Back to Journals » Lung Cancer: Targets and Therapy » Volume 9
Silicosis and lung cancer: current perspectives
Authors Sato T ![]() , Shimosato T
, Shimosato T ![]() , Klinman DM
, Klinman DM
Received 15 June 2018
Accepted for publication 31 August 2018
Published 26 October 2018 Volume 2018:9 Pages 91—101
DOI https://doi.org/10.2147/LCTT.S156376
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Sai-Hong Ou
Takashi Sato,1 Takeshi Shimosato,2 Dennis M Klinman3
1Department of Pulmonology, Yokohama City University Graduate School of Medicine, Yokohama 236-0004, Japan; 2Department of Interdisciplinary Genome Sciences and Cell Metabolism, Institute for Biomedical Sciences, Shinshu University, Nagano 399-4598, Japan; 3Cancer and Inflammation Program, National Cancer Institute, Frederick, MD 21702, USA
Abstract: “Silica” refers to crystalline particles formed by the combination of silicon with oxygen. Inhalation of silica particles promotes the development of pulmonary fibrosis that over prolonged periods increases the risk of lung cancer. The International Agency for Research on Cancer (IARC) classified crystalline silica as a human carcinogen in 1997. This categorization was questioned due to 1) the absence of dose–response findings, 2) the presence of confounding variables that complicated interpretation of the data and 3) potential selection bias for compensated silicosis. Yet, recent epidemiologic studies strongly support the conclusion that silica exposure increases the risk of lung cancer in humans independent of confounding factors including cigarette smoke. Based on this evidence, the US Occupational Safety and Health Administration (OSHA) lowered the occupational exposure limit for crystalline silica from 0.1 to 0.05 mg/m3 in 2013. Further supporting the human epidemiologic data, murine models show that chronic silicosis is associated with an increased risk of lung cancer. In animals, the initial inflammation induced by silica exposure is followed by the development of an immunosuppressive microenvironment that supports the growth of lung tumors. This work will review our current knowledge of silica-associated lung cancers, highlighting how recent mechanistic insights support the use of cutting-edge approaches to diagnose and treat silica-related lung cancer.
Keywords: silicosis, lung cancer, inflammation, fibrosis, occupational lung disease
Introduction
Silicosis is an irreversible and incurable lung disease caused by the inhalation of dust containing crystalline silica particles. Silicosis is one of the most important occupational diseases in the world.1–3 Approximately 2 million workers in USA,4,5 2 million workers in Europe,6 0.5 million workers in Japan7 and more than 23 million workers in China8 are estimated to have been exposed with 4.2% of deaths among industrial workers in China being attributed to silica dust exposure.9 Occupational exposure to crystalline silica occurs in mining, construction, stone works/quarries, pottery manufacturing and denim sandblasting.10–13 The greatest number of exposures is in the construction industry; however, workers in the mining industry experience the highest risk of lung cancer due to the magnitude and duration of their silica exposure.14,15
Silicosis is classified into phases: acute (developing within weeks to a few years), accelerated (developing within 10 years) and chronic (developing more than 10 years after initial exposure). The onset of disease is influenced by both the concentration and duration of exposure.4 A diagnosis of acute silicosis is supported in exposed individuals who experience the rapid onset and/or worsening of symptoms including dyspnea, cough, fever and pleuritic pain.1 More difficult is the diagnosis of chronic silicosis, which can be asymptomatic or may present with only mild-to-moderate exertional dyspnea.4 To aid in diagnosis, regular clinical examination accompanied by chest radiography is recommended for subjects having occupational exposure to silica dust. Unfortunately, the International Labor Organization 12-point grading system for radiographic opacities fails to identify silicosis in approximately half of all individuals with disease as determined by subsequent pathologic examination.16
There is little doubt that prolonged inhalation of crystalline silica can cause silicosis. Mechanistically, inhaled silica particles under 10 µm can reach and persist in the peripheral lung where they are phagocytosed by macrophages and/or taken up by epithelial cells, which results in the generation of reactive oxygen species (ROS).17,18 ROS causes injury to bronchial/alveolar epithelial cells and initiates a repair/regeneration process that supports fibrogenesis and carcinogenesis.18
Epidemiologic studies show that silica exposure is associated with an increased risk of esophageal, stomach, skin and most importantly lung cancer.19–21 The carcinogenicity of silica was reviewed by the International Agency for Research on Cancer (IARC), which concluded in 1997 that silica inhalation caused human lung cancer. This resulted in crystalline silica being classified as group 1 carcinogen.1 There is strong and consistent evidence of a dose–response relationship between silica exposure and lung cancer risk, although the RR posed by silica is lower than certain other pulmonary carcinogens such as cigarette smoke or asbestos. An IARC meta-analysis of data collected after 1997 confirmed that silica was a pulmonary carcinogen.22 In response, the US Occupational Safety and Health Administration (OSHA) lowered the permissible occupational exposure of silica dust from 0.1 mg/m3 (0.25 mg/m3 for the construction industry) to 0.05 mg/m3 in 2013.23 Whether this technically attainable change achieved the predicted reduction in lung cancer cases attributable to silicosis by two to threefold is under evaluation.5
Therapy of silicosis
Silicosis is a progressive disease characterized by fibrotic changes in the lungs. There is no known cure for silicosis. Supportive therapy involving the prevention of infection, use of bronchodilators and oxygen supplementation are the mainstays of treatment.4 Physicians should also consider that silicosis increases susceptibility to other disorders such as tuberculosis, autoimmunity and chronic renal disease.24 However, lung pathology dominates the clinical picture of silicosis, with enormous effort being expended toward elucidating the mechanisms underlying the inflammation triggered by silica and identifying additional therapeutic strategies. In animal studies, intratracheal administration of silica particles induces inflammatory cell infiltration, the accumulation of chemokines and cytokines (including tumor necrosis factor-α [TNF-α], IL-1 and IL-6) and the generation of ROS in the lungs. These factors cause lung cells to undergo apoptosis resulting in the destruction of pulmonary tissue.25,26
One mechanism that contributes to disease progression involves the activation of the ROS-derived mitogen-activated protein kinase system.27,28 As this system can be downregulated via the extracellular signal-regulated kinase (ERK) pathway, several studies explored whether upregulating the production of antioxidants or ERK could protect against silica-induced ROS.25,29 Sato et al reported that heme oxygenase (HO)-1, a strong antioxidant that is the rate-limiting enzyme in heme catabolism, could attenuate the progression of silicosis in mice.30 HO-1 reduces silica-induced ROS levels by scavenging ROS via the heme degradation products, bilirubin and carbon monoxide.31 This benefits the host not only by reducing the direct harm caused by ROS but also by inhibiting ROS-dependent ERK activation that supports epithelial cell proliferation and tumorigenesis.32–34 Of interest, these findings suggest that serum HO-1 levels might be markers of disease severity in individuals exposed to silica.35
There is also interest in the use of Probucol, an HO-1 inducer, for the treatment of silicosis. Probucol was developed to treat atherosclerosis but failed in human clinical trials because of its unfavorable characteristic of decreasing high-density lipoprotein levels.36,37 As Probucol was found to upregulate HO-1 levels in the blood, it may be useful in patients with silicosis.38 In another approach, Shigemori et al developed a genetically modified form of Lactococcus lactis capable of secreting biologically active HO-1. In animal models, this biologic approach showed promise for the treatment of inflammatory diseases characterized by oxidative stress and could be useful in high-risk silicosis patients with low serum HO-1 levels.39
Anti-inflammatory agents may also be used to treat silicosis. In a study of 34 individuals with silicosis complicated by COPD, systemic treatment with prednisolone led to a significant improvement in pulmonary function (as measured by increased diffusing capacity and oxygenation) when compared with patients receiving supportive therapy alone.40,41 To date, there have been no large randomized clinical trials evaluating the effect of systemic glucocorticoid therapy in patients with silicosis.
