Back to Journals » International Journal of Nanomedicine » Volume 13
Silica nanoparticle-induced oxidative stress and mitochondrial damage is followed by activation of intrinsic apoptosis pathway in glioblastoma cells
Authors Kusaczuk M, Krętowski R, Naumowicz M ![]() , Stypułkowska A, Cechowska-Pasko M
, Stypułkowska A, Cechowska-Pasko M
Received 29 November 2017
Accepted for publication 1 February 2018
Published 12 April 2018 Volume 2018:13 Pages 2279—2294
DOI https://doi.org/10.2147/IJN.S158393
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Magdalena Kusaczuk,1 Rafał Krętowski,1 Monika Naumowicz,2 Anna Stypułkowska,1 Marzanna Cechowska-Pasko1
1Department of Pharmaceutical Biochemistry, Medical University of Białystok, 2Institute of Chemistry, University of Białystok, Białystok, Poland
Introduction: Recently, the focus of oncological research has been on the optimization of therapeutic strategies targeted at malignant diseases. Nanomedicine utilizing silicon dioxide nanoparticles (SiNPs) is one such strategy and is rapidly developing as a promising tool for cancer diagnosis, imaging, and treatment. Nevertheless, little is known about the mechanisms of action of SiNPs in brain tumors.
Materials and methods: Here, we explored the effects of 5–15 nm SiNPs in the human glioblastoma cell line LN229. In this respect, MTT assays, microscopic observations, flow cytometry analyses, and luminescent assays were performed. Moreover, RT-qPCR and Western blot analyses were done to determine gene and protein expressions.
Results: We demonstrated that SiNPs triggered evident cytotoxicity, with microscopic observations of the nuclei, annexin V–fluorescein isothiocyanate/propidium iodide staining, and elevated caspase 3/7 activity, suggesting that SiNPs predominantly induced apoptotic death in LN229 cells. We further showed the occurrence of oxidative stress induced by enhanced reactive oxygen-species generation. This effect was followed by deregulated expression of genes encoding the antioxidant enzymes SOD1, SOD2, and CAT, and impaired mitochondria function. SiNP-induced mitochondrial dysfunction was characterized by membrane-potential collapse, ATP depletion, elevated expression of BAX, PUMA, and NOXA with simultaneous downregulation of BCL2/BCL2L1, and activation of caspase 9. Moreover, RT-qPCR and Western blot analyses demonstrated increased levels of the endoplasmic reticulum stress markers GRP78, GRP94, and DDIT3, as well as strongly increased expressions of the IL1B and COX2 genes, suggesting activation of endoplasmic reticulum stress and a proinflammatory response.
Conclusions: Altogether, our data indicate that in LN229 cells, SiNPs evoke cell death via activation of the intrinsic apoptosis pathway and suggest that other aspects of cellular function may also be affected. As such, SiNPs represent a potentially promising agent for facilitating further progress in brain cancer therapy. However, further exploration of SiNP long-term toxicity and molecular effects is necessary prior to their widespread application.
Keywords: mitochondrial membrane potential, nanomedicine, ER stress, nanotoxicity, silica nanoparticles
Introduction
Synthetic silicon dioxide nanoparticles (SiNPs) are broadly used in many fields of science, and are well known for their significant industrial and commercial applications.1 Their broad utilization in the field of nanomedicine has recently attracted a great deal of attention. In biomedical and biotechnological sciences, the use of SiNPs is becoming increasingly accepted for diagnosis, imaging, drug delivery, gene therapy, biomolecule detection, and photodynamic therapy.2,3 As a consequence of their wide exploitation, SiNPs were named among the top five widely applied NPs by the Organization for Economic Cooperation and Development and put on the priority list for toxicity evaluation.4 Given this, cytotoxic effects of SiNPs have already been reported for many in vitro studies.1,5–7 However, the mechanisms underlying SiNP cytotoxicity in vivo might be different, and require further exploration. To date, SiNPs have been shown to induce oxidative and endoplasmic reticulum (ER) stress,5,6 inflammatory response,1 cell-cycle arrest,8 apoptosis,7 and necrosis9 in a variety of cell lines.
Although significant progress has been made in clarifying the effects of SiNPs in normal cells, knowledge of SiNP effects on tumor cells is still limited and unclear. In contrast to normal cells, the cytotoxic effects of NPs may be beneficial for killing tumor cells and controlling cancer progression. Many anticancer therapies focus on activating apoptotic death of transformed cells, which can be triggered through various pathways. As such, identification of the molecular mechanisms underlying NP-triggered cell death is particularly important, and may have implications for determination of their drug-carrier potential, possible functionalization, or application with cotherapeutic drugs.
Many reports on SiNP-mediated apoptosis demonstrate activation of the intrinsic pathway after NP exposure.10–12 Indeed, Ahamed et al reported that treatment of A431 and A549 cells with SiNPs resulted in excessive generation of reactive oxygen species (ROS) and oxidative stress, which in turn led the upregulation of CASP9 and CASP3 genes and initiation of mitochondria-mediated apoptosis.11 Accordingly, Ahmad et al demonstrated the upregulation of the BAX and CASP3 genes together with downregulation of the antiapoptotic BCL2 gene in human liver cell line HEPG2.12 In contrast, Tokgun et al suggested that SiNP-dependent apoptosis occurs via death receptor-mediated pathways in the A549 cell line,7 and studies have demonstrated necrotic cell death after treatment with SiNPs.9,13 Exposure of human umbilical vein endothelial cells to 304 nm and 310 nm SiNPs has resulted in enhanced necrosis, while exposure of alveolar macrophages to the same NPs evoked 80% apoptosis and 20% necrosis.9 Moreover, Corbalan et al demonstrated that after penetrating plasma membrane in endothelial cells, silica NPs caused the release of cytoprotective NO and marked overproduction of cytotoxic ONOO, leading to increased nitroxidative/oxidative stress and subsequent endothelial inflammation and necrosis.13 Preliminary reports on the application of SiNPs in cancer treatment are promising, with increased data suggesting antiproliferative effects in cancer cells compared to normal cells.10,14 Following SiNP treatment, Lu et al demonstrated increased expression of p53 and caspase 3 and decreased expression of Bcl2 and procaspase 9 in human HEPG2 hepatoma cells, while none of these effects was observed in normal human L02 hepatocytes.10 Likewise, our own research has revealed higher cytotoxicity in SiNP-treated glioblastoma LN18 and LBC3 cell lines, with only slight cytotoxic effects in normal skin fibroblasts.14
NP-dependent cytotoxicity may be of particular importance in cases of incurable cancers, such as glioblastoma multiforme, where new modalities of therapeutic strategies are highly desired. Unlike other cancers, brain tumors are particularly inaccessible to chemotherapeutics, due to the blood–brain barrier. A number of other factors, such as molecular heterogeneity, anaplastic cancer cells, and difficulties in targeting therapeutics specifically to transformed cells, are among the limitations halting development of effective glioblastoma therapies.15–17 To address this need for new therapeutic strategies, the field of nanomedicine is currently being explored in the management of brain malignancies.17 To date, several reports illustrating the utility of SiNPs for brain-tumor treatment have been published. Zhang et al demonstrated that mesoporous SiNPs enhanced the radiosensitivity of valproic acid in rat glioma C6 cells and human glioma U87 cells.18 Wan et al investigated SiNPs as cancer-targeted carriers to deliver siRNA against MRP1 into glioblastoma cells, showing that siRNA-loaded SiNPs downregulated mRNA and protein expression of MRP1, inducing cancer-cell death.8 Another report indicated that treatment of U87 cells with SiNPs decreased cell survival, with subsequent alterations in expression of mitochondrial DNA-encoded cytochrome Cox2, ND6, and the cell-signaling protein ERK and its phosphorylated forms.19
While promising, existing data on SiNPs in glioblastoma are limited, and little is known about their toxicological effects in this disease.13 In order to broaden this knowledge, we investigated the mechanisms of silicon dioxide nanotoxicity in the human glioblastoma LN229 cell line. In this respect, we studied the influence of SiNPs on apoptosis, ER, oxidative stress, mitochondrial damage, and inflammatory response. Although many aspects of LN229 cellular physiology were altered by SiNP exposure, further studies are necessary to fully understand the role of SiNPs in glioblastoma and to use them successfully as a potential brain cancer treatment.
Materials and methods
Reagents
DMEM containing glucose at 4.5 mg/mL (25 mmol/L) with GlutaMax, streptomycin, penicillin, and trypsin–EDTA were provided by Thermo Fisher Scientific (Waltham, MA, USA). A high-capacity RNA-to-cDNA kit was purchased from Thermo Fisher Scientific. The ReliaPrep RNA Cell Miniprep system, Caspase-Glo 3/7 assay, Caspase-Glo 9 assay, CellTiter-Glo luminescent cell-viability assay, ROS-Glo H2O2 assay, and Caspase-Glo 1 inflammasome assay were provided by Promega (Fitchburg, WI, USA). FBS Gold was from Thermo Fisher Scientific, a fluorescein isothiocyanate (FITC)–annexin V apoptosis-detection kit from BD Biosciences (San Jose, CA, USA), and radioimmunoprecipitation-assay lysis buffer and BCA protein-assay kit from Thermo Fisher Scientific. SigmaFast BCIP/NBT reagent and molecular-grade purity water were provided by Sigma-Aldrich (St Louis, MO, USA). The polyclonal (mouse) anti-KDEL antibody was purchased from Enzo Biochem (Farmingdale, NY, USA). Alkaline phosphatase-conjugated antimouse IgG was from Rockland Immunochemicals (Limerick, PA, USA). Alkaline phosphatase-conjugated antirabbit IgG and polyclonal (rabbit) anti-β-tubulin antibody were provided by Cell Signaling Technology (Boston, MA, USA). Silicon dioxide nanopowders (7 nm, 5–15 nm, and 10–20 nm) were purchased from Sigma-Aldrich.
