Back to Journals » International Journal of Nanomedicine » Volume 12
Simple nanoliposomes encapsulating Lycium barbarum polysaccharides as adjuvants improve humoral and cellular immunity in mice
Authors Bo R, Sun Y, Zhou S, Ou N, Gu P, Liu Z, Hu Y, Liu J ![]() , Wang D
, Wang D ![]()
Received 12 March 2017
Accepted for publication 8 August 2017
Published 28 August 2017 Volume 2017:12 Pages 6289—6301
DOI https://doi.org/10.2147/IJN.S136820
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Dongwoo Khang
Ruonan Bo,1 Yaqin Sun,1 Shuzhen Zhou,2 Ning Ou,1 Pengfei Gu,1 Zhenguang Liu,1 Yuanliang Hu,1 Jiaguo Liu,1 Deyun Wang1
1Institute of Traditional Chinese Veterinary Medicine, College of Veterinary Medicine, Nanjing Agricultural University, Nanjing, 2Foshan City Nanhai Eastern Along Pharmaceutical Co., Ltd, Foshan, China
Abstract: The success of subunit vaccines has been hampered by the problems of weak or short-term immunity and the lack of availability of nontoxic, potent adjuvants. It would be desirable to develop safe and efficient adjuvants with the aim of improving the cellular immune response against the target antigen. In this study, the targeting and sustained release of simple nanoliposomes containing Lycium barbarum polysaccharides (LBP) as an efficacious immune adjuvant to improve immune responses were explored. LBP liposome (LBPL) with high entrapment efficiency (86%) were obtained using a reverse-phase evaporation method and then used to encapsulate the model antigen, ovalbumin (OVA). We demonstrated that the as-synthesized liposome loaded with OVA and LBP (LBPL-OVA) was stable for 45 days and determined the encapsulation stability of OVA at 4°C and 37°C and the release profile of OVA from LBPL-OVA was investigated in pH 7.4 and pH 5.0. Further in vivo investigation showed that the antigen-specific humoral response was correlated with antigen delivery to the draining lymph nodes. The LBPL-OVA were also associated with high levels of uptake by key dendritic cells in the draining lymph nodes and they efficiently stimulated CD4+ and CD8+ T cell proliferation in vivo, further promoting antibody production. These features together elicited a significant humoral and celluar immune response, which was superior to that produced by free antigen alone.
Keywords: Lycium barbarum polysaccharide liposome, adjuvant, ovalbumin, draining lymph nodes, antigen-specific humoral response
Introduction
The recent history of adjuvant development, in part coupled with recent technological developments, has transformed the field of vaccines with safer subunit vaccines, novel delivery systems, and rationally designed adjuvants and immunomodulators.1–5 Immunologic adjuvants can be used to address the problem of subunit vaccines with low immunogenicity. Although several potent adjuvants are available, toxicity and adverse reactions have limited their use in vaccine formulations. Alum is a conventional adjuvant and the only one widely licensed for human use. Despite being used in vaccines for more than 80 years,6 alum has some disadvantages, including side effects and safety concerns.7 Moreover, alum is not effective for the induction of cell-mediated immunity and subsequent cytotoxic T lymphocyte (CTL) responses, and it can cause allergic reactions in some cases.8,9 These disadvantages necessitate the development of new adjuvants for subunit vaccines.
Particulate antigen delivery systems (viz, liposomes, microspheres, immune-stimulating complexes [ISCOMs], and virosomes) are very potent adjuvants that have been widely used against various infectious diseases.10,11 Liposomes – closed bilayer vesicles made up of lipids surrounding an aqueous core – are considered to be the best class of organic nanoparticles for the treatment of many diseases.12–14 More than 12 of these drugs are in routine clinical use for a range of indications (nine of which are approved by the US Food and Drug Administration).15 Liposomes elicit both antigen-specific humoral and cell-mediated immunity.16 They have been used as a vaccine delivery system/adjuvant with various antigens, and these formulations have proved to be better than Freund’s adjuvant or alum.17 Owing to their unique self-closed structures, liposomes can entrap hydrophilic agents in the aqueous compartment and hydrophobic agents in the lipid bilayer.18 Liposomes protect the loaded drug from the external environment, avoiding both degradation of the drug and undesirable exposure of the environment to the drug action. The presence of specific lipid species such as cholesterol or rigid saturated lipids12,19–21 stabilizes the lipid bilayer against attack by plasma proteins and reduces drug leakage, and thus most of the drug-loaded liposomes reported to date have incorporated cholesterol in the liposomal membrane.
Polysaccharides are macromolecules with repeating carbohydrate units and are widely distributed throughout nature, for example, in the cell wall of fruit cells.22 Over the past decade, plant polysaccharides have attracted a great deal of attention in the biomedical field due to their broad spectrum of therapeutic properties and low toxicity.23 Lycium barbarum L., also known as goji berry or wolfberry, belongs to the genus Lycium of the family Solanaceae, and is a well-known traditional Chinese medicine that has been used as a tonic food for thousands of years.24 Modern scientific research and experiments have proved that polysaccharides are the main bioactive constituents in L. barbarum.25 It has also been proved that L. barbarum polysaccharides (LBP) possess various bioactivities, including immunomodulatory,26–28 antioxidant,29 antitumor,30,31 and hypoglycemic activities.32 However, LBP also have some disadvantageous characteristics, such as low bioavailability and limited formulation methods, which limit its development for future clinical applications.
As an alternative to systemic immune activation, secondary lymphoid tissues such as the lymph nodes (LNs) have been proposed as intriguing sites for targeted immunotherapy.33 Targeted delivery of the antigen and adjuvant to LNs is increasingly being explored in vaccination34–36 as well as transplantation-associated37 immunotherapy, given the role of LNs in supporting adaptive immune cell priming responses.38
We hypothesized that this vaccine carrier has the capacity to deliver antigen to the draining LNs in a sustained manner and elicit a significant antigen-specific humoral immune response. The mice were immunized three times at 2-week intervals via subcutaneous injection, and the role of liposomes as antigenic depot is probably important to sustain substantial activation through successive restimulations. Targeted delivery of antigen to the draining LNs may be adequate to make dendritic cells (DCs) active. T cells were activated in the draining nodes following DC migration, thereby initiating and regulating Th1 and Th2 immune responses.
To test this hypothesis, we present a versatile vaccine delivery platform based on liposomes consisting of soybean phospholipids and cholesterol encapsulating polysaccharides. The kinetics of antigen releasing from LBPL at physiological (pH 7.4) and endosomal pH (pH 5.0) were investigated. Antigen transport to the draining LNs and activation of LN-resident DCs were measured. We investigated ovalbumin (OVA)-specific CD4+/CD8+ T cell activation and explored OVA-specific antibody responses and Th1 and Th2 cytokine secretion.
Materials and methods
Reagents
LBP (purchased from Shanxi Ciyuan Biotechnology Co. Ltd., Shanxi, China; 98% ultraviolet [UV], No CY131207) and LBP liposomes (LBPL) were synthesized based on the reverse-phase evaporation method according to a previous publication.39 OVA was obtained from Sigma-Aldrich Co. (St Louis, MO, USA) as a model antigen. The bicinchoninic acid (BCA) Protein Assay Kit (E112-01) was purchased from Vazyme Biotech (Nanjing, China) and used with a bovine serum albumin (BSA) standard (725041). Cell culture-grade media and fetal bovine serum (FBS) were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Lipopolysaccharide (LPS) and phytohemagglutinin (PHA) were obtained from Sigma-Aldrich Co. to serve as B cell mitogen and T cell mitogen, respectively. Flow cytometry staining buffer (311114) and 4% paraformaldehyde (LK-F0001) were purchased from MultiSciences Biotech Co., Ltd. (Hangzhou, China). The antibodies, fluorescein isothiocyanate (FITC)-CD11c, antigen-presenting cell (APC)-CD86, phycoerythrin (PE)-cyanine 7 (Cy7)-CD80, PE-major histocompatibility complex II (MHCII), PE-cyanine5-CD3e, FITC-CD4, and PE-CD8a, were purchased from eBioscience (San Diego, CA, USA).
