Back to Journals » International Journal of Nanomedicine » Volume 14
Simultaneous Intramuscular And Intranasal Administration Of Chitosan Nanoparticles–Adjuvanted Chlamydia Vaccine Elicits Elevated Protective Responses In The Lung
Authors Li Y, Wang C, Sun Z, Xiao J, Yan X, Chen Y, Yu J, Wu Y
Received 5 June 2019
Accepted for publication 11 September 2019
Published 8 October 2019 Volume 2019:14 Pages 8179—8193
DOI https://doi.org/10.2147/IJN.S218456
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Yumeng Li,1 Chuan Wang,1 Zhenjie Sun,1 Jian Xiao,1 Xiaoliang Yan,1 Yuqing Chen,1 Jian Yu,2 Yimou Wu1
1Hunan Provincial Key Laboratory for Special Pathogens Prevention and Control, Institution of Pathogenic Biology, Hengyang Medical College, University of South China, Hengyang 421001, People’s Republic of China; 2Department of Experimental Zoology, Hengyang Medical College, University of South China, Hengyang 421001, People’s Republic of China
Correspondence: Yimou Wu
Hunan Provincial Key Laboratory for Special Pathogens Prevention and Control, Institution of Pathogenic Biology, Hengyang Medical College, University of South China, 28 West Changsheng Road, Hengyang, Hunan 421001, People’s Republic of China
Tel +86 734-13707340050
Fax +86-734-8282907
Email [email protected]
Background: Chlamydia psittaci is a zoonotic bacteria closely associated with psittacosis/ornithosis. Vaccination has been recognized as the best way to inhibit the spread of C. psittaci due to the majority ignored of infections. The optimal Chlamydia vaccine was obstructed by the defect of single immunization route and the lack of availability of nontoxic and valid adjuvants.
Methods: In this study, we developed a novel immunization strategy, simultaneous (SIM) intramuscular (IM) and intranasal (IN) administration of a C. psittaci antigens (Ags) adjuvanted with chitosan nanoparticles (CNPs). And SIM-CNPs-Ags were used to determine the different types of immune response and the protective role in vivo.
Results: CNPs-Ags with zeta-potential values of 13.12 mV and of 276.1 nm showed excellent stability and optimal size for crossing the mucosal barrier with high 71.7% encapsulation efficiency. SIM-CPN-Ags mediated stronger humoral and mucosal responses by producing meaningfully high levels of IgG and secretory IgA (sIgA) antibodies. The SIM route also led to Ags-specific T-cell responses and increased IFN-γ, IL-2, TNF-α and IL-17A in the splenocyte supernatants. Following respiratory infection with C. psittaci, we found that SIM immunization remarkably reduced bacterial load and the degree of inflammation in the infected lungs and made for a lower level of IFN-γ, TNF-α and IL-6. Furthermore, SIM vaccination with CNPs-Ags had obviously inhibited C. psittaci disseminating to various organs in vivo.
Conclusion: SIM immunization with CNPs-adjuvanted C. psittaci Ags may present a novel strategy for the development of a vaccine against the C. psittaci infection.
Keywords: Chlamydia psittaci, vaccine, chitosan nanoparticles, immunization route, respiratory infection
Introduction
Chlamydia psittaci is the cause of an infectious disease psittacosis/ornithosis in poultry and birds. It can also lead to severe infection by transferring to humans, which occur mainly via the inhalation of contaminated aerosols originating from faeces, urine, or other excretions from infected birds.1,2 Thus, the largest burden of disease from C. psittaci is in pet breeder, veterinarian, and poultry keeper, where untreated respiratory infections may result in serious complications such as bacteremia, encephalitis and myocarditis.3 Moreover, a recent study reported that chlamydial lung infection may contribute to increase the risk of co-infection with other pathogens including H9N2.4 Obviously, vaccination is the most effective measure to prevent C. psittaci infection and control chlamydial diseases.5 Up to now, however, no effective vaccine has been developed.
Appropriate candidate antigens (Ags) are one of the crucial factors for the development of Chlamydia vaccine.6 There are many studies that have been done to search protective antigens in animal models, such as C. psittaci major outer membrane protein (MOMP) and plasmid-encoded protein.7–9 Although the protective effects of subunit protein Ags have already been confirmed, the complexity of protection and stability made them far from ideal candidate vaccines. Therefore, we designed a new multi-epitope peptide Ags based on CPSIT_p6 and MOMP to against C. psittaci infection in our previous study.10 Due to the advantages of peptide-based vaccines, such as well-targeted immunity and few side effects, the multi-epitope peptide Ags is more suitable as a candidate than the protein immunogens.
An effective immunization strategy can combine several delivery routes to influence both the immune profile and the persistence of vaccine Ags.11 Regarding the immune response, it is universally accepted that an optimal chlamydial vaccine will need to elicit both cell-mediated immunity and mucosal immunity.12,13 Several chlamydial studies showed that CD4 T-cells can play a significant role by decreasing the initial chlamydial load through neutralization and possible complement activation.14 And it also has been determined that the presence of secretory IgA (sIgA) correlated with accelerated clearance of chlamydia in pulmonary- and genital-infected animals.15 Hence, the choice of immunization routes is highly relevant when determining the effect of the immune response against chlamydial infection. Previous studies demonstrated that intramuscular (IM) vaccination can induce the production of a stronger, local antigen-specific immunity and cell-mediated immune response against chlamydial challenge.16 However, it failed to induce an effective mucosal immunity. Nasal mucosal immunization not only induces strong mucosal immunity in the respiratory tract but also enhances immune response at other mucosal systems.17 Thus, intranasal (IN) vaccination that targets the mucosal immune system can provide an effective protection in respiratory infections. According to the facts above, the combination of IN and IM immunization routes may be a specific strategy to elicit both mucosal and cell-mediated immunity to prevent pulmonary chlamydial infection.
Nanoparticle (NP) delivery systems provide an innovative strategy of mucosal vaccines due to their advantages, such as maintaining antigen release in the mucosal sites, inhibiting the antigen from degradation, and potentiating the co-deliver of antigen and adjuvant.18,19 As a promising antigen delivery system, chitosan possesses well-defined properties, including cationic nature and mucosal adhesion,20 which prolonged and sustained the antigen retention time in different mucosal systems.21 In addition, chitosan nanoparticle (CNP) has the properties of biodegradable, high aqueous solubility, high surface to volume ratio and stability over a range of ionic conditions, which makes the spectrum of its applicability much broader.22 Chitosan nanoparticle-entrapped antigen is shown to enhance mucosal IgA response in the respiratory tract and confers valid protection in an infected animal.23 In another study, the encapsulation of antigen in chitosan nanoparticle also elicited the strong IgG and secretory IgA response in mice.24 Therefore, chitosan nanoparticles have drawn most attention for mucosal immunization through IN route.
In the present study, we used CNPs for the adsorption of C. psittaci Ags (CNPs-Ags) and performed a specific strategy combination of IM and IN administration in BALB/c mice. Our results demonstrated that the simultaneous (SIM) IM and IN administration of CNPs-Ags provided an improved mucosal IgA response in the respiratory tract and genital tract. It also elicited stronger humoral and cell-mediated immune responses against C. psittaci infection, resulting in a reduced lung bacterial load, inflammation in the pulmonary parenchyma and a less dissemination of Chlamydia in other tissues.
