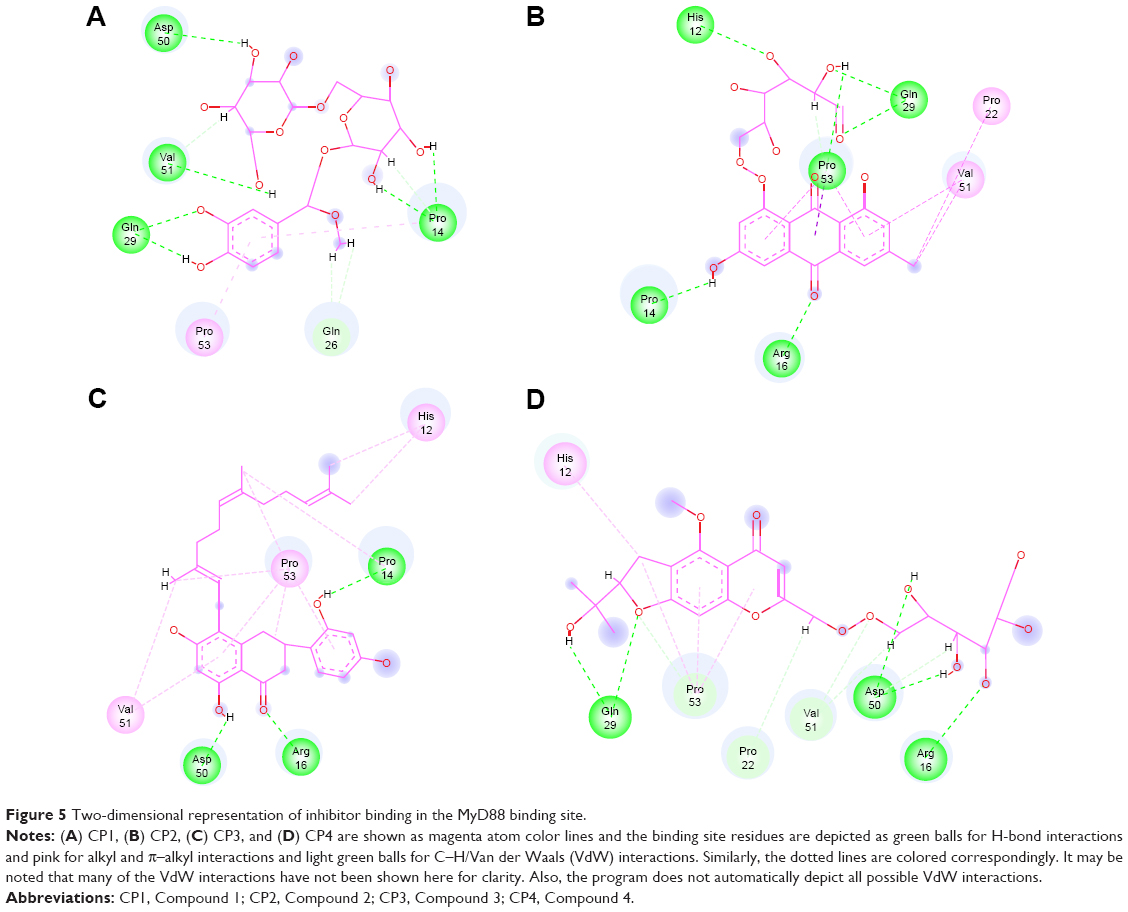 | Figure 5 Two-dimensional representation of inhibitor binding in the MyD88 binding site. |
While comparing the binding points of all four compounds in the Nur77 binding site (Figure 6A–D), the H-bonds with Ala102, Lys259, Asp263, Thr264, and Glu109 were found to be common in CP1, CP2, and CP4, while the critical Ser110 H-bond interaction was present only in CP1 and CP4. Further, the amide–π-stacked bond between Asp263 and the various rings of the compounds appeared to be common in compounds CP1, CP2, and CP4. Next, the VdW interactions involving the residues Asp103, Ile260, Asp105, Gly186, and Arg184 were found to be present in CP1, CP2, and CP4 but not in CP3 where there was a different set of residues making VdW interactions with the long alkyl side chain, including Ala111, Glu109, and Leu187, as discussed previously.