Animals
BALB/c mice were purchased from the Comparative Medicine Center of Yangzhou University, Yangzhou, China. Five-week-old mice weighing 18–20 g were used for this study and allowed to acclimatize for 1 week prior to the experiments. All protocols involving animal subjects were approved by the University Ethics Committee for the humane care and use of experimental animals, and each mouse was used once and treated according to the National Institutes of Health guidelines for the care and use of laboratory animals.
Synthesis and characterization of LBPL loaded with OVA
To encapsulate OVA in the LBPL, 100 μg/mL OVA was added to the aqueous phase consisting of LBP dissolved in 4 mL of phosphate-buffered saline (PBS; pH 7.4). The optimal preparation conditions for the liposome loaded with OVA and LBP (LBPL-OVA) nanoparticles were as follows: a lipid to drug ratio (w/w) of 25:1, a ultrasound duration of 14 min, and a soybean phospholipids to cholesterol ratio (w/w) of 2.4:1. Three freeze–thaw cycles were used to improve protein encapsulation efficiency (EE). The nanoparticles were stored at 4°C prior to use.
To determine the stability of the LBPL loaded with OVA, the liposomes and LBPL-OVA were stored at 4°C for 45 days. The particle size distribution and polydispersity index (PDI) of the nanoparticles were determined using a Zetasizer Nano ZS90 instrument (Malvern Instruments, Malvern, UK) on days 7, 15, 30, and 45. The samples were diluted 10-fold in the same PBS used to prepare the liposomes, and the measurements were repeated three times for each sample.
Quantification of encapsulation stability and in vitro release profile of OVA
The EEs of OVA into LBPL and blank liposomes (BL) were evaluated at 4°C and 37°C over 44 days. We prepared LBPL-OVA and BL loaded with OVA (BL-OVA), and the particles were removed and centrifuged (12,000 rpm, 1 h) at predetermined time intervals. The supernatant was removed and assayed for protein content (Cs) using the BCA Protein Assay Kit. Samples of LBPL-OVA were ruptured using 10% Triton X-100, and the total protein content (Ct) was measured. The EE of OVA was calculated according to the following formula:
|
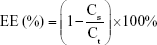
To investigate the release behavior of OVA from LBPL-OVA, the LBPL-OVA formulation was prepared as previously described and incubated at 37°C under mild agitation (100 rpm). The release mediums of OVA were PBS (pH 7.4) and PBS (pH 5.0), respectively. At different time points, 500 μL aliquot of release medium was removed and centrifuged at 12,000 rpm for 1 h. The amount of protein released, present in the supernatant, was measured by a BCA Protein Assay Kit.
In vivo immunization study
Subcutaneous immunization protocol
BALB/c mice were randomly divided into six groups with 20 mice in each group. The mice were immunized three times at 2-week intervals via subcutaneous injection with 200 μL of various OVA-based materials as follows: 20 μg OVA in 200 μL LBPL (2 mg/mL); 20 μg OVA in 200 μL BL; 20 μg OVA in 200 μL LBP (2 mg/mL); 20 μg OVA in 200 μL PBS; 20 μg OVA in 200 μL Freund’s complete adjuvant (FCA); 200 μL PBS (blank control [BC]). At 14 and 28 days after the third immunization, splenocytes were collected for in vitro proliferation and flow cytometric assays. Blood samples were harvested 7, 14, 21, and 28 days after the third immunization. Sera were separated and stored at −70°C for subsequent analysis.
Determination of available antigen in draining LNs by immunohistochemistry
BALB/c mice (n=4) were subcutaneously vaccinated as described in the “Subcutaneous immunization protocol” section. The mice were sacrificed at the indicated time point, and draining LNs were resected to detect the expression of OVA. Paraffin sections of 3–4 μm thickness were prepared and stained by a two-step method using Envision kits (Dako Denmark A/S, Glostrup, Denmark). Antigens were prepared in 0.01 M ethylenediaminetetraacetic acid (EDTA) buffer (pH 9.0) at high temperature for 2 min. Exposure to 3% H2O2 in PBS solution for 10 min was used to block endogenous peroxidase activity. The primary antibody used in the immunostaining was goat anti-OVA (LifeSpan BioSciences, Inc., Seattle, WA, USA). The secondary antibody from the PicTure-PV6000 kit (Zymed Laboratories Inc., South San Francisco, CA, USA) and diaminobenzidine (ZSGB-Bio, Beijing, China) were used for color development according to the manufacturer’s instructions. Sections were examined using a BX20 fluorescence microscope camera (Olympus Corporation, Tokyo, Japan) at 200× magnification. Yellow and brown granules were positive in the cytoplasm under microscopic observation. The positive results were mainly located in the lymphoid tissue within the phagocytic group.
Determination of DC phenotype in draining LNs
The draining LNs of the mice in the experimental groups were harvested at 24 h and 48 h after the first subcutaneous injection. The LNs were ground up and then washed with PBS. The cell suspensions were centrifuged at 3,000 rpm for 10 min, and the supernatants were discarded. After washing again, the cell samples were stained to analyze their phenotype by flow cytometry. The anti-mouse antibodies, FITC-CD11c, APC-CD86, PE-Cy7-CD80, and PE-MHCII, were added to each sample, and four single standards and background were obtained. The cells were mixed with antibodies and incubated for 30 min at 4°C in the dark. The cell pellets were washed twice with staining buffer and centrifuged at 10,000 rpm for 5 min. The treated cells were fixed with 4% formaldehyde solution and then examined using flow cytometry (BD FACSCalibur, San Jose, CA, USA).
Splenocyte stimulation index (SI) assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to determine the SI of T and B lymphocytes after OVA (50 μg/mL) restimulation or treatment with PHA (10 μg/mL) or LPS (5 μg/mL). Four mice from each group were sacrificed at weeks 2 and 4 after the final booster, at the same time, as the challenge experiment. The spleens were aseptically removed, and the lymphocytes were isolated based on previously described methods.40 The cells were washed and resuspended in complete medium (Roswell Park Memorial Institute [RPMI] 1640 supplemented with fetal calf serum) and seeded at 5.0×106 cells/mL in 96-well flat-bottom plates (Corning Incorporated, Corning, NY, USA). Each sample was replicated in 16 wells with a volume of 80 μL per well, to which was added 20 μL OVA (four wells), 20 μL PHA (four wells), 20 μL LPS (four wells), or 20 μL RPMI-1640 medium as the BC (four wells). PHA and LPS were used to stimulate T lymphocyte and B lymphocyte proliferation, respectively. After incubation at 37°C and 5% CO2 for 60 h, 30 μL of MTT (5 mg/mL in PBS; Amresco Co., Solon, OH, USA) was added to each well, and the samples were incubated for another 4 h. The supernatant in each well was gently discarded, and then 100 μL of dimethyl sulfoxide (DMSO) was added to each well to completely dissolve the formazan crystals while shaking on a micro-oscillator for 5 min. The absorbance was measured at 570 nm using enzyme-linked immunosorbent assay (ELISA), and the SI was calculated according to the following formula:
|

OVA-specific CD3+ and CD4+/CD8+ T cell activation
To assess the potential of LBPL to prime antigen-specific CD3+ and CD4+/CD8+ T cells in vivo, an in vitro OVA restimulation experiment was performed. Flow cytometry was applied to measure the splenocytes collected from immunized mice 14 and 28 days after the third immunization. The splenocytes (5.0×106 cells/mL) were seeded in a 24-well plate and cultured in RPMI 1640 supplemented with 10% FBS and restimulated with OVA (50 μg/mL) for 60 h. Cell suspensions were then stained with the following set of fluorophore-conjugated anti-mouse antibodies: PE-cyanine5-CD3e, FITC-CD4, and PE-CD8a (eBioscience). After washing twice with staining buffer, the cell samples were examined by flow cytometry, and the data were analyzed using the Summit software.