Materials And Methods
Preparation Of The Recombinant Ags-Absorbed Nanoparticles
Ultrapure chitosan (85% deacetylated, mol wt 60–170 kDa) was purchased from Golden-Shell Biochemical Inc (Zhejiang, China). The recombinant Ags of C. psittaci were synthesized by Life Tein Biotechnology Company (Beijing, China) as described previously.10 The production of chitosan nanoparticles and the recombinant Ags (1 mg/mL) was done by using a physical adsorption technique.21,25 Briefly, nanoparticles were resuspended in 25 mM acetate buffer (pH 5.7) and incubated with the recombinant Ags (at a concentration of 1000 µg/mL) for 15, 30, 45, 60 mins at room temperature. Unabsorbed antigen was collected in the supernatant by centrifugation at 10,000 rpm for 20 mins and quantified by bicinchoninic acid (BCA) protein assay, and absorbance was read at 570 nm using a microplate reader (Multiskan MK-3, Finnpipette, FI). The recombinant Ags adsorption efficiency (AE) was calculated using the following formula:
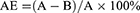
Here, A is the total amount of the recombinant Ags (µg/mL) and B is the unbound amount of the antigens (µg/mL). These results were performed three times.
The morphological analysis of the chitosan nanoparticles incubated antigen was observed by transmission electron microscopy (TEM) (Tecnai G2 Spirit TWIN). The particle size, distribution and zeta potential were measured by photon correlation spectroscopy at 26°C using Zetasizer Nano-ZS instrument.
Ethics Statement
All animal experiments were approved by the Animal Welfare Committee of the University of South China and conducted in accordance with the regulations of the institution and all efforts were made to minimize the animals' suffering.
Mice And Immunization
Eight- to six-week-old female BALB/c mice were purchased from Hunan SJA Laboratory Animal Co. Ltd (Animal Production License No. SCXK 2016-0002) and allowed to acclimatize for at least 4 days before entering experiments. Mouse care and monitoring followed a protocol approved by the University of South China Institutional Animal Use and Ethics Committee (Hengyang, China). A total of six groups of mice with 6 animals in each group received three immunizations at 2-week intervals (Scheme 1). Experimental group mice were immunized with 40 μL of recombinant Ags (1 mg/mL) adsorbed on 40 μL chitosan nanoparticles (CNPs-Ags). Vaccines were given either IM or IN routes in a total volume of 80 μL CNPs-Ags or both routes simultaneously as SIM-CNPs-Ags group. IM, IN with CNPs-PBS and SIM with CNPs-unabsorbed Ags (SIM-Ags) were used as control groups. Two weeks after the final immunization (day 42), serum samples were harvested by centrifugation (3000 rpm, 10 mins) and stored at −80°C for the detection of specific antibody production. Nasal washes and vaginal washes were collected from the immunized mice to determine the mucosal responses. Spleens from each group were homogenized in RPMI 1640 (Hyclone, Logan, Utah, USA) containing 10% fetal calf serum (FBS; Invitrogen, USA) for the detection of cytokine level and lymphocyte proliferation assay.
Bacteria And Infection
The C. psittaci 6BC (VR-125) was purchased from the ATCC and propagated in HeLa-229 cells (ATCC CCL-2.1). C. psittaci 6BC EBs were harvested, purified, and quantified as described previously.12,26 The C. psittaci 6BC EB stocks in sucrose-phosphate-glutamic acid (SPG) buffer (0.25 M sucrose, 10 mM sodium phosphate, 5 mM glutamic acid, pH 7.5) were stored at −80°C. To evaluate the effect of protective immunity, another 36 mice were also separated into 6 groups and received the same immunization as mentioned above. At the last day of week 6 (day 42), the mice were intranasally challenged with 5 × 105 IFU of C. psittaci in 30 μL of SPG buffer for protection assay (Scheme 1).
Determination Of Ag-Specific Antibodies And Secretory IgA Level
ELISA was performed to determine the levels of Ag-specific serum IgG IgG1, IgG2a, IgA and secretory IgA. In brief, 96-well plates coated with 10 μg/mL of the recombinant Ags (100 μL/well at 4°C overnight) were rinsed with PBS solution containing 0.05% Tween 20 (PBST) and then blocked with 250 μL blocking buffer (3% bovine serum albumin in PBST) at 37°C for 2 hrs. After being rinsed for 5 times with PBST, the plates were incubated with immune sera (diluted at 1:200 in blocking buffer, 100 μL/well), nasal washes or vaginal washes (diluted at 1:10 in blocking buffer, 100 μL/well) at 37°C for 1 hr. The plates were washed thoroughly with PBST and then incubated with 100 μL of horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG, IgG1, IgG2a, IgA (diluted at 1:3000 in blocking buffer, 100 μL/well) at 37°C for 1 hr. After being washed again, the plates were incubated with the substrate 3,3,5,5-tetramethylbenzidine (100 μL/well) at 37°C for 15 mins, and then 50 μL of 1 M H2SO4 was added to stop the reaction. The absorbance at 450 nm was measured by a microplate reader (Thermo Labsystems, FI).
Lymphocyte Proliferation Assay And Antigen-Specific Cytokine Analysis
The splenocytes harvested from each mouse were re-suspended in RPMI-1640 (1% antibiotics). The red blood cells were lysed using red blood lysis buffer (Biosharp) for 5 mins on ice. Then, the splenocytes were cultured in 96-well plates (1 × 105 cells/well) and stimulated with 1 mg/mL of the recombinant Ags (10 μL/well) or 1 μg/mL ConA (Sigma, USA) as a positive control. After the incubation for 48 hrs, spleen lymphocyte proliferative capacity was evaluated using a Cell Counting Kit-8 (Dojindo, Japan). Antigen-specific cytokine levels were detected by ELISA using methods described previously. Briefly, splenocytes were seeded into 24-well plates (1 × 106 cells/well) and stimulated with 1 mg/mL of the recombinant Ags (20 μL/well) or medium. Supernatants were harvested 48hrs after incubation, and secreted IFN-γ, IL-2, TNF-α, IL-4, IL-10 and IL-17A were assayed by Ready-SET-Go! kits (eBioscience Inc., CA, USA) as per the manufacturer's instruction. The levels of IFN-γ, IL-2, TNF-α, IL-4, IL-10 and IL-17A were quantified based on absorbance (OD) at 450 nm, and the sensitivities of the kits were in the ranges of 15–2000, 2–200, 8–1000, 4–500, 32–4000 and 4–500 pg/mL, respectively.
