Back to Journals » International Journal of General Medicine » Volume 15
Significant Biomarkers Identification Associated with Cutaneous Squamous Cell Carcinoma Progression
Authors Qiu CG, Shen B, Sun XQ
Received 4 January 2022
Accepted for publication 21 February 2022
Published 2 March 2022 Volume 2022:15 Pages 2347—2360
DOI https://doi.org/10.2147/IJGM.S357022
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Cheng-Gang Qiu,1 Bin Shen,1 Xiao-Qi Sun2
1Department of Burn, Affiliated Xiaoshan Hospital, Hangzhou Normal University, Hangzhou, 311200, Zhejiang, People’s Republic of China; 2Department of Plastic Surgery, Affiliated Xiaoshan Hospital, Hangzhou Normal University, Hangzhou, 311200, Zhejiang, People’s Republic of China
Correspondence: Xiao-Qi Sun, Department of Plastic Surgery, Affiliated Xiaoshan Hospital, Hangzhou Normal University, 728 Yucai North Road, Chengxiang Town, Xiaoshan District, Hangzhou, Zhejiang, 311200, People’s Republic of China, Email [email protected]
Background: This study aimed to identify significant genes associated with cutaneous squamous cell carcinoma (CSCC) initiation and development.
Methods: The overlapped differential expressed genes (DEGs) between normal and CSCC samples were firstly screened out, followed by KEGG analysis. The top 10 hub genes were then detected from the whole protein–protein interaction (PPI) network. Further, important biomarkers continuously associated with actinic keratosis (AK), CSCC, and CSCC invasion was successively filtrated. GSEA analysis was finally performed to reveal potential mechanisms associated with biomarkers.
Results: A total of 179 DEGs were identified, which were enriched in pathways in cancer, PI3K-Akt signaling pathway, and human papillomavirus infection. The 10 hub genes were firstly identified from the PPI network, and they were all highly expressed in AK tissues compared with normal tissues. Next, we found that 6 genes were overexpressed in CSCC compared with AK tissues. Further, we identified that the expression of 2 genes (MYBL2 and TK1) was higher in CSCC invasion groups compared with samples without invasion. Through a series of filtrations, we confirmed that MYBL2 and TK1 were the most significant biomarkers associated with CSCC initiation and progression. The pan-cancer analysis further supported their prognostic value in human cancers. GSEA analysis found that they positively correlated with N glycan biosynthesis and p53 signaling pathways.
Conclusion: MYBL2 and TK1 were proved to play a vital role in CSCC tumorigenesis and progression, which may act as promising biomarkers or therapeutic targets for accurate diagnosis and treatment of CSCC.
Keywords: cutaneous squamous cell carcinoma, DEGs, MYBL2, TK1
Introduction
Cutaneous squamous cell carcinoma (CSCC) is related to the uncontrolled proliferation of epidermal keratinocytes and has become the second most common skin cancer. According to the report, the incidence of CSCC increased from 18.6/100,000 persons in 2006 to 28.1 in 2015.1 CSCC predominantly affected the older generation with 5 years survival rates reported as <30%.2 Regarding the pathogenic mechanism of CSCC, actinic keratosis (AK) is generally considered as the precancerous lesion of CSCC, and the previous study has suggested that AK and CSCC exhibited highly similar clinical, histological, and molecular characteristics.3 It is estimated that 10% of AK will develop into CSCC in approximately 2 years.4 What was worse, the majority of metastases (28/48; 58%) were found within 3 months of the diagnosis of CSCC,5 and metastatic CSCC patients always presented a poor prognosis, as more than 70% will die of the disease within 3 years.6 Therefore, CSCC is regarded as a spectrum of diseases that arises from premalignant lesion AK to CSCC in situ (CSCCIS, Bowen’s disease) and finally to invasive and metastatic CSCC.
To reduce the development and metastasis risk of CSCC, it was important to identify potential novel molecular targets for the diagnosis and therapy of patients with CSCC. At present, several pieces of research focused on the investigations of clinically useful biomarkers for CSCC. Chen et al applied weighted gene co-expression network analysis (WGCNA) to find 7 hub genes that played important roles in CSCC tumorigenesis, and significantly correlated with the survival of various cancer types.7 Zheng et al suggested that HIF1A, MAPK8, mTOR, BCL2L1, and RAB23 may be associated with CSCC of head and neck (CSCCHN) and serve as potential therapeutic targets in cSCCHN with clinical perineural invasion (PNI).8 Xu et al revealed 3 core genes (AGO4, E2F1, and CCND) and one associated lncRNAHLA-F-AS1, which may be provided as potential therapeutic targets for CSCC.9 It was necessary to detect more biomarkers to reveal potential mechanisms in CSCC.
The present study aimed to explore potential targets involved in the initiation and development of CSCC. Differentially expressed genes (DEGs) between normal and CSCC samples were firstly detected, and hub genes continuously associated with AK, CSCC, and CSCC invasion were successively filtrated. Regarding the identified biomarkers, we performed the GSEA analysis to explore the potential regulatory mechanism. This study revealed several important targets and contributed to further revealing the mechanism behind CSCC.