OVA-specific antibodies
OVA-specific immunoglobulin G (IgG), IgG1, and IgG2a in the serum were quantitatively determined by ELISA according to a protocol described previously.41 In brief, 96-well microtiter plates (Costar, Corning, NY, USA) were coated overnight with 100 μL of OVA solution (5 μg/mL in 0.05 M carbonate buffer, pH 9.6) at 4°C. After removal of the unbound antigen by washing six times with PBST (0.01 M PBS containing 0.1% Tween-20, pH 7.4), the plates were blocked with 2% (w/v) BSA in PBST for 90 min at room temperature. After washing with PBST, serial dilutions of the serum (100 μL/well) in buffer (PBST containing 0.1% [w/v] BSA) were added to the plates and incubated for 1 h at 37°C. After subsequent washing steps, the plates were incubated with 100 μL of horseradish peroxidase (HRP)-conjugated anti-mouse IgG, IgG1, or IgG2a antibodies for 30 min at 37°C. Finally, HRP was quantified by adding the 3,3′,5,5′-tetramethylbenzidine (TMB) substrate solution and incubating for 20 min at room temperature. After stopping the reaction by adding 50 μL of 2 M H2SO4 to each well, the optical density (OD, 450 nm) was measured with a microplate reader. Samples were measured in duplicate.
Interferon gamma (IFN-γ), tumor necrosis factor alpha (TNF-α), and interleukin 4 and 6 (IL-4 and IL-6) secretion
To evaluate the levels of the cytokines, IFN-γ, TNF-α (Th1), IL-4, and IL-6 (Th2), in the collected serum (previously stored at −70°C), samples were assayed using commercially available cytokine ELISA kits (R&D Systems, Inc., Minneapolis, MN, USA) according to the manufacturer’s instructions. Briefly, all reagents and samples were equilibrated to room temperature before use. Then, 100 μL of assay diluent RD1W and 100 μL of standard, sample, or control were added to each well. The plates were incubated at room temperature for 2 h and then washed four times. Subsequently, 200 μL of conjugate was added to each well and incubated for 2 h at room temperature. The plates were then washed again and incubated at room temperature for 20 min in the dark after adding 200 μL of substrate solution. The intensity of the developed color in each well was monitored, and the enzyme–substrate reaction was stopped by the addition of 50 μL of stop solution. The absorbance at 450 nm was measured using a microplate reader within 30 minutes.
Statistical analysis
Data are represented as mean ± standard error of the mean (SEM). For the statistical analysis, a p-value of <0.05 was considered statistically significant. Duncan’s multiple range test was used to determine the differences between groups using the IBM SPSS Statistics 20.0 software (IBM Corporation, Armonk, NY, USA) for the analysis.
Results
Characterization of LBPL loaded with OVA
Transmission electron microscopy (TEM) provided the evidence for the spherical shape of LBPL, as previously reported.39 The particle size distribution and PDI of the nanoparticles were determined (Figure 1A and B) using a Zetasizer Nano ZS90 instrument on days 7, 15, 30, and 45. The mean particle size of LBPL (119.3±0.19 nm) was slightly increased after encapsulation of OVA (LBPL-OVA, 121.5±0.2 nm). The size of BL (111.67±0.3 nm) after encapsulation of OVA (BL-OVA, 121.13±0.37 nm) increased by a slightly greater amount. Over the 45 days of investigation, the particle size of LBPL-OVA only increased by 17.53 nm (from 121.5 nm to 139.03 nm); meanwhile, that of BL-OVA increased by 33.77 nm (from 121.13 nm to 154.9 nm). These results indicated that LBPL-OVA was more stable than BL-OVA. As shown in Figure 1B, at the beginning of the experiment, all the nanoparticles remained well dispersed with PDI values below 0.25. After 45 days, only small-scale fluctuations were observed for all the nanoparticles. The colloidal stability of LBPL-encapsulated antigen was further demonstrated.
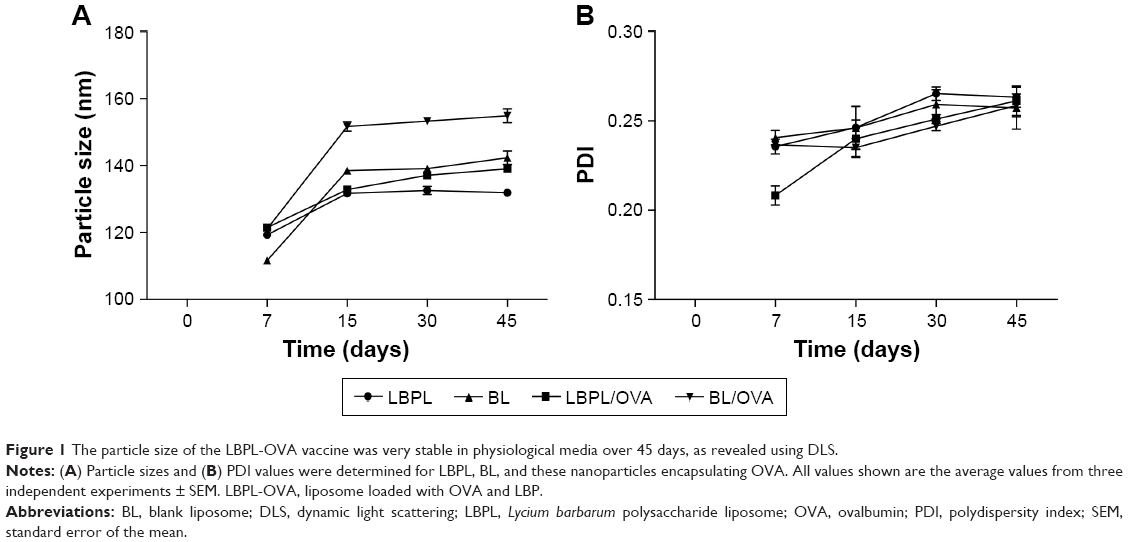 | Figure 1 The particle size of the LBPL-OVA vaccine was very stable in physiological media over 45 days, as revealed using DLS. |
Stability of antigen encapsulation and OVA release profiles
We next further studied the stability of the antigen encapsulation, as shown in Figure 2A and B at 4°C and 37°C, respectively. On day 0, the EEs for OVA within LBPL and BL were 71.45% and 72.4%, respectively. The slightly lower value for LBPL may be associated with the simultaneous encapsulation of polysaccharides and antigens. Over the subsequent 44 days, the extent of encapsulation was observed to decline to a certain extent for all the formulations. The entrapment rate at 4°C exhibited a slower decrease than that observed at 37°C, and remained above 60% throughout the duration of the experiment. This proved that storing the liposome-encapsulated antigen at 4°C maintained good colloidal stability and higher EE of OVA.