Evaluation Of Specific Cellular Responses By Flow Cytometry
Splenocytes were collected from vaccinated mice at day 42 (Scheme 1), and 1 × 106 splenocytes were stimulated for 6 hrs at 37°C with 15 μL of the recombinant Ags (1 mg/mL). Following stimulation, the cells were intracellular stained (BD Pharmingen, USA) in accordance with the BD manual. Briefly, after being washed with FACS buffer (PBS + 1% bovine serum albumin) and incubated with Fc receptor blocking antibody (CD16/CD32 mAb), the cells were stained with surface markers-CD3a-FITC and -CD4-APC for 30 mins at 4°C. Then, the cells were permeabilized with Cytofix/Cytoperm™ kit (BD Pharmingen) and stained intracellularly for 30 mins at 4°C for cytokines IL-4-PE and IFN-γ-PE (BD Pharmingen, USA). After being washed, sample acquisition (10,000 events were harvested) was performed with a BD FACS Canto II flow cytometer (Becton Dickinson, USA) and analyzed using a FACS Aria flow cytometer (BD Biosciences, USA) with FACS Diva software (BD Biosciences, USA).
Quantification Of C. Psittaci Burden And Cytokine In The Lungs
The mice were sacrificed 10 days after the challenge, and the lung tissues were homogenized with SPG buffer (1.2 mL/100 mg) as described.26 The lung homogenates were centrifuged (4°C, 15 mins, 6000 rpm) and inoculated on HeLa 229 cell monolayers after being serially diluted of the supernatants determining the C. psittaci titers. Intracellular inclusions of C. psittaci in the lungs were detected by fluorescence microscopy as described previously.7 The lungs from each mouse were embedded in paraffin for a histopathological assay, sectioned and stained with hematoxylin-eosin (H&E) and an UltraSensitive™SP (Rabbit) IHC Kit (Maixin, China) for the evaluation of inflammatory invasion. Image-Pro Plus 6.0 software (Media Cybernetics Inc, USA) was used to assess the C. psittaci positive count of IHC results. Meanwhile, the supernatants of lung homogenates were harvested as described above for cytokine level measurements. The concentrations of the cytokines were assayed in duplicate by using Ready-SET-Go! kits (eBioscience Inc., CA, USA) in accordance with the manufacturer's instruction. The sensitivities of the IFN-γ, TNF-α, IL-4, IL-6, IL-10 and IL-12 detection kits were in the ranges of 15–2000, 8–1000, 4–500, 4–500, 32–4000, and 32–2000 pg/mL, respectively.
Extraction And Purification Of C. Psittaci DNA
According to the manufacturer’s instructions, genomic DNA (gDNA) extraction was performed in blood and tissues were collected from C. psittaci-infected mice using the Qiagen QIAamp DNA Mini Kit (Qiagen, Shanghai, China). DNA was quantified using a Beckman Coulter DU 730 Life Science UV/Vis Spectrophotometer (Beckman Coulter Canada, Mississauga, ON, Canada).
Quantitative PCR
Quantitative real-time PCR was used to determine the gDNA extractions of tissues in the infected mice. Primers used for C. psittaci 16s rRNA and mouse β-actin were as described by Wen et al.27 All primers were synthesized by Sangon Biotech (Shanghai, China). Quantitative real-time PCR was performed in 20 μL reaction volumes according to the manufacturer’s instruction (Qiagen, Shanghai, China) under the following conditions: 16s rRNA (initial denaturation at 95°C for 8 mins, amplification by cycling 40 times at 95°C for 15 s, 56°C for 20 s, and 72°C for 30 s, final denaturation at 95°C for 15 s, melting curve at 60°C to 95°C for 20 mins); β-actin (initial denaturation at 95°C for 10 mins, amplification by cycling 40 times at 95°C for 15 s, 55°C for 20 s, and 72°C for 40 s, final denaturation at 95°C for 15 s, melting curve at 60°C to 95°C for 20 mins). A standard curve was drawn for 16s rRNA and β-actin and each assay was run with controls according to the previous study.28
Statistical Analysis
Differences in antibody, cytokine levels, SI and chlamydial burden between the groups were compared statistically by one-way analysis of variance (ANOVA) with the Student–Newman–Keuls test. P < 0.05 was considered significant. All calculations were performed with SPSS 18.0 software.
Results
Physical–Structural Characterization Of Chitosan Nanoparticles
TEM techniques were used to assess the morphology and the size of chitosan-based nanoparticles. A schematic of the adsorption of the recombinant Ags on CNPs is depicted in Figure 1A. By TEM analysis, the images of CNPs-Ags (Figure 1B) revealed the appearance of nanoparticles to be obviously more spherical with nanorange sizes (100–300nm), and all nanoparticles appeared to be in similar sizes. Further, as shown in Figure 1C, the adsorption efficiency of the recombinant Ags was present in significantly higher incubated for 30 mins (71.7±5.03%) than those of 15 mins (55.3±8.39%), but no significant difference to incubated for 45 min and 60 min (67.0±5.29 and 69.3±3.79%). Thus, confirming the optimal adsorption time.
Mean particle diameter, zeta potential and polydispersity index (PDI) of CNPs-Ags or CNPs-PBS are summarized in Table 1. Analysis of physicochemical characterization in the present study showed the long-term stability of CNPs-Ags with a zeta potential of +13.12±0.41 mV as compared with +11.62±0.27 mV for CNPs-PBS, with small differences in PDI (0.30–0.42), and average sizes of 225.2±29.1 nm (CNPs-PBS) and 276.1±36.3 nm (CNPs-Ags). The recombinant Ags release from CNPs was a sustained slow process with a gradual release of 48% over a 20-day period (data not shown).
 |
Table 1 Nanoparticle Size Distribution, Zeta Potential, And Polydispersity Index |
Assessment Of Humoral Responses And Mucosal Responses
Previous studies showed that both IgG and secretory IgA play a crucial role during the respiratory chlamydial challenge. To investigate whether it was possible to elicit stronger humoral responses and mucosal responses in the immunized mice by IN and IM routes simultaneously, the levels of IgG, IgG1 and IgG2a in the serum, which were collected from immunized mice 14 days after the last vaccination, were assessed by ELISA. It is very clear that the SIM immunization strategy (SIM-CNPs-Ags) induced significantly higher levels of antigen-specific IgG, IgG1 and IgG2a antibodies than the IN (IN-CNPs-Ags), IM (IM-CNPs-Ags) immunization routes and CNPs-unabsorbed group (SIM-Ags) (P < 0.05). In addition, the levels of serum antibodies in SIM-Ags, IN-CNPs-Ags and IM-CNPs-Ags group were higher in comparison with those of IN-CNPs-PBS and IM-CNPs-PBS group (Figure 2A).
To compare the mucosal responses induced by the SIM, IN and IM immunization strategy, next we detected the levels of IgA in the serum, nasal and vaginal secretions. As shown in Figure 2B, after the immunization, the serum IgA induced in the SIM-CNPs-Ags group was also significantly higher than those induced in the SIM-Ags, IN-CNPs-Ags and IM-CNPs-Ags groups (P < 0.05). Equal levels of nasal and vaginal IgA were assessed in SIM-CNPs-Ags and IN-CNPs-Ags groups, while significantly higher secretory IgA level was induced in the SIM- and IN-immunized mice compared to the IM immunized mice (P < 0.05). Thus, SIM immunization mediated stronger humoral responses than the IN and IM immunization strategy, but there were comparable mucosal responses to the IN route.