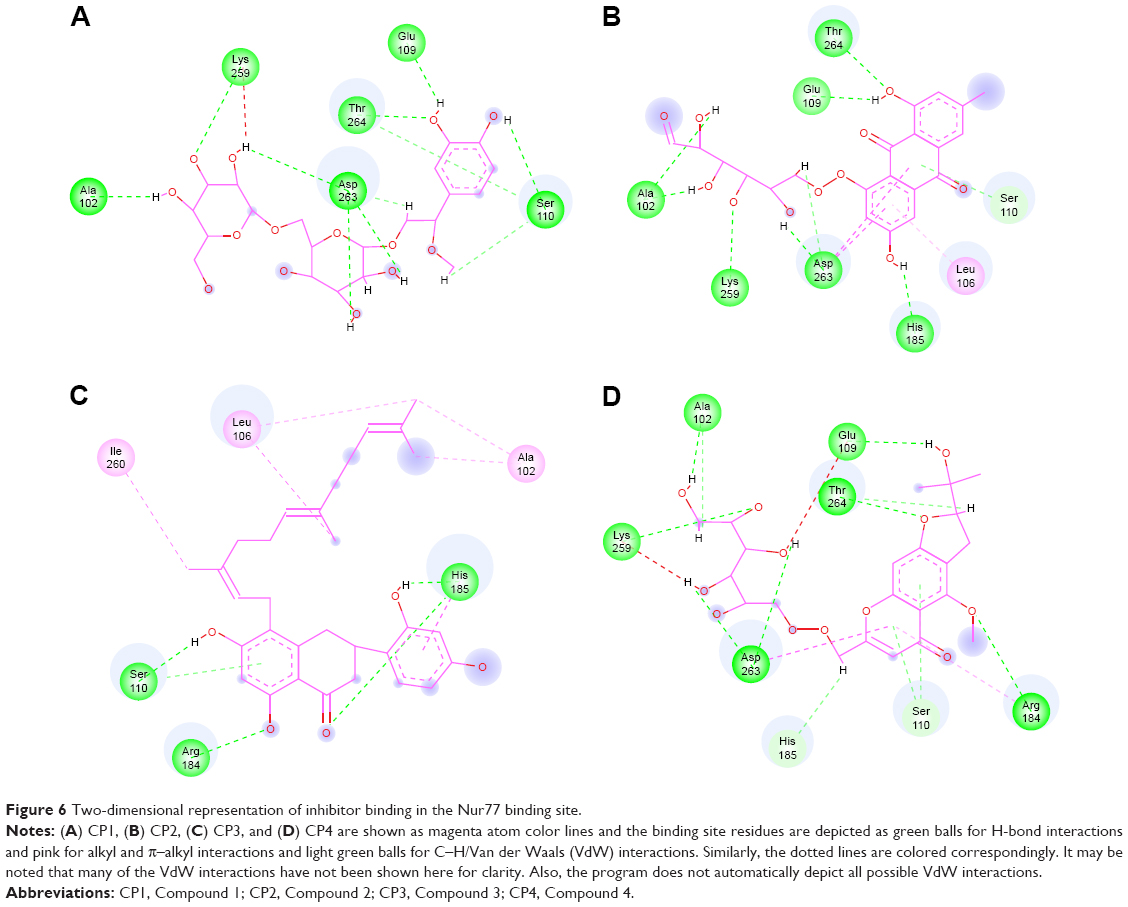 | Figure 6 Two-dimensional representation of inhibitor binding in the Nur77 binding site. |
The next important task was to select only those compounds from the four that share only the common features between them, although this step was not mandatory considering that all four compounds were binding to both the receptors with good affinity. However, for our next step, which was to develop a common pharmacophore model, we needed to weed out any compound that did not represent the architecture of both the binding sites, namely MyD88 and Nur77. Our interest was to select those compounds that showed pharmacophore points common in both MyD88 and Nur77 binding sites so that one model could represent the binding pattern of both the targets. This led us to focus on the feature derivation, where we could select the compounds common in both targets displaying similar interaction profiles. So, we started manually analyzing the common features of the compounds with respect to binding with both the targets. Among the four, the first to be expelled was CP3 due to its absence in few features like the absence of H-bonds with Gln29 in MyD88. In Nur77, it lacked the amide–π-stacked bond with Asp263 and various other important H-bonds with binding-site residues including that with Thr264. The next compound that was verified for expulsion was CP2. Though it could have been considered for retention, however, we realized that the interaction with Asp50, which is an important BB-loop residue in the MyD88 binding, was too important to be avoided, and since CP2 does not have any donor atom in the vicinity of Asp50, it could not make this important interaction at all. Similarly, the interaction with Ser110, a key LSD motif residue in the Nur77 binding site, was absent in CP2. This led us to suggest that CP2 might not be a strong candidate for inhibition for the dual targets and for further pharmacophore development. Hence, the final two compounds that were retained were CP1 and CP4.
Pharmacophore points of CP1 and CP4 in the MyD88 binding site
In order to elaborate the picture of pharmacophore mapping, we numbered all atoms of the compounds. So, we correlate the atom name and numbers in Figure 7A–D.