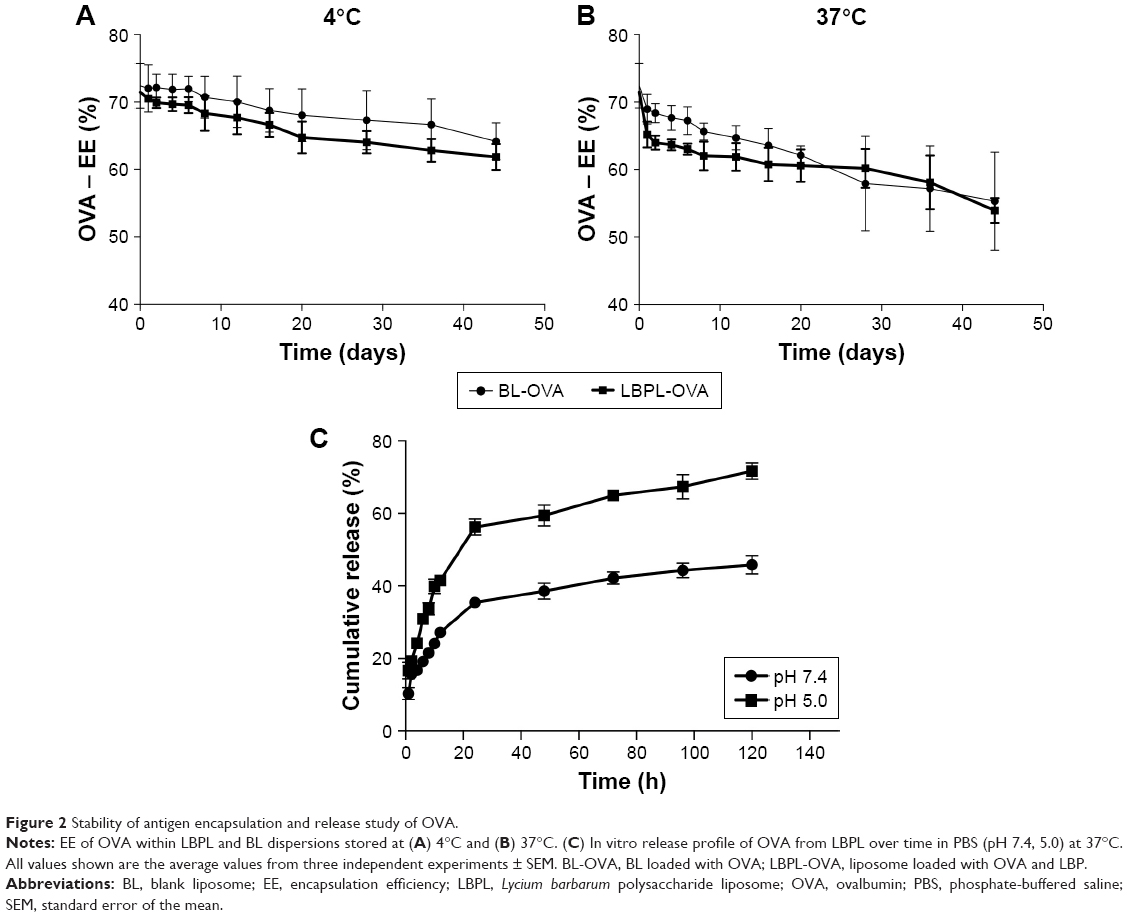 | Figure 2 Stability of antigen encapsulation and release study of OVA. |
The cumulative release profiles of LBPL-OVA at physiological (pH 7.4) and endosomal (pH 5.0) pH were shown in Figure 2C. After 24 h of incubation, a burst release of approximately 60% was observed in pH 5.0 medium. By contrast, only a few of OVA released from the LBPL-OVA during pH 7.4 medium. Moreover, antigen of LBPL-OVA in pH 5.0 medium released above 70% at 120 h. While the remaining OVA of LBPL-OVA in pH 7.4 medium was gradually released over 4 days.
Antigen transport into draining LNs
To determine whether increased accumulation of OVA in LN-resident APCs and prolonged DC activation occurred upon subcutaneous administration of LBPL-OVA, we measured antigen transport to the draining LNs by immunohistochemistry. As shown in Figure 3, the yellow and brown granules were positive in the cytoplasm under microscopic examination. Seven days after the first immunization, abundant levels of the antigen were detected in the LNs of mice immunized with LBPL-OVA. The positive results were mainly located in the lymphoid tissue within the phagocytic group. These results suggested that LBPL could provide not only adequate antigen exposure in the LNs but also long-term antigen persistence.
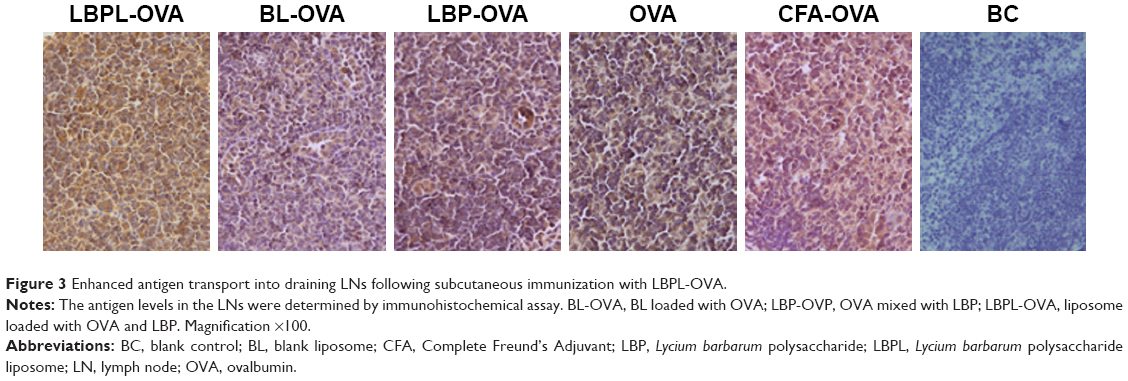 | Figure 3 Enhanced antigen transport into draining LNs following subcutaneous immunization with LBPL-OVA. |
LBPL promotes activation of LN-resident DCs
We first investigated whether the increased accumulation of OVA in the APCs would alter DC activation compared to soluble OVA. In this experiment, we mixed soluble OVA with BL, LBP, or LBPL as adjuvant. The results for the samples taken at 24 hours or 48 hours after subcutaneous immunization showed comparable increased frequencies of activated LN MHCII+ in CD11c+ DCs for each treatment group relative to naive mice (Figure 4A). BLs or LBP also elicited a modest level of MHCII upregulation, which was intermediate between the results for soluble OVA and DCs from naive mice. This upregulation may reflect internalization of the smallest particles in the BL preparation leading to inflammasome activation.42 At 2 days after injection, DCs with a mature phenotype were decreasing toward naive levels in the soluble OVA groups. In contrast, LBPL-OVA generally mediated increasing frequencies of activated DCs compared with the other formulations and compared with the same vaccine at day 1. Moreover, the LBPL-OVA group always exceeded the positive control group (Complete Freund’s Adjuvant [CFA]-OVA). The numbers of mature phenotype, CD80+ and CD86+, in these subpopulations decreased slightly from day 1 to day 2 (Figure 4B and C). Meanwhile, the LBPL-OVA group was still the highest and showed superior results to the soluble OVA groups (p<0.05). These results suggest that the liposome-encapsulated LBP promoted more enduring DC activation, in a manner that required sustained LBP release and was not simply the sum of responses triggered by the liposomes and LBP.