Characterization of silica NPs
SiNPs were evaluated using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK), as already described in our previous work.14 Samples for transmission electron microscopy (TEM) analysis were prepared by dropping aliquots of SiNP solutions on 400-mesh carbon-coated copper grids (SPI Supplies, West Chester, PA, USA). Prior to measurements, the films on the TEM grids were allowed to air-dry. To characterize the size and shape of SiNPs, images were collected using an FEI Tecnai G2 X-Twin 200 kV microscope (Thermo Fisher Scientific). The accelerating voltage during measurements was 200 kV.
Cell culture and exposure to silica NPs
The human glioblastoma cell line LN229 (American Type Culture Collection [ATCC]) was a kind gift from Professor Cezary Marcinkiewicz from the Department of Neuroscience, Temple University, Philadelphia. Cells were cultured in high-glucose DMEM with 10% of heat-inactivated FBS Gold, penicillin (100 U/mL), streptomycin (100 μg/mL), and 2 mmol/L L-glutamine. Cells were cultured in Falcon flasks (BD Biosciences) in a 5% CO2 incubator (Galaxy S+; Eppendorf, Hamburg, Germany) at 37°C. Cells reaching subconfluence were detached from the culture plates using 0.05% trypsin 0.02%–EDTA in calcium-free PBS and counted in a Scepter cell counter (Merck Millipore, Billerica, MA, USA). Directly before experiments, stock solutions of SiNPs in deionized water were sonicated for 10 minutes to avoid particle aggregation. The final concentrations of SiNPs were obtained by diluting stock solutions with culture medium containing 10% FBS.
Cell viability
The viability of LN229 cells was evaluated according to methods in Carmichael et al.20 Briefly, cells were seeded in 24-well plates at a density of 5×104/well. Confluent cells were then cultured with three sizes of SiNPs (7 nm, 5–15 nm, and 10–20 nm) at concentrations of 5–1,800 μg/mL for 24 hours and 48 hours. Next, cells were washed twice with PBS and incubated with 1 mL MTT solution (0.25 mg/mL in PBS) at 37°C in a humidified 5% CO2 atmosphere for 3 hours. The medium was removed and formazan products solubilized in 1 mL of 0.1 mmol/L HCl in absolute isopropanol. Absorbance of a converted dye in living cells was read on a microplate reader (Tecan, Männedorf, Switzerland) at a wavelength of 570 nm. The viability of SiNP-treated LN229 cells was calculated as a percentage of control untreated cells. All experiments were run in triplicates.
Cell morphological analysis
To visualize morphological characteristics of glioblastoma LN229 cells exposed to 5–15 nm SiNPs, cells were double-stained with acridine orange (AO) and ethidium bromide (EtBr). Staining was followed by fluorescence-microscopy observations. AO is able to enter both dead and viable cells. It emits red fluorescence when bound to single-stranded DNA, found predominantly in dead cells, and green fluorescence when bound to double-stranded DNA observed in viable cells. EtBr is actively excreted from living cells.21 LN229 cells at a density of 2.5×105 were seeded into six-well plates and incubated with 50 μg/mL and 100 μg/mL SiNPs at 37°C in a humidified atmosphere containing 5% CO2 for 24 hours and 48 hours. After incubation, cells were stained with a mixture of AO (10 μmol/mL) and EtBr (10 μmol/mL). Cells were visualized using fluorescence microscope (CKX 41; Olympus, Tokyo, Japan) at 100× magnification.
Detection of apoptosis
Apoptosis of LN229 cells was evaluated using the FITC–annexin V apoptosis-detection kit followed by flow-cytometry analysis. The cells were seeded in a 6-well plates at a density of 2.5×105 per well (in 2 mL of medium) and cultured until they reached confluence. The cells were grown in high-glucose DMEM with 50 μg/mL and 100 μg/mL of 5–15 nm SiNPs, for 24 hours and 48 hours. Next, cells were trypsinized and resuspended in DMEM and then in a binding buffer. The cells were stained using FITC–annexin V and propidium iodide (PI) for 15 min in the dark, according to the manufacturer’s manual. Flow cytometry analysis was performed using the FACSCanto II cytometer (BD FACSCanto II, San Diego, CA, USA). Analysis of data was performed using the FACSDiva software (BD, San Diego, CA, USA). The dead cells were discriminated on the basis of forward- and side-scatter parameters; annexin V+/PI− were identified as early apoptotic and annexin V+/PI+ as late apoptotic cells. A sum of Q2 and Q4 quadrant populations of analyzed cells was presented as the percentage of apoptotic cells.
Caspase 3/7 and caspase 9 activities
Measurement of caspase 3/7 and caspase 9 activities after SiNP treatment was performed using the luminescent Caspase-Glo 3/7 and Caspase-Glo 9 assays following the manufacturer’s instructions. Briefly, LN229 cells were seeded in white-walled 96-well culture plates (Nunclon; Thermo Fisher Scientific) at a density of 104/well. Subsequently, cells were incubated with medium containing 5–15 nm SiNPs at concentrations of 50 μg/mL and 100 μg/mL for 24 hours and 48 hours. After incubation, 100 μL Caspase-Glo 3/7 or Caspase-Glo 9 reagent was added to each sample. Cells were mixed using a plate shaker at 300 rpm for 45 seconds and left in the dark at room temperature for 40 minutes, followed by measurement of luminescence with a microplate reader (Tecan). The experiment was run in triplicate.
Reactive oxygen-species generation
Generation of ROS was detected using the luminescent ROS-Glo H2O2 assay. LN229 cells were plated at a density of 2×104 per well in 80 μL DMEM in 96-well white-walled plates (Nunclon), as recommended by the manufacturer. Briefly, cells were allowed to attach to the plates at 37°C in a CO2 incubator, and then growth medium was replaced with DMEM containing 50 μg/mL and 100 μg/mL 5–15 nm SiNPs for 24 hours and 48 hours. Substrate solution was added to cells in a final concentration of 25 μmol/mL. Then, cells were returned to the incubator (5% CO2, 37°C) and cultured for 6 hours. After this, 100 μL ROS-Glo detection solution was added to each well for 20 minutes at room temperature, and then relative luminescence units were recorded using the microplate reader. The experiment was run in triplicate.
Mitochondrial membrane-potential assessment
LN229 cells were seeded at a density of 2×105 per well in six-well plates with 2 mL DMEM. After 24 hours, the medium was removed and substituted with DMEM containing SiNPs in concentrations of 50/100 μg/mL and further incubated for 24 hours and 48 hours. Cells were detached and resuspended in PBS (106 cells/mL). Next, the disruption of mitochondrial membrane potential (ΔΨm) was evaluated using a MitoScreen kit (BD Biosciences). Briefly, cells were washed in PBS and resuspended in PBS containing 10 mg/mL lipophilic cationic probe JC1. Subsequently, cells were incubated for 15 minutes at 37°C, then washed and resuspended in PBS. Flow cytometry (FACSCanto II; BD Biosciences) was used for further analysis of the samples and FACSDiva software utilized to calculate the percentage of cells with disrupted ΔΨm.
Determination of cellular ATP levels
Measurement of cellular ATP levels in control and SiNP-treated LN229 cells was determined using the CellTiter-Glo assay following the supplier’s specifications. Briefly, LN229 cells were seeded in a white-walled 96-well culture plate (Nunclon) at a density of 104/per well. Cells were allowed to attach and then incubated with medium containing 5–15 nm SiNPs in concentrations of 50 μg/mL and 100 μg/mL at 37°C for 24 hours and 48 hours. After incubation, 100 μL staining solution (CellTiter-Glo reagent) was added to each well and mixed for 2 minutes on an orbital shaker to induce cell lysis. Cells were incubated at room temperature for 10 minutes to stabilize the luminescence signal, which was recorded using the microplate reader. The experiment was run in triplicate.
RNA isolation and gene-expression analysis
Total RNA was isolated using the ReliaPrep system with DNase I treatment according to the manufacturer’s instructions. Spectrophotometric measurements were performed to evaluate the quality and quantity of the extracted RNA (NanoPhotometer; Implen, Munich, Germany). Synthesis of cDNA was performed using the high-capacity RNA-to-cDNA Kit following the supplier’s recommendations. Briefly, 1 μg purified total RNA was used in a 20 μL reaction mixture containing oligo(dT)16 primers, random octamers, dNTPs and murine leukemia virus reverse transcriptase (RT). cDNA (2 μL) served as a template for real-time RT quantitative polymerase chain reaction (qPCR). Amplification of the product was performed using 2× HS-PCR Master Mix SYBR A (A & A Biotechnology, Gdynia, Poland). Primer sequences for DDIT3 (CHOP), BAX, BCL2L11 (BIM), BCL2, BCL2L1 (BCL-XL), PUMA, NOXA, and housekeeping RPL13A have been described in our previous work.15,16 Sequences of the other PCR primers were previously described as: HSPA5 (GRP78),22 SOD1,23 SOD2,23 CAT,23 COX2,24 and IL1B.24 Additional evaluation of primer accuracy was done using Primer-BLAST software. The following reaction parameters were applied: initial denaturation at 95°C for 3 minutes, followed by 40 cycles of 95°C for 1 minute, 60°C–69°C for 30 seconds, and 72°C for 45 second. The CFX Connect real-time PCR system (Bio-Rad Laboratories, Hercules, CA, USA) was used to perform a real-time qPCR assay. Reactions were run in triplicates and expressions were analyzed using the relative quantification method modified by Pfaffl.25
Protein assays
LN229 cells were seeded in six-well plates and treated as previously described. After treatment, cells were washed with cold PBS and solubilized in 100 μL radioimmunoprecipitation-assay lysis buffer per well. Cell lysates were then subjected to centrifugation (14,000 g at 4°C for 10 minutes), and supernatants were collected for protein evaluation. The BCA protein-assay kit was used to determine protein concentration in cell lysates. Protein assays were performed according to the method described by Smith et al.26 BSA was used as a standard.