Methods
Identification of DEGs
GSE42677 and GSE66359 which were obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) were used to identify the differential expressed genes (DEGs) between normal and SCC progression-related samples, respectively. The GSE42677 contained 10 normal skin tissues and 15 SCC progression-related tissues including 5 cases of actinic keratosis, 5 cases of in situ SCC, and 5 cases of invasive SCC, and GSE66359 included 5 normal human epidermal keratinocyte tissues and 8 CSCC tissues. Expression profiles of GSE42677 and GSE66359 were subjected to normalization through log2 (X+1) processing. The DEGs between CSCC and normal tissues were screened out by the limma method. Threshold of false discovery rate (FDR) < 0.05 and |log2 fold change| > 1 were used to identify the significant DEGs. Overlapped DEGs in 2 datasets were subsequently detected via Venn analysis.
PPI Network Construction and Enrichment Analyses on DEGs
A total of 179 overlapped DEGs were finally identified. The protein–protein interaction (PPI) network among DEGs was established through the String database (https://cn.string-db.org/) with setting the medium confidence as the significant threshold and then visualized by Cytoscape. The function of DGEs was explored through the KEGG Orthology Based Annotation System (KOBAS) database (http://kobas.cbi.pku.edu.cn/program.run.do) by performing GO annotation and KEGG analyses. The GO annotation consisted of biological process (BP), cellular component (CC), and molecular function (MF). KEGG analysis was performed to evaluate the pathways in which DEGs may be involved.
Identification of Significant Modules and Hub Genes in the PPI Network
In this study, Molecular Complex Detection (MCODE) analysis was utilized to screen out the significant modules within the whole PPI network. The network with a score >10 was regarded as the significant module. Potential KEGG pathways associated with genes in significant modules were predicted.
In addition, the top 20 hub genes in the whole network were screened out by the cytoHubba plugin by MCC, DMNC, and Degree methods, respectively. Finally, 10 overlapped hub genes were detected among 3 methods. Furthermore, the interaction between hub genes was also explored. In addition, the genetic alteration of 10 hub genes in CSCC was evaluated through the dataset of cutaneous squamous cell carcinoma (UCSF, 2021) (83 total samples), which was obtained from the cBioportal database (http://www.cbioportal.org/).
Target Genes Determination Associated with CSCC Initiation and Progression
CSCC can be regarded as a spectrum of diseases that commonly arise from precursor actinic keratosis (AK) and a SCC in situ to an invasive CSCC. Therefore, this study successively identified important genes associated with CSCC initiation and progression. Firstly, we detected the significance of hub genes associated with AK progression using GSE98780 which contained 18 AK and 20 sun-exposed normal skin samples. Then, we determined the importance of hub genes associated with CSCC development using GSE45216 dataset which contained 30 CSCC and 10 AK samples. Subsequently, we evaluated the vital role of hub genes associated with CSCC progression using GSE86544 which contained 9 CSCC, 7 CSCC with incidental perineural invasion (PNI), and 8 CSCC with clinical PNI samples. Through a series of analysis, the target genes continuously associated with AK, CSCC, and CSCC deterioration were filtrated. We further disclosed the clinical importance of target genes in cancer progression by pan-cancer analysis in the TISIDB database (http://cis.hku.hk/TISIDB/) which integrates multiple heterogeneous data types.
Expression and Potential Regulation of Target Genes in CSCC
The detailed expression of target genes in CSCC and normal tissues was assessed by merging the GSE66359 and GSE42677 datasets. Their protein expressions in CSCC were further evaluated in the Human Protein Atlas (HPA) database (https://www.proteinatlas.org/). GSEA analysis was then performed to reveal the potential regulatory mechanism involved in CSCC. For GSEA analysis, we merged the GSE42677, GSE66359, and GSE45216 datasets to obtain a larger sample size containing 51 CSCC samples. In addition, it has been reported that p53 signaling pathway participated in CSCC PNI progression, therefore, the regulation of target genes on the p53 pathway was also verified.
Statistical Analysis
The SPSS 23.0 was used to perform statistical analysis. The expression of hub genes between the 2 groups was compared via independent samples t-test or Mann–Whitney U-test. The one-way ANOVA or Kruskal–Wallis H-test was used for multiple groups comparison. The combat method in the R-sva package was used to remove batch effects among different datasets. P<0.05 was regarded as the statistical significance.
Results
Identification and Enrichment Analyses of DEGs
A total of 2879 and 677 DEGs were identified in GSE42677 and GSE66359, respectively. The up- and down-regulated DEGs in 2 datasets were presented with volcano plots (Figure 1A). The Venn analysis detected 179 overlapped DEGs between 2 datasets (Figure 1B). The PPI network of 179 DEGs was constructed, which contained 123 nodes and 354 edges (Figure 2A).