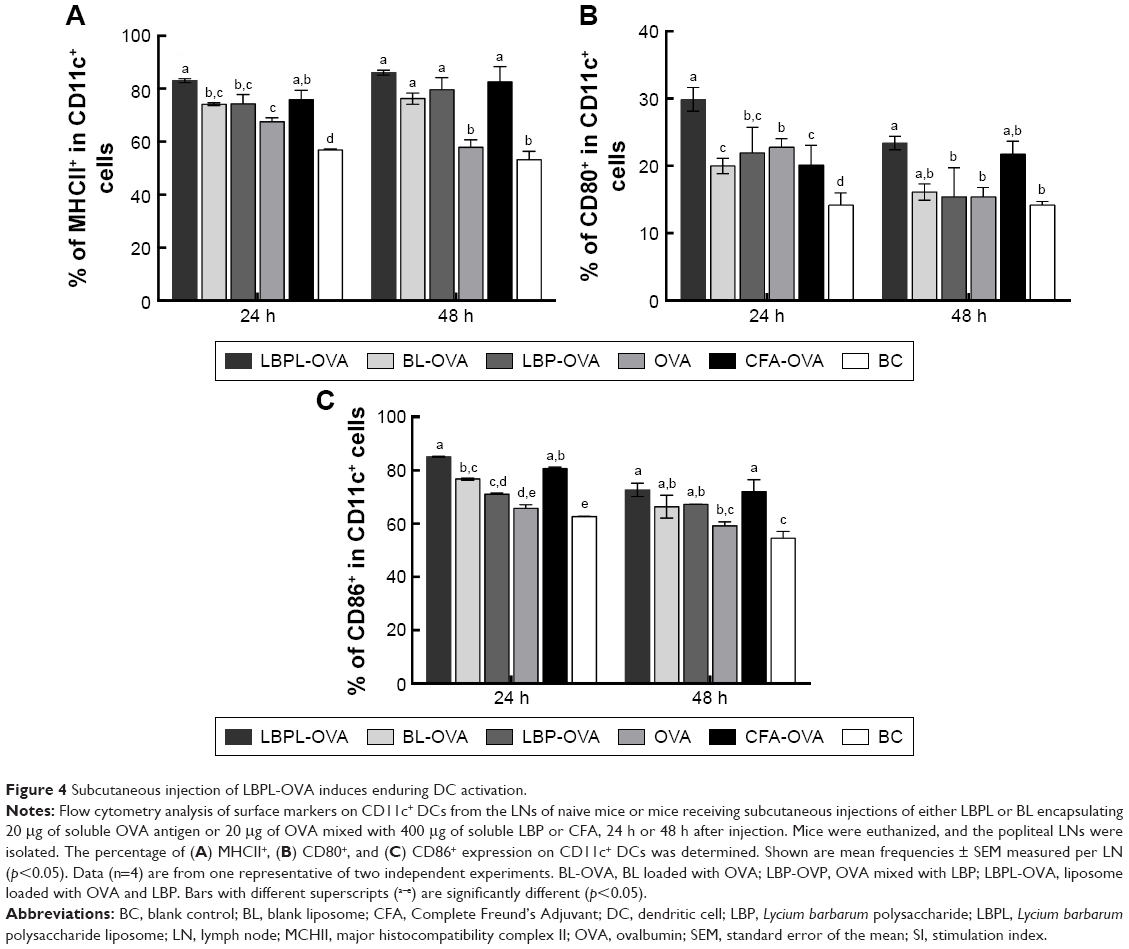 | Figure 4 Subcutaneous injection of LBPL-OVA induces enduring DC activation. |
Splenocyte proliferation assay
An ex vivo splenocyte proliferation assay was performed 14 days and 28 days after the third immunization to assess the influence of various antigen–nanoparticle formulations on the splenocyte proliferative responses. The SI was used as an indicator of lymphocyte proliferation. As shown in Figure 5A, under the restimulation of OVA, splenocytes collected from mice immunized with LBPL-encapsulated antigen vaccine formulation proliferated more efficiently than those collected from mice immunized with BL-encapsulated antigen, soluble antigen mixed with either LBP or CFA, or soluble antigen alone (p<0.05). In addition, the SI of the LBPL-OVA group exhibited a greater increase from day 14 to day 28 compared with the other formulations. To further investigate the splenocyte proliferation, LPS (Figure 5B) and PHA (Figure 5C) were used to stimulate B and T lymphocytes, respectively. The data revealed that the LBPL-encapsulated antigen vaccine formulation still led to more efficient proliferation of the B and T lymphocytes than the other groups (p<0.05). Hence, these results show that the LBPL-encapsulated antigen vaccine formulation led to more potent and sustained antigen-specific immune responses than the other formulations.
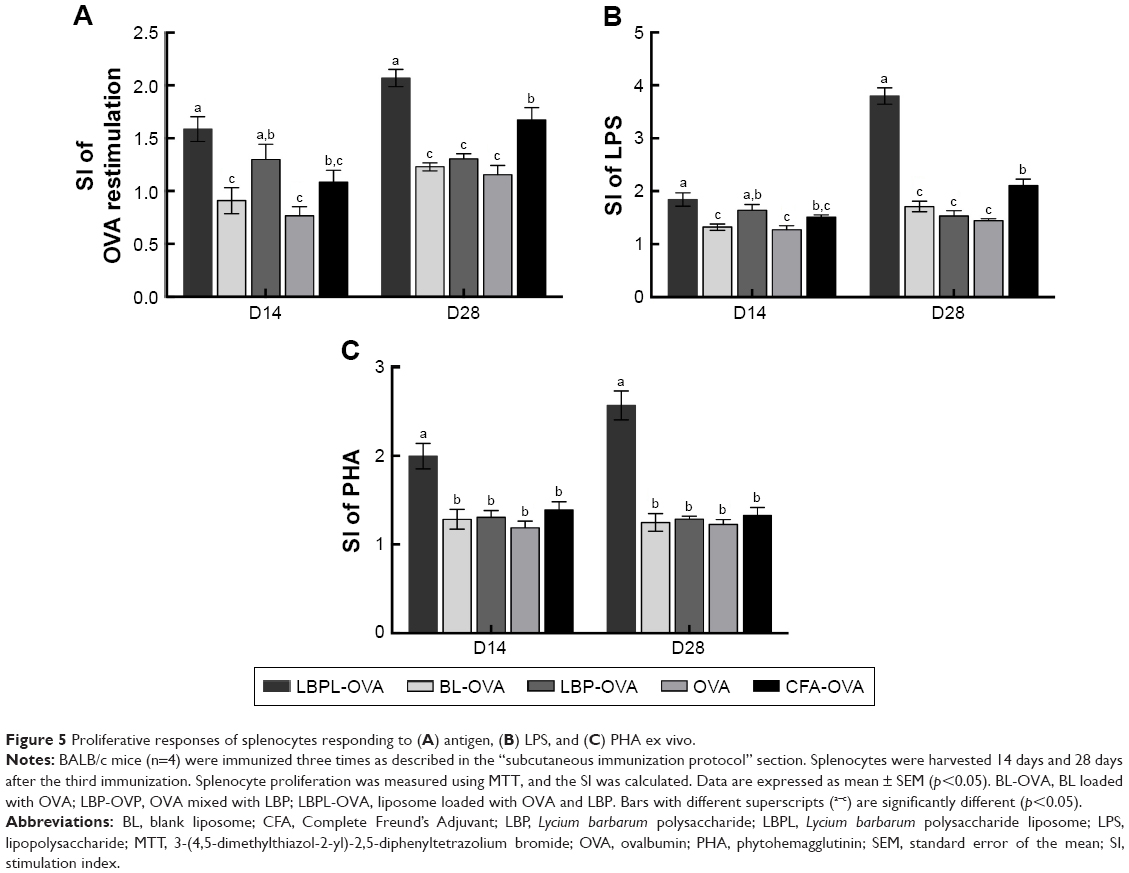 | Figure 5 Proliferative responses of splenocytes responding to (A) antigen, (B) LPS, and (C) PHA ex vivo. |
OVA-specific CD3+ and CD4+/CD8+ T cell activation
In this study, we tested whether the OVA-based vaccines could induce the activation of antigen-specific CD4+/CD8+ T cells after 2 weeks and 4 weeks following the final immunization. Lymphocytes were isolated from the spleens of the animals and then incubated with 50 μg/mL of OVA for 60 h. As shown in Figure 6A, LBPL containing 20 μg OVA was found to stimulate CD3+ T cell proliferation, the level of which was higher than that of the other groups (p<0.05). Importantly, the amount of CD4+/CD8+ T cells (Figure 6B) increased significantly for the group of mice immunized with LBPL-OVA compared with the group immunized with CFA-OVA (positive control group). From 14 days to 28 days, a prolonged antigen-specific CD3+ and CD4+/CD8+ T cell activation was elicited by LBPL-OVA, suggesting that LBPL possesses a sustained-release effect.
