Back to Journals » Journal of Multidisciplinary Healthcare » Volume 10
Sickle cell retinopathy: improving care with a multidisciplinary approach
Authors Menaa F ![]() , Khan BA
, Khan BA ![]() , Uzair B
, Uzair B ![]() , Menaa A
, Menaa A
Received 11 April 2017
Accepted for publication 20 July 2017
Published 30 August 2017 Volume 2017:10 Pages 335—346
DOI https://doi.org/10.2147/JMDH.S90630
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Farid Menaa,1,2 Barkat Ali Khan,3 Bushra Uzair,4 Abder Menaa2
1Department of Pharmaceutical Sciences and Nanomedicine, California Innovations Corporation, San Diego, CA, USA; 2Departments of Clinical Medicine and Laser Therapy, Centre Médical des Guittières, Saint-Philbert-de-Grand-Lieu, Loire-Atlantique, France; 3Faculty of Pharmacy and Alternative Medicine, The Islamia University of Bahawalpur, Bahawalpur, 4Department of Bioinformatics and Biotechnology, International Islamic University, Islamabad, Pakistan
Abstract: Sickle cell retinopathy (SCR) is the most representative ophthalmologic complication of sickle cell disease (SCD), a hemoglobinopathy affecting both adults and children. SCR presents a wide spectrum of manifestations and may even lead to irreversible vision loss if not properly diagnosed and treated at the earliest. Over the past decade, multidisciplinary research developments have focused upon systemic, genetic, and ocular risk factors of SCR, enabling the clinician to better diagnose and manage these patients. In addition, newer imaging and testing modalities, such as spectral domain-optical coherence tomography angiography, have resulted in the detection of subclinical retinopathy related to SCD. Innovative therapy includes intravitreal injection of an anti-vascular endothelial growth factor (eg, Lucentis® [ranibizumab] or Eylea® [aflibercept]) which appears comparatively safe and efficient, and may be combined with laser photocoagulation (LPC) for proliferative SCR. The effect of LPC alone does not significantly lead to the regression of advanced SCR, although it helps in avoiding hemorrhage and sight loss. This comprehensive article is based on 10-years retrospective (2007–2017) studies. It aims to present advances and recommendations in SCR theranostics while pointing out the requirement of combinatorial approaches for better management of SCR patients. To reach this goal, we identified and analyzed randomized original and review articles, clinical trials, non-randomized intervention studies, and observational studies using specified keywords in various databases (eg, Medline, Embase, Cochrane, ClinicalTrials.gov).
Keywords: sickle cell disease, retinopathy, medicine practice, translational medicine, personalized medicine, theranostics, omics, innovations, combinatorial clinical approaches
Introduction
SCD is the first and the most common group of hemoglobinopathies in the world.1 Manifestations of SCD involve complex processes and pathways such as bioavailability of NO, endothelial activation, inflammation, blood cells adhesiveness, and oxidative stress.1,2 In the world, due to its associated significant morbidity and mortality,3 SCD remains a major public health concern affecting millions of people, especially those of African descent (0.4%).4 Hence, there is an urgent need for these people to have easier access to comprehensive team care.5 In the USA, SCD affects nearly 100,000 individuals5,6 with a frequency of up to 0.003% in African-American births.5,7 In Brazil, the frequency reaches up to 0.06% depending on the different regions,8 whereas in Africa, the frequency is the highest in the world, varying from 5% to 40%.9,10
Within the SCD leading group, SCA is associated with mutations in the normal β-globin gene which is inherited through an autosomal recessive pattern (ie, single point mutation at the sixth position in the β-globin chain that substitutes the amino acid valine for the amino acid glutamic acid resulting in HbS).1 In USA, the homozygous HbS (ie, SS) genotype represents a severe common (65%) form of SCD and occurs in ~0.2% of African-American children.1 In addition to the SS genotype, SCA includes HbS/β0-thalassemia heterozygous genotypes which account for 0.03% among African-Americans in North America.4 Although SCA is characterized by the same point mutation, the causes of its phenotypic heterogeneity remain unclear.1 This substantial phenotypic variability manifests with different clinical outcomes (eg, retinopathy, stroke, vaso-occlusive episodes, priapism, leg ulcers, pulmonary hypertension, acute chest syndrome, avascular necrosis)1 (Figure 1). The complications arising from this life-threatening genetic disease lead to poor quality of life and may cause premature death.1 Our current investigations, among others, strongly suggest that it could depend on genetic modifiers (eg, CNVs, single-nucleotide polymorphisms) and environmental effects.1,2,11
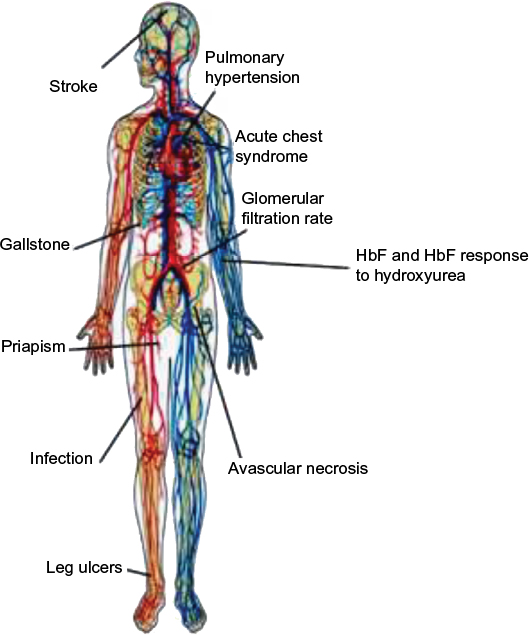 | Figure 1 Main clinical complications in patients with sickle cell anemia. Note: Copyright ©2009. Genomics Insights. Adapted from Driss A, Asare KO, Hibbert JM, Gee BE, Adamkiewicz TV, Stiles JK. Sickle cell disease in the post genomic era: a monogenic disease with a polygenic phenotype. Genomics Insights. 2009;2009(2):23–48.2 Abbreviation: HbF, fetal hemoglobin. |
Other common but relatively more benign conditions of SCD include HbSC and HbAS. HbSC results from coinheritance of HbS with another abnormal β-globin chain variant (ie, HbC),1,12 and occurs in about 3% of USA’s black population.13 HbSC patients often present blood flow abnormalities due to hypercoagulable Hb,14 and are associated with advanced SCR although visual impairment is SCD genotype independent.9,15,16 Besides, SCT is considered benign but can become a pathologic risk factor for marked retinopathy (ie, vaso-occlusive features and retinal proliferative changes). SCR in HbAS patients can occur in few weeks under conditions of stress, concomitant systemic diseases, or trauma (eg, blunt ocular trauma, scarring) leading to hypoxia, independent of raised intraocular pressure.3,17,18 In USA, over two million people (ie, about 8%–8.5% of USA’s population of African ancestry) have SCT,19,4 and the incidence of AS genotype among African-American is about 2%.4
Nowadays, the high prevalence of SCR, due to increased human longevity and its associated potentially severe complications (eg, maculopathy, CRAO, PSCR), justifies a comprehensive medical workup. SCR screening tests should be performed in SCA, heterozygous SCD, as well as in certain SCT patients with the aid of a very coordinative multidisciplinary team in the setting of a referral center.17,20 This team should include hematologists, geneticists, and molecular biologists to diagnose SCD and assess its inheritance risk; ophthalmologists–optometrists to diagnose and treat SCR; a family physician, internists, and nurses for regular checking and coordination; cardiovascular specialists and phlebotomists to avoid any further blood flow issues, particularly after chronic blood transfusions; pain management specialists; pharmacists to minimize adverse effects and optimize the efficacy of the therapeutic regimen; and biomedical researchers to find cost-effective and innovative theranostics.