Specific T-Cell Responses In Spleen
Specific splenocyte proliferation was detected at day 14 post-third boost in 6 mice per group. Immunization with CNPs-adsorbed Ags via the SIM route generated an antigen-specific proliferation response in the splenocytes, significantly greater than SIM-Ags, IN-CNPs-Ags and IM-CNPs-Ags groups (P < 0.01), but displayed a lower stimulation index compared with the positive control (Figure 3A).
To determine the phenotypic characteristics of T-cell populations activated after the immunization via the SIM, IN and IM immunization strategy, multi-parameter intracellular flow cytometry staining (IFCS) analysis was used to determine the Ags-specific T-cell response according to the previous study.29 Briefly, splenocytes from 6 mice per group were stimulated by the recombinant Ags in the presence of brefeld in for 5 hrs. Figure 3B–D shows that SIM-CNPs-Ags group (26.1±3.09%) secreted comparable CD4+/IFN-γ level to the IN-CNPs-Ags and IM-CNPs-Ags groups (23.8±3.27 and 22.4±3.14%), but significantly higher than control groups (SIM-Ags, IN-CNPs-PBS and IM-CNPs-PBS groups) (18.7±1.42, 13.7±1.96 and 15.4±2.08%, respectively, P < 0.05). However, the secretion of CD4+/IL-4 in SIM-CNPs-Ags group showed no significant difference compared with other groups. Therefore, SIM-CNPs-Ags group could induce an enhanced activation of T-cells, but there was parallel specific T-cell response comparable to that of IN- and IM-immunized mice.
Cytokine Responses In The Vaccinated Mice
The levels of cytokines were measured to determine the cell-mediated immune response induced by different immunization strategies. Then, several typical cytokines of IFN-γ, IL-2, TNF-α, IL-4, IL-10, and IL-17A secreted by splenocytes were next detected by ELISA. As shown in Figure 4A and B, the secretion of IFN-γ and IL-2 (Th1 cytokines) in the splenocytes of SIM-CNPs-Ags group (477±39.1 and 79.7±9.03 pg/mL) was increased compared to the SIM-Ags (270±31.0, 41.0±5.20 pg/mL), IN-CNPs-Ags (375±36.8, 56.0±6.29 pg/mL) and IM-CNPs-Ags groups (344±43.0, 64.7±4.80 pg/mL) (P < 0.05). However, the levels of the IL-4 and IL-10 (Th2 cytokines) in the SIM-CNPs-Ags group showed no differences compared with those in the other groups (Figure 4D and E). The results showed that the simultaneous vaccine administration could induce a stronger Th1-type cell response compared to the IN and IM immunization strategy alone. Furthermore, compared to control groups, immunization by IN and IM combination with CNPs also resulted in significantly higher TNF-α (Innate immune cytokine)30 and IL-17A (Th17 cytokine) levels (287±26.9 and 143±19.9 pg/mL, respectively, P < 0.05) (Figure 4C and F), indicating that SIM-CNPs immunization obviously enhanced the cell-mediated immune response.
Results Of Lung Infection In The Mice Model
The efficacy of protective immune responses against C. psittaci was evaluated by lung challenge. After optimization of the challenge with C. psittaci 6BC strain, mice were sacrificed to determine the C. psittaci load in the lungs on day 10. As seen in Figure 5, the result revealed a decreased chlamydial burden in the lungs of the SIM-CNPs-Ags group compared to both the IN-, IM-immunized and SIM-Ags group (P < 0.05), indicating a greater ability of chlamydial clearance induced by simultaneous vaccine administration with CNPs. Next, we investigated the levels of cytokines in the supernatants of the lung homogenates, which were harvested from the immunized mice following C. psittaci intranasal infection. The levels of IFN-γ (341±52.1 pg/mL, P < 0.01), TNF-α and IL-6 (87.2±21.5 and 62.8±20.3 pg/mL, respectively, P < 0.05) in the lungs of SIM-CNPs-immunized mice were significantly lower than the control immunizations (Figure 6A, B and D). In contrast, there were no significant differences in the levels of IL-4, IL-10, and IL-12 between the SIM-CNPs-Ags group and other controls (Figure 6C, E and F).
We further evaluated whether the SIM-CNPs strategy would result in decreased inflammatory pathologies of the mouse lungs by hematoxylin-eosin (H&E) and immunohistochemistry (IHC) analysis. Similar to the above results, inflammatory infiltrates in C. psittaci-infected mice immunized by SIM route with CNPs were remarkably reduced compared to those in the IN-, IM-immunized mice and SIM-Ags group (Figure 7). And severe lesions including the loss of pulmonary alveoli and diffuse inflammatory cell infiltration were observed in IN-CNPs-PBS and IM-CNPs-PBS groups. The evaluation of lung tissues by IHC analysis showed that SIM-CNPs-immunized group gave rise to a lower chlamydial burden (brown granule) than the IN-, IM-immunized and CNPs-unabsorbed groups (Figure 8). Accordingly, immunization by simultaneous vaccine administration with CNPs induced a stronger protective efficacy against C. psittaci infection compared to other routes.
The Dissemination Of Chlamydia In Vivo
We continued to investigate whether C. psittaci in the lung would disseminate to other tissues. Quantitative real-time PCR (qPCR) was used to evaluate the C. psittaci load in lung, blood, liver, spleen and kidney on day 10 post-infection. As shown in Figure 9, the C. psittaci burden in lung, blood and liver of the mice immunized by SIM route with CNPs was significantly lower than that of the IN-, IM-immunized mice and SIM-Ags groups (P < 0.05). Interestingly, SIM-CNPs-immunized mice revealed a decreased level of the C. psittaci burden in the spleen, but there were no significant differences compared to SIM-Ags, IN-CNPs-Ags and IM-CNPs-Ags groups (Figure 9D). Furthermore, the chlamydial burden in the kidney of SIM-CNPs-Ags group showed no differences compared with those in the other groups (Figure 9E). Overall, the above experiments demonstrate that SIM-CNPs immunization is an effective vaccine strategy against C. psittaci dissemination in vivo of the infected mice, which is better than immunization by the IN and IM strategy alone or immunized with CNPs-unabsorbed Ags.
Discussion
It is becoming increasingly clear that an effective vaccine to prevent Chlamydia infection has not been developed partly due to the absence of an effective delivery system and optimal administration route.31 Previous studies showed that the route of immunization influenced the effectiveness of each vaccine antigens, particularly against respiratory infections.32,33 IM administration is one of the main delivery approaches that can induce antigen-specific immune responses.16 However, major limitations of the IM immunization are forming persistent precipitates, resulting in being dissolved and re-absorbed relatively slowly. In addition, the low level of cell transfection and the single type of immune responses made it far from ideal as an optimal administration route. Thus, immunization via mucosae would be a feasible alternative to avoid some of the injectable vaccine disadvantages. Recent studies reported that IN immunization could induce mucosal immune responses in the respiratory and genital tracts, which played a protective role through mucosal surfaces. But again, it also has some imperfection, such as it cannot induce immune responses by itself and it failed to elicit strong cell-mediated immunity in vivo.34,35 Thus, the combinations of different delivery routes for immunization strategies can modulate not only the type but also the degree and duration of immune responses.