 |
Figure 1 Differential expressed genes (DEGs) selection. (A) Volcano plots of DEGs. Blue indicated downregulated genes and red indicated upregulated genes. (B) Venn analysis for selecting consistent DEGs. |
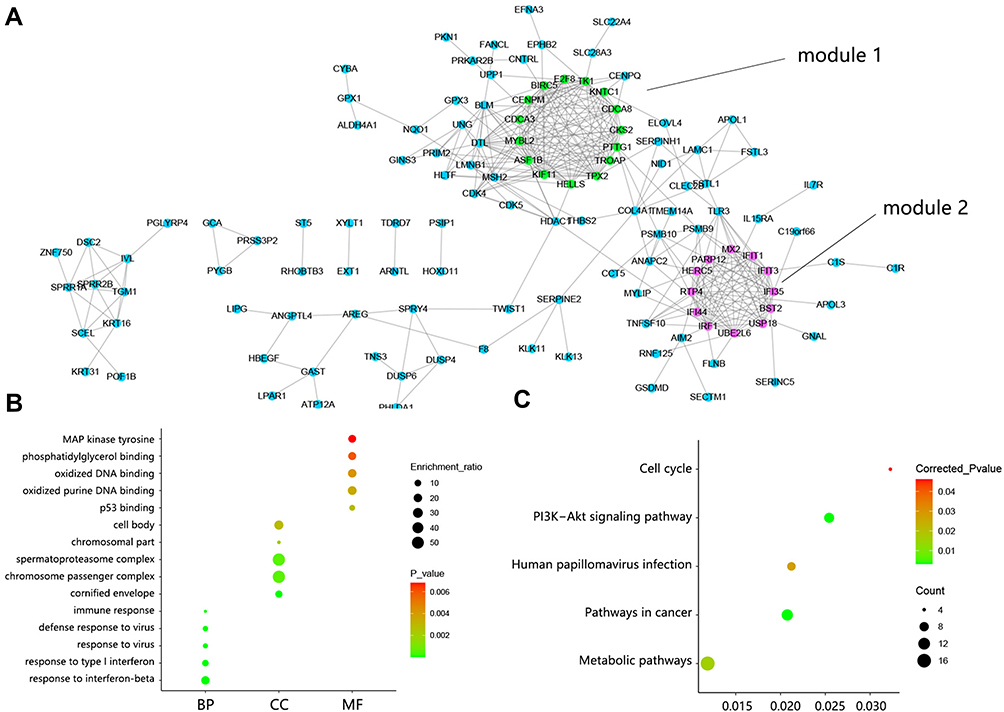 |
Figure 2 PPI network construction and function analysis on DEGs. (A) PPI network among 179 DEGs. Green indicated DEGs within module 1 and fuchsine indicated DEGs within module 2. (B) The top 5 GO annotation terms. (C) Significant KEGG pathways. Abbreviations: DEGs, differentially expressed genes; BP, biological process; CC, cellular component; MF, molecular component. |
The enrichment analysis on DEGs was performed to reveal their potential function. The top 5 GO annotation terms are shown in Figure 2B. The biological process was mainly involved in stress response such as response to the virus and immune response. Primary cellular component referred to spermatoproteasome complex and chromosome passenger complex. The oxidized DNA binding was the main molecular function. Significant KEGG pathways contained Pathways in cancer, PI3K-Akt signaling pathway, Human papillomavirus infection, Metabolic pathway, and Cell cycle (Figure 2C).
Significant Module and Hub Genes Determination in PPI Network
Among the whole PPI network, 2 significant modules were chosen by MCODE analysis. The module 1 network possessed a 12.571 score, containing 15 nodes and 88 edges (Figure 3A). KEGG analysis subsequently showed that these genes within module 1 participated in Human T-cell leukemia virus 1 infection, Apoptosis, Pathways in cancer, and Platinum drug resistance. Module 2 network showed 11.638 score, containing 12 nodes and 64 edges (Figure 3B). Pathway analysis indicated that these genes within module 2 correlated with Hepatitis C, Prolactin signaling pathway, and Pertussis.
 |
Figure 3 The significant module analysis. (A) DEGs determination within Module 1 and associated KEGG pathways. (B) DEGs determination within Module 2 and associated KEGG pathways. |
From the whole PPI network, 10 consistent hub genes were identified among MCC, DMNC, and Degree methods (Figure 4A), namely IFI35, RTP4, IFIT3, IFIT1, MYBL2, TPX2, PTTG1, TK1, BST2, and BIRC5. The expression heat map of 10 hub genes is presented in Figure 4B. Venn analysis determined 5 hub genes that were contained in module 1, namely BIRC5, MYBL2, PTTG1, TK1, and TPX2 (Figure 4C). The interaction between these 5 hub genes is shown in Figure 4D. It followed that the regulation level of BIRC5 ranked first because of its regulation on other 4 hub genes, and TPX2 ranked last. Another 5 hub genes were included in module 2 (Figure 4E), namely BST2, IFI35, IFIT1, IFIT3, and RTP4. The interaction between 5 hub genes in module 2 is shown in Figure 4F. Similarly, BST2 was ranked first on the regulation level and RTP4 ranked last.
 |
Figure 4 Hub genes determination. (A) Venn analysis for screening the top 10 hub genes. (B) The expression heat map of 10 hub genes between CSCC and normal samples in GSE42677 and GSE66359 datasets. (C) 5 hub genes identification within module 1. (D) The interaction between 5 hub genes within module 1. (E) 5 hub genes identification within module 2. (F) The interaction between 5 hub genes within module 2. |
It should be noted that a CSCC commonly has a high mutational burden (eg, TP53, CDKN2A, Ras, and NOTCH1). Regarding the 10 hub genes, we also detected their genetic alteration in CSCC (Figure 5). The analysis found no genetic alteration regarding RTP4, TK1, and BIRC5. MYBL2 showed the highest alteration frequency among 10 hub genes with the main characteristic of missense mutation.