In this article, we present a comprehensive and retrospective overview of advances in SCR pathogenesis, theranostic options, while providing our opinion and recommendations for better management of SCR patients, particularly those at a higher risk for vision loss.
Classification and epidemiology of SCR
In SCD, ocular lesions result from stasis and occlusion of small eye vessels by sickled erythrocytes.21
SCR is mainly classified as NPSCR or PSCR. PSCR is based on the presence of vascular proliferation (ie, neoangiogenesis which can be followed by VH and retinal detachment).21
Goldberg and Penman classifications are used to grade retinopathy severity (stage I–V) in SCD patients.16,20,22 Four decades ago, Goldberg classified PSCR in five stages:23
- peripheral arterial occlusion;
- peripheral arteriovenous anastomoses, representing dilated preexisting capillaries (hairpin loop);
- neovascular and fibrous proliferation (sea fan) occurring at the posterior border of non-perfusion. The subsequent white sea fan appearance is due to auto-infarction of the neovasculature. This stage III PSCR has been further divided by other authors24 into five new grades (A–E) considering size (grade A) with flat sea fan with leakage <1 macular photocoagulation study disc area, hemorrhage (grade B), partial fibrosis (grade C) with elevated sea fan, complete fibrosis (grade D) with elevated sea fan, and visible/well-defined vessels (grade E);
- VH;
- TRD.
As is notable, RRD was not mentioned. Yet, RRD is the most common type of retinal detachment, which is characterized by an accumulation of subretinal fluid in the space between the neurosensory retina and the underlying retinal pigment epithelium. In RRD, vitreous traction causes a tear in the retina (ie, retinal breaks), and its most common cause is degeneration of the vitreous body.25 Thus, we suggest the inclusion of RRD within the fifth stage of Goldberg and Penman’s classification. Further, this classification was updated by Penman about two decades ago based on a comparison of the peripheral retinal vascular bed in subjects with SS and SCD using normal hemoglobin (AA) genotype as control.26 Therefore, the vascular patterns were classified as qualitatively normal (type I) or abnormal (type II). A normal vascular border, if posterior, results from gradual modification of the capillary bed, and is an indicator of low risk of proliferative disease. An abnormal border does not revert to normal, and is subject to continuous evolution until development of proliferative lesions.
Importantly, it is estimated that the prevalence of retinopathy in USA is about 15%–20% among SS patients and 33%–40% among SC patients.13 Also, the incidence of PSCR is highest in patients with SC or S-Thal (33% and 14%, respectively), while patients with SS have a 3% incidence of PSCR.13,4 Moreover, there is an increase with age in the incidence and prevalence rates of all ocular complications of SCD. Thus, the highest prevalence of PSCR in SS patients occurs between 25 and 39 years in both men and women, while in SC patients it occurs earlier in men (15–24 year old) and women (20–39 years old).27 This is illustrated by a study published a decade ago showing a higher prevalence of SCR among SC patients (43%) who developed PSCR by the age of 24 years compared to that of SS patients (14%).28 Concordantly, in a recent retrospective study20 involving SS, SC, and AS adult patients, about thrice more SC patients (54.6%) developed grade 3–5 PSCR compared to that of SS patients (18.1%). Also, in the same study, the prevalence of severe SCR forms was significantly higher among SS men (21.7%) than among SS women (15.5%), whereas systemic and ocular complications were infrequently seen in AS patients. Besides, in a very small number of patients with HbSE (another heterozygous state of HbS,29 albeit still rare and slowly rising worldwide),30,31 NPSCR was most common.30
Taken together, PSCR is rarer than NPSCR in SCD patients, but PSCR is diagnosed more frequently in SC patients than in SS patients.9,15,16,26,27
Pathogenesis, clinical manifestations, and complications of SCR
The pathophysiology of SCD is not limited to abnormal red blood cells. Current knowledge about ocular manifestations in SCD patients revealed that virtually every vascular bed in the eye can be affected by vaso-occlusive events (eg, relatively high occlusion indices of peripheral retinal vessels).17 In SCR patients, who are often asymptomatic, the most significant ocular changes occur in the fundus and the retinal vascular damages can progressively cause ischemic retinopathy, proliferative lesions in both the anterior and posterior segments of the eye, and visual impairment, that is, corrected distance visual acuity of 20/40 or poorer.17,21,22,33 It is worth noting that SCR patients display a variable phenotype, even among individuals with the same genotype.32 For instance, damages of the anterior segment would be more common in SC patients.33
NPSCR, more commonly observed in SS patients, is manifested by typical alterations depending on the retinal location. Hence, the retinal periphery of patients with NPSCR is frequently characterized by precocious bilateral changes, peripheral closure/anastomoses, salmon-patch hemorrhages (Figure 2), iridescent spots, and black sunbursts.34 The central part of the retina usually reveals arteriovenous tortuosity, enlargement of the foveal avascular (non-perfused) zone (FAZ), CRAO (eye stroke), as well as peripapillary and peri-macular arterial occlusions.34 It is worthwhile to mention that CRAO is a rare but a potentially devastating cause of acute blindness in SCD.35
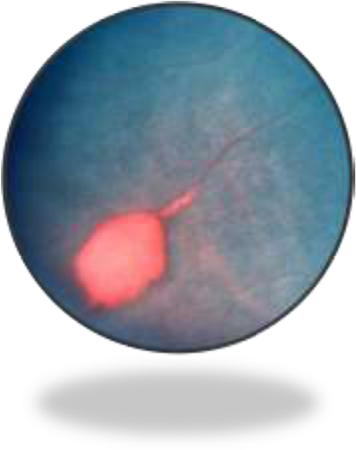 | Figure 2 Salmon-patch hemorrhage in sickle cell retinopathy. Notes: This image was originally published in the ASRS Retina Image Bank by Larry Halperin, MD, Retina Group of Florida. Sickle Salmon-Patch Hemorrhage. 2012; image number, 1789. ©American Society of Retina Specialists. http://eyewiki.aao.org/Sickle_Cell_Retinopathy.91 |
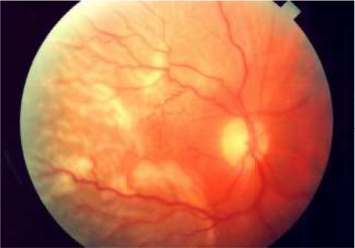
PSCR, more commonly observed in SC patients, is due to the high prevalence of a qualitatively abnormal peripheral retinal vasculature.26 Peripheral changes secondary to PSCR are generally associated with thinning of macular inner retinal layers and ganglion cell complex focal loss, as well as thickening of the central fovea.36–39 For some unknown reasons, PSCR is uncommonly manifested by simultaneous bilateral macular occlusive events.40 These retinal vascular occlusions cause tissue ischemia and release of angiogenic mediators that promote retinal neovascularization (ie, neovascular proliferation).41–43 Then, this neovascularization, a hallmark in PSCR patients, grows anteriorly from the perfused to non-perfused (avascular) retina to reach the posterior ischemic retina, and is characterized by flat vessels denominated sea fan structures (Figure 3) for their close resemblance to the marine invertebrate Gorgonia flabellum (Venus sea fan).44 The sea fan is a thick caliber preretinal fibrovascular membrane involving primarily the retinal nerve fiber and ganglion cell layers.45 The repetition of hemorrhages can cause TRD (Figure 4) or RRD (Figure 5), which are severe complications of PSCR.23,28,46 Visual impairments, such as vision loss, occur in 5%–20% of affected eyes of PSCR patients.32,33 The reason why most patients maintain good vision 2 years after PSCR development is explained by the fact that the ocular damages occur in the retinal periphery, and that any associated sea fan structures have a high tendency to spontaneously regress (20%–60% of cases) through the development of atrophic lesions or auto-infarction.27,28,32
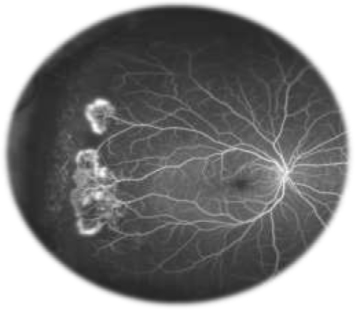 | Figure 3 Sea fan formation with neovascularization. Notes: Fluorescein angiogram image of an individual with sickle cell retinopathy showing sea fan formation with neovascularization. This image was taken using an Optos P200MA ultrawide-field imaging device. This image was originally published in the ASRS Retina Image Bank by Michael P Kelly, FOPS Director, Duke Eye Center Labs, Duke University Hospital. Sickle Cell Retinopathy. 2012; image number, 721. ©American Society of Retina Specialists. http://eyewiki.aao.org/Sickle_Cell_Retinopathy.92 |