Murine studies were performed to evaluate the use of bone marrow-derived mononuclear or mesenchymal cells to treat silicosis. Results showed that such cells could reduce pulmonary inflammation and improve lung function.17,42–44 Based on those findings, two clinical trials involving cell-based therapy were conducted.45,46 Morales et al performed a pilot safety study using bone marrow-derived mononuclear cells from five patients with silicosis and documented good tolerability.46 In a second study involving mesenchymal cells from four patients genetically modified to express hepatocyte growth factor results showed that treatment could 1) ameliorate symptoms, 2) improve pulmonary function and 3) partially reverse radiographically detectable fibrotic changes.45
Another potential strategy for limiting the production of inflammatory cytokines and ROS following silica exposure involves treatment with immunosuppressive oligodeoxynucleotides (ODNs).26 Immunosuppressive ODNs contain repetitive TTAGGG motifs that inhibit the production of inflammatory cytokines and p47 phagocytic oxidase (p47phox, the primary component of NADPH oxidase) when murine macrophages are exposed to silica in vitro.26 Importantly, the upregulation of p47phox contributes to the inflammatory pathology observed in many diseases including cancer, raising the possibility that immunosuppressive ODNs might reduce the risk of inflammation-associated tumorigenesis.47,48 Immunosuppressive ODNs were found to inhibit ERK activation in human lung cancer cells, leading to cell cycle arrest in G1 and a marked reduction in proliferation.49 Thus, immunosuppressive ODNs have the potential to reduce ERK-mediated tumorigenesis.32,33
There is recent evidence that silica-induced lung fibrosis mediated by the infiltration of macrophages and Th17 cells is promoted by the “mineral dust-induced gene (mdig)”.50 Impairment of mdig gene function ameliorated fibrogenic changes by reducing the infiltration of macrophages and Th17 cells into the lungs while enhancing the accumulation of immune suppressive regulatory T cells. These findings raise the possibility that a treatment strategy involving targeting of mdig could be successful.
Silica and lung cancer risk: experimental evidence
In rodents, the intra-pulmonary delivery of silica particles induces a strong inflammatory response characterized by cellular infiltration of the lungs. The main cell types involved are macrophages and neutrophils (Figure 1). Upon activation, these cells produce a variety of chemokines, cytokines and ROS.25,51,52 Lung tumors subsequently develop in mice and rats into which silica particles are delivered by inhalation or instillation.53–58 Inflammation is considered the primary mechanism underlying this carcinogenicity, although ROS may also have a toxic effect on lung epithelial cells (Figure 1).25,59 A recent study by Wang et al documented the strong association between inflammation and carcinogenesis by generating mice in which GPRC5A (the G protein-coupled receptor family C group 5 type A that is predominantly expressed in lung epithelial cells) was knocked out.60 GPRC5A-KO mice exposed to silica had increased sensitivity to both lung injury and tumorigenesis, with neoplastic lesions in the epithelium correlating with the appearance of intense inflammatory/fibrotic lesions in the lungs.60 Silica can influence the process of epithelial cell damage and subsequent carcinogenesis through several pathways as shown in Figure 1. 1) Activated neutrophils and macrophages cause persistent inflammation that damages epithelial cells; 2) oxidants produced by macrophages indirectly damage epithelial cells and 3) oxidants produced as a consequence of particle uptake by epithelial cells cause direct injury. All these pathways provide inflammatory stimuli and place cells at increased risk of genotoxic (DNA) damage that increases susceptibility to tumor formation.22,61,62
 | Figure 1 Proposed mechanism of silica-induced pulmonary toxicity. Notes: Studies in the rat model suggest that silica can influence the process of epithelial cell damage and subsequent carcinogenesis via several pathways. 1) Impairment of particle clearance by alveolar macrophages due to silica-dependent toxicity and/or inflammation, 2) persistence of silica particles in the lungs activating macrophages/neutrophils to produce cytokines/chemokines/oxidants known to facilitate cancer development and 3) oxidant-induced epithelial cell genotoxicity/injury/proliferation. The impact of these pathways on tumorigenesis is described in the text. |
Focusing on the contribution of chronic inflammation to DNA damage, Zheng et al examined the role of autotaxin (ATX). Silica exposure upregulates ATX, a phosphodiesterase that mediates lysophosphatidic acid production.62 Rising levels of ATX in pulmonary epithelial cells causes DNA damage to accumulate (an effect reversed by agents that inhibit ATX activation). Assessing ATX levels might, therefore, aid in monitoring silica-mediated carcinogenesis. The tumor-promoting activity of silica particles internalized by epithelial cells may also reflect their ability to act as scaffolds that anchor DNA close to oxygen radicals, as reported by Saffiotti et al63
Once neoplastic transformation occurs, local inflammation characterized by chronic production of inflammatory cytokines/chemokines may enable tumor survival/growth by 1) supporting replication, 2) improving resistance to growth inhibition, 3) reducing cell death, 4) enhancing angiogenesis and 5) promoting tumor cell extravasation and metastasis.64,65
Short-term pulmonary exposure to high concentrations of silica can be achieved in rodents but is rarely observed in humans. Thus, it is atypical for patients to present with acute silicosis, although there is a case report showing that strong anti-inflammatory therapy involving systemic glucocorticoids can be life-saving when acute silicosis occurs.66 The histopathologic findings of acute silicosis in humans are characterized by the accumulation of foamy macrophages containing silica particles in the alveolar spaces and thickened interstitium with inflammatory cells as seen in rodent models.67,68
In mice, the acute inflammatory changes induced by silica resolve over time despite alveolar macrophages failing to clear many of the particles through 12–24 weeks of follow-up.30 The persisting particles lead to the development of chronic inflammation followed by fibrosis.69,70 In humans, chronic uncomplicated silicosis is commonly seen in individuals with 15–20 years of mild-to-moderate exposure.70 The chronic accumulation of silica particles leads to radiological changes characterized by nodular formations in the upper lung and air trapping/emphysematous changes in the lower lung.71 The typical symptoms associated with long-term lung inflammation and fibrosis are chronic cough and dyspnea on exertion.72 The pathology of this disease is mediated by the persistence of particles that escape clearance despite the presence of phagocytic macrophages leading to lung tissue/epithelial cell damage.70 In patients with chronic silicosis, systemic glucocorticoid therapy was evaluated in a small clinical trial. Results indicate that treatment led to improved pulmonary function and oxygenation.40