Sodium dodecyl sulfate–polyacrylamide-gel electrophoresis
Samples of the lysates containing 20 μg of protein were subjected to sodium dodecyl sulfate–polyacrylamide-gel electrophoresis as described by Laemmli.27 Electrophoresis was run for 40–45 minutes using a 7.5% polyacrylamide-gel. A constant current of 25 mA was applied.
Immunoblotting
Resolved proteins were transferred to nitrocellulose membranes and preincubated with Tris-buffered saline (TBS) containing 0.05% Tween 20 (TBS-T) and 5% nonfat dry milk for 2 hours. Membranes were soaked in a mixture of monoclonal (mouse) anti-KDEL antibody (1:1,000) and polyclonal (rabbit) anti-β-tubulin antibody (1:1,000) in 5% dried milk in TBS-T at 4°C for 16 hour. Next, 1-hour incubation with secondary alkaline phosphatase-conjugated antibody against mouse or rabbit IgG at 1:2,500 dilution was carried out. Finally, the nitrocellulose membranes were washed five times with TBS-T and exposed to SigmaFast BCIP/NBT reagent.
Caspase 1-activity assessment
Inflammasome formation was determined using the bioluminescent Caspase-Glo 1 inflammasome assay. LN229 cells were seeded at 104/well in 100 μL DMEM in 96-well white-walled plates according to the manufacturer’s instructions. Briefly, 100 μL Caspase-Glo 1 reagent (containing MG132 inhibitor in the final concentration of 60 μmol/L) or Caspase-Glo 1 YVAD-CHO reagent (containing Ac-YVAD-CHO inhibitor at a final concentration of 1 μmol/L) was added to the 96-well plate containing 100 μL of blank reaction, negative control cells, or treated cells in culture medium. Next, plates were covered with a lid and well contents were gently mixed using a plate shaker at 300 rpm for 30 seconds. Plates were incubated at room temperature for 1 hour to allow stabilization of the luminescent signal. Luminescence was recorded using the microplate reader.
Statistical analysis
Results are presented as mean ± SD from three independent experiments run in triplicate. Statistica Data Miner (Dell, Round Rock, TX, USA) was used to perform statistical analyses. One-way analyses of variance were carried out for comparisons between control and treated groups. Pairwise comparisons were made by post hoc Tukey’s test. Differences were considered significant for P<0.05.
Results
Effect of SiNPs on cell viability
Antiproliferative effects of SiNPs on LN229 glioblastoma cells were assessed using MTT assays. Cells were exposed to increasing concentrations of SiNPs– 7 nm, 5–15 nm, and 10–20 nm, for 24 hours and 48 hours. For all three sizes, concentrations of 5–1,800 μg/mL caused dose- and time-dependent reductions in LN229-cell viability (Figure 1). The 7 nm NPs had relatively low ability to limit LN229-cell proliferation, with only 50% of cells losing viability at the highest (1,800 μg/mL) concentration (Figure 1A), and 5–15 nm SiNPs reduced cell viability to a similar extent after 24 hours and 48 hours of treatment, with the greatest differences observed at 50 μg/mL and 100 μg/mL concentrations. Cells stimulated with 50 μg/mL SiNPs were 11.73%±5.31% unviable after 24 hours, and 34.57%±5.64% unviable after 48 hours. At 100 μg/mL, these values were 12.68%±4.28% vs 42.33%±6.11% for 24 hours and 48 hours, respectively (Figure 1B). In comparison, cells exposed to larger 10–20 nm SiNPs showed more pronounced cytotoxic effects, approaching nearly 80% unviable cells, at the highest concentration after 48 hours of treatment. In this case, time-dependent loss of viability was most apparent (Figure 1C). Based on these MTT results, we chose to proceed with 5–15 nm NPs for further examination. These SiNPs were considered the most suitable for studying cellular and molecular effects, as they showed median cytotoxicity levels and covered almost all particle sizes applied in the study. Doses of 50 μg/mL and 100 μg/mL were chosen in order to examine sublethal concentrations of SiNPs.
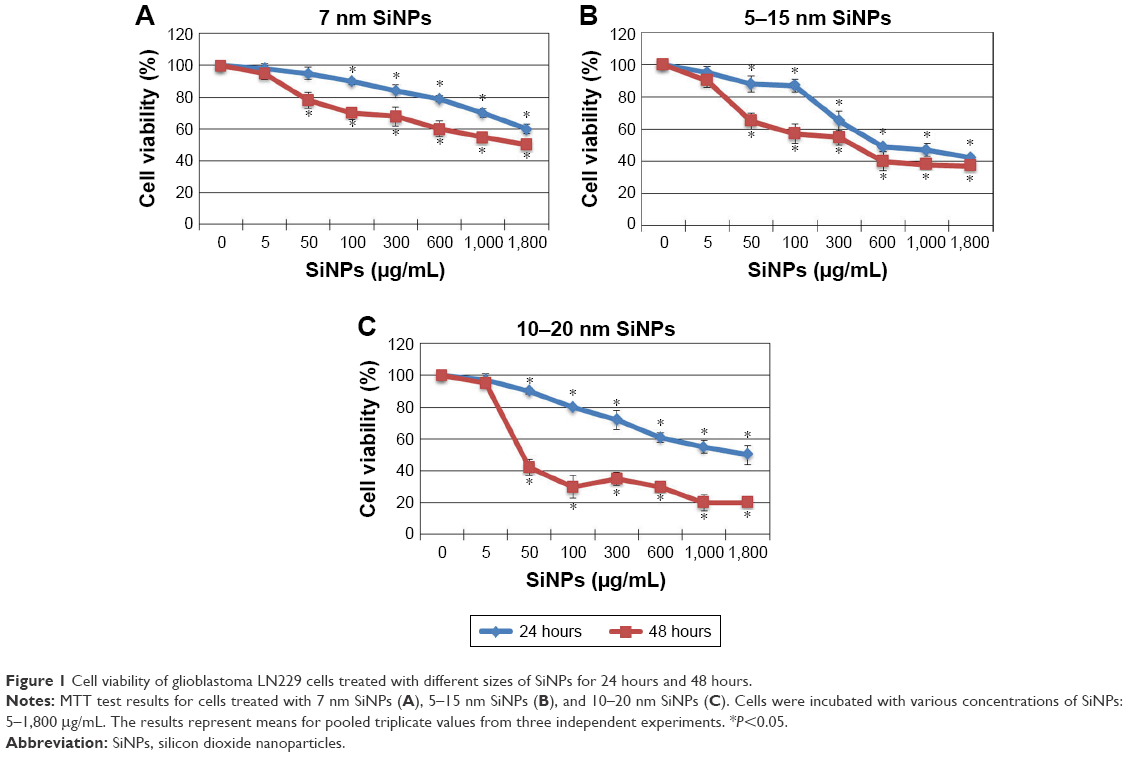 | Figure 1 Cell viability of glioblastoma LN229 cells treated with different sizes of SiNPs for 24 hours and 48 hours. |
Characterization of SiNPs
Despite the fact that commercially available silica nanopowders have undergone preliminary evaluation of physicochemical properties, further characterization of NPs is necessary to confirm the indicated features.28 The morphology of 5–15 nm SiNPs, either in dry form (Figure 2A and B) or dispersed in water (Figure 2C and D) and DMEM (containing 10% FBS) (Figure 2E and F), was visualized by TEM and is presented in Figure 2. SiNPs displayed irregularly shaped NPs prone to forming agglomerates. Moreover, in culture medium, single NPs seemed larger than the declared size, which may be attributed to adsorption of protein corona on the particle surface (comprehensively described by Sikora et al)29 (Figure 2E and F). TEM observations were in general agreement with our previous results from Zetasizer measurements showing that in water, SiNP dispersions were stable, achieving ζ-potentials below −30 mV.14 However, in DMEM supplemented with 10% FBS, ζ-potentials changed significantly to between −8.11 mV and −8.96 mV,14 suggesting that SiNPs were more prone to form aggregates in culture medium, which is in line with findings from Breznan et al.28
 | Figure 2 TEM images of synthetic SiNPs in dry form (A, B), dispersed in water (C, D), and dispersed in DMEM containing 10% FBS (E, F). |
Effect of SiNPs on apoptosis
Microscopic observations were carried out to determine whether the reduced viability of SiNP-treated LN229 cells was accompanied by alterations in cellular morphology and increased cell death. Staining of LN229 cells with AO–EtBr after 48 hours incubation revealed an increased number of cells with red-stained nuclei, indicative of augmented apoptosis (Figure 3A). To confirm these staining results, we performed flow-cytometry analysis. Treatment with SiNPs at 50 μg/mL or 100 μg/mL for 24 hours resulted in markedly elevated levels of early apoptotic cells (23.43% and 36.8%, respectively) (Figure 3B and C). Late apoptosis was less commonly observed after 24 hours of incubation (8.6% and 12.3% for 50 μg/mL and 100 μg/mL SiNPs, respectively) (Figure 3B and C). After 48 hours, the proapoptotic effect became more pronounced, with nearly 85% of cells undergoing apoptosis at either SiNP dose. At this time point, the majority of cells were in late apoptosis (59.4% and 65.2% for 50 μg/mL and 100 μg/mL SiNPs, respectively) (Figure 3B and C). According to these results, apoptosis was the primary cause of cell death in LN229 cells, while necrosis was marginal (Figure 3B).