 | Figure 4 Patient with a central retinal vein occlusion complicated by neovascularization at the disc with subsequent tractional retinal detachment. Notes: Image reprinted with permission from Lihteh Wu, MD, Ophthalmologist, Costa Rica Vitreo and Retina Macular Associates, published by Medscape Drugs & Diseases (http://emedicine.medscape.com/), Tractional Retinal Detachment, 2017, available at: http://emedicine.medscape.com/article/1224891-overview#a5.93 |
 | Figure 5 Clinical picture of a rhegmatogenous retinal detachment. Notes: Notice that the macula is involved and that the retina is corrugated and has a slightly opaque color. Image reprinted with permission from Lihteh Wu, MD, Ophthalmologist, Costa Rica Vitreo and Retina Macular Associates, published by Medscape Drugs & Diseases (http://emedicine.medscape.com/), Tractional Retinal Detachment, 2017, available at: http://emedicine.medscape.com/article/1224891-overview#a5.93 |
Prevention and diagnosis of SCR
In the recent years, health care has dramatically increased the life expectancy of SCD patients, meantime contributing to the emergence of ocular complications related to ischemic retinopathy22 (eg, maculopathy,33,47,48 CRAO,35 hyphema,49 retinal neovascularization [PSCR],49 vitreoretinal complications50).
The hallmark of primary prevention should consist in reducing the prevalence of SCD by controlling the spread and perpetuation of the HbS gene pool. This can only be done by efforts at genetic counseling before marriage and child birth.
Furthermore, most of the studies, led in different populations of patients with SCR, confirm the importance of periodic eye monitoring. Retinal examination should be done not only in homozygous (ie, SS) or double heterozygous patients (eg, SC, SE, Sβ0-thalassemia) but also in patients with SCT (ie, AS) when additional systemic vascular conditions are present.17,51
SCD patients should be screened from early childhood (usually 9–10 years for SC genotype patients and at 13 years for SS and Sβ0-thalassemia genotype patients) to timely identify retinal lesions, visual impairment (eg, visual loss), and prevent the progression of NPSCR to severe stages (ie, PSCR with VH or retinal detachment).16,21,30,32,33,52 Serial examinations may be done biennially for eyes with normal findings51 in order to decrease morbidity.53 SCD patients with retinopathy suspicion or predisposed to develop SCR should undergo complete ophthalmologic examination and be followed up as necessary.
Thus, electrophoretic1 or spectroscopic54 confirmation of SCD remains the first step. Then, systemic and genetic risk factors of SCR could be evaluated (see “Clinical, systemic, and genetic risk factors of SCR” section). Ophthalmologic screening of SCD patients should involve a number of basic exams as well as the use of state-of-the-art retinal imaging techniques when available/possible due to their relative high cost.9,16,20,47,48,55,56 Thus, routine screening of ocular abnormalities for earliest detection and intervention of retinopathy should include visual acuity testing (to measure the clarity or sharpness of vision at a distance of 20 feet), complete dilated funduscopy, as well as the use of slit-lamp biomicroscopy (to observe any ocular abnormalities in the anterior portion of the eye and eventual retinal detachments). In case of abnormal funduscopy,16,34 patient cases with ischemic retinopathy often undergo FA9,16,17,51,56,57 or OCTA. Goldberg and Penman classifications are eventually employed to grade the retinopathy severity.34,48
FA is more sensitive in the diagnosis of retinopathy when compared to indirect ophthalmoscopy.34 Ultrawide-field FA enables one to demonstrate macular arteriolar occlusive disease,55 visualize peripheral vascular remodeling,40 calculate peripheral ischemic index,57 or evidence retinal microvascular occlusions.58 AOSLO-FA is an emerging high-resolution technique useful for noninvasive in vivo visualization of the human retinal microvasculature, measurements of blood flow velocity, and microvascular network mapping.59 Although relatively well-tolerated in patients with SCR, indocyanine green angiography is rarely used but can show anomalies of the choroidal circulation, clearly delimit the black spots and the photocoagulation scars which appear larger than in FA.60
Spectral domain-OCT is essential in diagnosing retinal rather than optic nerve disease (eg, glaucoma), and can be used to confirm multifocal electroretinography.40 OCT is useful for assessing bilateral temporal inner retinal hyper reflectivity,40 inner retinal thinning temporal to the fovea even normal in FA and clinical examination,61 and major peripheral changes secondary to PSCR (eg, peripapillary RNFL thinning, macular ganglion cell complex thinning, and total macular thicknesses).36,37,47,57,58 Interestingly, the presence of temporal macular atrophy is strongly associated with peripheral neovascularization,48 and OCT demonstrated a positive linear relationship between temporal macular thickness and global peripapillary RNFL thickness.58 Nevertheless, it has been recently reported that scanning laser ophthalmoscope microperimetry, a sensitive measurement of macular function in SCD patients, is better than OCT for measuring focal macular thinning.55 Importantly, for accurate differential diagnosis, OCT is able to identify in vivo morphological changes (eg, vitreous adhesions, membrane-retinal layers twist) between neovascularization in PSCR and proliferative diabetic retinopathy.45 Interestingly, in a recent clinical trial assessing ocular changes in an African-American population with SCD, the authors used Spectralis HRA + OCT, an advanced multimodal imaging technology, enabling them to show individual layers of the retina which helped to 1) determine whether there were underlying changes in the gross ophthalmic posterior segment, 2) understand the ocular disease progression.62
OCTA is an improved version of OCT (ie, OCT + angiography). It is a gold standard in identifying retinal ischemia in patients with SCD.61 It represents a relatively new noninvasive imaging system for monitoring ocular pathology and detecting early disease.63 OCTA is able to show and compare both structural and blood flow information. Interestingly, when compared with AOSLO-FA, OCTA is able to generate volumetric data of retinal and choroidal layers (eg, retinal thinning, lumen diameter, FAZ perimeter, acircularity index).53,56,63,64 Furthermore, its ease of use, short acquisition time, and avoidance of potentially phototoxic blue light, can quantitatively and qualitatively better evidence retinal microvascular occlusions (eg, abnormalities in the perifoveal region of the macula such as reduced flow within the superficial and deep capillary plexus, vascular density, choroidal neovascularization, and areas of non-perfusion) than any other current FA tool.32,39,53,56,61,63,64 OCTA also appeared more sensitive than FA techniques in detecting macular microangiopathy56 and macular capillary non-perfusion65 in asymptomatic patients. A vital component of OCTA, the split-spectrum amplitude-decorrelation angiography algorithm, helps to decrease the signal-to-noise ratio of flow detection, subsequently enhancing the visualization of retinal vasculature using motion contrast.53 However, although blood vessels obscured by leakage are visible only on OCTA, microaneurysms, fine structure of vessel loops, leakage, and some vessel segments are invisible on OCTA and the use of AOSLO-FA is then required.53 This suggests the need of employing OCTA and/or FA depending on the cases. In a newly published clinical trial, a retrospective observational study led in African-Canadian patients with SCT confirmed that OCTA is a valuable noninvasive office procedure that can be used to show macular vascular occlusions that usually occur in SC patients.66
Clinical, systemic, and genetic risk factors of SCR
In addition to patient screening and complete eye examination, it would be helpful for the health care professional to earlier assess the presence of factors and biomarkers that increase the chance of developing PSCR.