Oxidants also play a role in the pathology of chronic silicosis. Free radicals are produced by 1) epithelial cells that take up silica particles (an effect linked to direct genotoxicity) and 2) macrophages that phagocytize silica particles (Figure 1). Impaired particle clearance increases the persistence of silica in the lungs, which contributes to the development of lung cancer. The direct genotoxicity and epithelial cell injury caused by both the intra- and extracellular generation of free radicals could play a role in cancer generation (Figure 1).22 Although the association between inflammation and lung cancer is well documented in multiple animal models, susceptibility varies between species with rats being more susceptible to both the inflammatory and carcinogenic effects of crystalline silica than mice or hamsters.73
While this differential sensitivity may have many components, evidence suggests that silica-induced expression of regulatory growth factors plays a role.63 For example, alveolar type II epithelial cells adjacent to silicotic granulomas in rats but not mice or hamsters with silicosis produce high levels of the precursor to transforming growth factor-β1 that supports hyperplasia and malignant transformation.74
Freire et al showed that silica-mediated chronic inflammation created a microenvironment favoring tumor development and progression in mice several months after co-delivery. In brief, mice treated with the carcinogen N-nitrosodimethylamine (NDMA) plus silica generated significantly more adenomas/adenocarcinomas than those treated with NDMA alone.75 Histopathologic analysis of tumors from mice treated with the combination of NDMA plus silica accumulated more anergic and regulatory T cells (characterized by the expression of the programmed cell death protein 1 [PD-1] and forkhead box P3 [Foxp3] markers, respectively) than tumors from mice treated only with NDMA. The predicted reduction in tumoricidal T-cell activity associated with these changes is consistent with the escape of cancer cells from immune elimination.75 This led Freire et al to conclude that silica-induced chronic inflammation facilitates the development of preneoplastic lesions and subsequently lung cancer (Figure 2).
 | Figure 2 Proposed mechanism by which the immunosuppressive microenvironment induced by chronic silicosis supports tumor growth. Notes: In response to the chronic inflammation induced by prolonged silica exposure (upper portion of figure), physiological host processes designed to minimize ongoing immune stimulation are triggered. This results in the creation of an immunosuppressive microenvironment characterized by the presence of regulatory T cells and MDSC. Within this microenvironment, PD-1:PD-L1 interactions inhibit the activity of tumorigenic T cells, allowing for the growth of transformed cells. Abbreviations: MDSC, myeloid-derived suppressor cells; PD-1, programmed cell death protein 1; TNF-α, tumor necrosis factor-α; Treg, regulatory T cells. |
Similar changes were reported when Lewis lung carcinoma cells were instilled directly into the lungs of mice.76 In that model, tumor formation was accompanied by the accumulation Foxp3+ cells in the tumor and nearby lymphoid tissues. Treatment with immunostimulatory ODNs converted this immunosuppressive microenvironment into one that was pro-inflammatory, leading to improved tumoricidal T-cell activity.76
Silica-induced chronic inflammation supports tumor formation and progression in at least two ways. First, chronic inflammation facilitates the development of preneoplastic lesions by activating oncogenes (eg, K-RAS) through codon mutation (Figure 1). Second, prolonged inflammation triggers an immunosuppressive response by the host designed to minimize the damage caused by excessive inflammation/immune stimulation (Figure 2).77,78 By this mechanism, the inflammatory cytokine TNF-α shifts the host’s immune system toward immunosuppression by supporting the accumulation of myeloid-derived suppressor cells via signaling through TNF receptor-2 and FLICE-inhibitory protein (Figure 2).79,80 A similar process supports the generation of anergic T cells and regulatory T cells that help create and maintain an immunosuppressive microenvironment in the tumor bed (Figure 2).
In summary, silica-dependent carcinogenesis is well documented in experimental animals. The mechanisms involved include the induction of chronic inflammation mediated by the continuous activation of macrophages in their effort to clear the silica particles accompanied by the production of factors that cause epithelial cell genotoxicity/injury/proliferation. Once tumors arise in silicotic lungs, their survival/growth is enhanced by the creation of an immunosuppressive microenvironment that protects them from immune elimination.
Silica and lung cancer risk: evidence from human studies
The IARC evaluated the carcinogenicity of over 1,000 different agents and classified them into five groups (http://monographs.iarc.fr/ENG/Classification/). Those in Group 1 (120 agents) were found to be human carcinogens while those in Group 2A (81 agents) were classified as “probably” carcinogenic in humans. In 1995, Smith et al conducted a meta-analysis focusing on the risk of lung cancer among patients with silicosis. Based on the results showing an elevated RR, they concluded that the association between silicosis and lung cancer was causal.81 In 1997, the IARC reclassified crystalline silica from Group 2A to Group 1, while noting the absence of an exposure–response relationship in epidemiologic data available at that time.1
That concern was addressed by Steenland et al in 2001, who reported a significant positive relationship between cumulative silica exposure and lung cancer mortality in data pooled from 10 large cohorts totaling 65,980 silica-exposed workers that included 1,079 deaths from lung cancer.82 The RR of lung cancer increased in workers with a lifetime exposure to silica of >2.0 mg/m3 (with the RR rising with increased exposure). Although the intensity of exposure was more important than the duration, it was calculated that the existing OSHA occupational limit of 0.1 mg/m3 per year could result in an unacceptably increased risk of cancer in those working for 20 or more years. OSHA, therefore, lowered the yearly exposure limit by half (to 0.05 mg/m3) in 2013, consequently limiting total silica exposure over a working lifetime of 45 years to a level that was not associated with an increased risk of developing lung cancer.