 |
Figure 5 The genetic alteration of 10 hub genes in the dataset of cutaneous squamous cell carcinoma (UCSF, 2021) (83 total samples). |
Significant Biomarkers Identification Associated with CSCC Progression
Actinic keratosis (AK) is generally considered a precancerous lesion of CSCC. Therefore, we firstly identified the important biomarker associated with AK (Figure 6). The results showed that 10 hub genes were all highly expressed in the AK group than in the normal group, implying the involvement of these genes in AK progression.
 |
Figure 6 Expression difference of hub genes between actinic keratosis (AK) and normal skin samples in GSE98780. |
Many studies have found that AK and CSCC exhibit highly similar clinical, histological, and molecular characteristics, and it is estimated that 10% of AK will develop into CSCC in approximately 2 years. Therefore, the CSCC commonly arises from precursor AK. In this section, we further identified the important biomarker associated with CSCC development. The analysis indicated that among 10 hub genes, 6 hub genes showed higher expression in CSCC than in AK tissues (Figure 7), indicating the importance of 6 genes in the progression of CSCC. Expression difference was not observed between the 2 groups regarding the other 4 genes.
 |
Figure 7 Differential expression of significant hub genes between actinic keratosis (AK) and cutaneous squamous cell carcinoma (CSCC) in GSE45216. Among 10 hub genes, the expression of 4 genes showed no difference between 2 groups. |
It was estimated that the metastasis rate of primary CSCC is 3–5% and the prognosis of metastatic CSCC is poor, as more than 70% will die of the disease within 3 years. The CSCC finally develops into invasive and metastatic CSCC. Hence, we further identified the makers associated with CSCC migration and invasion. The result (Figure 8) showed that only the expression of MYBL2 and TK1 showed the difference among all groups. The expression of MYBL2 in CSCC-PNI samples was higher than in CSCC samples (all P<0.05), but the expression of TK1 in CSCC samples without PNI was higher than in CSCC samples.
 |
Figure 8 The differential expression of hub genes between cutaneous squamous cell carcinoma (CSCC) samples with or without perineural invasion (PNI) in GSE86544. *P<0.05. |
According to the above results, among 10 hub genes, only MYBL2 and TK1 were finally determined as the most significant biomarkers involved in the initiation and development of CSCC.
To further support the clinical significance of identified MYBL2 and TK1, we detected their expression and prognostic value in pan-cancer. The analysis showed that almost all the cancer types showed the high expression of MYBL2 and TK1 in tumor groups compared with the normal group (Figure 9A). Regarding prognosis analysis, MYBL2 high expression significantly shortened the survival time of patients in the majority of cancer types (black bar chart), and the same trend was also observed about TK1 (Figure 9B). However, TK1 high expression can prolong the survival time of CESC patients (red bar chart). These results further confirmed the important involvement of MYBL2 and TK1 in cancer progression.
 |