For the last four decades, after Goldberg’s seminal studies, risk factors (eg, clinical, genetic) for visual impairment have been reported in patients with SCD.22 Considering the Mendelian inheritance of SCD, pregnancy is a high-risk situation for both the mother and child; hence, a search of genetic factors in the family is relevant and contributes to preventive medicine.
Since SCR is more frequent and more severe in SC patients than in SS patients16,22 although visual impairment is not significantly influenced by hemoglobin genotype,22 several influencing risk parameters (eg, clinical predisposition, systemic factors, systemic gene polymorphisms influencing key proteins expression implicated specifically in the retinal pathogenesis) are more likely to be involved in SCR.
Pain crisis,7,67 male gender,7 and splenic sequestration7 in SCD patients are clinical risk factors that suggest complete eye examination. Importantly, eyes of SCD patients with iris atrophy or depigmentation are about twice more at risk of PSR than eyes without.68 Nevertheless, it is worthwhile noting that there is no evidence to support an association between cognitive function and retinopathy in SCD, although there is an association between lower cognitive function and older age as well as history of stroke in genotyped SS patients.1,69 In patients treated during 1 year of regular HU and transfusion therapy, SCR has been found to be correlated with time-averaged mean flow velocity in middle cerebral arteries but not in ophthalmic arteries.67 In SC patients, characteristics of PSCR (eg, neovascular and fibrous proliferations, the so-called “sea fan” structures, VH and retinal detachment) are associated with older age (>35 years),22 pulmonary disease, deafness or tinnitus, and no history of osteomyelitis. In SS patients, older age, male gender, and history of acute pyelonephritis are associated with the development of PSCR.21,70–72
Besides, some systemic factors can corroborate SCR diagnosis and may influence the prognosis of the disease. Thereby, high levels of Hb or hematocrit are associated with PSCR in SC genotyped patients, while high leukocyte count is associated with PSCR in SS or Sβ0-thalassemia patients.9 Interestingly, low (<15%) HbF level is strongly associated with retinopathy (7 fold in children) independently of the SCD genotype, and so, the high prevalence of SCR is negatively associated to HbF level.9,73 In one of our research studies involving Brazilian SCR patients, we showed that the significant decrease of soluble intercellular adhesion molecule and the significant increase of pigment epithelium-derived factor could contribute to the pathophysiology of retinal neovascularization.6 We also demonstrated that angiopoietin (Ang-1 and Ang-2) as well as IL-1β levels were significantly elevated in patients with SCR, and SS patients presented significantly higher levels of Ang-2 compared to that of SC patients.6 Interestingly, from autopsied eye tissues, it was shown that HIF-1α and VEGF are strongly expressed in the inner retina,43 but it remains undefined whether the expression of these factors from blood samples could be associated or correlated with clinicopathological features of retinopathy. In a lesser extent, glucose-6-phosphate dehydrogenase deficiency was insignificantly more common in SCR patients but could also deserve early screening.7
Recent advances in the post-human genome sequence era have opened the door to the identification in SCD patients of novel genetic modifiers (eg, gene polymorphisms, CNVs) mostly implicated in inflammation, NO biology, cell–cell interaction, and modulators of oxidant injury.1,2 Discovery of submicroscopic genomic alterations associated with SCR phenotypes may aid in establishing precocious diagnosis as well as targeted cures. Interestingly, an across-sectional study showed that IL-6-597G>A is associated with a higher likelihood of retinopathy in Brazilian SS patients, and may thus be considered as a genetic predictor of SCR.74 In another study that aimed to determine endothelial NO synthase genotype for T786C and G894T polymorphisms, the 786CC genotype was more common in SS and Sβ0 Greek patients with retinopathy.75 Moreover, 894TT SS and Sβ0 patients tended to have a higher hematocrit value than 894GG and GT ones, suggesting that they could be predisposed to higher risk of retinopathy.75 Nevertheless, there is still a paucity of reports and a lack of GWAS regarding genetic modifiers specifically involved in SCR phenotypes. In addition to genetic modifiers, one should remember that other contributors could be significantly involved in the etiology of the heterogeneous SCD and SCR phenotypes (eg, environmental components, genetic background of the population, socioeconomics, and psychology).2
To date, overall reports showed that the prevalence of PSCR increased mainly with age (over 35 years) and with systemic severity (ie, inflammation), particularly in SC genotyped patients.
Current and advanced therapeutic options for SCR
Over the past 20 years, improved therapeutic management has significantly increased the life expectancy of children with SCD. More than 90% of patients now reach the age of 20, and the median life expectancy of SCD patients reached over 50 years in countries with advanced health care systems. However, increase in the number of SCD adults is accompanied by frequent chronic ocular complications (eg, osteoarticular, renal, cardiorespiratory, cutaneous, and cerebral).
Treatment for SCR remains palliative, and the development of novel, noninvasive, and effective retinal drug delivery systems is hampered by the lack of studies on SCR pathophysiology, animal models that accurately mimic human SCR.6 Conventional therapies for SCR often include chemotherapy (eg, HU ± anti-VEGF), phlebotomy usually used in conjunction with long-term blood transfusions (eg, cell exchange transfusion) to mainly reduce total HbS red cells. In severe cases, laser-mediated photocoagulation (ie, repeated pan-retinal photocoagulation) and surgery (ie, PPV) are often required. In a lesser extent, alternative and complementary therapies such as HBOT, stem cells, and gene therapy are used. In general, experimental anti-sickling therapy and preoperative transfusion to prevent or treat eye lesions still need more investigations.49 Thus, we recommend that health care professionals undertake all necessary measures to reduce ischemia, and it becomes evident that a well-trained multidisciplinary team should take on severe cases.