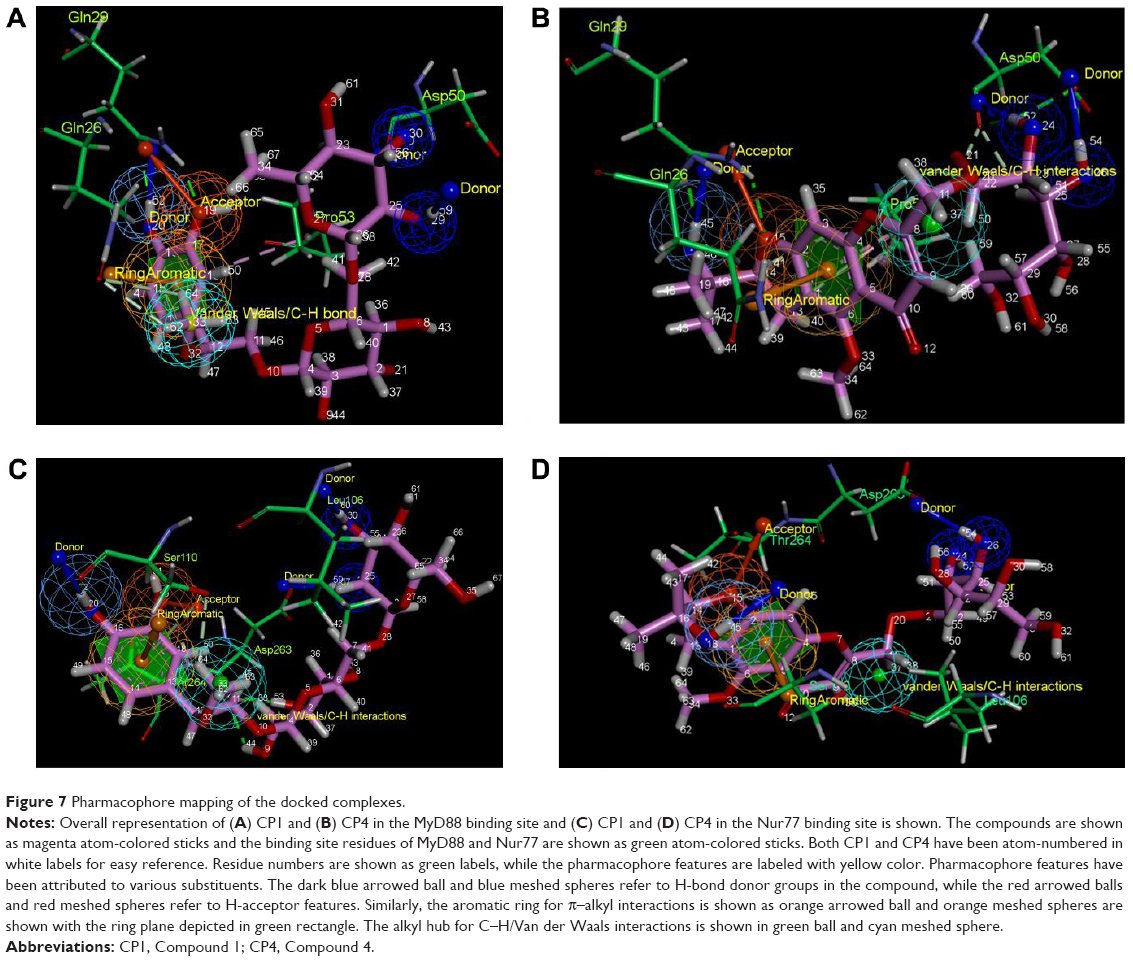 | Figure 7 Pharmacophore mapping of the docked complexes. |
In CP1, H-52 is making H-bond donor interactions with Gln29 (Figure 7A), while the O-19 in the vicinity is making H-bond acceptor interactions again with Gln29. Further, it has been observed that H-59 and H-60, further away from H-52, are making donor H-bonds with Asp50. Another feature is the presence of a phenyl ring that makes π–amide/π–alkyl interactions. In CP1, the ring atoms 13–18 are making π–alkyl interactions with Pro53 present at the binding site. CP1 makes multiple VdW interactions through its H62–H64 atom hub with many of the surrounding residues including Gln26 and Leu52.
Similar to CP1, CP4 also exhibits an exact replica of the ligand–receptor interactions with the binding site residues of MyD88 (Figure 7B). Among these, H-45 makes a donor H-bond with Gln29, along with an acceptor H-bond with Gln29 through H-15. As in CP1, H-54 and H-58 are acting as hydroxyl donors for Asp50. Again, the ring 1–6 is a site for π–alkyl interactions with the Pro53 residue. Next, the weak but numerous VdW interactions of H36–H38 specifically with residue Gln26 and Leu52 are consistent with those in CP1.
As a point of reference, the atom numbers of both compounds with their common binding partner amino acids are shown in Table 5, revealing how both compounds bind to the same residues of MyD88 binding site.