Chitosan nanoparticles were considered as an effective delivery system of the vaccine due to the advantages of biocompatible, biodegradable, mucoadhesive, polycationic, and immunomodulatory properties.36,37 In this study, we explored the effectiveness of CNPs as an immune adjuvant and vaccine delivery system by adsorbing the recombinant Ags on CNPs. The physicochemical qualities of nanoparticles may carry out a number of functions. NP size plays a crucial role to let the antigen cross the mucosal membrane.38 Our results revealed that CNPs-Ags were spherical with nanorange sizes at about 100–300nm, which were shown to be optimal for crossing the mucosal barrier and for being efficiently taken up by antigen-presenting cells (APCs).39,40 All the nanoparticles were positively charged with zeta potential higher than +9 mV which ensured the stability of NPs adjuvant and their adhesion for nasal mucosa. The optimal adsorption efficiency of Ags on CNPs formulation was 71.7%, comparable to the encapsulation efficiency of other Ags (67%) in CNPs.23 In addition, compared to the SIM-Ags group, CNPs could significantly enhance the immunogenicity and protective immunity of the chlamydial Ags (as shown in Figure 2–9), which is consistent with the adjuvant properties of CNPs in previous studies.24
It has previously been shown that the presence of specific antibody in the serum following vaccination is highly relevant with regard to the ongoing debate on the impact of humoral responses toward chlamydial infection.41–43 The IgA generated in the mucosal surface of the respiratory and vaginal tracts also could transcytose into the lumen to neutralize the infection in vivo by the polyclonal immunoglobulin receptor.44,45 We previously demonstrated that immunization by the recombinant Ags could result in stronger humoral immune responses, which play a significant role in preventing C. psittaci attachment.10 In the present study, we found that simultaneous use of the IN and IM immunization routes elicits high levels of IgG-, IgG1- and IgG2a-specific antibodies in serum, which is consistent with previous studies using the multi-epitope peptide as Ags.10 Unexpectedly, SIM immunization mediated stronger humoral responses to the IN and IM immunization strategies alone, but there were comparable nasal IgA and vaginal IgA levels to IN route. We speculate that the IN immunization has a more pronounced increase to the humoral and mucosal immunity. However, for IM vaccination, only a minor mucosal immunity has been induced after injection. Consequently, the stronger mucosal immune responses found in SIM-immunized mice were most likely induced by IN immunization.
The activation and expansion of T-cells are significant in preventing Chlamydia infection via cell-mediated responses.46,47 And it is also important for the Chlamydia vaccine's trigger of stronger T-cell proliferation.48,49 In this study, we found a robust antigen-specific proliferation response of the splenocytes in SIM-vaccinated group, which is consistent with previous studies using simultaneous immunization routes.50 In addition, we also found that high levels of Th1/Th17 cytokines and TNF-α were secreted by splenocytes following IN and IM immunization simultaneously, which are crucial against Chlamydia challenge via activation of phagocytes that limit the bacterial invasion.51,52 Moreover, we are not surprised that there were no changes in the levels of Th2 cytokines. This occurs primarily because the recombinant Ags synthesis in our previous studies is biased to induce a Th1-type reaction. Interestingly, different from the supernatant detection, the SIM immunization induces slightly stronger or similar peripheral CD4+/IFN-γ and CD4+/IL-4 levels compared to IN and IM immunization. To the best of our knowledge, it is partly due to IFN-γ in the supernatant that was secreted by CD4+ T-cells, NK cells and CD8+ T-cells, but in the IFCS analysis, we only detected the IFN-γ secreted by CD4+ T cells, which could explain the high levels of IFN-γ secretion in the supernatant, but there were no significant differences in the intracellular detection. Therefore, it is vital to determine the secretion of IFN-γ from other cells in future studies.
In the current study, SIM immunization that elicited significant levels of humoral, mucosal immunity and cell-mediated immunity against chlamydial respiratory tract infection, determined by obviously decreasing the chlamydial loading and pathological changes in the lungs of infected mice (As shown in Figure 5–8), which supported the recent findings that protects mice against a Chlamydia muridarum challenge by stimulating robust systemic and local mucosal immune responses.53 Nevertheless, another study found that simultaneous, nasal and subcutaneous immunized mice were equally well protected in Chlamydia trachomatis–infected mice.54 It is possible that C. psittaci is a respiratory tract–transmitted pathogen,55 and SIM immunization can play an anti-infective role at the preliminary respiratory tract invasion stage through IN-induced mucosal immunity and at the later stage of infection by the IM-induced humoral and cellular immunity. This provides an explanation for why SIM vaccine elicited a significant level of protection against C. psittaci infection. However, the specific role of mucosal immunity referring to the prevention against Chlamydia is unclear and must be balanced against the contribution of other types of immune responses.
C. psittaci is a highly invasive pathogen which causes respiratory infections, followed by disseminating to various organs throughout the body, including heart, liver, spleen, etc.,1,2,56 and therefore effective immunization needs to target the bacteria within the respiratory system to inhibit C. psittaci transfer into secondary sites. Analysis of the C. psittaci load in the lung, blood, liver, spleen, and kidney using qPCR showed that SIM vaccination obviously inhibited C. psittaci dissemination to other tissues compared to IN and IM strategies alone. In particular, the most significant decrease of the C. psittaci DNA concentration was observed in the blood and spleen, while the bacterial load was lower in the spleen and kidney of all groups. We speculate that only a smaller amount of Chlamydia may spread to various tissues during the relatively short time of acute infection. On the contrary, the Chlamydia burden may be significantly higher in the kidney, liver and other tissues during the chronic or persistent course than that observed in acute infection. However, these speculations need to be identified by further research.
Conclusion
In this study, we confirm a new immunization strategy for C. psittaci vaccines. Simultaneous intramuscular and intranasal immunizations of chitosan nanoparticles–adjuvanted vaccine induce robust humoral, mucosal immunity and cell-mediated immunity to C. psittaci, which prevent the respiratory chlamydial infections by decreasing the C. psittaci load and eliminating C. psittaci in mice. Furthermore, it is noteworthy that SIM is also an effective route against C. psittaci disseminating to other tissues after infection. Hence, our studies may provide all-sided protection of mice against a bacterial respiratory tract infection, further propelling the development of Chlamydia vaccine against challenge.