Figure 9 Clinical significance analysis on MYBL2 and TK1 in pan-cancer. (A) Differential expression of MYBL2 and TK1 between tumor and normal samples. (B) Associations between gene expression and overall survival across human cancers. Abbreviations: FDR, false discovery rate; log2 (FC), log2 (fold change); NS, no significance. |
Expression and Regulatory Mechanism Exploration on MYBL2 and TK1 in CSCC
The above results have confirmed the importance of MYBL2 and TK1 in cancer progression including CSCC. We further explored their detailed expression in CSCC. Due to the insufficient samples, we merged the expression profile of GSE66359 and GSE42677 to enlarge the sample size. Sample distribution before and after remove batch is presented in Figure 10A. Comparison analysis showed the higher expression of MYBL2 and TK1 in the CSCC group compared with that in the normal group (Figure 10B, all P<0.001). The protein expressions of MYBL2 and TK1 in CSCC are shown in Figure 10C.
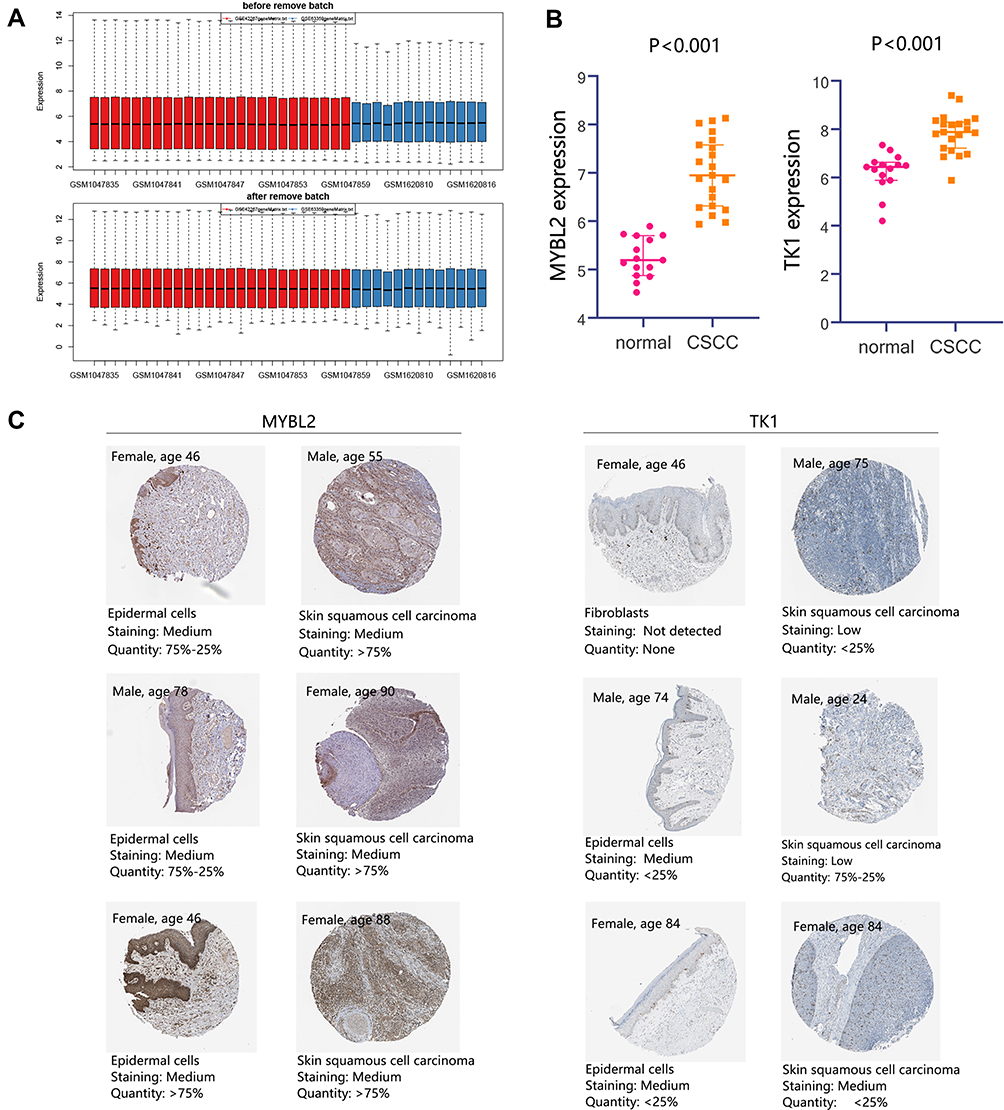 |
Figure 10 Detailed expression of MYBL2 and TK1 in CSCC. (A) Samples distribution before and after remove batch. The expression profile of GSE66359 and GSE42677 were merged, which contained 15 normal samples and 21 CSCC samples. (B) Differential expression analysis of MYBL2 and TK1. (C) Protein expression of MYBL2 and TK1. Abbreviation: CSCC, cutaneous squamous cell carcinoma. |
Due to the significance of MYBL2 and TK1 in CSCC, their potential regulatory mechanism was explored using GSEA analysis. We obtained the expression profile matrix for GSEA analysis by combining the GSE42677, GSE66359, and GSE45216 datasets. The sample distribution before and after remove batch is presented in Figure 11A. Pathway analysis in Figure 11B showed that Proteasome, Homologous recombination, DNA replication, Pentose phosphate pathway, N glycan biosynthesis pathways referred to MYBL2 high expression. The TK1 high expression correlated with Pathways in cancer, Complement, and coagulation cascades, and N glycan biosynthesis pathways. And TK1 low expression was associated with Glycine serine and threonine metabolism and Tyrosine metabolism. It followed that both MYBL2 and TK1 were related to N glycan biosynthesis pathways.
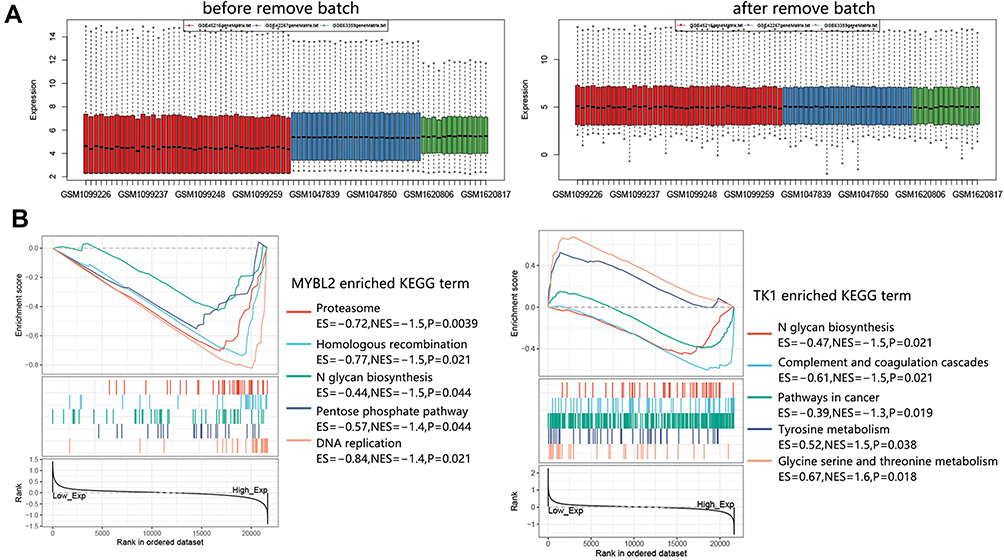 |
Figure 11 GSEA analysis on MYBL2 and TK1 in CSCC. (A) Sample distribution before and after remove batch. The expression profiles of CSCC samples in GSE42677, GSE66359, and GSE45216 were combined for GSEA analysis. A total of 51 CSCC samples were enrolled. (B) Enriched KEGG terms associated with MYBL2 and TK1. Abbreviation: CSCC, cutaneous squamous cell carcinoma. |
The previous study has indicated the activation of p53 in CSCC with PNI compared with tumors without PN1. Therefore, we further explored the potential regulation of MYBL2 and TK1 on the p53 signaling pathway. GSEA analysis showed a positive regulation between MYBL2/TK1 and the p53 pathway (Figure 12). Our result has found that MYBL2 was upregulated in CSCC PNI compared with the group without PNI, but TK1 was downregulated in the CSCC PNI group. According to these results, we speculated that MYBL2 might contribute to the activation of the p53 pathway in CSCC PNI progression, while CDK1 exerted inhibition effects.