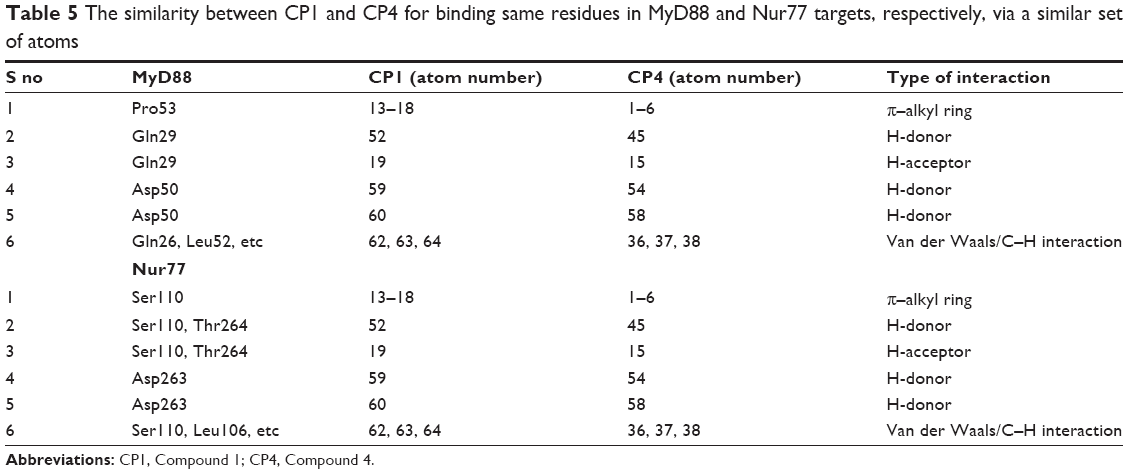 | Table 5 The similarity between CP1 and CP4 for binding same residues in MyD88 and Nur77 targets, respectively, via a similar set of atoms |
Pharmacophore points of CP1 and CP4 in the Nur77 binding site
Analysis of CP1 in the Nur77 binding site led us to highly interesting, though surprising, discoveries (Figure 7C). CP1 as in MyD88 is making H-bond donor interactions via its H-52 and acceptor interactions through its O-19 to both the Ser110 and Thr264 residues of the Nur77 binding site. Similarly, its H-60 and H-59 are making H-bond donor interactions with Asp263 of Nur77, which were H-bonded to Asp50 in the MyD88 binding site. The ring 13–18 was making similar π–alkyl interactions with Ser110 of Nur77 as with Pro53 in the MyD88 binding site. Again, the C–H/VdW interaction between H-62 and H-64 seems to be preserved with Ser110, His185, etc in Nur77 as with Gln26, Leu52, etc in the MyD88 binding site.
CP4, similar to CP1, has its pharmacophore points well conserved (Figure 7D). H-45 acts as a H-bond donor to both Ser110 and Thr264, while O-15 acts as H-bond donor also to Ser110 and Thr264 in Nur77, similar to donating and accepting Gln29 in the MyD88 binding site. Next, H-58 and H-54 act as H-bond donors to Asp263 just like they do to Asp50 in the MyD88 binding site. The ring 1–6 makes π–alkyl interactions with Ser110 similar to the one it makes with Pro53 in MyD88. Furthermore, H-36 to H-38 make consistent VdW interactions/C–H bonds with Ser110 and the surrounding residues as seen for Gln26 and others in the MyD88 binding site.
Common features among compounds as well as in both binding sites are clearly shown in Table 5.
Development of a common pharmacophore model
A common pharmacophore model has been constructed by combining the common features from both ligand–receptor complexes, as discussed earlier (Table 5 and Figure 8A and B). The pharmacophore model consists of a (1) ring system (orange color sphere) for making π–alkyl interactions with Pro53 of MyD88 and Ser110 of Nur77; (2) a H-bond donor group (blue centroid) substituted onto the ring (1) for donating H– to Gln29 in MyD88 and Ser110/Thr264 in Nur77; (3) an acceptor =O close to the ring (red sphere) for accepting H’s from Gln29 in MyD88 and Thr264/Ser110 in Nur77; (4) and (5) two donor H’s at some distance from (1)–(3) points (dark blue spheres), making strong H-bonds with Asp50 in MyD88 and Asp263 in Nur77; and (6) a centroid/spatial hub (cyan sphere) for VdW/C–H bonds with Gln26, Leu52, and the surrounding residues of MyD88 and Ser110, Leu106, and the neighboring residues of Nur77. Another interesting observation of these complexes led us to propose that the distances between all these pharmacophore points was quite similar for both compounds in both the targets. For example, the distance between the pharmacophore feature (1) and (3) is 2.7 Å, which is strictly consistent in all four complexes (Table 6). Hence we applied spatial constraints in the final model according to the tolerance level for each pharmacophore point. For example the spatial constraints for (1) ring feature is 2 Å, which shows that any virtual screening hit should have its ring feature within the 2 Å area of the ring plane of our pharmacophore query. Similarly, the spatial constraints for (2) donor point is 1.5 Å, (3) acceptor point is 1.5 Å, (4) and (5) donor points are 1 Å each, and (6) spatial hub is 1.5 Å. The spatial constraints would help the pharmacophore query to retrieve results not only with the suggested pharmacophore features but also with a specific conformation in the three-dimensional space.