 |
Scheme 1 The timeline of immunization and overall in vivo work through the terminal point on day 52. |
 |
Figure 1 Physical–structural characterization of chitosan nanoparticles. Notes: (A) Schematic representation of the recombinant Ags adsorbed on CNPs; (B) morphological characteristics of CNPs by TEM observation; (C) the adsorption efficiency of the recombinant Ags adsorbed on CNPs. Each bar indicates the mean ± SD from three independent experiments. *P < 0.05. Abbreviations: CNPs, chitosan nanoparticles; TEM, transmission electron microscopy; PDI, polydispersity index; SD, standard deviation. |
 |
Figure 2 Production of antigen-specific Ig antibodies and the sIgA antibodies in immunized mice. Notes: Antigen-specific Ig antibodies and the sIgA antibodies against the C. psittaci Ags were induced 14 days after the final immunization in vaccinated BALB/c mice. (A) The sera were collected from immunized mice to determine antigen-specific IgG antibody levels by ELISA at dilutions of 1:200. (B) The serum IgA was detected by ELISA at dilutions of 1:200. And the nasal washes and vaginal washes collected from immunized mice were determined of the sIgA antibodies' levels by ELISA at dilutions of 1:10. Each bar indicates the mean ± SD of triplicates from 6 mice per group. *P < 0.05; **P < 0.01. Abbreviations: sIgA, secretory IgA; PBS, phosphate-buffered saline; Ags, antigens; ELISA, enzyme-linked immunosorbent assay; SIM, simultaneous; IM, intramuscular; IN, intranasal; SD, standard deviation. |
 |
Figure 3 Ags-specific T-cell responses in spleen. Notes: Splenocytes from immunized mice were seeded into 24-well plates and incubated at 37°C in 5% CO2 for 48 hrs and assayed for Ags-specific CD4+ T-cell response. (A) Ags-specific proliferative responses in splenocytes were induced by different immunization routes. (B) Frequency of CD4+/IFN-γ cells was calculated according to IFCS. (C) Frequency of CD4+/IL-4 cells was calculated according to IFCS. (D) Ags-specific CD4+ T-cell response was determined by intracellular IL-4 and IFN-γ staining. The FACS plots are representative of the mean percentages of 6 mice in each group. Each bar indicates the mean ± SD of 6 mice per group from three independent results. *P < 0.05; **P < 0.01. Abbreviations: IFCS, intracellular flow cytometry staining; FACS, fluorescence-activated cell sorting; CD4, cluster of differentiation 4; IFN-γ, interferon gamma; IL, interleukin; SI, stimulation index; SD, standard deviation. |
 |
Figure 4 Cytokine secretion by splenocytes from vaccinated mice. Notes: (A) IFN-γ, (B) IL-2, (C) TNF-α, (D) IL-4, (E) IL-10 and (F) IL-17A levels were measured in the splenocyte supernatants of immunized mice by ELISA kits. Each bar indicates the mean ± SD of triplicates from 6 mice per group. *P < 0.05; **P < 0.01. Abbreviations: IFN-γ, interferon gamma; IL, interleukin; TNF-α, tumor necrosis factor alpha; ELISA, enzyme-linked immunosorbent assay; Th, helper T; SD, standard deviation. |
 |
Figure 5 The burden of C. psittaci in the lungs of immunized mice after the C. psittaci infection. Notes: The lung homogenates collected from immunized mice 10 days after the C. psittaci challenge were inoculated into HeLa 229 cell monolayers. Then, the chlamydial inclusions were detected by indirect immunofluorescence. Each bar indicates the mean ± SD of the C. psittaci titers (IFU/lung) in the lung homogenates from 6 mice per group in three independent results. *P < 0.05; **P < 0.01. Abbreviations: SIM, simultaneous; IM, intramuscular; IN, intranasal; IFU, inclusion-forming units; CNPs, chitosan nanoparticles; PBS, phosphate-buffered saline; SD, standard deviation. |
 |
Figure 6 Cytokine levels in the lungs of immunized mice after the C. psittaci challenge. Notes: (A) IFN-γ, (B) TNF-α, (C) IL-4, (D) IL-6, (E) IL-10 and (F) IL-12 levels were detected in the lung homogenates of immunized mice by ELISA kits after the C. psittaci infection. Each bar represents the mean ± SD of the cytokine levels (pg/mL) in the lung homogenates from 6 mice per group in three independent experiments. *P < 0.05; **P < 0.01. Abbreviations: IFN-γ, interferon gamma; IL, interleukin; TNF-α, tumor necrosis factor alpha; ELISA, enzyme-linked immunosorbent assay; SD, standard deviation. |
 |
Figure 7 Histopathological assessment of lung tissues after C. psittaci infection. Notes: H&E-stained sections of lung tissue from each group of mice (magnification 100× and 400×) are shown as (A) SIM-CNPs-Ags group, (B) SIM-Ags group, (C) IN-CNPs-Ags group, (D) IM-CNPs-Ags group, (E) IN-CNPs-PBS group and (F) IM-CNPs-PBS group. Abbreviations: H&E, hematoxylin-eosin; SIM, simultaneous; IM, intramuscular; IN, intranasal; CNPs, chitosan nanoparticles; PBS, phosphate-buffered saline. |
 |
Figure 8 C. psittaci burden and pathological assessment in lung tissue measured by IHC. Notes: (A) The infected lungs were sectioned and stained with S-P immunohistochemistry using an UltraSensitive™SP (Rabbit) IHC Kit with rabbit anti-C. psittaci 6BC antibody used as the primary antibody. (a) SIM-CNPs-Ags group, (b) SIM-Ags group, (c) IN-CNPs-Ags group, (d) IM-CNPs-Ags group, (e) IN-CNPs-PBS group and (f) IM-CNPs-PBS group. Brown granules (black arrow) indicate C. psittaci inclusions in the nuclei of the lung tissue cells. (B) C. psittaci positive count per 100× field. (C) C. psittaci positive count per 400× field. Each bar indicates the mean ± SD from three independent experiments. *P < 0.05; **P < 0.01. Abbreviations: IHC, immunohistochemistry; S-P, streptavidin-peroxidase; SIM, simultaneous; IM, intramuscular; IN, intranasal; CNPs, chitosan nanoparticles; PBS, phosphate-buffered saline. |
 |
Figure 9 The dissemination of Chlamydia in vivo. Notes: C. psittaci burden was evaluated using qPCR to measure C. psittaci DNA concentrations in (A) lung, (B) blood, (C) liver, (D) spleen, and (E) kidney. Points correspond to that separately extracted from each animal. Horizontal lines represent mean values. *P < 0.05; **P < 0.01. Abbreviations: qPCR, quantitative real-time polymerase chain reaction; SIM, simultaneous; IM, intramuscular; IN, intranasal; CNPs, chitosan nanoparticles; rRNA, ribosomal ribonucleic acid; DNA, deoxyribonucleic acid. |
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 81671986, Grant No. 31872643, Grant No. 31800162), Hunan Provincial Key Laboratory for Special Pathogens Prevention and Control Foundation (Grant No. 2014-5), Hunan Province Cooperative Innovation Center for Molecular Target New Drug Study (Grant No. 2015-351).