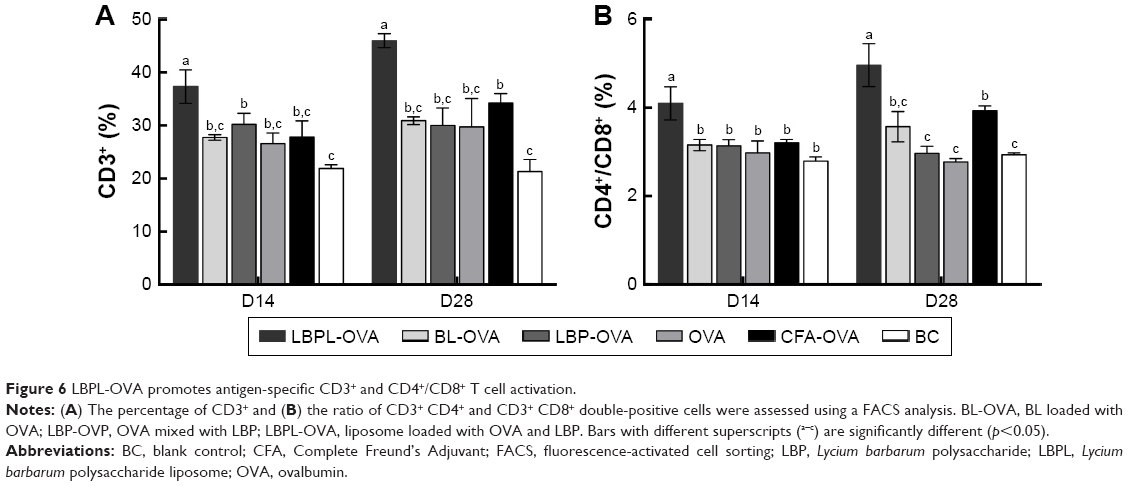 | Figure 6 LBPL-OVA promotes antigen-specific CD3+ and CD4+/CD8+ T cell activation. |
OVA-specific antibody responses
To investigate the effects of the various formulations on antibody response, serum samples were collected over time from mice subcutaneously immunized with particle-encapsulated antigen (LBPL-OVA and BL-OVA), soluble antigen alone (OVA), and soluble antigen mixed with either LBP (OVA mixed with LBP [LBP-OVA]), CFA (CFA-OVA), or PBS. The serum levels of IgG, IgG1, and IgG2a were then evaluated by ELISA. As shown in Figure 7A, the LBPL-OVA vaccine formulation induced significantly higher antigen-specific IgG than all the other formulations from day 7 to day 21 after the third immunization. On day 28, the serum IgG levels elicited by LBPL-OVA declined, although they still remained significantly higher than those elicited by soluble antigen alone or antigen mixed with CFA and those observed for the control naive mice. IgG2a antibody production revealed a Th1-polarized immune response, and the ratio of IgG2a/IgG1 was indicative of a Th1-biased immune response.43 The LBPL-OVA vaccine formulation generated the highest IgG2a/IgG1 ratios, which were significantly higher than those for all other formulations (Figure 7B).
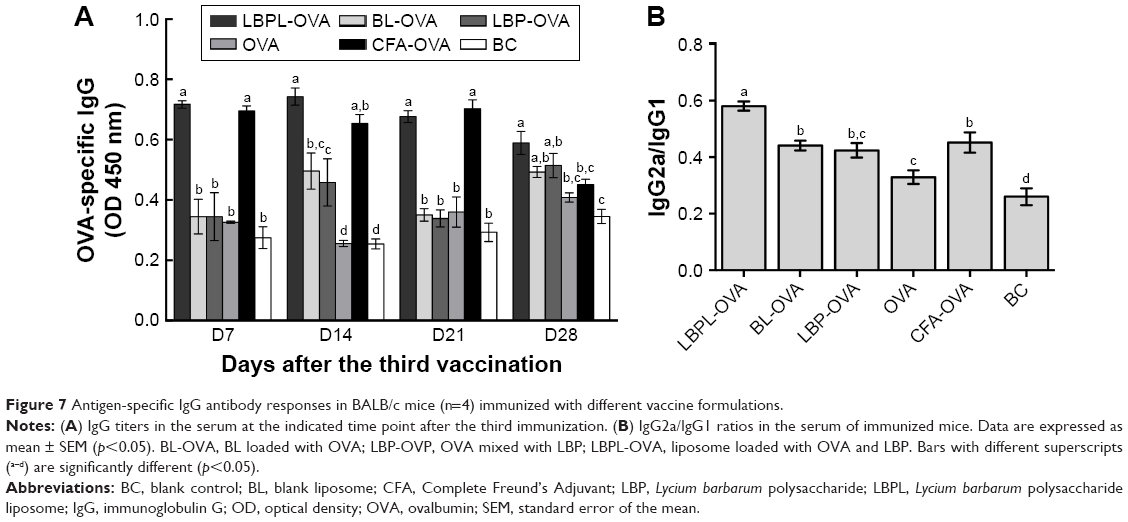 | Figure 7 Antigen-specific IgG antibody responses in BALB/c mice (n=4) immunized with different vaccine formulations. |
Cytokine levels secreted ex vivo
To measure the levels of IFN-γ, TNF-α (Th1), IL-4, and IL-6 (Th2) in the mice, serum samples were collected on days 14 and 28 after the third immunization. As shown in Figure 8A and B, the LBPL-OVA vaccine formulation induced similar increases in the levels of IFN-γ and TNF-α. From day 14 to day 28, the levels of IFN-γ and TNF-α in the LBPL-OVA group increased and were always significantly higher than those elicited by all other formulations. As for Th2 cytokine secretion (Figure 8C and D), the results revealed that the levels of IL-4 and IL-6 in mice immunized with LBPL-OVA vaccine formulation were significantly higher than those for the other groups, although they were low compared with the positive control group (CFA-OVA). On the whole, the LBPL-OVA vaccine formulation induced higher levels of both Th1 and Th2 cytokine secretion, indicating stronger immune responses. We also observed a Th1-biased immune response, which was consistent with the antibody response results.
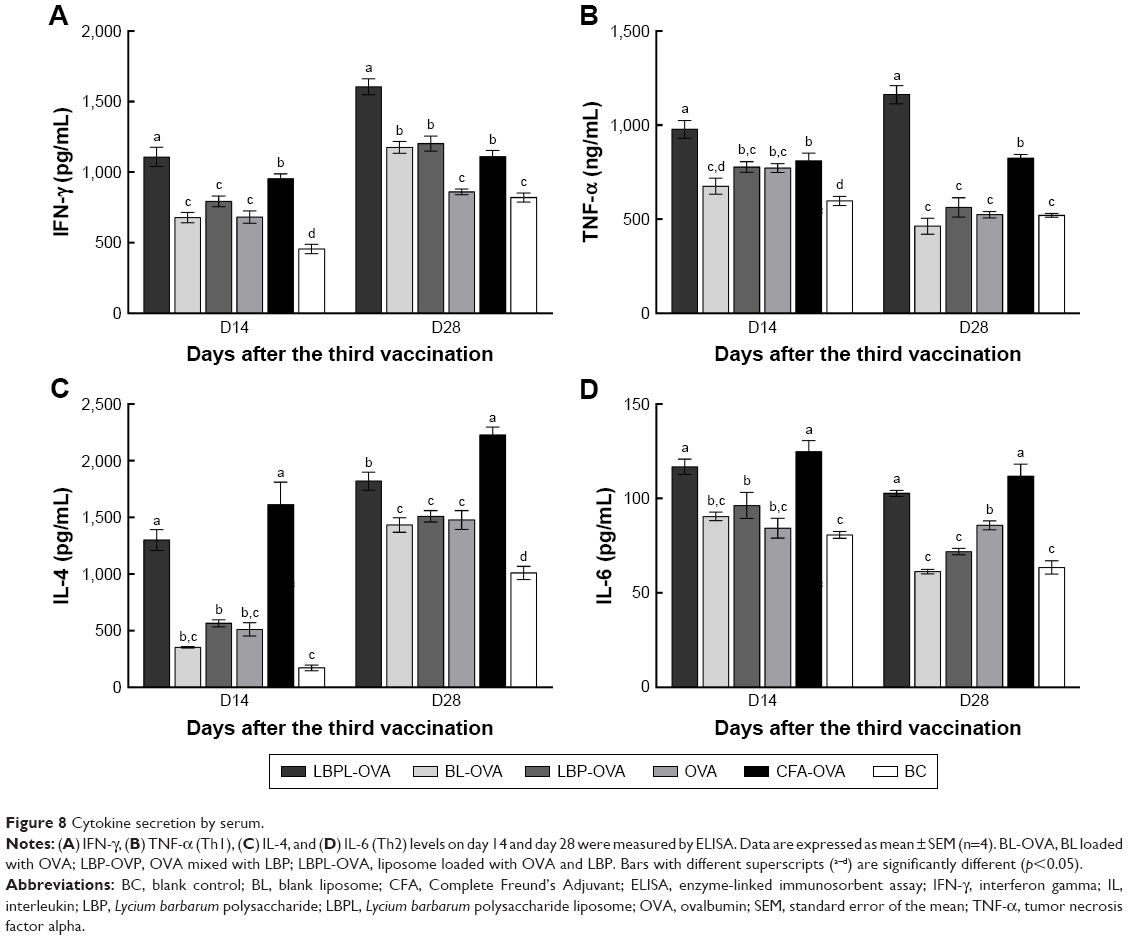 | Figure 8 Cytokine secretion by serum. |
Discussion
Nanoparticle delivery of protein subunit vaccines to the LNs allows the antigens to interact directly with the immune system. A delivery vector that traffics quickly and efficiently to the draining LNs would be beneficial for delivering antigens to B cells and other APCs resident in the LNs.44 After migrating to the draining LNs and presenting an antigen fragment to T lymphocytes with high expression levels of MHC class I and II, adhesion, and co-stimulatory molecules and secretion of inflammatory cytokines,45 DCs activate T cells, thereby initiating and regulating Th1 and Th2 immune responses.46,47
In this study, we chose liposomes as the vaccine and adjuvant vesicle, encapsulating LBP as the adjuvant and OVA as the model vaccine. The colloidal stability of LBPL-OVA was studied by measuring the particle size, PDI, and EE of OVA within LBPL and BL at 4°C and 37°C for 44 days. From the data shown in Figure 1A and B, we found that the LBPL particle size only increased by 17.53 nm upon encapsulation of OVA and the PDI changed from 0.208 to 0.261. The EE of OVA (Figure 2A and B) in LBPL-OVA declined by about 10% and 17.5% at 4°C and 37°C, respectively, yet still remained above 60% at 4°C. The results indicated that LBPL-OVA possessed a high integrity and remained stable at 4°C. The cumulative release profiles of LBPL-OVA at physiological (pH 7.4) and endosomal (pH 5.0) pH suggested that antigen releasing from LBLP in the physical environment was in a sustained manner.