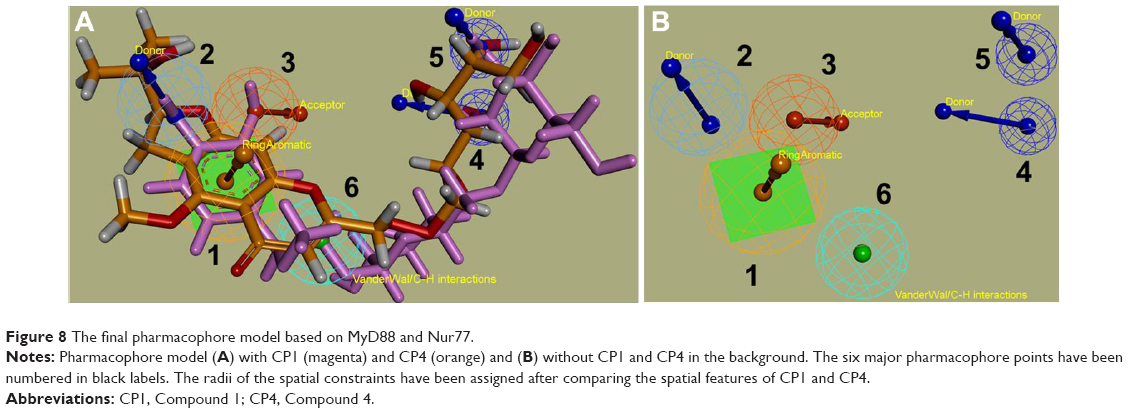 | Figure 8 The final pharmacophore model based on MyD88 and Nur77. |
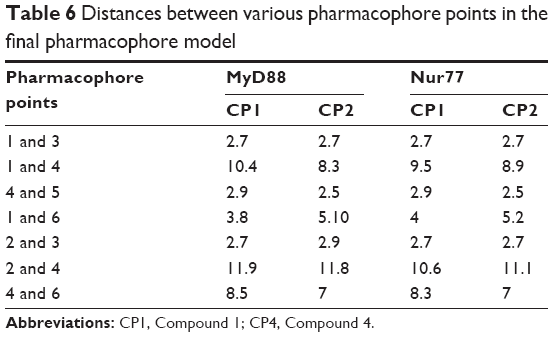 | Table 6 Distances between various pharmacophore points in the final pharmacophore model |
Materials and methods
Structural modeling via docking of the MyD88 homodimeric complex
To model the MyD88 homodimeric complex, we used the monomeric crystal structures of MyD88 (PDB: 4DOM and 4EO7). We chose these structures from other published structures because of their high resolution. Molecular docking was performed using the protein–protein docking software GRAMM-X,23 which is one of the most widely used rigid-body protein–protein docking programs for predicting and assessing the interactions between the protein complexes. GRAMM-X was employed for protein–protein docking where 4DOM was provided as the MyD88 receptor protein and 3EO7 was provided as the MyD88 ligand protein PDB file. The GRAMM-X program uses a smoothed Lennard–Jones potential on a fine grid during the global search fast Fourier transformation stage, followed by refinement optimization in continuous coordinates and rescoring with several knowledge-based potential terms. We examined the top 100 docked configurations to select most plausible configurations.
To further test the reliability of GRAMM-X prediction, a different and more advanced docking program, ClusPro 2.0, was used to predict the homodimerization interface of the MyD88 protein.24–27 ClusPro is a docking program that not only uses rigid geometry and energy-based solutions but also attempts to find the native site under the assumption that it will have a wide free-energy attractor with the largest number of results. It works in three main steps. First, it runs PIPER, a rigid-body docking program, based on a novel fast Fourier transform docking method with pairwise potentials. Second, by using a clustering technique for the detection of near-native conformations and by eliminating some of the non-native clusters, the 1,000 best energy conformations are clustered and the 30 largest clusters are retained for refinement. Third, by short Monte Carlo simulations, stability of these clusters is analyzed, and by the medium-range optimization method SDU (semi-definite programming-based underestimation), the structures are refined. Input of both proteins as receptor and ligand proteins in the ClusPro server resulted in a file containing four categories of predicted models: 1) balanced, 2) electrostatically favored, 3) hydrophobically favored, and 4) VdW + electrical. This approach provides another alternative for the clustering of possible docking solutions. Models in all the categories were ranked by the cluster size. The solutions from the ClusPro program were quite similar to the previous docking solution from the GRAMM-X programs.
Virtual screening
The total number of compounds in the TCM database, Taiwan, was 61,000. This is the largest TCM database28 in the world, and has 2D and 3D chemical structures. The bioactivity of 61,000 compounds extracted from TCM herbs can be searched through the database portal. The TCM Database@Taiwan application has been investigated for treatments of insomnia,29 pigmentary disorders,30 and Parkinson’s disease,31 as well as epidermal growth factor receptor (EGFR) inhibition,32 pain relief,33 and viral infections.34–38 It is helpful to screen TCM compounds via a cloud computing platform.39,40 Individual screens were carried out using the modeled MyD88 structure and the 4RZG crystal structure of Nur77.20 Next, the docking study for each receptor was carried out by specifying the probable binding sites for each. In case of Nur77, we provided the coordinates of the bound PDNPA inhibitor binding to the LSD motif as the probable binding site. We specified the conserved LPG motif residue coordinates from the BB loop as the binding site in MyD88 dimer. Both screens returned many potential hits, among which we analyzed the top 25.