Similar to erythrocytapheresis or exchange blood transfusion, hydroxycarbamide, commonly called HU, is now considered a preventive treatment for SCD due to its relative acceptable safety and efficacy.1,9,76,77 A multicenter, placebo-controlled, Phase III clinical trial supports the safety and potentially salutary effects of HU treatment for patients with HbSC disease.73,77 Thus, HU treatment is associated with a reduced absolute reticulocyte count, a stable Hb concentration, and an increased HbF known to exert protective benefit against retinopathy.73 HU therapy is strongly recommended for SCD adults with at least three severe vaso-occlusive crises during any 12-month period, with pain or chronic anemia interfering with daily activities, or with severe or recurrent episodes of acute chest syndrome.6,77 A recommendation of moderate strength suggests offering treatment with HU for infants, children, and adolescents with SCD without regard to the presence of symptoms.6 Interestingly, oral administration of the antioxidant fumaric acid ester monomethyl fumarate, a known inducer of nuclear factor erythroid 2-related factor 2-antioxidant response element signaling pathway, is able to ameliorate retinopathy at the dose of 15 mg/mL for 5 months by inducing γ-globin expression and HbF production in the validated humanized Townes mouse model of SR.78
Concomitantly, anti-VEGF biotherapy by intravitreal injection of Lucentis® (ranibizumab; Roche, Basel, Switzerland) or the more recent Eylea® (aflibercept; Regeneron, Tarrytown, NY, USA) is widely used to treat retinopathy. In practice, both of these FDA-approved drugs are used to treat SCR with VH. Interestingly, in a first case report of PSCR, ranibizumab improved the visual acuity, resolution of VH, and regression of neovascularization with no recurrence or adverse effects.79 Another FDA-approved drug Avastin® (bevacizumab; Genentech, South San Francisco, CA, USA) is now restricted to the treatment of various types of cancer, hence its “off-label” use in ophthalmology. Bevacizumab, albeit efficient in preoperative injection,80 was associated with significant secondary effects such as secondary hyphema few days postinjection in the eye of a PSCR patient with VH.81 Clinical trials aiming to compare efficacy and safety of ranibizumab versus aflibercept, in patients with SCR versus patients with diabetic retinopathy, would be an asset.
Also, iron-chelation therapy in patients with SCR presenting hemochromatosis (ie, iron overload) can be moderately recommended.1,6 However, particular awareness is required when Exjade® (Novartis International AG, Basel, Switzerland), Desirox® (Cipla Limited, Mumbai, India), Defrijet® (CIMS, Mumbai, India), Rasiroxpine® (Hikma Pharmaceuticals, Amman, Jordan), Jadenu® (Novartis International AG) (deferasirox) are used. Although deferasirox is an EMA and FDA-approved iron-chelating agents, a case study implicating a child with a history of SCA revealed that the substitution of Desferal® (Novartis Pharma Stein AG, Stein, Switzerland) (deferoxamine mesylate) by deferasirox for 2 years can cause retinopathy and related manifestations (eg, decreased visual acuity, electrophysiological abnormalities, and mild funduscopic changes) were completely reversed after deferasirox therapy cessation.82
Preoperative transfusion therapy is strongly recommended to increase Hb levels to 10 g/dL for many individuals with SCD.6 In SS patients undergoing long-term transfusion therapy, it is moderately recommended to maintain the HbS level below 30% prior to the next transfusion.6 In SCR patients, there were only few attempts of exchange transfusions (ie, blood transfusion) when apheresis (ie, collection of donor blood constituents) is not used. A case study reported the successful use of chronic red cell exchange transfusion to preserve vision and stabilize recurrent retinopathy untreatable by LPC and vitrectomy in a child with PSCR.83 Further, exchange transfusion was successful in an SS adult patient with unilateral paracentral occlusive retinopathy, an uncommon manifestation of SCD characterized by macular ischemia which can lead to vision loss if not reversed on time.84
Besides, despite warnings from the FDA for its use, HBOT could improve the acuity visual of SCD patients with CRAO.85 Further, strong recommendations for patients with PSCR include referral to expert specialists for consideration of LPC, the most common treatment against vision loss and VH.10,43,86 Clinical practitioners generally use argon arc rather than xenon arc for LPC in order to quench the expression of HIF-1-driven angiogenic mediators (eg, VEGF).43 It is then recommended that pan photocoagulation is preferentially realized with an argon arc beam, which should be well-placed at the ischemic areas, in order to avoid PPV in some PSCR eyes with VH.79 Although SS-LPC to ischemic areas was reported to be effective after 6 months in achieving regression of peripheral neovascularization,30 FV-LPC prevented occurrence of VH with lesser adverse effects.43,86 In two randomized controlled trials comparing efficacy and safety of LPC to no therapy in children and adults with PSCR, 1) no difference in the complete PSCR regression was observed after an averaged follow-up period of 21–32 months when SS-LPC was compared to the control group; 2) SS-LPC or FV-LPC may prevent loss of vision in eyes with PSCR; 3) occurrence prevention of VH is possible either with argon or xenon laser but xenon arc was associated with a significantly higher risk of choroidal neovascularization; 4) FV-LPC displayed greater protective effects when compared to SS-LPC; 5) minimal adverse effects (ie, very low incidence of retinal tear, no induction of retinal detachment, vision loss was uncommon within at least 3 years of follow-up). It is concluded that in the absence of further evidence, laser treatment for SCR should be considered as one of the therapeutic options for preventing visual loss and VH.86 To the best of our knowledge, randomized controlled clinical trials aiming to assess the benefit/risk ratio of LPC combined with intravitreal injection of ranibizumab or aflibercept are lacking in PSCR patients.