 |
Figure 12 The regulation of MYBL2 and TK1 on p53 pathway in CSCC. Abbreviations: NES, normalized enrichment score; FDR, false discovery rate; CSCC, cutaneous squamous cell carcinoma. |
Discussion
This study firstly identified 179 differential expressed genes (DEGs) between normal and cutaneous squamous cell carcinoma (CSCC) samples, and these genes mainly correlated with PI3K-Akt signaling pathway, Human papillomavirus infection, and Pathway in cancer. The PI3K-Akt signaling pathway was significantly associated with CSCC progression10 and metastasis.11 The pathway also participated in the suppression effect of lapatinib on mesenchymal transition (EMT) in CSCC treatment.12 In addition, PI3K-Akt activation caused Stat3 activation, which played a critical role in the proliferation and survival of human CSCC.13 It followed that activation of PI3K-dependent signaling pathways has a key role in CSCC. In terms of human papillomavirus (HPV) infection, it was commonly known for its causal relationship with cervical cancer.14 However, the papillomavirus family consisted of numerous human cutaneous types, potentially involved in skin cancer development, and especially CSCC contained HPV DNA (30–60%).15 It has been reported that officially recognized HPV-174 was isolated from CSCC.16 Several studies have indicated the association between cutaneous HPV infections and SCC development. Other CSCC-related pathways were also determined. For example, the CD95 signaling pathway was involved in apoptosis in human CSCC.17 Targeting the mTOR signaling pathway can reduce the growth of CSCC cells.18 The other important pathways associated with CSCC development need further investigation.
Among 179 DEGs, we determined the significant 10 hub genes, which may play a vital role in CSCC. CSCC commonly has a high mutational burden such as TP53 and NOTCH1.19 We found that 7 hub genes showed the genetic alteration and mutation in CSCC. Accumulation of these mutations may contribute to the uncontrolled proliferation of keratinocytes and the formation of CSCC and disease progression.20 Our results indicated the possibility of hub genes as risk targets in CSCC. Apart from genetic factors, the occurrence of CSCC was also related to many risk factors, such as cumulative ultraviolet exposure, old age, immunosuppression, and smoking.21 Actinic keratosis (AK) occurred on sun-exposed skin and may progress to invasive SCC.22 In this study, 10 hub genes were all upregulated in AK compared with sun-exposure normal tissues, suggesting their important role in process of CSCC precursor AK. CSCC can be regarded as a spectrum of diseases that commonly arise from precursor AK and a SCC to an invasive CSCC to a metastatic SCC with a distant spread.23 As a pre-cancerous condition, AK has an up to 20% risk of progression to CSCC.24 This stimulated our study to define the potential genes that are associated progression of CSCC. Among 10 hub genes, 6 hub genes were further upregulated in CSCC compared with AK samples. Namely, these 6 hub genes were significantly associated with the initiation and development of CSCC. Moreover, CSCC finally develops into invasive CSCC. The invasion such as perineural invasion (PNI) has been recognized as a high-risk feature affecting the cure of CSCC.25 Hence, we also detected the targets associated with CSCC deterioration. We found that only MYBL2 and TK1 were differentially expressed between CSCC samples with or without invasion. These results confirmed that MYBL2 and TK1 were the most significant biomarkers associated with CSCC initiation and progression. The pan-cancer analysis further supported their clinical significance from expression level and prognostic value.
GSEA analysis found that both MYBL2 and TK1 correlated with N glycan biosynthesis pathways. CSCC has been typically characterized by the over-expression of the tumor suppressor protein p53.26 A previous study showed that CSCC with clinical PNI may be more likely to contain alterations in the p53 pathway.27 The regulation of MYBL2 and TK1 on the p53 pathway was also evaluated finally. We found a positive correlation between these 2 genes and the p53 signaling pathway. We speculated that MYBL2 may promote progression of CSCC to occur PNI through altering the p53 pathway, but TK1 exerted inhibition effects.
MYBL2, a cell-cycle-regulated transcription factor, is a member of the Myb gene family involved in the control of cell growth, differentiation, and apoptosis. Unlike other members, MYBL2 is ubiquitously expressed. Maruyama et al suggested that MYBL2 plays an important role in the maintenance of the undifferentiated phenotype of keratinocytes in the basal epidermal layer.28 Cicchillitti et al reported that MYBL2 may be an important factor in the pathway(s) regulating collagen production in scleroderma fibroblasts.29 MYBL2 is overexpressed in prostate cancer30 and basal-like breast cancer,31 and it is involved in the poor prognosis of these diseases. The significance of MYBL2 in cancer progression has been revealed by more and more researchers.