Another important study involved a meta-analysis of four cohorts and six case–control studies performed by Lacasse et al,83 That analysis confirmed that the risk of lung cancer increased significantly when an individual’s cumulative exposure to silica exceeded 1.8 mg/m3, with the risk nearly doubling among workers exposed to >6.0 mg/m3 of silica. Although several of the reports included in that meta-analysis did not separate out the effects of cigarette smoke as a confounding covariable, the RRs were similar regardless of smoking history in those that did.22
The correlation between silica exposure and lung cancer was verified in several additional recent studies.15,84 Liu et al examined a cohort of 34,018 Chinese workers. They found the RR of lung cancer increased monotonically with silica exposure such that an increased risk was detected even among individuals with cumulative exposures <1 mg/m3.84 Their cohort included 12,177 individuals who never smoked, of whom 77 developed lung cancer. By analyzing just those individuals, the confounding effect of smoking on lung cancer could be excluded. Results showed that the RR of lung cancer rose by 1.6-fold in subjects with high cumulative exposure (1.12 mg/m3 years or more) compared to those with low exposure. That study provides clear evidence that silica exposure alone is enough to increase the risk of developing lung cancer.
Poinen-Rughooputh et al conducted the largest meta-analysis to date, including data from 85 studies published from January 1982 through April 2016 selected in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analysis guidelines.85 Despite considerable heterogeneity across the studies, the relationship between occupational exposure to silica and risk of lung cancer was confirmed.15
To investigate whether low-level silica exposure could increase lung cancer risk, Kachuri et al performed a case–control study using the Canadian National Enhanced Cancer Surveillance System.86 In that work, 1,681 lung cancer cases and 2,053 controls were analyzed after adjustment for confounding factors including smoking, second-hand smoke and other occupational exposures. Results showed that 1) those exposed to silica at any time in life had significantly higher odds of lung cancer (OR =1.27, 95% CI: 1.09–1.49) compared to those without, 2) silica exposure of >30 years duration was significantly associated with increased lung cancer risk (OR =1.67, 1.21–2.24) and 3) this included those exposed to low levels of silica for >30 years after adjustment for other confounding variables (OR =1.63, 1.19–2.23). However, those exposed to low levels of silica for <30 years were not found to be at increased risk.86 Thus, available evidence supports the conclusion that crystalline silica, particularly at high cumulative doses, is a human carcinogen. That finding is reinforced by the combined pooled/meta-analyses and case–control studies summarized in Table 1.
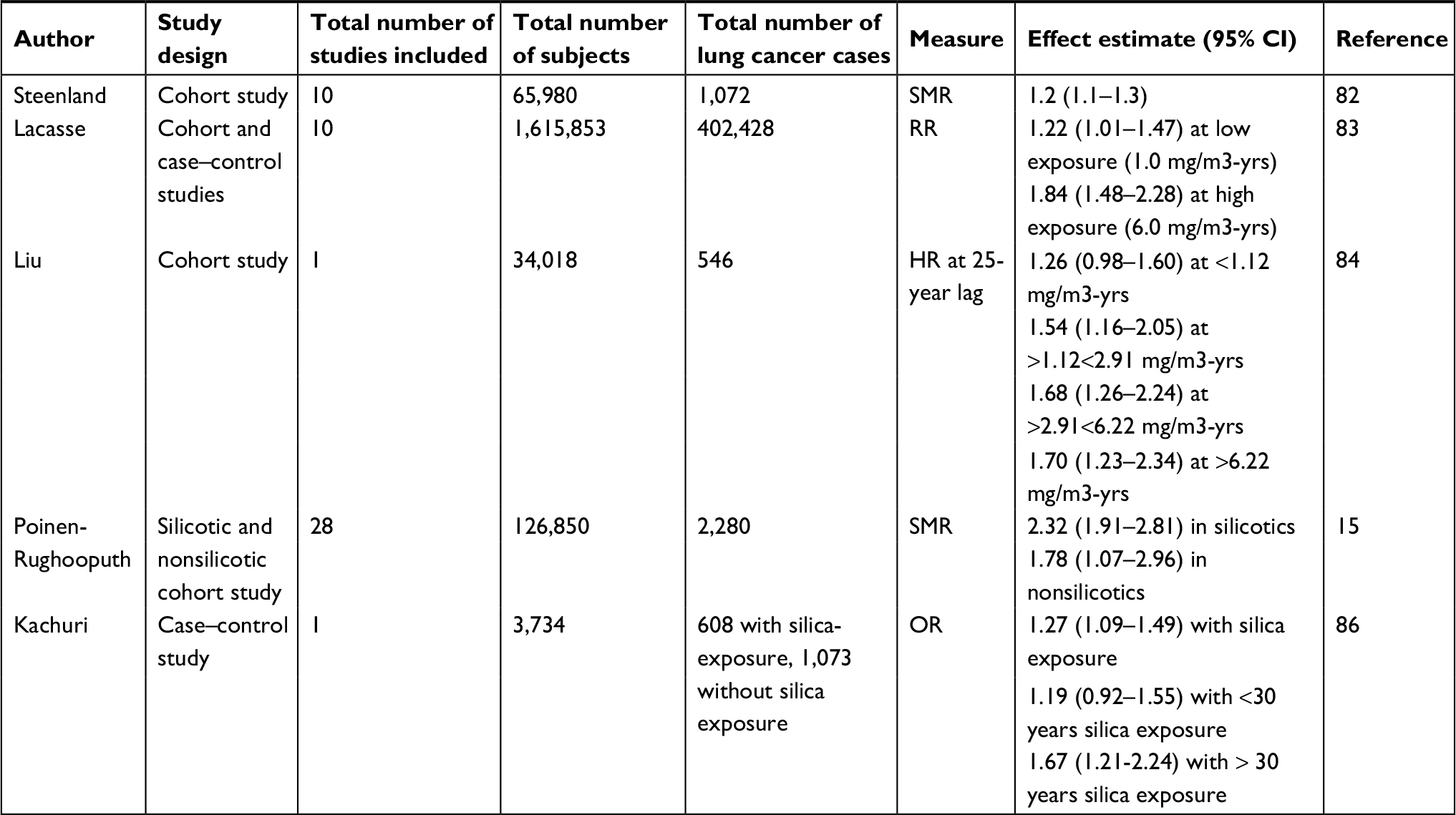 | Table 1 Representative human studies elucidating the association between silica and lung cancer risk since 2000 Abbreviation: SMR, standardized mortality ratio. |
Effect of smoking in patients with silicosis
Smoking is the single most important risk factor contributing to the development of lung cancer and must be considered when evaluating the effect of occupational exposure to other carcinogens. For example, the risk of lung cancer among workers exposed to asbestos is significantly increased by smoking.87 Liu et al found that the HR of lung cancer death among smokers exposed to silica was 5.07 (3.41–7.52) vs 1.60 (1.01–2.55) among those exposed to the same cumulative dose of silica (>1.12 mg/m3) who did not smoke.84 Among those exposed to low levels of silica, smoking increased the RR for lung cancer to 3.43 (much higher than workers in the high silica exposure group who never smoked). Based on these findings, Liu et al concluded that the joint effect of silica and smoking on lung cancer was more than additive and close to multiplicative.84 Silica-exposed workers should be advised that smoking cessation should substantially reduce their risk of lung cancer, as would lowering their total exposure to silica.