Eventually, vitrectomy can be applied if SCR presents vitro-retinal complications.83 Usually, PSCR responds to vitrectomy procedures when sea fan proliferation is segmented.50 PPV (Figure 6) is growing in popularity as a first-line procedure for primary RRD, especially in pseudophakic patients.87 PPV presents advantages (eg, better visualization of retinal breaks, less painful intervention) over the longer and well-established scleral buckling, another common reattachment procedure.87 A recent retrospective, interventional case series of SCR patients managed with PPV over a 12-year period at a single institution showed full success in improving vision postoperatively in patients with VH or epiretinal membrane.88 Nevertheless, moderate success was noticed in patients with recurrent TRD or RRD and so, these patients required a second operation.88 Also, severe SCR patients with a hemorrhagic disease (eg, Glanzmann thrombasthenia) can find vitrectomy difficult.89 Also, it is worthwhile to mention that vitro-retinal complications may spontaneously regress and reappear after vitrectomy or even LPC.83
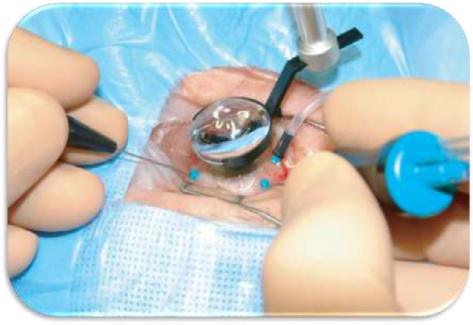 | Figure 6 Pars plana vitrectomy (PPV) for rhegmatogenous retinal detachment, utilizing wide-field imaging and small-gauge transconjunctival sutureless instrumentation. Note: Copyright ©2008. Dove Medical Press. Reproduced from Schwartz SG, Flynn HW. Pars plana vitrectomy for primary rhegmatogenous retinal detachment. Clin Ophthalmol. 2008;2(1):57–63.87 |
General conclusion and perspectives
SCD is the most common and the most severe hemoglobinopathy. Retinopathy represents the most important ocular complication of SCD. SC genotype is associated with SCR. SCR is characterized by the vaso-occlusion of capillary beds (ie, interaction between sickled cells and the vascular endothelium) that subsequently leads to inflammation, retinal hypoxia, ischemia, infarction, hemolysis, neovascularization, and fibrovascularization. Classically, NPSCR or PSCR is classified according to posterior segment changes. In NPSCR, the retinal changes do not involve neovascularization as they do in PSCR. Early-stage diagnosis of SCD patients with risk of retinopathy is recommended to prevent the progression of the disease through regular retinal examinations and adapted treatment as indicated. Retinal examination must be done in homozygous, double heterozygous patients or when the sickle trait is present with additional systemic vascular conditions. Diagnosis modalities (eg, OCTA, FA) and multi-approach therapy (eg, chemotherapy combined or not to LPC) should contribute to the prevention and palliative care of SCD patients. Although, photocoagulation to ischemic areas is effective in achieving regression of neovascularization, its relative efficacy and safety should be better investigated and compared to anti-VEGF therapy in PSCR patients. Various measures to reduce ischemia and hemorrhage are recommended before surgical procedure. Multidisciplinary teams should then take on severe cases. Furthermore, GWAS, clinical trials, meta-analyses of SCR are still required to clarify the predominant risk factors and better screen, manage, and monitor individuals with SCR in a personalized fashion. Considering that advances in theranostics are not always available in some parts of the world, the global benefit–risk ratio of all current theranostic tools and procedures in various populations with or susceptible to develop SCR may be an asset for SCR patients. Moreover, development of animal models mimicking human SCR to better understand the pathophysiology of SCR is needed. Eventually, clear outcomes of gene and cell therapy experiments in SCR animal models (eg, HbF targeting, ex vivo gene editing of CD34+ hematopoietic stem/progenitor cells to correct Hb mutation using the CRISPR-Cas system,90 intravitreal injection of bone marrow-derived stem cells) remain a challenge before translating the findings from bench-to-bedside.
Abbreviations
AOSLO:adaptive optics scanning light ophthalmoscopy
CNV:copy number variation
CRAO:central retinal artery obstruction
CRISPR-Cas:clustered regularly interspaced short palindromic repeats-associated nuclease 9
EMA:European Medicines Agency
FA:fluorescein angiography
FV-LPC:feeder vessel LPC
FDA:US Food and Drug Administration
FV-LPC:feeder vessel laser photocoagulation
GWAS:genome wide association studies
Hb:hemoglobin
HbC:hemoglobin C
HbF:fetal hemoglobin
HbS:sickle hemoglobin
HbSC:sickle hemoglobin C disease
HbAS:sickle trait
HBOT:hyperbaric oxygen therapy
HiF-1α:hypoxia-inducible factor-1α
HRA:Heidelberg retina angiography
HU:hydroxyurea
LPC:laser photocoagulation
NO:nitric acid
NPSCR:non-proliferative sickle cell retinopathy
OCT:optical coherence tomography
OCTA: optical coherence tomography angiography
PPV:pars plana vitrectomy
PSCR:proliferative sickle cell retinopathy
RNFL:retinal nerve fiber layer
RRD:rhegmatogenous retinal detachment
SCA:sickle cell anemia
SCD:sickle cell disease
SCR:sickle cell retinopathy
SCT:sickle cell trait
Sβ0:sickle beta-zero
SS-LPC:sectoral scatter laser photocoagulation
TRD: tractional retinal detachment
VEGF:vascular endothelial growth factor
VH:vitreous hemorrhage
Disclosure
The authors report no conflicts of interest in this work.
References
Menaa F. Stroke in sickle cell anemia patients: a need for multidisciplinary approaches. Atherosclerosis. 2013;229(2):496–503. | ||
Driss A, Asare KO, Hibbert JM, Gee BE, Adamkiewicz TV, Stiles JK. Sickle cell disease in the post genomic era: a monogenic disease with a polygenic phenotype. Genomics Insights. 2009;2009(2):23–48. | ||
Reynolds SA, Besada E, Winter-Corella C. Retinopathy in patients with sickle cell trait. Optometry. 2007;78(11):582–587. | ||
Fekrat SG, Morton F. Sickle Retinopathy. New York: Thieme Medical Publishers; 1998. | ||
Centers for Disease Control and Prevention. Sickle cell disease. [Updated August 31, 2016]. Available from: https://www.cdc.gov/ncbddd/sicklecell/data.html. Accessed April 1, 2017. | ||
Cruz PR, Lira RP, Pereira Filho SA, et al. Increased circulating PEDF and low sICAM-1 are associated with sickle cell retinopathy. Blood Cells Mol Dis. 2015;54(1):33–37. | ||
Rosenberg JB, Hutcheson KA. Pediatric sickle cell retinopathy: correlation with clinical factors. J AAPOS. 2011;15(1):49–53. | ||
Lervolino LG, Baldin PE, Picado SM, Calil KB, Viel AA, Campos LA. Prevalence of sickle cell disease and sickle cell trait in national neonatal screening studies. Rev Bras Hematol Hemoter. 2011;33(1):49–54. | ||
Dembélé AK, Toure BA, Sarro YS, et al. Prévalence et facteurs de risque de la rétinopathie drépanocytaire dans un centre de suivi drépanocytaire d’Afrique subsaharienne. [Prevalence and risk factors for sickle retinopathy in a sub-Saharan comprehensive sickle cell center]. Rev Med Interne. Epub 2017 Feb 22. French. | ||