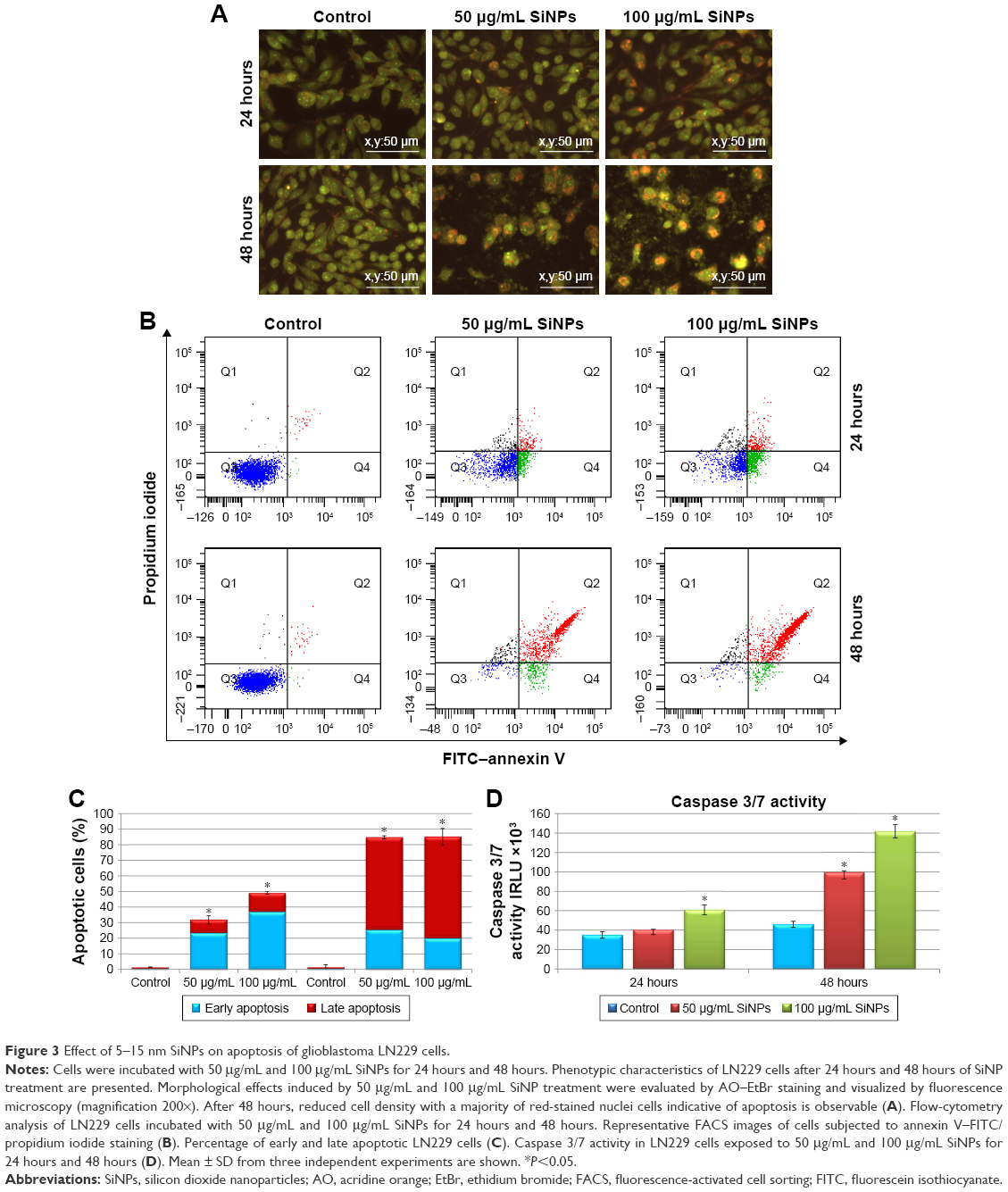 | Figure 3 Effect of 5–15 nm SiNPs on apoptosis of glioblastoma LN229 cells. |
To explore the mechanism of apoptotic death in LN229 cells, we assayed the cells for evidence of caspase-dependent apoptosis. After 24 hours of treatment, significant elevation in caspase 3/7 activity was observed only in cells treated with 100 μg/mL SiNPs (Figure 3D). However, after 48 hours of SiNP incubation at either 50 μg/mL or 100 μg/mL, we observed nearly twofold and threefold increases in caspase 3/7 activity (Figure 3D).
Effect of SiNPs on ER stress
To determine whether SiNPs induced ER stress in LN229 cells, two markers of this process – molecular chaperone GRP78 (HSPA5) and proapoptotic transcription factor CHOP (DDIT3) – were assayed (Figure 4). RT-qPCR analysis of SiNP-stimulated LN229 cells revealed altered expression of HSPA5 and DDIT3 at the mRNA level (Figure 4A and B). Interestingly, after 24 hours of incubation, HSPA5 was significantly upregulated (more than double) only at the highest SiNP concentration (100 μg/mL) (Figure 4A). After 48 hours, HSPA5 showed marked upregulation at 50 μg/mL SiNPs and marked down-regulation at 100 μg/mL SiNPs (Figure 4A). These results were also confirmed at the protein level by immunoblot detection of GRP78 using anti-KDEL antibody, which simultaneously bound to another ER-resident chaperone – GRP94 (Figure 4C). GRP78 and GRP94 were overexpressed after treatment with 100 μg/mL SiNPs for 24 hours and 50 μg/mL SiNPs for 48 hours (Figure 4C). Interestingly, expression levels of these chaperone proteins in LN229 cells were lower than in control cells after 48 hours of incubation with 100 μg/mL SiNPs. Accordingly, the results obtained for DDIT3 demonstrated greater than double upregulation of DDIT3 transcript in SiNP-stimulated cells after 24 hours of treatment and nearly triple upregulation in cells exposed to 50 μg/mL, but not 100 μg/mL, of SiNPs after 48 hours (Figure 4B). These data suggest that treatment with 5–15 nm SiNPs may disrupt ER homeostasis and initiate an ER-stress state in LN229 cells.
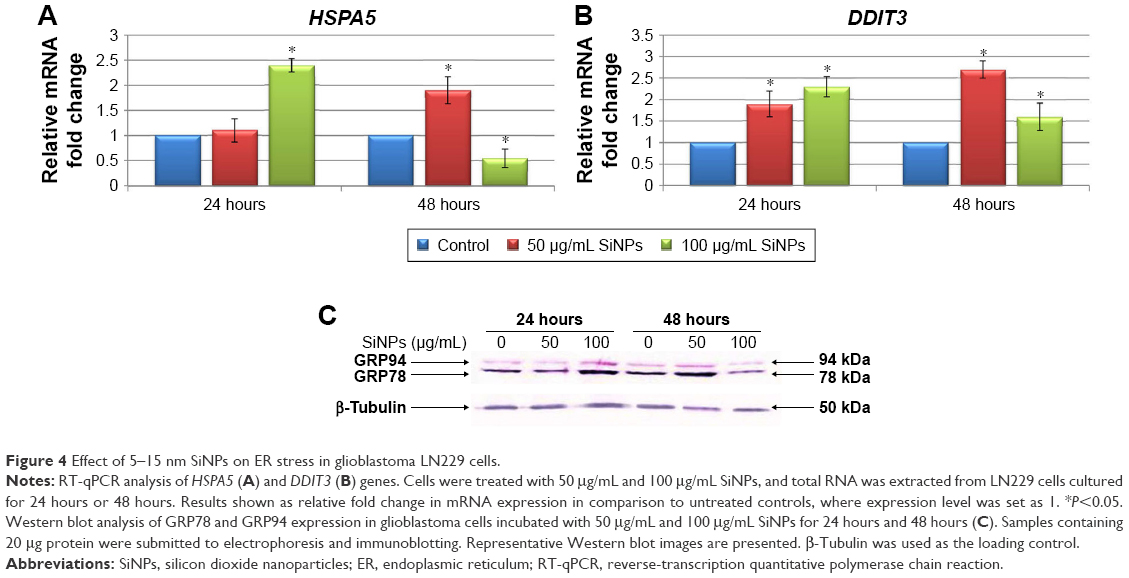 | Figure 4 Effect of 5–15 nm SiNPs on ER stress in glioblastoma LN229 cells. |
Effect of SiNPs on mitochondrial dysfunction and oxidative stress
To gain insight into the nanotoxicity of SiNPs in LN229 cells, we examined ROS levels. A variety of ROS generated in cell cultures included hydroxyl radical, superoxide, singlet oxygen, and hydrogen peroxide, although many of these are converted into H2O2. Because of this and the long half-life and versatile character of H2O2, measurement of this molecule is a convenient proxy for assaying overall ROS levels in cells.30 Our results demonstrated that SiNP-induced intracellular ROS generation changed in a dose- and time-dependent manner. As presented in Figure 5A, ROS production was significantly increased after exposure to 100 μg/mL SiNPs for 24 hours. However, ROS levels were markedly increased after 48 hours at both applied SiNP concentrations (Figure 5A). Overproduction of ROS disturbed the balance between oxidative and antioxidative systems, resulting in reduced antioxidative capacity. To determine whether enhanced ROS production was accompanied by deregulated expression of key antioxidative enzymes, real-time PCR analysis of SOD1, SOD2, and CAT was performed (Figure 5B and C). Indeed, we noticed markedly disrupted expression of the analyzed genes. Interestingly, SOD1 and CAT were significantly downregulated, especially after 48-hour exposure to SiNPs (nearly double decrease in each gene, independently of SiNP concentration), while the SOD2 transcript was intensely upregulated after 24-hour and 48-hour treatments, showing more than a fourfold increase at either 50 μg/mL or 100 μg/mL of SiNPs (Figure 5B and C).