Two large case–control studies of lung cancer were conducted in Canada.86,88 Vida et al combined and analyzed results from studies conducted from 1979–1986 and from 1996–2001. Despite the differences in duration, both studies showed that the combined effects of silica exposure and smoking were at least additive and perhaps multiplicative. Kachuri et al analyzed a population-based case–control study conducted in 1994–1997 and found a multiplicative effect of smoking on those exposed to silica.79 Workers exposed to silica for >30 years who smoked more than 40 pack-years had the highest risk of lung cancer (OR =42.53, 23.54–76.83), while those with the same smoking history who were never exposed to silica had a lower risk (OR =18.82, 13.93–25.43).86 After adjusting for confounding variables including other occupational exposures, workers with intermediate smoking histories (10–40 pack years) exposed to silica had a higher risk of lung cancer than those without silica exposure (OR =1.65, 1.14–2.40). These findings led Kachuri et al to conclude that the interaction between silica exposure and smoking was multiplicative. Prudent patient management would thus support the cessation of smoking in patients with silicosis.
Effect of other carcinogens on patients with silicosis
Few studies have explored whether carcinogens other than cigarette smoke influence susceptibility to silica-induced cancer. Arsenic is a wellknown cause of lung cancer.89 Ferreccio et al conducted a case–control study in northern Chile that showed that subjects exposed to low vs high levels of arsenic had ORs of 1.0 vs 6.0 for lung cancer risk, while those co-exposed to low vs high arsenic plus silica had ORs of 1.6 vs 13. These findings led him to conclude that there was a greater than additive interaction between exposure to silica plus arsenic.
Radon is another known occupational carcinogen. Sogl et al analyzed a cohort of 58,677 German uranium miners who were exposed to silica.90 Of these, 86% were also exposed to radon. While an exposure–response relationship between silica and lung cancer risk was found, it was markedly decreased when adjusted for radon exposure. Based on this analysis, Sogl et al concluded that the combined effects of silica and radon exposure were more likely to be additive than multiplicative.
Etiopathogenesis of silica-induced lung cancer
Silicosis is an inflammatory disease of the lungs induced by exposure to crystalline silica particles. As described above, multiple studies show that high cumulative levels of silica increase an individual’s risk of developing lung cancer. These findings raise the question of whether the inflammation characteristic of silicosis is a necessary precursor to the development of lung cancer or whether lung cancer can arise solely as a result of subsequent fibrotic/destructive changes.91
To address that issue, Liu et al evaluated the frequency with which lung cancer developed in subjects with and without radiological evidence of silicosis.84 Subjects with the highest cumulative exposure to silica (N=3,907; 6.22 mg/m3 years or more) showed an HR of 1.7 (95% CI 1.23–2.34) for lung cancer death vs the reference group without silica exposure. Subjects without radiological evidence of silicosis (N=1,970) showed the same HR of 1.7 (1.15–2.52). Although this finding suggests that the radiological changes associated with silicosis may not be integral to the development of lung cancer, it could alternatively be interpreted as showing that conventional radiography fails to identify the presence of silicosis in approximately half of all individuals with pathologic evidence of silicosis.16 Thus, physicians should recognize that signs/symptoms of silicosis are just one marker of excessive silica exposure.84 Such an interpretation is consistent with results from the meta-analysis conducted by Poinen-Rughooputh et al showing that the risk of lung cancer was more prominent in the presence of silicosis.15
The severity of silicosis in patients is generally evaluated by following X-ray changes over time. However, some patients with moderate-to-severe silicosis (as determined histologically by biopsy) were not diagnosed radiologically due to the low sensitivity of chest X-ray.4,16 This would explain why workers exposed to silica with no radiological findings still have an elevated risk of lung cancer. Smokers exposed to silica develop clinical silicosis more frequently than equally exposed nonsmokers.91,92 The National Comprehensive Cancer Network (NCCN) guidelines recommend that individuals with significant occupational exposure to a carcinogen be screened by computed tomography (CT) beginning at the age of 50 years if they have a smoking history of >20 pack-years. A review of the literature suggests that CT should be used to evaluate workers with high silica exposure, irrespective of radiological findings, to enhance the early detection of lung cancer. In selecting appropriate therapy, physicians should consider the mechanisms underlying lung cancer development in silicosis, including the development of an immunosuppressive microenvironment. This would support the use of immunotherapy involving PD-1/PD-L1 immune checkpoint inhibitors in the treatment of silica-mediated lung cancer. Evidence indicates that checkpoint inhibitor therapy can increase the 5-year survival of patients with advanced non-small-cell lung cancer (unrelated to silica exposure) by fourfold, emphasizing the benefit of such a treatment strategy.93,94
Conclusion