Diallo JW, Sanfo O, Blot I, et al. Étude épidémiologique et facteurs pronostiques de la rétinopathie drépanocytaire à Ouagadougou (Burkina Faso). [Epidemiology and prognostic factors for sickle cell retinopathy in Ouagadougou (Burkina Faso)]. J Fr Ophtalmol. 2009;32(7):496–500. French. | ||
Menaa F, Bezerra MA, Araujo AS, Ananina G, Costa FF, de Melo MB. Copy-number variation of the UGT2B28 gene is strongly associated with stroke complication in Brazilian sickle cell anemia patients: importance for drug discovery and therapy? Fourth International Conference on Drug Discovery and Therapy, Dubai, UAE, 12–15 February 2012. Curr Med Chem. 2012;19(1). | ||
Lemonne N, Billaud M, Waltz X, et al. Rheology of red blood cells in patients with HbC disease. Clin Hemorheol Microcirc. 2016;61(4):571–577. | ||
Gualandro SF, Fonseca GH, Yokomizo IK, Gualandro DM, Suganuma LM. Cohort study of adult patients with haemoglobin SC disease: clinical characteristics and predictors of mortality. Br J Haematol. 2015;171(4):631–637. | ||
Colella MP, de Paula EV, Machado-Neto JA, et al. Elevated hypercoagulability markers in hemoglobin SC disease. Haematologica. 2015;100(4):466–471. | ||
Freitas LG, Isaac DL, Tannure WT, et al. Alterações retinianas apresentadas em pacientes portadores de hemoglobinopatia falciforme atendidos em um Serviço Universitário de Oftalmologia. [Retinal manifestations in patients with sickle cell disease referred to a University Eye Hospital]. Arq Bras Oftalmol. 2011;74(5):335–337. Portuguese. | ||
Tran TH, Mekinian A, Godinaud M, Rose C. Rétinopathie drépanocytaire chez les adultes de la région Nord-Pas-de-Calais. [Screening for sickle cell disease retinopathy in the north of France]. J Fr Ophtalmol. 2008;31(10):987–992. French. | ||
Morel C. Atteinte rétinienne des hémoglobinopathies. [Retinal involvement in hemoglobinopathy]. J Fr Ophtalmol. 2001;24(9):987–992. French. | ||
Pandey N. Unusual presentation of ocular trauma in sickle cell trait. Indian J Ophthalmol. 2015;63(9):738–740. | ||
Demeritt MJ, Schechtman DL, Reynolds SA. Spotlight on sickle cell retinopathy: a thorough patient history and appropriate laboratory testing are essential to treat and diagnose this hereditary blood disorder. Review of Optometry. 2014. Available from: https://www.reviewofoptometry.com/article/spotlight-on-sickle-cell-retinopathy. Accessed April 01, 2017. | ||
Leveziel N, Lalloum F, Bastuji-Garin S, et al. Rétinopathie drépanocytaire : analyse rétrospective portant sur 730 patients suivis dans un centre de référence. [Sickle-cell retinopathy: retrospective study of 730 patients followed in a referral center]. J Fr Ophtalmol. 2012;35(5):343–347. French. | ||
El-Ghamrawy MK, El Behairy HF, El Menshawy A, Awad SA, Ismail A, Gabal MS. Ocular manifestations in Egyptian children and young adults with sickle cell disease. Indian J Hematol Blood Transfus. 2014;30(4):275–280. | ||
Saidkasimova S, Shalchi Z, Mahroo OA, et al. Risk factors for visual impairment in patients with sickle cell disease in London. Eur J Ophthalmol. 2016;26(5):431–435. | ||
Goldberg MF. Classification and pathogenesis of proliferative sickle retinopathy. Am J Ophthalmol. 1971;71(3):649–665. | ||
Sayag D, Binaghi M, Souied EH, et al. Retinal photocoagulation for proliferative sickle cell retinopathy: a prospective clinical trial with new sea fan classification. Eur J Ophthalmol. 2008;18(2):248–254. | ||
Feltgen N, Walter P. Rhegmatogenous retinal detachment—an ophthalmologic emergency. Dtsch Arztebl Int. 2014;111(1–2):12–21. | ||
Penman AD, Talbot JF, Chuang EL, Thomas P, Serjeant GR, Bird AC. New classification of peripheral retinal vascular changes in sickle cell disease. Br J Ophthalmol. 1994;78(9):681–689. | ||
Elagouz M, Jyothi S, Gupta B, Sivaprasad S. Sickle cell disease and the eye: old and new concepts. Surv Ophthalmol. 2010;55(4):359–377. | ||
Downes SM, Hambleton IR, Chuang EL, Lois N, Serjeant GR, Bird AC. Incidence and natural history of proliferative sickle cell retinopathy: observations from a cohort study. Ophthalmology. 2005;112(11):1869–1875. | ||
Mais DD, Gulbranson RD, Keren DF. The range of hemoglobin A(2) in hemoglobin E heterozygotes as determined by capillary electrophoresis. Am J Clin Pathol. 2009;132(1):34–38. | ||
Baciu P, Yang C, Fantin A, Darnley-Fisch D, Desai U. First reported case of proliferative retinopathy in hemoglobin SE disease. Case Rep Ophthalmol Med. 2014;2014:782923. | ||
Knox-Macaulay HH, Ahmed MM, Gravell D, Al-Kindi S, Ganesh A. Sickle cell-haemoglobin E (HbSE) compound heterozygosity: a clinical and haematological study. Int J Lab Hematol. 2007;29(4):292–301. | ||
Scott AW. Ophthalmic manifestations of sickle cell disease. South Med J. 2016;109(9):542–548. | ||
Fadugbagbe AO, Gurgel RQ, Mendonça CQ, Cipolotti R, dos Santos AM, Cuevas LE. Ocular manifestations of sickle cell disease. Ann Trop Paediatr. 2010;30(1):19–26. | ||
Santos AM, Faro GB, Amaral MV, Mendonça Cde Q, Leal BC, Cipolotti R. Alterações retinianas em jovens portadores de anemia falciforme (hemoglobinopatias) em hospital universitário no nordeste do Brasil. [Retinal impairment in young individuals with sickle cell anemia (hemoglobin SS disease) in university hospital in Northeastern of Brazil]. Arq Bras Oftalmol. 2012;75(5):313–315. Portuguese. | ||
Liem RI, Calamaras DM, Chhabra MS, Files B, Minniti CP, Thompson AA. Sudden-onset blindness in sickle cell disease due to retinal artery occlusion. Pediatr Blood Cancer. 2008;50(3):624–627. | ||
Brasileiro F, Martins TT, Campos SB, et al. Macular and peripapillary spectral domain optical coherence tomography changes in sickle cell retinopathy. Retina. 2015;35(2):257–263. | ||
Murthy RK, Grover S, Chalam KV. Temporal macular thinning on spectral-domain optical coherence tomography in proliferative sickle cell retinopathy. Arch Ophthalmol. 2011;129(2):247–249. | ||
Rajagopal R, Apte RS. Full-thickness macular hole in a patient with proliferative sickle cell retinopathy. Retina. 2010;30(5):838–839. | ||
Han IC, Tadarati M, Pacheco KD, Scott AW. Evaluation of macular vascular abnormalities identified by optical coherence tomography angiography in sickle cell disease. Am J Ophthalmol. 2017;177:90–99. | ||
Cusick M, Toma HS, Hwang TS, Brown JC, Miller NR, Adams NA. Binasal visual field defects from simultaneous bilateral retinal infarctions in sickle cell disease. Am J Ophthalmol. 2007;143(5):893–896. | ||
Mantovani A, Figini I. Sickle cell-hemoglobin C retinopathy: transient obstruction of retinal and choroidal circulations and transient drying out of retinal neovessels. Int Ophthalmol. 2008;28(2):135–137. | ||
Scott AW, Lutty GA, Goldberg MF. Hemoglobinopathies: retinal vascular diseases. In: Ryan SJ, editor. Retina. Philadelphia: Elsevier; 2013: 1071–1082. | ||