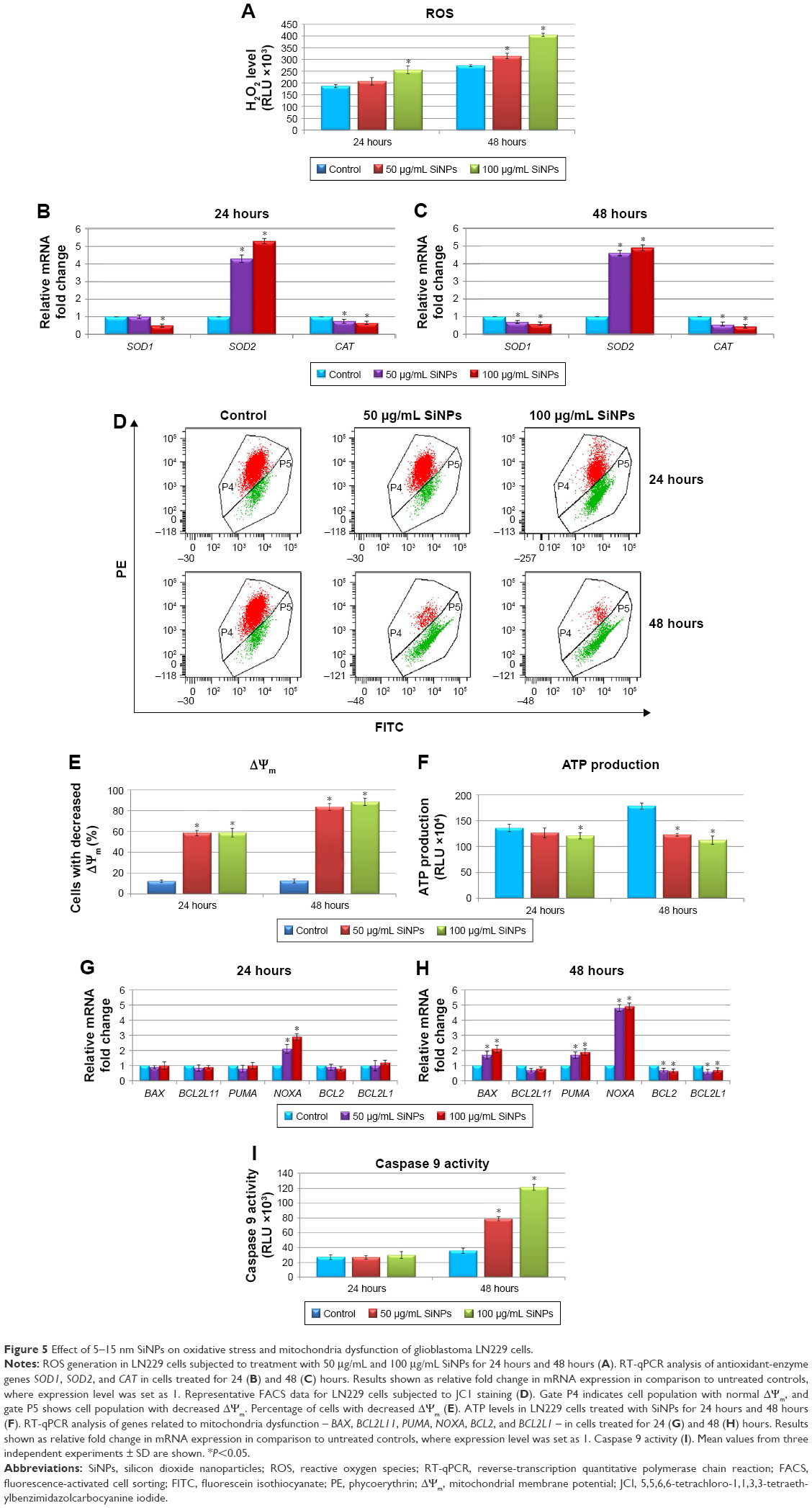 | Figure 5 Effect of 5–15 nm SiNPs on oxidative stress and mitochondria dysfunction of glioblastoma LN229 cells. |
Next, we investigated whether SiNP exposure impaired mitochondria function. In this respect, ΔΨm and ATP production in LN229 cells were assessed. ΔΨm is a key indicator of membrane integrity. Stimulation of LN229 cells with SiNPs led to a significant decrease in ΔΨm in comparison to control cells (Figure 5D and E). After 24 hours of treatment, approximately 58% of cells showed decreased ΔΨm independently of NP dosage (Figure 5D and E). This effect was even more pronounced after 48 hours, where 81.9% and 88.5% of cells treated with 50 μg/mL and 100 μg/mL of SiNPs, respectively, showed decreased ΔΨm (Figure 5D and E). To confirm this mitochondrial toxicity, ATP evaluation was performed. These results showed decreased ATP levels in LN229 cells as early as 24 hours after treatment with SiNPs at high concentration (100 μg/mL) (Figure 5F). Importantly, after 48 hours of incubation with SiNPs at both dosages, ATP generation was significantly attenuated, with 31.1% and 36.8% decreases observed in cells treated with 50 μg/mL or 100 μg/mL SiNPs, respectively (Figure 5F).
Expression of genes related to mitochondria function – BAX, BCL2L11, PUMA, NOXA, BCL2, and BCL2L1 – was analyzed by real-time qPCR (Figure 5G and H). SiNP treatment for 24 hours resulted in significant upregulation of the proapoptotic NOXA, while no significant changes were detected in other gene transcripts (Figure 5G). However, after 48-hour SiNP treatment, a shift toward a proapoptotic transcriptional profile was observed, with significant up-regulation of BAX, PUMA, and NOXA, accompanied by simultaneous downregulation of antiapoptotic BCL2 and BCL2L1 (Figure 5H). To determine whether these perturbations induced mitochondrial pathway apoptosis, we evaluated activity of caspase 9, an indicator of mitochondria-dependent cell death. We demonstrated that 24-hour SiNP exposure was insufficient to activate caspase 9. However, after 48 hours of treatment, levels of caspase 9 activity were markedly elevated in both utilized SiNP concentrations (Figure 5I). These results indicated that SiNPs induced oxidative stress via ROS overproduction, followed by disruption of the oxidant–antioxidant system balance, impaired mitochondria function, and finally activation of the intrinsic apoptosis pathway.
Effect of SiNPs on inflammation
Given the increasing frequency of reports on the proinflammatory potential of SiNPs in several cell- and animal-based studies,1,31–33 we sought to determine whether 5–15 nm SiNPs can evoke a proinflammatory response in LN229 cells. To this end, expression of the proinflammatory mediators IL1B and COX2 was assayed along with inflammasome formation (Figure 6). Inflammasome formation was determined using the Caspase-Glo 1 inflammasome assay, while IL1B and COX2 expression was analyzed by RT-qPCR. Results showed that mRNA expression of IL1B and COX2 was markedly upregulated in a manner dependent on both the dose and time of SiNP treatment (Figure 6A and B). The most pronounced effects were visible after 48 hours of treatment, where SiNP-treated cells showed greater than threefold and sevenfold upregulation of IL1B at 50 μg/mL and 100 μg/mL SiNPs, respectively (Figure 6A). Likewise, changes in COX2 expression were also dose- and time-dependent, with more than sixfold and elevenfold upregulation observed in cells treated for 48 hours with 50 μg/mL and 100 μg/mL SiNPs, respectively (Figure 6B).