TK1 is an enzyme in the pyrimidine salvage pathway and catalyzes the phosphorylation of thymidine monophosphate.32 The level of TK1 is very low in non-proliferating cells but increases dramatically at the late G1 to late S-phase/early G2 phase during the cell cycle in proliferating cells and tumor cells. This makes TK1 a noteworthy marker for cell proliferation and tumor growth. In patients with malignancies, >95% of the TK1 activity in serum (S-TK1) is derived from malignant cells.33 S-TK1 activity has been used to monitor the metastasis and prognosis in acute leukemia, bladder carcinoma, and cervical carcinoma.34 Wu et al suggested that TK1 might be involved in the deep lymphatic dissemination and progression of melanoma metastasis.34 These researches all supported the importance of TK1 in cancer progression.
The present study finally revealed MYBL2 and TK1 as the crucial biomarker involved in CSCC initiation and progression. Unfortunately, there was no related clinical prognosis data on CSCC samples for performing the further verification. But, we performed pan-cancer analysis to predict their potential clinical value and found the unfavorable prognostic impacts of MYBL2 and TK1 high expression in the majority of cancer types. Their prognostic value in CSCC needs further investigation by scholars through clinical trials.
Conclusions
The present study aimed to explore the important biomarkers associated with CSCC initiation and progression. A total of 179 DEGs were obtained, and 10 hub genes were first identified to play important roles in CSCC tumorigenesis. Among them, MYBL2 and TK1 were proved to play continuous roles in AK, CSCC, and CSCC invasion. Moreover, the pan-cancer analysis showed that these 2 genes shortened the overall survival time of patients in the majority of cancer types, suggesting that MTBL2 and TK1 may serve as promising prognostic biomarkers and therapeutic targets in the future. Although N glycan biosynthesis and p53 signaling pathways associated with them were revealed, the detailed mechanism was deserved further investigation.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding authors upon reasonable request.
Ethics Statement
The need for ethics approval was waived by Affiliated Xiaoshan Hospital, Hangzhou Normal University.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Korhonen N, Ylitalo L, Luukkaala T, et al. Characteristics and trends of cutaneous squamous cell carcinoma in a patient cohort in Finland 2006–2015. Acta Derm Venereol. 2019;99:412–416.
2. Petit A, Hourbeigt K, Servagi-Vernat S, Visseaux L, Grange F. Factors associated with inoperable cutaneous squamous cell carcinoma. Eur J Dermatol. 2021;31(3):396–402. doi:10.1684/ejd.2021.4048
3. Filosa A, Filosa G. Actinic keratosis and squamous cell carcinoma: clinical and pathological features. Giornale italiano di dermatologia e venereologia. 2015;150(4):379–384.
4. Fuchs A, Marmur E. The kinetics of skin cancer: progression of actinic keratosis to squamous cell carcinoma. Dermatol Surg. 2007;33(9):1099–1101. doi:10.1111/j.1524-4725.2007.33224.x
5. Korhonen N, Ylitalo L, Luukkaala T, et al. Recurrent and metastatic cutaneous squamous cell carcinomas in a cohort of 774 patients in Finland. Acta Derm Venereol. 2020;100(8):adv00121. doi:10.2340/00015555-3479
6. Knuutila J, Riihilä P, Kurki S, Nissinen L, Kähäri V. Risk factors and prognosis for metastatic cutaneous squamous cell carcinoma: a cohort study. Acta Derm Venereol. 2020;100(16):adv00266. doi:10.2340/00015555-3628
7. Chen H, Yang J, Wu W. Seven key hub genes identified by gene co-expression network in cutaneous squamous cell carcinoma. BMC Cancer. 2021;21(1):852. doi:10.1186/s12885-021-08604-y
8. Zheng LQ, Li SY, Li CX. Expression profiling analysis of autophagy-related genes in perineural invasion of cutaneous squamous cell carcinoma. Oncol Lett. 2018;15(4):4837–4848. doi:10.3892/ol.2018.7971
9. Xu Y, Dong Y, Deng Y, et al. Identifying an lncRNA-related ceRNA network to reveal novel targets for a cutaneous squamous cell carcinoma. Biology. 2021;10(5):432. doi:10.3390/biology10050432
10. Mei XL, Zhong S. Long noncoding RNA LINC00520 prevents the progression of cutaneous squamous cell carcinoma through the inactivation of the PI3K/Akt signaling pathway by downregulating EGFR. Chin Med J. 2019;132(4):454–465. doi:10.1097/CM9.0000000000000070
11. Sun Y, Tu Y, He LI, Ji C, Cheng BO. High mobility group box 1 regulates tumor metastasis in cutaneous squamous cell carcinoma via the PI3K/AKT and MAPK signaling pathways. Oncol Lett. 2016;11(1):59–62. doi:10.3892/ol.2015.3843
12. Yao M, Shang YY, Zhou ZW, et al. The research on lapatinib in autophagy, cell cycle arrest and epithelial to mesenchymal transition via Wnt/ErK/PI3K-AKT signaling pathway in human cutaneous squamous cell carcinoma. J Cancer. 2017;8(2):220–226. doi:10.7150/jca.16850
13. Bito T, Sumita N, Ashida M, et al. Inhibition of epidermal growth factor receptor and PI3K/Akt signaling suppresses cell proliferation and survival through regulation of stat3 activation in human cutaneous squamous cell carcinoma. J Skin Cancer. 2011;2011:874571. doi:10.1155/2011/874571
14. Nair S, Pillai MR. Human papillomavirus and disease mechanisms: relevance to oral and cervical cancers. Oral Dis. 2005;11(6):350–359. doi:10.1111/j.1601-0825.2005.01127.x
15. Struijk L, Ter Schegget J, Bouwes Bavinck JN, Feltkam P. [Human papillomavirus in the aetiology of skin cancer]. Ned Tijdschr Geneeskd. 2005;149(10):518–522. Dutch.