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Knittler MR, Berndt A, Bocker S, et al. Chlamydia psittaci: new insights into genomic diversity, clinical pathology, host-pathogen interaction and anti-bacterial immunity. Int J Med Microbiol. 2014;304(7):877–893. doi:10.1016/j.ijmm.2014.06.010
2. Knittler MR, Sachse K. Chlamydia psittaci: update on an underestimated zoonotic agent. Pathog Dis. 2015;73(1):1–15. doi:10.1093/femspd/ftu007
3. Beeckman DS, Vanrompay DC. Zoonotic Chlamydophila psittaci infections from a clinical perspective. Clin Microbiol Infe. 2009;15(1):11–17. doi:10.1111/j.1469-0691.2008.02669.x
4. Chu J, Zhang Q, Zhang T, et al. Chlamydia psittaci infection increases mortality of avian influenza virus H9N2 by suppressing host immune response. Sci Rep. 2016;6:29421. doi:10.1038/srep29421
5. Longbottom D, Livingstone M. Vaccination against chlamydial infections of man and animals. Vet J. 2006;171(2):263–275. doi:10.1016/j.tvjl.2004.09.006
6. Farris CM, Morrison RP. Vaccination against Chlamydia genital infection utilizing the murine C. muridarum model. Infect Immun. 2011;79(3):986–996. doi:10.1128/IAI.00881-10
7. Liang M, Wen Y, Ran O, et al. Protective immunity induced by recombinant protein CPSIT_p8 of Chlamydia psittaci. Appl Microbiol Biot. 2016;100(14):6385–6393. doi:10.1007/s00253-016-7494-8
8. Zhou J, Qiu C, Cao XA, Lin G. Construction and immunogenicity of recombinant adenovirus expressing the major outer membrane protein (MOMP) of Chlamydophila psittaci in chicks. Vaccine. 2007;25(34):6367–6372. doi:10.1016/j.vaccine.2007.06.031
9. Tan Y, Li Y, Zhang Y, et al. Immunization with Chlamydia psittaci plasmid-encoded protein CPSIT_p7 induces partial protective immunity against chlamydia lung infection in mice. Immunol Res. 2018;66(4):471–479. doi:10.1007/s12026-018-9018-3
10. Li Y, Zheng K, Tan Y, et al. A recombinant multi-epitope peptide vaccine based on MOMP and CPSIT_p6 protein protects against Chlamydia psittaci lung infection. Appl Microbiol Biot. 2019;103(2):941–952. doi:10.1007/s00253-018-9513-4
11. Samanta GP. Mathematical analysis of a Chlamydia epidemic model with pulse vaccination strategy. Acta Biotheor. 2015;63(1):1–21. doi:10.1007/s10441-014-9234-8
12. Bode J, Dutow P, Sommer K, et al. A new role of the complement system: C3 provides protection in a mouse model of lung infection with intracellular Chlamydia psittaci. PloS One. 2012;7(11):e50327. doi:10.1371/journal.pone.0050327
13. Moore T, Ekworomadu CO, Eko FO, et al. Fc receptor-mediated antibody regulation of T cell immunity against intracellular pathogens. J Infect Dis. 2003;188(4):617–624. doi:10.1086/377134
14. Farris CM, Morrison SG, Morrison RP. CD4+ T cells and antibody are required for optimal major outer membrane protein vaccine-induced immunity to Chlamydia muridarum genital infection. Infect Immun. 2010;78(10):4374–4383. doi:10.1128/IAI.00622-10
15. Jaffar Z, Ferrini ME, Herritt LA, Roberts K. Cutting edge: lung mucosal Th17-mediated responses induce polymeric Ig receptor expression by the airway epithelium and elevate secretory IgA levels. J Immunol. 2009;182(8):4507–4511. doi:10.4049/jimmunol.0900237
16. Badamchi-Zadeh A, McKay PF, Holland MJ, et al. Intramuscular immunisation with chlamydial proteins induces Chlamydia trachomatis specific ocular antibodies. PLoS One. 2015;10(10):e0141209. doi:10.1371/journal.pone.0141209
17. Neutra MR, Kozlowski PA. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol. 2006;6(2):148–158. doi:10.1038/nri1777
18. Zariwala MG, Bendre H, Markiv A, et al. Hydrophobically modified chitosan nanoliposomes for intestinal drug delivery. Int J Nanomed. 2018;13:5837–5848. doi:10.2147/IJN.S166901
19. Hassan UA, Hussein MZ, Alitheen NB, Yahya Ariff SA, Masarudin MJ. In vitro cellular localization and efficient accumulation of fluorescently tagged biomaterials from monodispersed chitosan nanoparticles for elucidation of controlled release pathways for drug delivery systems. Int J Nanomed. 2018;13:5075–5095. doi:10.2147/IJN.S164843
20. Frank LA, Sandri G, D’Autilia F, et al. Chitosan gel containing polymeric nanocapsules: a new formulation for vaginal drug delivery. Int J Nanomed. 2014;9:3151–3161.
21. Bento D, Staats HF, Goncalves T, Borges O. Development of a novel adjuvanted nasal vaccine: C48/80 associated with chitosan nanoparticles as a path to enhance mucosal immunity. Eur J Pharm Biopharm. 2015;93:149–164. doi:10.1016/j.ejpb.2015.03.024
22. Malik A, Gupta M, Gupta V, Gogoi H, Bhatnagar R. Novel application of trimethyl chitosan as an adjuvant in vaccine delivery. Int J Nanomed. 2018;13:7959–7970. doi:10.2147/IJN.S165876
23. Dhakal S, Renu S, Ghimire S, et al. Mucosal immunity and protective efficacy of intranasal inactivated influenza vaccine is improved by chitosan nanoparticle delivery in pigs. Front Immunol. 2018;9:934. doi:10.3389/fimmu.2018.00934
24. Malik A, Gupta M, Mani R, Gogoi H, Bhatnagar R. Trimethyl chitosan nanoparticles encapsulated protective antigen protects the mice against anthrax. Front Immunol. 2018;9:562. doi:10.3389/fimmu.2018.00562
25. Da Silva RL, Da Silva JR, Junior APD, et al. Adsorption of vi capsular antigen of salmonella typhi in chitosan-poly (methacrylic acid) nanoparticles. Polymers. 2019;11(7):E1226. doi:10.3390/polym11071226
26. Dutow P, Fehlhaber B, Bode J, et al. The complement C3a receptor is critical in defense against Chlamydia psittaci in mouse lung infection and required for antibody and optimal T cell response. J Infect Dis. 2014;209(8):1269–1278. doi:10.1093/infdis/jit640
27. Wen Y, Chen Y, Li L, et al. Localization and characterization of a putative cysteine desulfurase in Chlamydia psittaci. J Cell Biochem. 2019;120(3):4409–4422. doi:10.1002/jcb.27727
28. Zheng K, Xu M, Xiao Y, et al. Immunogenicity and protective efficacy against Treponema pallidum in New Zealand rabbits immunized with plasmid DNA encoding flagellin. Emerg Microbes Infec. 2018;7(1):177. doi:10.1038/s41426-018-0176-0
29. Stepanova LA, Kotlyarov RY, Kovaleva AA, et al. Protection against multiple influenza A virus strains induced by candidate recombinant vaccine based on heterologous M2e peptides linked to flagellin. PLoS One. 2015;10(3):e0119520. doi:10.1371/journal.pone.0119520
30. Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1(2):135–145. doi:10.1038/35100529