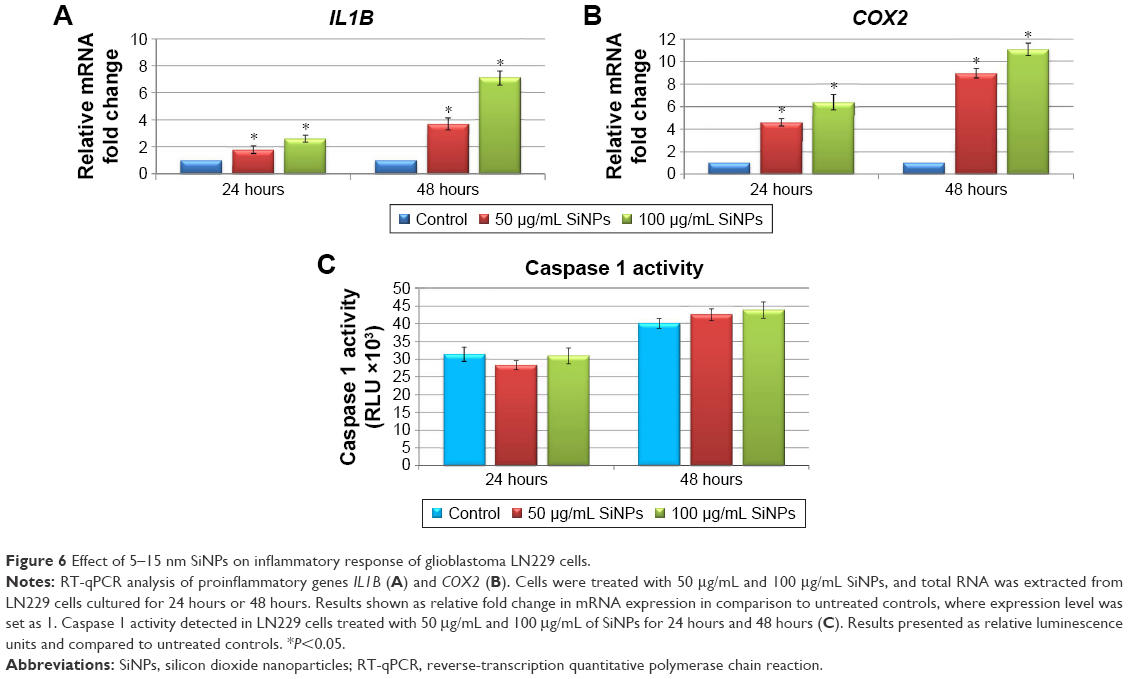 | Figure 6 Effect of 5–15 nm SiNPs on inflammatory response of glioblastoma LN229 cells. |
Inflammasomes are multiprotein complexes, of which caspase 1 is an essential component, responsible for cleavage of IL1β and IL18.34 To investigate whether the observed transcriptional changes in IL1B and COX2 were accompanied by inflammasome formation, we assessed levels of active caspase 1. Despite observation of slightly increased caspase 1 activity in cells following stimulation with SiNPs, these increases failed to reach statistical significance (Figure 6C). These data suggest that SiNP treatment provokes proinflammatory changes in LN229 cells without direct activation of inflammasomes.
Discussion
The field of nanomedicine has opened new possibilities for improved diagnosis and treatment of malignant diseases.35 Over the last two decades, the application of nanotechnology for cancer therapy has been widely investigated; nevertheless, the use of nanomedical approaches for treatment of brain cancers has only just begun and requires further exploration.
Studies carried out over the last 10 years have demonstrated that the physicochemical properties of nanomaterials are crucial factors determining their further utilization. In this respect, SiNPs present favorable characteristics, including wide biodistribution, chemical stability, cellular internalization, and tumor penetration.12 In addition, studies report that SiNPs can trigger cytotoxic and genotoxic effects, causing generation of ROS, aberrant aggregation of nucleoplasmic proteins, DNA damage, and finally apoptotic death in treated cells.36 These cell-damaging effects have shifted the attention of many researchers toward application of these SiNPs in cancer therapy.36 Preliminary reports suggest that SiNPs are capable of crossing the blood–brain barrier, making them potentially attractive agents for glioblastoma treatment.37–40 Moreover, Ducray et al showed that two sets of modified SiNPs (Si–indocyanine green/polycaprolactone–polylactic acid–rhodamine-doped and indocyanine green/polycaprolactone–rhodamine-doped NPs) did not impair cell viability or induce neuroinflammation in primary hippocampal cultures.41 Additionally, animal models have demonstrated the biocompatibility and relative safety of SiNPs for in vivo use in mice and rats.42,43 This relatively low toxicity in normal brain cells and high cytotoxicity in malignant cells make SiNPs an attractive agent for glioblastoma treatment, either as a single therapeutic or a drug carrier.
On these premises, we investigated the cytotoxic effect of SiNPs on glioblastoma LN229 cells. Since NP size is an important factor influencing NP-dependent cellular effects, we studied small SiNPs (7–20 nm). Lu et al showed that decreasing NP size (<70 nm) increased their diffusivity,10 while other studies have suggested that small inorganic NPs (<100 nm or <20 nm) readily accumulate at the tumor site, resulting in an enhanced permeation and retention effect.44,45 Indeed, we observed time- and dose-dependent reduction in viability of SiNP-treated glioblastoma cells. The growth-inhibitory effect was visible regardless of NP size, although some differences in nanotoxicity were observed. This is in general agreement with the results of Breznan et al, who tested similar size ranges of SiNPs (5–15 nm, 10–20 nm, 12 nm), and demonstrated that 5–15 nm and 10–20 nm SiNPs exerted similar cytotoxicity in A549, THP1, and J774A.1 cell lines.28 Therefore, we utilized 5–15 nm SiNPs for further in-depth analysis in glioblastoma cells.
It is known that SiNPs can evoke apoptosis, necrosis, and autophagy in various in vitro experiments,4,11,12,14 and these cell-damaging effects are beneficial in the context of developing anticancer therapies. Since impaired apoptosis is often involved in the development of cancers,14 induction of apoptotic cell death is a key focus in the discovery of a novel anticancer drugs. In this study, we demonstrated that apoptotic death and not necrosis is the principal outcome of 5–15 nm SiNP exposure in LN229 cells. Microscopic observations of nuclei and annexin V–FITC/PI staining of LN229 cells showed prominent induction of apoptosis after SiNP treatment, and enhanced activity of caspase 3/7 was observed, with a considerable percentage of cells in late-phase apoptosis. This encouraged us to explore further the molecular mechanisms underlying apoptosis in glioblastoma LN229 cells.
Molecular mechanisms activated in response to NP treatment are strictly dependent on the cellular uptake of these particles, and it is increasingly recognized that SiNPs can easily enter cells by endocytosis, localizing to the cytoplasm or crucial cellular organelles, such as the ER46 and mitochondria.14 Indeed, SiNPs have been shown to interfere with the ER, affecting homeostasis and causing a state known as ER stress.5,6,46 Downstream of ER stress, the unfolded protein response is initiated, with one of two potential outcomes: activation of prosurvival pathways via increased expression of chaperone proteins GRP78, GRP94, and ORP150, or activation of a proapoptotic pathway through enhanced expression of CHOP.15,16 Therefore, increased levels of ER chaperones and CHOP are considered markers of ER stress. In this study, we observed altered expression of GRP78 and GRP94 at mRNA and protein levels in a manner dependent on time and dose of SiNPs. After 24 hours, significant overexpression of GRP78/GRP94 was visible in cells cultured with 100 μg/mL SiNPs, while after 48 hours, cultures containing 50 μg/mL SiNPs showed marked increase in GRP78/GRP94 expression. Interestingly, in comparison to controls, evident decrease in expression of these proteins was observed in cells cultured with 100 μg/mL SiNPs for 48 hours. The results obtained for CHOP expression demonstrated statistically relevant up-regulation of DDIT3 transcript in SiNPs-stimulated cells after 24 hours of treatment, while 48-hour exposition resulted in marked DDIT3 up-regulation only in cells exposed to 50 μg/mL but not 100 μg/mL of SiNPs. These results might suggest that SiNPs initially disrupt ER homeostasis and cause ER stress in LN229 cells. Interestingly, in cells treated with 100 μg/mL SiNPs for 48 hours, there was relatively modest overexpression of DDIT3, which be attributed to activation of the executor phase of apoptosis connected with the activation of effector caspases 3 and 9. Our results suggest that ER stress is one of the factors contributing to SiNP-dependent apoptosis in LN229 cells. Consistent with these conclusions, Christen and Fent demonstrated upregulation of ATF4, HSPA5, XPB1, PPP1R15A, and DDIT3 in Huh7 cells.5,6 Also, Phukan et al demonstrated increased expression of ER-stress markers, such as GRP78, p-PERK, and eIF2α, without activation of CHOP, suggesting a CHOP-independent pathway of apoptosis in HEK293 cells treated with silica-coated magnetic NPs.47 While these data suggest that ER stress is responsible for cytotoxic effects of SiNPs in many cancer cell lines,5,6,47 our data are the first to demonstrate this in glioblastoma cells. Although it seems that ER homeostasis is affected in LN229 cells, the inherent heterogeneity of glioblastoma cells necessitates further comprehensive analysis of ER function to resolve the complexity of ER-mediated signaling pathways.