There is strong and consistent evidence of a causal relationship between silica exposure and lung cancer in both animal models and humans. Animal studies show that silica induces acute inflammation followed by the development of an immunosuppressive microenvironment that supports tumorigenesis. Future research should focus on the cells and genes activated by silica exposure to identify potential therapeutic targets. As the RR for lung cancer in silica-exposed workers tends to be greater in patients with silicosis, physicians should be aware of the low sensitivity of chest X-ray surveillance and pay increased attention to workers with minimal radiographic changes. Furthermore, it appears that cumulative silica exposure correlates directly with lung cancer risk, supporting the decision to reduce the occupational exposure limit to 0.05 mg/m3. It is further recommended that workers with a cumulative silica exposure of >30 years be assessed by chest CT to enhance the likelihood of detecting lung cancer as early as possible. Additional research is needed to clarify the optimal frequency and evaluate the efficacy of such screening. If tumors are detected, physicians should examine the expression of PD-L1 to determine whether treatment with immune checkpoint inhibitors is likely to be efficacious.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization. IARC monographs on the evaluation of carcinogenic risks to humans: Silica, some silicates, coal dust, and para-aramid fibrils. Geneva, Switzerland: International Agency for Research on Cancer, 1997:337. | ||
Hessel PA, Gamble JF, Gee JB, et al. Silica, silicosis, and lung cancer: a response to a recent working group report. J Occup Environ Med. 2000;42(7):704–720. | ||
Wong O. The epidemiology of silica, silicosis and lung cancer: some recent findings and future challenges. Ann Epidemiol. 2002;12(5):285–287. | ||
Leung CC, Yu IT, Chen W. Silicosis. Lancet. 2012;379(9830):2008–2018. | ||
Steenland K, Ward E. Silica: a lung carcinogen. CA Cancer J Clin. 2014;64(1):63–69. | ||
Maciejewska A. Occupational exposure assessment for crystalline silica dust: approach in Poland and worldwide. Int J Occup Med Environ Health. 2008;21(1):1–23. | ||
Ministry of Health, Labour and Welfare. Pneumoconiosis health management implementation status report in Japan 2016. Tokyo, Japan; 2016. | ||
Ministry of Health C. China’s Health Statistics Yearbook 2011. Beijing, China: Peking Union Medical College Press; 2011. | ||
Chen W, Liu Y, Wang H, et al. Long-term exposure to silica dust and risk of total and cause-specific mortality in Chinese workers: a cohort study. PLoS Med. 2012;9(4):e1001206. | ||
Hnizdo E, Sluis-Cremer GK. Silica exposure, silicosis, and lung cancer: a mortality study of South African gold miners. Br J Ind Med. 1991;48(1):53–60. | ||
Croteau GA, Flanagan ME, Camp JE, Seixas NS. The efficacy of local exhaust ventilation for controlling dust exposures during concrete surface grinding. Ann Occup Hyg. 2004;48(6):509–518. | ||
Ulm K, Gerein P, Eigenthaler J, Schmidt S, Ehnes H. Silica, silicosis and lung-cancer: results from a cohort study in the stone and quarry industry. Int Arch Occup Environ Health. 2004;77(5):313–318. | ||
Güngen AC, Aydemir Y, Çoban H, Düzenli H, Tasdemir C. Lung cancer in patients diagnosed with silicosis should be investigated. Respir Med Case Rep. 2016;18:93–95. | ||
Brown T. Silica exposure, smoking, silicosis and lung cancer--complex interactions. Occup Med (Lond). 2009;59(2):89–95. | ||
Poinen-Rughooputh S, Rughooputh MS, Guo Y, Rong Y, Chen W. Occupational exposure to silica dust and risk of lung cancer: an updated meta-analysis of epidemiological studies. BMC Public Health. 2016;16(1):1137. | ||
Hnizdo E, Murray J, Sluis-Cremer GK, Thomas RG. Correlation between radiological and pathological diagnosis of silicosis: an autopsy population based study. Am J Ind Med. 1993;24(4):427–445. | ||
Lopes-Pacheco M, Bandeira E, Morales MM. Cell-based therapy for silicosis. Stem Cells Int. 2016;2016:5091838. | ||
Mossman BT, Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. Am J Respir Crit Care Med. 1998;157(5 Pt 1):1666–1680. | ||
Yu IT, Tse LA, Wong TW, Leung CC, Tam CM, Chan AC. Further evidence for a link between silica dust and esophageal cancer. Int J Cancer. 2005;114(3):479–483. | ||
Cocco P, Ward MH, Buiatti E. Occupational risk factors for gastric cancer: an overview. Epidemiol Rev. 1996;18(2):218–234. | ||
Partanen T, Pukkala E, Vainio H, Kurppa K, Koskinen H. Increased incidence of lung and skin cancer in Finnish silicotic patients. J Occup Med. 1994;36(6):616–622. | ||
IARC monograph. Silica dust, Crystalline, in the Form of Quartz or Cristobalite. A Review of Human Carcinogens: Arsenic, Metals, Fibers and Dusts. Vol 100C. Lyon, France: International Agency for Research on Cancer; 2012:356–405. | ||
US Occupational Health and Safety Administration. Proposed rule: occupational exposure to respirable crystalline silica. Federal Register. 2013;78:56273–56504. | ||
National Institute for Occupational Safety and Health. Health effects of occupational exposure to respirable crystalline silica. DoHaH Services, editor. Cincinnati OH; 2002. | ||
Fubini B, Hubbard A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic Biol Med. 2003;34(12):1507–1516. | ||
Sato T, Shimosato T, Alvord WG, Klinman DM. Suppressive oligodeoxynucleotides inhibit silica-induced pulmonary inflammation. J Immunol. 2008;180(11):7648–7654. | ||
Ding M, Shi X, Dong Z, et al. Freshly fractured crystalline silica induces activator protein-1 activation through ERKs and p38 MAPK. J Biol Chem. 1999;274(43):30611–30616. | ||
Vallyathan V, Castranova V, Pack D, et al. Freshly fractured quartz inhalation leads to enhanced lung injury and inflammation. Potential role of free radicals. Am J Respir Crit Care Med. 1995;152(3):1003–1009. | ||
Papaiahgari S, Kleeberger SR, Cho HY, Kalvakolanu DV, Reddy SP. NADPH oxidase and ERK signaling regulates hyperoxia-induced Nrf2-ARE transcriptional response in pulmonary epithelial cells. J Biol Chem. 2004;279(40):42302–42312. | ||