Interaction profiling and pharmacophore development
The UCSF Chimera software41 was used to visualize the basic information of the macromolecules, whereas most of the interaction profiling and pharmacophore development were done manually using the Discovery Studio (DS) Visualizer version 4.5.42 DS 4.5 was used to track atom–atom bonds and distances between ligands and receptors, as well as to build the major pharmacophore points in the binary complexes.
Discussion
In the new phase of re-evaluating the targets for the available drugs, discovery of multi- or dual-target inhibitors bring both to the same page. Although the dual-target inhibitors provide a new opportunity for treating complex diseases, reassessment of their key binding features with respect to both the targets is both complicated and critical.
The idea of dual targeting came to our mind after critically analyzing the roles of both MyD88 and Nur77 in the NF-kB activation leading to the triggering of inflammatory response. NF-κB signaling is complex due to its involvement in the modification of various signaling proteins leading to many regulatory events in inflammation.43 MyD88 is an innate adaptor protein that binds to the TIR domain to coordinate the cellular response to different agonists. Upon binding with one of the TIR receptor complexes, MyD88 homodimers activate the IRAK complex, which phosphorylates downstream kinases and transcription factors including NF-kB, further triggering the inflammatory response. On the other hand, Nur77 had long been perceived to have anti-inflammatory property in various cell models in which elevation of its expression reduced the expression of several cytokines and chemokines in macrophages in response to LPS or tumor necrosis factor-α stimulation. However, the latest and a more specific role of Nur77 in inflammation has been identified very recently by Li et al,20 where they strongly suggest that blocking the Nur77–p38α interaction by targeting the LBD of Nur77 could restore the suppression of the hyperinflammatory response through Nur77 inhibition of NF-kB. Since both MyD88 and Nur77 are an integral part of LPS-induced, NF-kB-mediated inflammatory response, we decided to pull off these targets for dual-target drug discovery. In order to do so, we first checked out the structures of both the targets. While Nur77 had a high-resolution crystal structure available in PDB (4RZG), MyD88 structure was available only for a monomeric unit. In a recent study published by Olson et al,18 they reported the binding site of the MyD88 inhibitors and suggested that they bind at the interface of the homodimer hosting the BB-loop region. Any inhibitor targeting this interface would be able to disrupt the dimerization of MyD88 and further inflammatory signaling. Following their modeling protocol, we also developed our homodimer MyD88 model, which was close to that developed by them in respect of the positioning of the monomer units in the three-dimensional space as well as in the conformational space of the conserved BB loop that was docking perfectly at the dimer interface. Next, we docked the Chinese medicinal compounds library known for a myriad range of inhibitors including but not limited to insomnia, pigmentary disorders, Parkinson’s disease prevention, EGFR inhibition, pain relief, antivirals, etc. The TCM database of 61,000 compounds was used to screen for compounds binding both MyD88 and Nur77. This resulted in retrieval of diverse compounds with various scaffolds. However, since we were interested in only those high-scoring compounds that were common to both the targets, we only selected four from the top 25 compounds from each hit list of MyD88 and Nur77 that were binding to both the targets. Further, all these four compounds were individually analyzed for ligand–receptor interactions with each of the target. Although there had been many similarities between the binding modes of these inhibitors among themselves and in both the targets, we strictly looked for the common features among them; these specifically included all H-bond donors, H-bond acceptors, π–alkyl bonds, C–H bonds as well as VdW forces. Also, we were well aware of the critical binding residues in both the targets, eg, BB loop residues in MyD88 and the LBD residues in Nur77, both of which had been experimentally proved to be indispensable for the ligand binding. Hence we moved ahead keeping these features in mind, which were the commonness among compounds and their binding with key binding site residues in both the targets. We observed that, although the binding score of CP2 and CP3 was quite good in both the targets, they lacked some essential pharmacophore points in terms of binding with critical residues in the MyD88 and Nur77 binding sites. On the other hand, CP1 and CP4 were consistent in all major critical interactions in the binding sites of both targets. This led us to discard CP2 and CP3 and move further with CP1 and CP4. After closely examining CP1 and CP4, we derived a common pharmacophore model that was good enough for both the targets. This model, which includes six pharmacophore points, is a robust prototype for screening anti-inflammatory inhibitors against MyD88 and Nur77. It has (1) an aromatic ring for Pro53 (MyD88) and Ser110 (Nur77) interaction, (2) a donor point for acceptor residues Gln29 (MyD88) and Ser110/Thr264 (Nur77), (3) an acceptor point for Gln29 (MyD88) and Ser110/Thr264 (Nur77), (4) and (5) H-bond donor points for Asp50 (MyD88) and Asp263 (Nur77), and (6) VdW/C–H interaction hub points for Gln26, Glu30, Leu52, Val51, etc (MyD88) and Ser110, His185, Gly186, and Ile260 (Nur77). Further, on calculating the distances between all these features for both compounds in both the binding sites, we came up with this critical and at the same time highly interesting observation that all these follow similar distance patterns among the various pharmacophore points. Since the two compounds do not share any common scaffold between them, this is a novel finding that also suggests that these compounds attain a common three-dimensional conformation in both MyD88 and Nur77 binding sites in order to bind the respective key residues. Hence, we introduced spatial constraints in the final pharmacophore model so that hits with not only the common features but also with specific spatial arrangement could be retrieved once this model is queried for a virtual screen. The developed model is complete in itself for representing potent dual-target inhibitors for both MyD88 and Nur77. Our strategy, though simple and straightforward, is complete in itself to reveal the intricacy of the dual-binding ligand–receptor interactions in MyD88 and Nur77. Our next step is to synthesize these novel compounds to further test them in vitro and cell-based assay systems. To our knowledge, this is the first ever study on dual-target inhibitors against inflammation for MyD88 and Nur77, considering that the structural aspects of inhibition of the latter has only recently been discovered. We are confident that our study and the developed pharmacophore would be a good starting point for drug discovery against inflammation and related diseases.