Apoptosis can be activated by death receptors located on the surface of the cell or by intrinsic signals from mitochondria. Although induction of the extrinsic apoptosis pathway together with caspase 8 activation has been demonstrated in human lung carcinoma A549 cells after exposure to 6 nm SiNPs,7 the majority of anticancer therapeutics are believed to initiate apoptosis via the intrinsic apoptosis pathway.48,49 We hypothesized that SiNPs, as a potential anticancer biomaterial, might also trigger mitochondria-mediated apoptosis. Indeed, we demonstrated elevated activity of mitochondria-related caspase 9 after 48 hours of treatment, where mostly late apoptotic cells were observed, confirming SiNP-mediated apoptosis can occur via the intrinsic pathway. This route of apoptosis has also been identified in other in vitro studies exploring SiNP mode of action.10,50
NPs can localize in the mitochondria, organelles with high sensitivity to exogenous compounds, and lead to induction of structural damage through oxidative stress.51 ATP biogenesis is an important function of mitochondria, and through this process a percentage of oxygen is left incompletely reduced, causing the formation of superoxide anion radicals and subsequent generation of other oxygen-containing radicals. As such, these organelles are also the main source of cellular ROS, a byproduct of cellular oxidative metabolism.51
Excessive production of ROS can induce oxidative stress followed by lipid peroxidation, DNA damage, and inhibition of antioxidant activities, which may result in imbalanced physiological redox-dependent functions.52 Indeed, after 48 hours of SiNP treatment, we observed markedly increased levels of H2O2 in cultured LN229 cells. Moreover, we identified decreased expressions of the main antioxidant enzymes, SOD1 and CAT, with a concomitant increase in SOD2 expression. Since SOD2 is a mitochondrial protein, we speculate that its elevated expression is likely due to enhanced mitochondria efforts to overcome NP invasion. In line with our results, Guo et al reported enhanced SOD activity and increased SOD2 expression after exposure to SiNPs,4 and Alarifi et al demonstrated increased activity of SOD and CAT during nickel NP-induced oxidative stress in human skin A431 epidermal cells.53 These studies and ours confirm that oxidative stress is accompanied by deregulated antioxidant-enzyme expression. Although activity of some antioxidant enzymes may be elevated, this is likely insufficient to counteract the oxidative stress and thus still results in ROS overproduction. Furthermore, ROS can induce mitochondrial dysfunction, leading to opening of the mitochondrial permeability transition pore, ATP depletion, and finally cell death.4 Consistently with our previous work,14 incubation of LN229 cells with 50 μg/mL and 100 μg/mL SiNPs caused significant depolarization of ΔΨm and decreased ATP production. Interestingly, we also identified upregulation of the proapoptotic BCL2 family members BAX, PUMA, and NOXA, with simultaneous downregulation of the antiapoptotic BCL2 family members BCL2 and BCL2L1. The proapoptotic members of the BCL2 family, such as BAX, BAK, and BAD, activate the release of cytochrome C from mitochondria, while antiapoptotic members of the same family, BCL-2 and BCLXL, antagonize it. The key role of BCL2/BCLXL in mitochondrial permeability and ΔΨm loss is well established,4 and thus depleted ΔΨm with a concomitant decrease in the BCL2:BAX ratio suggests SiNP treatment activates mitochondria-dependent apoptosis. These observations are in agreement with previous reports showing overexpression of BAX and declined expression of BCL2 in human umbilical vein endothelial cells and the human liver cell line HEPG2 after exposure to SiNPs.4,12 Altogether, these results indicate that SiNPs increase oxidative stress, leading to mitochondrial dysfunction and low energy status. This, together with deregulated expression of key genes related to mitochondria functioning, ultimately leads to the activation of mitochondria-mediated apoptosis in glioblastoma LN229 cells.
Oxidative stress has not only been linked to impaired cell function but also to modulation of inflammatory responses, and new data increasingly support excessive ROS production as a factor triggering proinflammatory response.54,55 Likewise, a growing body of evidence suggests that SiNPs may also induce proinflammatory effects in cancer cells,4,56,57 To date, the mechanisms underlying the cross talk between inflammatory response and SiNP exposure are not fully understood. One possible link between ROS overproduction and inflammation may be through SOD2 activation. Manganese SOD (SOD2) is located in the mitochondrial matrix, and reacts with O2• produced by mitochondria to generate the less toxic byproduct H2O2. H2O2 has the ability to cross the outer membrane of the mitochondria to access cytosolic targets, leading to diverse functional outcomes, including activation of redox-dependent transcription factors, such as NFκB and HIF1α, inflammasome formation, and subsequent downstream stimulation of proinflammatory cytokines.54
Caspase 1 is an essential component of the multiprotein inflammasome complexes,34 and in its active form can cleave the proinflammatory cytokines IL1β and IL18 into their bioactive forms, causing pyroptosis, a type of inflammation-dependent cell death.34,54 Since caspase 1 activity was not significantly increased in SiNP-treated LN229 cells, pyroptosis was not likely responsible for the cell death observed in our study. However, we did identify elevated levels of H2O2 and upregulation of SOD2 together with increased expressions of IL1B and COX2 mRNAs. IL1β is a key regulator of inflammatory response,4 and COX2 is a downstream target of cytokines primarily responsible for prostanoid production in acute and chronic inflammatory conditions.58 Overproduction of any of these factors may impair cellular homeostasis and be harmful to cells. In line with our results, Guo et al reported the upregulation of key proinflammatory mediators, including IL6, IL1B, IL8, TNFA, ICAM1, VCAM1, and MCP1, in human vascular endothelial cells, suggesting the occurrence of robust proinflammatory response after SiNP treatment.4 Corbalan et al also demonstrated the ability of SiNPs to induce proinflammatory response in human endothelial cells, as evidenced by increased transcription of COX2, IL6, and IL8.13 Moreover, Kusaka et al studied the relationship between of amorphous silica size and macrophage inflammatory activity, demonstrating efficient internalization of SiNPs by mouse bone marrow-derived macrophages and induction of caspase 1 activity, irrespective of NP size.59 They found that 30–1,000 nm-diameter silica particles induced IL1β secretion, lysosomal destabilization, and cell death in tested cells.59 Despite the considerable amount of existing data, the exact role of inflammation in glioblastoma is still unclear and requires further exploration. Therefore, in order to properly utilize NPs, we suggest that future studies of SiNPs in glioblastoma include examination of inflammatory status, as well as other aspects of cellular physiology.
Conclusion
According to our findings, SiNPs cause cell-damaging effects involving various aspects of cellular function in glioblastoma LN229 cells. SiNP exposure results in ROS overproduction, mitochondrial damage, ER stress, and activation of inflammatory response, which together contribute to apoptotic death of treated cells. These findings suggest that SiNPs might be considered potential antiglioblastoma agents. Nevertheless, profound molecular analysis of the outcomes of SiNP treatment at the organism level is required to solve the nature of these NPs in anticancer therapy comprehensively.
NPs will definitively continue to be a popular research topic, with the possibility of some day becoming a favorable treatment option for people afflicted with brain tumors. However, prior to the widespread use of NPs, further exploration of their long-term toxicity and molecular effects will be necessary. In-depth analysis of the molecular mechanisms of NP-mediated apoptosis is crucial before application as an anticancer agent. Based on this knowledge, determination of drug-carrier potential, adequate functionalization, or potential application of NPs with cotherapeutic drugs will be possible to maximize drug efficiency and minimize adverse effects. Therefore, SiNPs might potentially be a promising agent facilitating further progress in brain cancer therapy.
Acknowledgments
We kindly thank Urszula Klekotka from the Department of Physicochemical Methods, Institute of Chemistry, University of Białystok, Białystok, Poland, for her help in performing TEM analyses. This study was conducted with the use of equipment purchased by the Medical University of Białystok as part of the OP DEP 2007–2013, Priority Axis I.3, contract POPW.01.03.00-20-001/12.
Disclosure
The authors report no conflicts of interest in this work.