Draining LNs are the primary site of action for initiating adaptive immunity, where T and B cells, the major cell types involved in the humoral immune response, meet the antigens or APCs.44 The efficient delivery of vaccine components into LNs is critical for mounting effective immune responses, because antigens that fail to reach the lymphoid organs may be effectively ignored by the immune system.48 Therefore, we first investigated whether antigen transport to the draining LNs and increased accumulation of OVA in APCs would alter DC activation. The amount of antigen delivery to the draining LNs was measured by immunohistochemistry. As clearly shown in Figure 3, the most abundant antigen was detected in the LNs of mice immunized with the LBPL-OVA compared with other formulations. We also found that the maturation markers CD80, CD86, and MHCII of DCs in the LNs increased in a dose-dependent fashion in response to LBPL-OVA vaccine formulations, suggesting that LBPL-OVA promotes the activation of LN-resident DCs.
Lymphocyte proliferation reflects the situation of the body’s immune response. The lymphocyte proliferative response depends on the mitogen used.40 To evaluate the immune response of LBPL encapsulating the OVA adjuvant, we analyzed the restimulation using OVA (Figure 5A), LPS (Figure 5B), and PHA (Figure 5C) toward lymphocytes. The results showed that the SI of the LBPL-OVA group mediated larger increasing frequencies compared with the other formulations from day 14 to day 28. In particular, under the stimulation of LPS, a twofold increase was observed for the LBPL-OVA group on day 28. Cell-mediated immunity allows the recognition and elimination of non-normal cells such as tumor cells or virus-infected cells. The activation of antigen-specific CD4+ T and CD8+ CTLs is a major feature of the cell-mediated immune response. The activated CD4+ T cells secrete many nonoverlapping sets of cytokines to mediate CTL and B cell functions.49 Previous results have shown that the ratio of CD4+ T cells to CD8+ T cells in the peripheral blood of tissues obtained from cancer patients decreases as the cancer becomes more severe.50 In this study, the ratio of CD3+ CD4+ and CD3+ CD8+ double-positive cells was determined (Figure 6B). We found that the amount of CD4+/CD8+ T cells increased significantly in the group of mice immunized with LBPL-OVA compared with the group immunized with CFA-OVA, indicating that the CD4+ T cells could proliferate to regulate the B cell and CTL activity after immunization with LBPL-OVA.
Based on the results in the current study, we therefore proposed the LBPL-OVA vaccine formulation to act as a vaccine adjuvant to promote antigen-specific humoral immune responses. We also analyzed the IgG1 and IgG2a antibody subtypes as markers to evaluate the Th2 and Th1 immune response. Our results showed that immunization with LBPL-OVA produced a high antibody level relative to CFA-OVA, and this level remained high until day 28 (Figure 7A). Moreover, the LBPL-OVA vaccine formulation generated the highest IgG2a/IgG1 ratios, which were significantly higher than those for all other groups (Figure 7B). This suggested that the LBPL-OVA vaccine formulation promoted a Th1-biased immune response. The enhanced antibody levels induced by immunization with antigen-containing liposomes may be attributable to physical association of the antigen with the liposome. We speculate that since soybean phospholipid contains both hydrophobic and hydrophilic residues, the protein might be incorporated in such a way that hydrophobic residues spanned the bilayer. Moreover, cholesterol may impart greater bilayer stability, resulting in liposome persistence for a longer period in vivo. Thus, these particles may be more visible to the immune system and be actively phagocytosed by APCs, resulting in effective presentation by MHCII molecules and activation of CD4 T cells.16 Coincidentally, the significantly higher levels of IFN-γ and TNF-α induced by LBPL-OVA indicate polarization toward a Th1 immune response.
Conclusion
This study investigated the effect of an antigen encapsulated within liposome formulations on the immune responses elicited by liposome-adjuvanted vaccines. The enhanced immune responses elicited by the LBPL-OVA vaccine formulation may be attributable to various factors, including the colloidal stability of LBPL-OVA, high and stable antigen EE, abundant antigen accumulation in LNs, efficient induction of DC activation, long-term antigen persistence, and efficient CD4+ and CD8+ T cell proliferation. Understanding the effect of the LBPL-OVA formulations on the resulting immune responses may have significant implications for rational vaccine design.
Acknowledgments
This project was supported by the National Natural Science Foundation of China (Grant nos 31672596, 31372472), the Special Fund for Agro-scientific Research in the Public Interest (Grant nos 201303046, 201403051), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). We are grateful to all the other staff members at the Institute of Traditional Chinese Veterinary Medicine of Nanjing Agricultural University for their assistance in this study.
Disclosure
The authors report no conflicts of interest in this work.