Sato T, Takeno M, Honma K, et al. Heme oxygenase-1, a potential biomarker of chronic silicosis, attenuates silica-induced lung injury. Am J Respir Crit Care Med. 2006;174(8):906–914. | ||
Nakashima K, Sato T, Shigemori S, Shimosato T, Shinkai M, Kaneko T. Regulatory role of heme oxygenase-1 in silica-induced lung injury. Respir Res. 2018;19(1):144. | ||
Thrane EV, Schwarze PE, Thoresen GH, Låg M, Refsnes M. Persistent versus transient map kinase (ERK) activation in the proliferation of lung epithelial type 2 cells. Exp Lung Res. 2001;27(4):387–400. | ||
Vicent S, López-Picazo JM, Toledo G, et al. ERK1/2 is activated in non-small-cell lung cancer and associated with advanced tumours. Br J Cancer. 2004;90(5):1047–1052. | ||
Choudhary S, Sood S, Donnell RL, Wang HC. Intervention of human breast cell carcinogenesis chronically induced by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine. Carcinogenesis. 2012;33(4):876–885. | ||
Sato T, Saito Y, Inoue S, et al. Serum heme oxygenase-1 as a marker of lung function decline in patients with chronic silicosis. J Occup Environ Med. 2012;54(12):1461–1466. | ||
Lönn ME, Dennis JM, Stocker R. Actions of “antioxidants” in the protection against atherosclerosis. Free Radic Biol Med. 2012;53(4):863–884. | ||
Saso L, Firuzi O. Pharmacological applications of antioxidants: lights and shadows. Curr Drug Targets. 2014;15(13):1177–1199. | ||
Thomas RA, Czopek A, Bellamy CO, Mcnally SJ, Kluth DC, Marson LP. Hemin preconditioning upregulates heme oxygenase-1 in deceased donor renal transplant recipients: a randomized, controlled, Phase IIB trial. Transplantation. 2016;100(1):176–183. | ||
Shigemori S, Watanabe T, Kudoh K, et al. Oral delivery of Lactococcus lactis that secretes bioactive heme oxygenase-1 alleviates development of acute colitis in mice. Microb Cell Fact. 2015;14:189. | ||
Sharma SK, Pande JN, Verma K. Effect of prednisolone treatment in chronic silicosis. Am Rev Respir Dis. 1991;143(4 Pt 1):814–821. | ||
Rose C Silicosis, Hollingsworth H, ed [homepage on the Internet]. UpToDate. Waltham, MA: UpToDate Inc. Available from: https://www.uptodate.com/home/about-us. Accessed March 21, 2018. | ||
Lassance RM, Prota LF, Maron-Gutierrez T, et al. Intratracheal instillation of bone marrow-derived cell in an experimental model of silicosis. Respir Physiol Neurobiol. 2009;169(3):227–233. | ||
Maron-Gutierrez T, Castiglione RC, Xisto DG, et al. Bone marrow-derived mononuclear cell therapy attenuates silica-induced lung fibrosis. Eur Respir J. 2011;37(5):1217–1225. | ||
Zhao MM, Cui JZ, Cui Y, et al. Therapeutic effect of exogenous bone marrowderived mesenchymal stem cell transplantation on silicosis via paracrine mechanisms in rats. Mol Med Rep. 2013;8(3):741–746. | ||
Liu WW, Wang HX, Yu W, et al. Treatment of silicosis with hepatocyte growth factor-modified autologous bone marrow stromal cells: a non-randomized study with follow-up. Genet Mol Res. 2015;14(3):10672–10681. | ||
Morales MM, Souza SA, Loivos LP, et al. Pilot safety study of intrabronchial instillation of bone marrow-derived mononuclear cells in patients with silicosis. BMC Pulm Med. 2015;15:66. | ||
Teufelhofer O, Parzefall W, Kainzbauer E, et al. Superoxide generation from Kupffer cells contributes to hepatocarcinogenesis: studies on NADPH oxidase knockout mice. Carcinogenesis. 2005;26(2):319–329. | ||
Bode C, Kinjo T, Alvord WG, Klinman DM. Suppressive oligodeoxynucleotides reduce lung cancer susceptibility in mice with silicosis. Carcinogenesis. 2014;35(5):1078–1083. | ||
Takahashi R, Sato T, Klinman DM, Shimosato T, Kaneko T, Ishigatsubo Y. Suppressive oligodeoxynucleotides synergistically enhance antiproliferative effects of anticancer drugs in A549 human lung cancer cells. Int J Oncol. 2013;42(2):429–436. | ||
Thakur C, Wolfarth M, Sun J, et al. Oncoprotein mdig contributes to silica-induced pulmonary fibrosis by altering balance between Th17 and Treg T cells. Oncotarget. 2015;6(6):3722–3736. | ||
Davis GS, Pfeiffer LM, Hemenway DR. Interferon-gamma production by specific lung lymphocyte phenotypes in silicosis in mice. Am J Respir Cell Mol Biol. 2000;22(4):491–501. | ||
Huaux F, Arras M, Tomasi D, et al. A profibrotic function of IL-12p40 in experimental pulmonary fibrosis. J Immunol. 2002;169(5):2653–2661. | ||
Dagle GE, Wehner AP, Clark ML, Buschbom RL. Chronic inhalation exposure of rats to quartz. In: Goldsmith DF, Winn DM, Shy CM, editors. Silica, Silicosis and Cancer. Controversy in Occupational Medicine. New York: Praeger; 1985. | ||
Holland L WJM, Tillery M. Lung cancer in rats exposed to fibrogenic dusts. New York: Praeger; 1986. | ||
Holland LM GM, Wilson JS. Pulmonary effects of shale dusts in experimental animals. Butterworth: Boston MA; 1983. | ||
Johnson NF, Smith DM, Sebring R, Holland LM. Silica-induced alveolar cell tumors in rats. Am J Ind Med. 1987;11(1):93–107. | ||
Spiethoff A, Wesch H, Wegener K, Klimisch HJ. The effects of Thorotrast and quartz on the induction of lung tumors in rats. Health Phys. 1992;63(1):101–110. | ||
Muhle H, Takenaka S, Mohr U, Dasenbrock C, Mermelstein R. Lung tumor induction upon long-term low-level inhalation of crystalline silica. Am J Ind Med. 1989;15(3):343–346. | ||
Knaapen AM, Borm PJ, Albrecht C, Schins RP. Inhaled particles and lung cancer. Part A: Mechanisms. Int J Cancer. 2004;109(6):799–809. | ||
Wang X, Xu D, Liao Y, et al. Epithelial neoplasia coincides with exacerbated injury and fibrotic response in the lungs of Gprc5a- knockout mice following silica exposure. Oncotarget. 2015;6(37):39578–39593. | ||
Valdés-Rives SA, González-Arenas A. Autotaxin-lysophosphatidic acid: from inflammation to cancer development. Mediators Inflamm. 2017;2017:9173090. | ||
Zheng H, Högberg J, Stenius U. ATM-activated autotaxin (ATX) propagates inflammation and DNA damage in lung epithelial cells: a new mode of action for silica-induced DNA damage? Carcinogenesis. 2017;38(12):1196–1206. | ||