References
Guo C, Xia Y, Niu P, et al. Silica nanoparticles induce oxidative stress, inflammation, and endothelial dysfunction in vitro via activation of the MAPK/Nrf2 pathway and nuclear factor-κB signaling. Int J Nanomedicine. 2015;10:1463–1477. | ||
Yang Y, Li J. Lipid, protein and poly(NIPAM) coated mesoporous silica nanoparticles for biomedical applications. Adv Colloid Interface Sci. 2014;207:155–163. | ||
Mai WX, Meng H. Mesoporous silica nanoparticles: a multifunctional nano therapeutic system. Integr Biol (Camb). 2013;5:19–28. | ||
Guo C, Yang M, Jing L, et al. Amorphous silica nanoparticles trigger vascular endothelial cell injury through apoptosis and autophagy via reactive oxygen species-mediated MaPK/Bcl-2 and PI3K/Akt/mTOR signaling. Int J Nanomedicine. 2016;11:5257–5276. | ||
Christen V, Camenzind M, Fent K. Silica nanoparticles induce endoplasmic reticulum stress response, oxidative stress and activate the mitogen-activated protein kinase (MAPK) signaling pathway. Toxicol Rep. 2014;1:1143–1151. | ||
Christen V, Fent K. Silica nanoparticles induce endoplasmic reticulum stress response and activate mitogen activated kinase (MAPK) signaling. Toxicol Rep. 2016;3:832–840. | ||
Tokgun O, Demiray A, Kaya B, et al. Silica nanoparticles can induce apoptosis via dead receptor and caspase 8 pathway on A549 cells. Adv Food Sci. 2015;37:65–70. | ||
Wan Y, Apostolou S, Dronov R, Kuss B, Voelcker NH. Cancer-targeting siRNA delivery from porous silicon nanoparticles. Nanomedicine (Lond). 2014;9:2309–2321. | ||
Joshi GN, Knecht DA. Silica phagocytosis causes apoptosis and necrosis by different temporal and molecular pathways in alveolar macrophages. Apoptosis. 2013;18:271–285. | ||
Lu X, Qian J, Zhou H, et al. In vitro cytotoxicity and induction of apoptosis by silica nanoparticles in human HepG2 hepatoma cells. Int J Nanomedicine. 2011;6:1889–18901. | ||
Ahmed M. Silica nanoparticles-induced cytotoxicity oxidative stress and apoptosis in cultured A431 and A549 cells. Hum Exp Toxicol. 2013;32:186–195. | ||
Ahmad J, Ahamed M, Akhtar MJ, et al. Apoptosis induction by silica nanoparticles mediated through reactive oxygen species in human liver cell line HepG2. Toxicol Appl Pharmacol. 2012;259:160–168. | ||
Corbalan JJ, Medina C, Jacoby A, Malinski T, Radomski MW. Amorphous silica nanoparticles trigger nitric oxide/peroxynitrite imbalance in human endothelial cells: inflammatory and cytotoxic effects. Int J Nanomedicine. 2011;6:2821–2835. | ||
Krętowski R, Kusaczuk M, Naumowicz M, Kotyńska J, Szynaka B, Cechowska-Pasko M. The effects of silica nanoparticles on apoptosis and autophagy of glioblastoma cell lines. Nanomaterials (Basel). 2017;7:E230. | ||
Kusaczuk M, Krętowski R, Stypułkowska A, Cechowska-Pasko M. Molecular and cellular effects of a novel hydroxamate-based HDAC inhibitor – belinostat – in glioblastoma cell lines: a preliminary report. Invest New Drugs. 2016;34:552–564. | ||
Kusaczuk M, Krętowski R, Bartoszewicz M, Cechowska-Pasko M. Phenylbutyrate – a pan-HDAC inhibitor – suppresses proliferation of glioblastoma LN-229 cell line. Tumor Biol. 2016;37:931–942. | ||
Tzeng SY, Green JJ. Therapeutic nanomedicine for brain cancer. Ther Deliv. 2013;4:687–704. | ||
Zhang H, Zhang W, Zhou Y, Jiang Y, Li S. Dual functional mesoporous silicon nanoparticles enhance the radiosensitivity of VPA in glioblastoma. Transl Oncol. 2017;10:229–240. | ||
Lai JC, Ananthakrishnan G, Jandhyam S, et al. Treatment of human astrocytoma U87 cells with silicon dioxide nanoparticles lowers their survival and alters their expression of mitochondrial and cell signaling proteins. Int J Nanomedicine. 2010;5:715–723. | ||
Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res. 1987;47:936–942. | ||
Ho K, Yazan LS, Ismail N, Ismail M. Apoptosis and cell cycle arrest of human colorectal cancer cell line HT-29 induced by vanillin. Cancer Epidemiol. 2009;33:155–160. | ||
Yoo SA, You S, Yoon HJ, et al. A novel pathogenic role of the ER chaperone GRP78/BiP in rheumatoid arthritis. J Exp Med. 2012;209:871–886. | ||
Ahmad TAFT, Jubri Z, Rajab NF, et al. Gelam honey protects against gamma-irradiation damage to antioxidant enzymes in human diploid fibroblasts. Molecules. 2013;18:2200–2211. | ||
Nhu QM, Shirey K, Teijaro JR, et al. Novel signaling interactions between proteinase-activated receptor 2 and Toll-like receptors in vitro and in vivo. Mucosal Immunol. 2010;3:29–39. | ||
Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. | ||
Smith PK, Krohn RI, Hermanson GT, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. | ||
Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. | ||
Breznan D, Das DD, O’Brien JS, et al. Differential cytotoxic and inflammatory potency of amorphous silicon dioxide nanoparticles of similar size in multiple cell lines. Nanotoxicology. 2017;11:223–235. | ||
Sikora A, Shard AG, Minelli C. Size and ζ-potential measurement of silica nanoparticles in serum using tunable resistive pulse sensing. Langmuir. 2016;32:2216–2224. | ||
Alfadda A, Sallam R. Reactive oxygen species in health and disease. J Biomed Biotechnol. 2012;2012:936486. | ||
Park HJ, Sohn JH, Kim YJ, et al. Acute exposure to silica nanoparticles aggravate airway inflammation: different effects according to surface characteristics. Exp Mol Med. 2015;47:e173. | ||
Park EJ, Park K. Oxidative stress and pro-inflammatory responses induced by silica nanoparticles in vivo and in vitro. Toxicol Lett. 2009;184:18–25. | ||
Di Cristo L, Movia D, Bianchi MG, et al. Proinflammatory effects of pyrogenic and precipitated amorphous silica nanoparticles in innate immunity cells. Toxicol Sci. 2016;150:40–53. | ||
Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signaling. Nat Rev Immunol. 2016;16:407–420. | ||
Piktel E, Niemirowicz K, Wątek M, Wollny T, Deptuła P, Bucki J. Recent insights in nanotechnology-based drugs and formulations designed for effective anti-cancer therapy. Nanobiotechnology. 2016;14:39. | ||
Mu Q, Hondow NS, Krzemiński L, Brown AP, Jeuken LJ, Routledge MN. Mechanism of cellular uptake of genotoxic silica nanoparticles. Part Fibre Toxicol. 2012;9:29. | ||
Zhou M, Xie L, Fang CJ, et al. Implications for blood-brain-barrier permeability in vitro oxidative stress and neurotoxicity potential induced by mesoporous silica nanoparticles: effects of surface modification. RCS Adv. 2016;6:2800–2890. | ||
Hanada S, Fujioka K, Inoue Y, Kanaya F, Manome Y, Yamamoto K. Cell-based in vitro blood–brain barrier model can rapidly evaluate nanoparticles’ brain permeability in association with particle size and surface modification. Int J Mol Sci. 2014;15:1812–1825. | ||
Baghirov H, Karaman D, Viitala T, et al. Feasibility study of the permeability and uptake of mesoporous silica nanoparticles across the blood-brain barrier. PLoS One. 2016;11:e0160705. | ||
Liu D, Lin B, Shao W, Zhu Z, Ji T, Yang C. In vitro and in vivo studies on the transport of PEGylated silica nanoparticles across the blood-brain barrier. ACS Appl Mater Interfaces. 2014;6:2131–2136. | ||
Ducray AD, Stojiljkovic A, Möller A, et al. Uptake of silica nanoparticles in the brain and effects on neuronal differentiation using different in vitro models. Nanomedicine. 2017;13:1195–1204. | ||
Chan WT, Liu CC, Chiau JS, et al. In vivo toxicologic study of larger silica nanoparticles in mice. Int J Nanomedicine. 2017;12:3421–3432. | ||
Ivanov S, Zhuravsky S, Yukina G, Tomson V, Korolev D, Galagudza M. In vivo toxicity of intravenously administered silica and silicon nanoparticles. Materials. 2012;5:1873–1889. | ||
Sarin H, Kanevsky AS, Wu HT, et al. Physiologic upper limit of pore size in the blood-tumor barrier of malignant solid tumors. J Transl Med. 2009;7:51. | ||
Neuwelt EA, Varallyay P, Bago AG, Muldoon LL, Nesbit G, Nixon R. Imaging of iron oxide nanoparticles by MR and light microscopy in patients with malignant brain tumours. Neuropathol Appl Neurobiol. 2004;30:456–471. | ||
Parveen A, Rizvi SHM, Mahdi F, et al. Silica nanoparticles mediated neuronal cell death in corpus striatum of rat brain: implication of mitochondrial, endoplasmic reticulum and oxidative stress. J Nanopart Res. 2014;16:2664. | ||
Phukan G, Shin TH, Shim JS, et al. Silica-coated magnetic nanoparticles impair proteasome activity and increase the formation of cytoplasmic inclusion bodies in vitro. Sci Rep. 2016;6:29095. | ||
Lu W, Wan J, Zhang Q, She Z, Jiang X. Aclarubicin-loaded cationic albumin-conjugated PEGylated nanoparticle for glioma chemotherapy in rats. Int J Cancer. 2007;120:420–431. | ||
Grad JM, Cepero E, Boise LH. Mitochondria as targets for established and novel anti-cancer agents. Drug Resist Updat. 2001;4:85–91. | ||
Ahamed M. Silica nanoparticles-induced cytotoxicity, oxidative stress and apoptosis in cultured A431 and A549 cells. Hum Exp Toxicol. 2013;32:186–195. | ||
Sun L, Li Y, Liu X, et al. Cytotoxicity and mitochondrial damage caused by silica nanoparticles. Toxicol In vitro. 2011;25(8):1619–1629. | ||
Fu PP, Xia Q, Hwang HM, Ray PC, Yu H. Mechanisms of nanotoxicity: generation of reactive oxygen species. J Food Drug Anal. 2014;22:64–75. | ||
Alarifi S, Ali D, Alakhtani S, Al Suhaibani ES, Al-Qahtani AA. Reactive oxygen species-mediated DNA damage and apoptosis in human skin epidermal cells after exposure to nickel nanoparticles. Biol Trace Elem Res. 2014;157:84–93. | ||
Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20:1126–1167. | ||
Choi J, Zheng Q, Katz HE, Guilarte TR. Silica-based nanoparticle uptake and cellular response by primary microglia. Environ Health Perspect. 2010;118:589–595. | ||
Mendoza A, Torres-Hernandez JA, Ault JG, Pedersen-Lane JH, Gao D, Lawrence DA. Silica nanoparticles induce oxidative stress and inflammation of human peripheral blood mononuclear cells. Cell Stress Chaperones. 2014;19:777–790. | ||
Yang L, Yan QQ, Zhao J, et al. The role of potassium channel in silica nanoparticle-induced inflammatory effect in human vascular endothelial cells in vitro. Toxicol Lett. 2013;223:16–24. | ||
Minghetti L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J Neuropathol Exp Neurol. 2004;63:901–910. | ||
Kusaka T, Nakayama M, Nakamura K, Ishimiya M, Furusawa E, Ogasawara K. Effect of silica particle size on macrophage inflammatory responses. PLoS One. 2014;9:e92634. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.