31. van Drunen Littel-van Den Hurk S, Gerdts V, Loehr BI, et al. Recent advances in the use of DNA vaccines for the treatment of diseases of farmed animals. Adv Drug Deliver Rev. 2000;43(1):13–28. doi:10.1016/S0169-409X(00)00074-0
32. Nogueira CV, Zhang X, Giovannone N, Sennott EL, Starnbach MN. Protective immunity against Chlamydia trachomatis can engage both CD4+ and CD8+ T cells and bridge the respiratory and genital mucosae. J Immunol. 2015;194(5):2319–2329. doi:10.4049/jimmunol.1402675
33. Tammiruusu A, Penttila T, Lahesmaa R, Sarvas M, Puolakkainen M, Vuola JM. Intranasal administration of chlamydial outer protein N (CopN) induces protection against pulmonary Chlamydia pneumoniae infection in a mouse model. Vaccine. 2007;25(2):283–290. doi:10.1016/j.vaccine.2006.07.043
34. Ciabattini A, Prota G, Christensen D, Andersen P, Pozzi G, Medaglini D. Characterization of the antigen-specific CD4(+) T cell response induced by prime-boost strategies with CAF01 and CpG adjuvants administered by the intranasal and subcutaneous routes. Front Immunol. 2015;6:430. doi:10.3389/fimmu.2015.00430
35. Christensen D, Mortensen R, Rosenkrands I, Dietrich J, Andersen P. Vaccine-induced Th17 cells are established as resident memory cells in the lung and promote local IgA responses. Mucosal Immunol. 2017;10(1):260–270. doi:10.1038/mi.2016.28
36. Mehrabi M, Montazeri H, Mohamadpour Dounighi N, Rashti A, Vakili-Ghartavol R. Chitosan-based nanoparticles in mucosal vaccine delivery. Arch Razi Inst. 2018;73(3):165–176. doi:10.22092/ari.2017.109235.1101
37. Sun T, Zhan B, Zhang W, et al. Carboxymethyl chitosan nanoparticles loaded with bioactive peptide OH-CATH30 benefit nonscar wound healing. Int J Nanomed. 2018;13:5771–5786. doi:10.2147/IJN.S156206
38. van der Lubben IM, Verhoef JC, Borchard G, Junginger HE. Chitosan for mucosal vaccination. Adv Drug Deliver Rev. 2001;52(2):139–144. doi:10.1016/S0169-409X(01)00197-1
39. Mahapatro A, Singh DK. Biodegradable nanoparticles are excellent vehicle for site directed in-vivo delivery of drugs and vaccines. J Nanobiotechnol. 2011;9:55. doi:10.1186/1477-3155-9-55
40. Mishra N, Goyal AK, Tiwari S, et al. Recent advances in mucosal delivery of vaccines: role of mucoadhesive/biodegradable polymeric carriers. Expert Opin Ther Pat. 2010;20(5):661–679. doi:10.1517/13543771003730425
41. Khan SA, Polkinghorne A, Waugh C, et al. Humoral immune responses in koalas (Phascolarctos cinereus) either naturally infected with Chlamydia pecorum or following administration of a recombinant chlamydial major outer membrane protein vaccine. Vaccine. 2016;34(6):775–782. doi:10.1016/j.vaccine.2015.12.050
42. Pickering H, Teng A, Faal N, et al. Genome-wide profiling of humoral immunity and pathogen genes under selection identifies immune evasion tactics of Chlamydia trachomatis during ocular infection. Sci Rep. 2017;7(1):9634. doi:10.1038/s41598-017-09193-2
43. De Clercq E, Devriendt B, Yin L, Chiers K, Cox E, Vanrompay D. The immune response against Chlamydia suis genital tract infection partially protects against re-infection. Vet Res. 2014;45:95. doi:10.1186/s13567-014-0095-6
44. Fahrbach KM, Malykhina O, Stieh DJ, Hope TJ. Differential binding of IgG and IgA to mucus of the female reproductive tract. PLoS One. 2013;8(10):e76176. doi:10.1371/journal.pone.0076176
45. Renegar KB, Jackson GD, Mestecky J. In vitro comparison of the biologic activities of monoclonal monomeric IgA, polymeric IgA, and secretory IgA. J Immunol. 1998;160(3):1219–1223.
46. Zhu C, Lin H, Tang L, Chen J, Wu Y, Zhong G. Oral Chlamydia vaccination induces transmucosal protection in the airway. Vaccine. 2018;36(16):2061–2068. doi:10.1016/j.vaccine.2018.03.015
47. Karunakaran KP, Yu H, Foster LJ, Brunham RC. Development of a Chlamydia trachomatis T cell Vaccine. Hum Vaccines. 2010;6(8):676–680. doi:10.4161/hv.6.8.12299
48. Pal S, de la Maza LM. Mechanism of T-cell mediated protection in newborn mice against a Chlamydia infection. Microbes Infect. 2013;15(8–9):607–614. doi:10.1016/j.micinf.2013.04.010
49. Quispe Calla NE, Vicetti Miguel RD, Mei A, Fan S, Gilmore JR, Cherpes TL. Dendritic cell function and pathogen-specific T cell immunity are inhibited in mice administered levonorgestrel prior to intranasal Chlamydia trachomatis infection. Sci Rep. 2016;6:37723. doi:10.1038/srep37723
50. Ralli-Jain P, Tifrea D, Cheng C, Pal S, de la Maza LM. Enhancement of the protective efficacy of a Chlamydia trachomatis recombinant vaccine by combining systemic and mucosal routes for immunization. Vaccine. 2010;28(48):7659–7666. doi:10.1016/j.vaccine.2010.09.040
51. Jupelli M, Guentzel MN, Meier PA, Zhong G, Murthy AK, Arulanandam BP. Endogenous IFN-gamma production is induced and required for protective immunity against pulmonary chlamydial infection in neonatal mice. J Immunol. 2008;180(6):4148–4155. doi:10.4049/jimmunol.180.6.4148
52. Rottenberg ME, Gigliotti Rothfuchs AC, Gigliotti D, Svanholm C, Bandholtz L, Wigzell H. Role of innate and adaptive immunity in the outcome of primary infection with Chlamydia pneumoniae, as analyzed in genetically modified mice. J Immunol. 1999;162(5):2829–2836.
53. Verma R, Sahu R, Dixit S, et al. The Chlamydia M278 Major Outer Membrane Peptide Encapsulated in the Poly (lactic acid)-Poly (ethylene glycol) Nanoparticulate Self-Adjuvanting Delivery System Protects Mice Against a Chlamydia muridarum Genital Tract Challenge by Stimulating Robust Systemic and Local Mucosal Immune Responses. Front Immunol. 2018;9:2369.
54. Wern JE, Sorensen MR, Olsen AW, Andersen P, Follmann F. Simultaneous subcutaneous and intranasal administration of a CAF01-adjuvanted Chlamydia vaccine elicits elevated IgA and protective Th1/Th17 responses in the genital tract. Front Immunol. 2017;8:569. doi:10.3389/fimmu.2017.00569
55. Reinhold P, Ostermann C, Liebler-Tenorio E, et al. A bovine model of respiratory Chlamydia psittaci infection: challenge dose titration. PLoS One. 2012;7(1):e30125. doi:10.1371/journal.pone.0030125
56. Hogerwerf L, De Gier B, Baan B, Van Der Hoek W. Chlamydia psittaci (psittacosis) as a cause of community-acquired pneumonia: a systematic review and meta-analysis. Epidemiol Infect. 2017;145(15):3096–3105. doi:10.1017/S0950268817002060
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.