Acknowledgments
This work was supported by the Department of Biotechnology (DBT), Government of India sponsored Ramalingaswami Fellowship (BT/HRD/35/02/2006) to MSB and the Department of Science and Technology (DST), Government of India sponsored Science and Engineering Research Board (SERB) Young Scientists Start-Up Research Grant (YSS/2015/001279) to US. The authors also gratefully acknowledge the Indian Institute of Technology Indore for providing facilities and other support.
Disclosure
The authors report no conflicts of interest in this work.
References
Hopkins AL, Mason JS, Overington JP. Can we rationally design promiscuous drugs? Curr Opin Struct Biol. 2006;16:127–136. | ||
Yang K, Ma WZ, Liang HH, Qi OY, Tang C, Lai LH. Dynamic simulations on the arachidonic acid metabolic network. PLoS Comput Biol. 2007;3:523–530. | ||
Chen Z, Wu Y, Liu Y, Yang S, Chen Y, Lai L. Discovery of dual target inhibitors against cyclooxygenases and leukotriene A4 hydrolyase. J Med Chem. 2011;54:3650–3660. | ||
Meng H, Liu Y, Zhai Y, Lai L. Optimization of 5-hydroxytryptamines as dual function inhibitors targeting phospholipase A2 and leukotriene A4 hydrolase. Eur J Med Chem. 2013;59:160–167. | ||
Li D, Han X, Tu Q, et al. Structure-based design and synthesis of novel dual-target inhibitors against cyanobacterial fructose-1,6-bisphosphate aldolase and fructose-1,6-bisphosphatase. J Agric Food Chem. 2013;61:7453–7461. | ||
Csermely P, Agoston V, Pongor S. The efficiency of multi-target drugs: the network approach might help drug design. Trends Pharmacol Sci. 2005;26:178–182. | ||
Huang S. Rational drug discovery: what can we learn from regulatory networks. Drug Discov Today. 2002;7:S163–S169. | ||
Borisy AA, Elliott PJ, Hurst NW, et al. Systematic discovery of multicomponent therapeutics. Proc Nat Acad Sci U S A. 2003;100: 7977–7982. | ||
Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. | ||
O’Neill LA. The role of MyD88-like adapters in toll-like receptor signal transduction. Biochem Soc Trans. 2003;31:643–647. | ||
Sun D, Ding A. MyD88-mediated stabilization of interferon-gamma-induced cytokine and chemokine mRNA. Nat Immunol. 2006;7:375–381. | ||
Kissner TL, Moisan L, Mann E, et al. A small molecule that mimics the BB-loop in the toll/IL-1 receptor domain of MyD88 attenuates staphylococcal enterotoxin B induced pro-inflammatory cytokine production and toxicity in mice. J Biol Chem. 2011;286:31385–31396. | ||
Hardiman G, Rock FL, Balasubramanian S, Kastelein RA, Bazan JF. Molecular characterization and modular analysis of human MyD88. Oncogene. 1996;13:2467–2475. | ||
Li C, Zienkiewicz J, Hawiger J. Interactive sites in the MyD88 Toll/interleukin (IL) 1 receptor domain responsible for coupling to the IL1beta signaling pathway. J Biol Chem. 2005;280:26152–26159. | ||
Jiang Z, Georgel P, Li C, et al. Details of toll-like receptor:adapter interaction revealed by germ-line mutagenesis. Proc Natl Acad Sci U S A. 2006;103:10961–10966. | ||
Kissner TL, Ruthel G, Alam S, et al. Therapeutic inhibition of pro-inflammatory signaling and toxicity to staphylococcal enterotoxin B by a synthetic dimeric BB-loop mimetic of MyD88. PLoS One. 2012;7:e40773. | ||
Dunne A, Ejdeback M, Ludidi PL, et al. Structural complementarity of toll/interleukin-1 receptor domains in toll-like receptors and the adaptors Mal and MyD88. J Biol Chem. 2003;278:41443–41451. | ||
Olson MA, Lee MS, Kissner TL, Alam S, Waugh DS, Saikh KU. Discovery of small molecule inhibitors of MyD88-dependent signaling pathways using a computational screen. Sci Rep. 2015;5:14246. | ||
Pei L, Castrillo A, Chen M, Hoffmann A, Tontonoz P. Induction of NR4A orphan nuclear receptor expression in macrophages in response to inflammatory stimuli. J Biol Chem. 2005;280:29256–29262. | ||