16. Kocjan BJ, Steyer A, Sagadin M, Hosnjak L, Poljak M. Novel human papillomavirus type 174 from a cutaneous squamous cell carcinoma. Genome Announc. 2013;1(4). doi:10.1128/genomeA.00445-13
17. Lin XY, He CD, Xiao T, et al. Acitretin induces apoptosis through CD95 signalling pathway in human cutaneous squamous cell carcinoma cell line SCL-1. J Cell Mol Med. 2009;13:2888–2898. doi:10.1111/j.1582-4934.2008.00397.x
18. Chaudhary SC, Kurundkar D, Elmets CA, Kopelovich L, Athar M. Metformin, an antidiabetic agent reduces growth of cutaneous squamous cell carcinoma by targeting mTOR signaling pathway. Photochem Photobiol. 2012;88(5):1149–1156. doi:10.1111/j.1751-1097.2012.01165.x
19. Chang D, Shain AH. The landscape of driver mutations in cutaneous squamous cell carcinoma. NPJ Genomic Med. 2021;6(1):61. doi:10.1038/s41525-021-00226-4
20. Fernandez Figueras MT. From actinic keratosis to squamous cell carcinoma: pathophysiology revisited. J Eur Acad Dermatol Venereol. 2017;31(Suppl 2):5–7. doi:10.1111/jdv.14151
21. Waldman A, Schmults C. Cutaneous squamous cell carcinoma. Hematol Oncol Clin North Am. 2019;33(1):1–12. doi:10.1016/j.hoc.2018.08.001
22. Fecker LF, Stockfleth E, Braun FK, et al. Enhanced death ligand-induced apoptosis in cutaneous SCC cells by treatment with diclofenac/hyaluronic acid correlates with downregulation of c-FLIP. J Invest Dermatol. 2010;130(8):2098–2109. doi:10.1038/jid.2010.40
23. Soura E, Gagari E, Stratigos A. Advanced cutaneous squamous cell carcinoma: how is it defined and what new therapeutic approaches are available? Curr Opin Oncol. 2019;31(5):461–468. doi:10.1097/CCO.0000000000000566
24. Zheng Y, Chi S, Li C. Identification of potential gene drivers of cutaneous squamous cell carcinoma: analysis of microarray data. Medicine. 2020;99(39):e22257. doi:10.1097/MD.0000000000022257
25. Yan F, Tillman BN, Nijhawan RI, et al. High-risk cutaneous squamous cell carcinoma of the head and neck: a clinical review. Ann Surg Oncol. 2021;28(13):9009–9030. doi:10.1245/s10434-021-10108-9
26. Black AP, Ogg GS. The role of p53 in the immunobiology of cutaneous squamous cell carcinoma. Clin Exp Immunol. 2003;132(3):379–384. doi:10.1046/j.1365-2249.2003.02159.x
27. Warren TA, Broit N, Simmons JL, et al. Expression profiling of cutaneous squamous cell carcinoma with perineural invasion implicates the p53 pathway in the process. Sci Rep. 2016;6(1):34081. doi:10.1038/srep34081
28. Maruyama H, Ishitsuka Y, Fujisawa Y, Furuta J, Sekido M, Kawachi Y. B-Myb enhances proliferation and suppresses differentiation of keratinocytes in three-dimensional cell culture. Arch Dermatol Res. 2014;306(4):375–384. doi:10.1007/s00403-014-1450-1
29. Cicchillitti L, Jimenez S, Sala A, Saitta B. B-Myb acts as a repressor of human COL1A1 collagen gene expression by interacting with Sp1 and CBF factors in scleroderma fibroblasts. Biochem J. 2004;378(2):609–616. doi:10.1042/bj20031110
30. Bar-Shira A, Pinthus JH, Rozovsky U, Goldstein M, Orr-Urtreger A. Multiple genes in human 20q13 chromosomal region are involved in an advanced prostate cancer xenograft. Cancer Res. 2002;62(23):6803–6807.
31. Thorner A, Hoadley K, Parker J, Winkel S, Millikan R, Perou C. In vitro and in vivo analysis of B-Myb in basal-like breast cancer. Oncogene. 2009;28(5):742–751. doi:10.1038/onc.2008.430
32. Ke P, Chang Z. Mitotic degradation of human thymidine kinase 1 is dependent on the anaphase-promoting complex/cyclosome-CDH1-mediated pathway. Mol Cell Biol. 2004;24(2):514–526. doi:10.1128/MCB.24.2.514-526.2004
33. Wu C, Yang R, Zhou J, et al. Production and characterisation of a novel chicken IgY antibody raised against C-terminal peptide from human thymidine kinase 1. J Immunol Methods. 2003;277(1–2):157–169. doi:10.1016/S0022-1759(03)00062-0
34. Wu B, Li W, Qian C, Ding W, Zhou Z, Jiang H. Increased serum level of thymidine kinase 1 correlates with metastatic site in patients with malignant melanoma. Tumor Biol. 2013;34(2):643–648. doi:10.1007/s13277-012-0591-0
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.