References
Garçon N, Van MM. Recent clinical experience with vaccines using mpl- and qs-21-containing adjuvant systems. Expert Rev Vaccines. 2011;10(4):471–486. | ||
Garçon N, Chomez P, Mechelen MV. GlaxoSmithKline adjuvant systems in vaccines: concepts, achievements and perspectives. Expert Rev Vaccines. 2007;6(5):723–739. | ||
Burdin N, Guy B, Moingeon DP. Immunological foundations to the quest for new vaccine adjuvants. BioDrugs. 2004;18(2):79–93. | ||
Guy B. The perfect mix: recent progress in adjuvant research. Nat Rev Microbiol. 2007;5(7):505–517. | ||
Black M, Trent A, Tirrell M, Olive C. Advances in the design and delivery of peptide subunit vaccines with a focus on toll-like receptor agonists. Expert Rev Vaccines. 2010;9(2):157–173. | ||
Marrack P, Mckee AS, Munks MW. Towards an understanding of the adjuvant action of aluminium. Nat Rev Immunol. 2009;9(4):287–293. | ||
Zaharoff DA, Rogers CJ, Hance KW, Schlom J, Greiner JW. Chitosan solution enhances both humoral and cell-mediated immune responses to subcutaneous vaccination. Vaccine. 2007;25(11):2085–2094. | ||
Chen X, Kim P, Farinelli B, et al. A novel laser vaccine adjuvant increases the motility of antigen presenting cells. PLoS One. 2010;5(10):e13776. | ||
Singh M, O’Hagan DT. Recent advances in vaccine adjuvants. Pharm Res. 2002;19(6):715–728. | ||
Behboudi S, Morein B, Rönnberg B. Isolation and quantification of Quillaja saponaria Molina saponins and lipids in iscom-matrix and iscoms. Vaccine. 1995;13(17):1690–1696. | ||
Zurbriggen R, Glück R. Immunogenicity of IRIV-versus alum-adjuvanted diphtheria and tetanus toxoid vaccines in influenza primed mice. Vaccine. 1999;17(11–12):1301–1305. | ||
Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36. | ||
Fan Y, Zhang Q. Development of liposomal formulations: from concept to clinical investigations. Asian J Pharm Sci. 2013;8(2):81–87. | ||
Al-Jamal WT, Kostarelos K. Liposomes: from a clinically established drug delivery system to a nanoparticle platform for theranostic nanomedicine. Acc Chem Res. 2011;44(10):1094–1104. | ||
Caracciolo G. Liposome-protein corona in a physiological environment: challenges and opportunities for targeted delivery of nanomedicines. Nanomedicine. 2015;11(3):543–557. | ||
Faisal SM, Yan WW, Mcdonough SP, Chang YF. Leptospira immunoglobulin-like protein A variable region (LigaVar) incorporated in liposomes and PLGA microspheres produces a robust immune response correlating to protective immunity. Vaccine. 2009;27(3):378–387. | ||
Mallick AI, Singha H, Chaudhuri P, et al. Liposomised recombinant ribosomal L7/L12 protein protects BALB/c mice against Brucella abortus 544 infection. Vaccine. 2007;25(18):3692–3704. | ||
Lin YY, Kao HW, Li JJ, et al. Tumor burden talks in cancer treatment with pegylated liposomal drugs. PLoS One. 2013;8(5):e63078. | ||
Barenholz Y. Doxil®; – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. | ||
Perche F, Torchilin VP. Recent trends in multifunctional liposomal nanocarriers for enhanced tumor targeting. J Drug Deliv. 2013;2013:Article ID 705265. | ||
Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001;53(2):283. | ||
Yu G, Duan Y, Fang G, Yan Z, Wang S. Polysaccharides from fruit calyx of Physalis alkekengi var. francheti: isolation, purification, structural features and antioxidant activities. Carbohydr Polym. 2009;77(2):188–193. | ||
Bao Y. Effect of methylation on the structure and radical scavenging activity of polysaccharides from longan (Dimocarpus longan lour.) fruit pericarp. Food Chem. 2010;118(2):364–368. | ||
Xie JH, Tang W, Jin ML, Li JE, Xie MY. Recent advances in bioactive polysaccharides from Lycium barbarum, l. Zizyphus jujuba, mill, Plantago, spp. and Morus, spp.: structures and functionalities. Food Hydrocolloid. 2016;60:148–160. | ||
Ke M, Zhang XJ, Han ZH, et al. Extraction, purification of Lycium barbarum, polysaccharides and bioactivity of purified fraction. Carbohydr Polym. 2011;86(1):136–141. | ||
Jin M, Huang Q, Zhao K, Shang P. Biological activities and potential health benefit effects of polysaccharides isolated from Lycium barbarum l. Int J Biol Macromol. 2013;54(1):16–23. | ||
Zhang X, Li Y, Cheng J, et al. Immune activities comparison of polysaccharide and polysaccharide-protein complex from Lycium barbarum l. Int J Biol Macromol. 2014;65(5):441. | ||
Su CX, Duan XG, Liang LJ, et al. Lycium barbarum, polysaccharides as an adjuvant for recombinant vaccine through enhancement of humoral immunity by activating Tfh cells. Vet Immunol Immunopathol. 2014;158(1–2):98–104. | ||
Zhang M, Wang F, Liu R, Tang X, Zhang Q, Zhang Z. Effects of superfine grinding on physicochemical and antioxidant properties of Lycium barbarum polysaccharides. LWT Food Sci Technol. 2014;58(2):594–601. | ||
Zhang M, Tang X, Wang F, Zhang Q, Zhang Z. Characterization of Lycium barbarum polysaccharide and its effect on human hepatoma cells. Int J Biol Macromol. 2013;61(10):270–275. | ||
Shen L, Du G. Lycium barbarum, polysaccharide stimulates proliferation of MCF-7 cells by the ERK pathway. Life Sci. 2012;91(9–10):353–357. | ||
Zhao Z, Luo Y, Li G, Zhu L, Wang Y, Zhang X. Thoracic aorta vasoreactivity in rats under exhaustive exercise: effects of Lycium barbarum polysaccharides supplementation. J Int Soc Sports Nutr. 2013;10(1):1–7. | ||
Hubbell JA, Thomas SN, Swartz MA. Materials engineering for immunomodulation. Nature. 2009;462(7272):449–460. | ||
Reddy ST, Aj VDV, Simeoni E, et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol. 2007;25(10):1159–1164. | ||
Thomas SN, Aj VDV, O’Neil CP, et al. Engineering complement activation on polypropylene sulfide vaccine nanoparticles. Biomaterials. 2011;32(8):2194. | ||
Ballester M, Nembrini C, Dhar N, et al. Nanoparticle conjugation and pulmonary delivery enhance the protective efficacy of ag85b and cpg against tuberculosis. Vaccine. 2011;29(40):6959–6966. | ||
Dane KY, Nembrini C, Tomei AA, et al. Nano-sized drug-loaded micelles deliver payload to lymph node immune cells and prolong allograft survival. J Control Release. 2011;156(2):154. | ||
Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol. 2005;5(8):617. | ||
Bo R, Ma X, Feng Y, et al. Optimization on conditions of Lycium barbarum, polysaccharides liposome by RSM and its effects on the peritoneal macrophages function. Carbohydr Polym. 2015;117:215–222. | ||
Fan Y, Ma L, Zhang W, et al. Liposome can improve the adjuvanticity of astragalus polysaccharide on the immune response against ovalbumin. Int J Biol Macromol. 2013;60(6):206–212. | ||
Yuan L, Wu L, Chen J, Wu Q, Hu S. Paclitaxel acts as an adjuvant to promote both th1 and th2 immune responses induced by ovalbumin in mice. Vaccine. 2010;28(27):4402–4410. | ||
Sharp FA, Ruane D, Claass B, et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the nalp3 inflammasome. Proc Natl Acad Sci U S A. 2009;106(3):870. | ||
Jusforgues-Saklani H, Uhl MN, Lemaitre F, et al. Antigen persistence is required for dendritic cell licensing and cd8+ t cell cross-priming. J Immunol. 2008;181(5):3067–3076. | ||
Mueller SN, Tian S, Desimone JM. Rapid and persistent delivery of antigen by lymph node targeting print nanoparticle vaccine carrier to promote humoral immunity. Mol Pharm. 2015;12(5):1356. | ||
Villadangos JA, Cardoso M, Steptoe RJ, et al. MHC class ii expression is regulated in dendritic cells independently of invariant chain degradation. Immunity. 2001;14(6):739–749. | ||
Fytianos K, Rodriguez-Lorenzo L, Clift MJ, et al. Uptake efficiency of surface modified gold nanoparticles does not correlate with functional changes and cytokine secretion in human dendritic cells in vitro. Nanomedicine. 2015;11(3):633–644. | ||
Bakhru P, Sirisaengtaksin N, Soudani E, Mukherjee S, Khan A, Jagannath C. Bcg vaccine mediated reduction in the MHC-II expression of macrophages and dendritic cells is reversed by activation of toll-like receptors 7 and 9. Cell Immunol. 2014;287(1):53–61. | ||
Jewell CM, López SCB, Irvine DJ. In situ engineering of the lymph node microenvironment via intranodal injection of adjuvant-releasing polymer particles. Proc Natl Acad Sci U S A. 2010;108(38):15745–15750. | ||
Zhu J, Paul WE. Cd4 t cells: fates, functions, and faults. Blood. 2008;112(5):1557. | ||
Liu Y, Jiao F, Qiu Y, et al. The effect of gd@c82(oh)22 nanoparticles on the release of Th1/Th2 cytokines and induction of TNF-alpha mediated cellular immunity. Biomaterials. 2009;30(23–24):3934. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.