Li L, Liu Y, Chen HZ, et al. Impeding the interaction between Nur77 and p38 reduces LPS-induced inflammation. Nat Chem Biol. 2015;11:339–346. | ||
Snyder GA, Cirl C, Jiang J, et al. Molecular mechanisms for the subversion of MyD88 signaling by TcpC from virulent uropathogenic Escherichia coli. Proc Natl Acad Sci U S A. 2013;110:6985–6990. | ||
Verstak B, Arnot CJ, Gay NJ. An alanine to proline mutation in the BB-loop of TLR3 toll/IL-1R domain switches signaling adaptor specificity from TRIF to MyD88. J Immunol. 2013;91:6101–6109. | ||
Tovchigrechko A, Vakser IA. GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 2006;34:W310–W314. | ||
Kozakov D, Beglov D, Bohnuud T, et al. How good is automated protein docking? Proteins. 2013;81:2159–2166. | ||
Kozakov D, Brenke R, Comeau SR, Vajda S. PIPER: an FFT-based protein docking program with pairwise potentials. Proteins. 2006;65:392–406. | ||
Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20:45–50. | ||
Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004;32:W96–W99. | ||
Chen CY. TCM Database@Taiwan: the world’s largest traditional Chinese medicine database for drug screening in silico. PLoS One. 2011;6:e15939. | ||
Chen HY, Chang SS, Chan YC, Chen CY. Discovery of novel insomnia leads from screening traditional Chinese medicine database. J Biomol Struct Dyn. 2014;32:776–791. | ||
Tang HC, Chen CYC. Investigation of the novel lead of melanocortin 1 receptor for pigmentary disorders. Evid Based Complement Alternat Med. 2014;2014:254678. | ||
Huang HJ, Lee CC, Chen CYC. Pharmacological chaperone design for reducing risk factor of Parkinson’s disease from traditional Chinese medicine. Evid Based Complement Alternat Med. 2014;2014:830490. | ||
Yang SC, Chang SS, Chen HY, Chen CYC. Identification of potent EGFR inhibitors from TCM database@Taiwan. PLoS Comput Biol. 2011;7:e1002189. | ||
Tou WI, Chang SS, Lee CC, Chen CYC. Drug design for neuropathic pain regulation from traditional Chinese medicine. Sci Rep. 2013; 3:844. | ||
Chang SS, Huang HJ, Chen CYC. High performance screening, structural and molecular dynamics analysis to identify H1 inhibitors from TCM database@Taiwan. Mol Biosyst. 2011;7:3366–3374. | ||
Lin CH, Chang TT, Sun MF, et al. Potent inhibitor design against H1N1 swine influenza: structure-based and molecular dynamics analysis for M2 inhibitors from traditional Chinese medicine database. J Biomol Struct Dyn. 2011;28:471–482. | ||
Chang SS, Huang HJ, Chen CYC. Two birds with one stone? Possible dual-targeting H1N1 inhibitors from traditional Chinese medicine. PLoS Comput Biol. 2011;7:e1002315. | ||
Chen CY, Chang YH, Bau DT, et al. Ligand-based dual target drug design for H1N1: swine flu – a preliminary first study. J Biomol Struct Dyn. 2009;27:171–178. | ||
Huang HJ, Jian YR, Chen CY. Traditional Chinese medicine application in HIV: an in silico study. J Biomol Struct Dyn. 2014;32:1–12. | ||
Tsai TY, Chang KW, Chen CYC. IScreen: world’s first cloud-computing web server for virtual screening and de novo drug design based on TCM database@Taiwan. J Comput Aided Mol Des. 2011;25:525–531. | ||
Chang KW, Tsai TY, Chen KC, et al. iSMART: an integrated cloud computing web server for traditional Chinese medicine for online virtual screening, de novo evolution and drug design. J Biomol Struct Dyn. 2011;29:243–250. | ||
Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. | ||
Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment, Release 4.5. San Diego: Dassault Systèmes; 2015. | ||
Baig MS, Zaichick SV, Mao M, et al. NOS1-derived nitric oxide promotes NF-κB transcriptional activity through inhibition of suppressor of cytokine signaling-1. J Exp Med. 2015;212:1725–1738. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.