Saffiotti U, Daniel LN, Mao Y, Shi X, Williams AO, Kaighn ME. Mechanisms of carcinogenesis by crystalline silica in relation to oxygen radicals. Environ Health Perspect. 1994;102 Suppl 10:159–163. | ||
Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30(7):1073–1081. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Goodman GB, Kaplan PD, Stachura I, Castranova V, Pailes WH, Lapp NL. Acute silicosis responding to corticosteroid therapy. Chest. 1992;101(2):366–370. | ||
Hoffmann EO, Lamberty J, Pizzolato P, Coover J. The ultrastructure of acute silicosis. Arch Pathol. 1973;96(2):104–107. | ||
Nugent KM, Dodson RF, Idell S, Devillier JR. The utility of bronchoalveolar lavage and transbronchial lung biopsy combined with energy-dispersive X-ray analysis in the diagnosis of silicosis. Am Rev Respir Dis. 1989;140(5):1438–1441. | ||
Hamilton RF Jr, Thakur SA, Holian A. Silica binding and toxicity in alveolar macrophages. Free Radic Biol Med. 2008;44(7):1246–1258. | ||
Pollard KM. Silica, silicosis, and autoimmunity. Front Immunol. 2016;7:97. | ||
Wade WA, Petsonk EL, Young B, Mogri I. Severe occupational pneumoconiosis among West Virginian coal miners: one hundred thirty-eight cases of progressive massive fibrosis compensated between 2000 and 2009. Chest. 2011;139(6):1458–1462. | ||
Cohen RA, Patel A, Green FH. Lung disease caused by exposure to coal mine and silica dust. Semin Respir Crit Care Med. 2008;29(6):651–661. | ||
Driscoll KE, Deyo LC, Carter JM, Howard BW, Hassenbein DG, Bertram TA. Effects of particle exposure and particle-elicited inflammatory cells on mutation in rat alveolar epithelial cells. Carcinogenesis. 1997;18(2):423–430. | ||
Williams AO, Flanders KC, Saffiotti U. Immunohistochemical localization of transforming growth factor-beta 1 in rats with experimental silicosis, alveolar type II hyperplasia, and lung cancer. Am J Pathol. 1993;142(6):1831–1840. | ||
Freire J, Ajona D, de Biurrun G, et al. Silica-induced chronic inflammation promotes lung carcinogenesis in the context of an immunosuppressive microenvironment. Neoplasia. 2013;15(8):913–924. | ||
Sato T, Shimosato T, Ueda A, Ishigatsubo Y, Klinman DM. Intrapulmonary delivery of CpG microparticles eliminates lung tumors. Mol Cancer Ther. 2015;14(10):2198–2205. | ||
Baniyash M, Sade-Feldman M, Kanterman J. Chronic inflammation and cancer: suppressing the suppressors. Cancer Immunol Immunother. 2014;63(1):11–20. | ||
Kanterman J, Sade-Feldman M, Baniyash M. New insights into chronic inflammation-induced immunosuppression. Semin Cancer Biol. 2012;22(4):307–318. | ||
Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M. Tumor necrosis factor-α blocks differentiation and enhances suppressive activity of immature myeloid cells during chronic inflammation. Immunity. 2013;38(3):541–554. | ||
Zhao X, Rong L, Zhao X, et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest. 2012;122(11):4094–4104. | ||
Smith AH, Lopipero PA, Barroga VR. Meta-analysis of studies of lung cancer among silicotics. Epidemiology. 1995;6(6):617–624. | ||
Steenland K, Mannetje A, Boffetta P, et al. Pooled exposure-response analyses and risk assessment for lung cancer in 10 cohorts of silica-exposed workers: an IARC multicentre study. Cancer Causes Control. 2001;12(9):773–784. | ||
Lacasse Y, Martin S, Gagné D, Lakhal L. Dose-response meta-analysis of silica and lung cancer. Cancer Causes Control. 2009;20(6):925–933. | ||
Liu Y, Steenland K, Rong Y, et al. Exposure-response analysis and risk assessment for lung cancer in relationship to silica exposure: a 44-year cohort study of 34,018 workers. Am J Epidemiol. 2013;178(9):1424–1433. | ||
Moher D, Liberati A, Tetzlaff J, Altman DG ; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA Statement. Open Med. 2009;3(3):e123–e130. | ||
Kachuri L, Villeneuve PJ, Parent ME, et al; Canadian Cancer Registries Epidemiology Group. Occupational exposure to crystalline silica and the risk of lung cancer in Canadian men. Int J Cancer. 2014;135(1):138–148. | ||
Gustavsson P, Nyberg F, Pershagen G, Schéele P, Jakobsson R, Plato N. Low-dose exposure to asbestos and lung cancer: dose-response relations and interaction with smoking in a population-based case-referent study in Stockholm, Sweden. Am J Epidemiol. 2002;155(11):1016–1022. | ||
Vida S, Pintos J, Parent ME, Lavoué J, Siemiatycki J. Occupational exposure to silica and lung cancer: pooled analysis of two case-control studies in Montreal, Canada. Cancer Epidemiol Biomarkers Prev. 2010;19(6):1602–1611. | ||
Ferreccio C, Yuan Y, Calle J, et al. Arsenic, tobacco smoke, and occupation: associations of multiple agents with lung and bladder cancer. Epidemiology. 2013;24(6):898–905. | ||
Sogl M, Taeger D, Pallapies D, et al. Quantitative relationship between silica exposure and lung cancer mortality in German uranium miners, 1946-2003. Br J Cancer. 2012;107(7):1188–1194. | ||
Checkoway H, Franzblau A. Is silicosis required for silica-associated lung cancer? Am J Ind Med. 2000;37(3):252–259. | ||
Hughes JM, Weill H, Checkoway H, et al. Radiographic evidence of silicosis risk in the diatomaceous earth industry. Am J Respir Crit Care Med. 1998;158(3):807–814. | ||
Aguiar PN Jr, de Mello RA, Barreto CMN, et al. Immune checkpoint inhibitors for advanced non-small cell lung cancer: emerging sequencing for new treatment targets. ESMO Open. 2017;2(3):e000200. | ||
Brahmer JHL, Jackman D. Five-year follow-up from the CA209-003 study of nivolumab in previously treated advanced non-small cell lung cancer (NSCLC): Clinical characteristics of long-term survivors. 2017 AACR Annual Meeting. Abstract CT077. Presented April 3, 2017; Washington, DC, USA. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.