Rodrigues M, Kashiwabuchi F, Deshpande M, et al. Expression pattern of HIF-1α and VEGF supports circumferential application of scatter laser for proliferative sickle retinopathy. Invest Ophthalmol Vis Sci. 2016;57(15):6739–6746. | ||
Welch RB, Goldberg MF. Sickle-cell hemoglobin and its relation to fundus abnormality. Arch Ophthalmol. 1966;75(3):353–362. | ||
Folgar FA, Reddy S. An in vivo morphologic comparison of retinal neovascularization in sickle cell and diabetic retinopathy. Retin Cases Brief Rep. 2012;6(1):99–101. | ||
Lutty GA, McLeod DS. Angiogenesis in sickle cell retinopathy. In: Penn JS, editor. Retinal and Choroidal Angiogenesis. Netherlands: Springer; 2008: 389–405. | ||
Kung JS, Leng T. West African crystalline maculopathy in sickle cell retinopathy. Case Rep Ophthalmol Med. 2015;2015:910713. | ||
Hood MP, Diaz RI, Sigler EJ, Calzada JI. Temporal macular atrophy as a predictor of neovascularization in sickle cell retinopathy. Ophthalmic Surg Lasers Imaging Retina. 2016;47(1):27–34. | ||
Charache S. Eye disease in sickling disorders. Hematol Oncol Clin North Am. 1996;10(6):1357–1362. | ||
Williamson TH, Rajput R, Laidlaw DA, Mokete B. Vitreoretinal management of the complications of sickle cell retinopathy by observation or pars plana vitrectomy. Eye (Lond). 2009;23(6):1314–1320. | ||
Gill HS, Lam WC. A screening strategy for the detection of sickle cell retinopathy in pediatric patients. Can J Ophthalmol. 2008;43(2):188–191. | ||
de Almeida Oliveira DC, Carvalho MO, do Nascimento VM, Villas-Bôas FS, Galvão-Castro B, Goncalves MS. Sickle cell disease retinopathy: characterization among pediatric and teenage patients from northeastern Brazil. Rev Bras Hematol Hemoter. 2014;36(5):340–344. | ||
Chalam KV, Sambhav K. Optical coherence tomography angiography in retinal diseases. J Ophthalmic Vis Res. 2016;11(1):84–92. | ||
Filho AC, Silveira L Jr, Yanai AL, Fernandes AB. Raman spectroscopy for a rapid diagnosis of sickle cell disease in human blood samples: a preliminary study. Lasers Med Sci. 2015;30(1):247–253. | ||
Chow CC, Genead MA, Anastasakis A, Chau FY, Fishman GA, Lim JI. Structural and functional correlation in sickle cell retinopathy using spectral-domain optical coherence tomography and scanning laser ophthalmoscope microperimetry. Am J Ophthalmol. 2011;152(4):704–711. | ||
Minvielle W, Caillaux V, Cohen SY, et al. Macular microangiopathy in sickle cell disease using optical coherence tomography angiography. Am J Ophthalmol. 2016;164:137–144. | ||
Ghasemi Falavarjani K, Scott AW, Wang K, et al. Correlation of multimodal imaging in sickle cell retinopathy. Retina. 2016;36(1):S111–S117. | ||
Chow CC, Shah RJ, Lim JI, Chau FY, Hallak JA, Vajaranant TS. Peripapillary retinal nerve fiber layer thickness in sickle-cell hemoglobinopathies using spectral-domain optical coherence tomography. Am J Ophthalmol. 2013;155(3):456–464. | ||
Chui TY, Mo S, Krawitz B, et al. Human retinal microvascular imaging using adaptive optics scanning light ophthalmoscopy. Int J Retina Vitreous. 2016;2:11. | ||
Diallo JW, Kuhn D, Haymann-Gawrilow P, Soubrane G. Apport de l’angiographie au vert d’indocyanine dans la rétinopathie drépanocytaire. [Contribution of indocyanine green angiography in sickle cell retinopathy]. J Fr Ophtalmol. 2009;32(6):430–435. French. | ||
Grover S, Sambhav K, Chalam KV. Capillary nonperfusion by novel technology of OCT angiography in a patient with sickle cell disease with normal fluorescein angiogram. Eur J Ophthalmol. 2016;26(5):e121–e123. | ||
Bowie EM. Spectralis HRA + OCT imaging of the retina with autofluorescence in sickle cell disease. 2010. Available from: https://clinicaltrials.gov/ct2/show/NCT01123369?term=sickle+cell+retinopathy&rank=3. ClinicalTrials.gov identifier: NCT01123369. Accessed on April 1, 2017. | ||
Mo S, Krawitz B, Efstathiadis E, et al. Imaging foveal microvasculature: optical coherence tomography angiography versus adaptive optics scanning light ophthalmoscope fluorescein angiography. Invest Ophthalmol Vis Sci. 2016;57(9):130–140. | ||
Han IC, Tadarati M, Scott AW. Macular vascular abnormalities identified by optical coherence tomographic angiography in patients with sickle cell disease. JAMA Ophthalmol. 2015;133(11):1337–1340. | ||
Sanfilippo CJ, Klufas MA, Sarraf D, Tsui I. Optical coherence tomography angiography of sickle cell maculopathy. Retin Cases Brief Rep. 2015;9(4):360–362. | ||
Michel SS. New, previously unknown, uses of optical coherence tomography angiography (OCTA). 2017. Available from: https://clinicaltrials.gov/ct2/show/NCT03068702?term=sickle+cell+retinopathy&rank=2. ClinicalTrials.gov identifier: NCT03068702. Accessed April 1, 2017. | ||
Tantawy AA, Andrawes NG, Adly AA, El Kady BA, Shalash AS. Retinal changes in children and adolescents with sickle cell disease attending a paediatric hospital in Cairo, Egypt: risk factors and relation to ophthalmic and cerebral blood flow. Trans R Soc Trop Med Hyg. 2013;107(4):205–211. | ||
Osafo-Kwaako A, Kimani K, Ilako D, et al. Ocular manifestations of sickle cell disease at the Korle-bu Hospital, Accra, Ghana. Eur J Ophthalmol. 2011;21(4):484–489. | ||
Oltra EZ, Chow CC, Wubben T, Lim JI, Chau FY, Moss HE. Cross-sectional analysis of neurocognitive function, retinopathy, and retinal thinning by spectral-domain optical coherence tomography in sickle cell patients. Middle East Afr J Ophthalmol. 2016;23(1):79–83. | ||
Rosenberg JB, Hutcheson KA. Pediatric sickle cell retinopathy: correlation with clinical factors. J AAPOS. 2011;15(1):49–53. | ||
Leveziel N, Bastuji-Garin S, Lalloum F, Querques G, Benlian P, Binaghi M. Clinical and laboratory factors associated with the severity of proliferative sickle cell retinopathy in patients with sickle cell hemoglobin C (SC) and homozygous sickle cell (SS) disease. Medicine (Baltimore). 2011;90(6):372–378. | ||
Lim JI. Ophthalmic manifestations of sickle cell disease: update of the latest findings. Curr Opin Ophthalmol. 2012;23(6):533–536. | ||
Estepp JH, Smeltzer MP, Wang WC, Hoehn ME, Hankins JS, Aygun B. Protection from sickle cell retinopathy is associated with elevated HbF levels and hydroxycarbamide use in children. Br J Haematol. 2013;161(3):402–405. | ||
Vicari P, Adegoke SA, Mazzotti DR, Cançado RD, Nogutti MA, Figueiredo MS. Interleukin-1β and interleukin-6 gene polymorphisms are associated with manifestations of sickle cell anemia. Blood Cells Mol Dis. 2015;54(3):244–249. | ||
Armenis I, Kalotychou V, Tzanetea R, et al. Prognostic value of T786C and G894T eNOS polymorphisms in sickle cell disease. Nitric Oxide. 2017;62:17–23. | ||
Girot R, Stankovic K, Lionnet F. New issues in adult sickle cell disease. Bull Acad Natl Med. 2008;192(7):1395–1409; discussion 1409–1411. | ||
Luchtman-Jones L, Pressel S, Hilliard L, et al. Effects of hydroxyurea treatment for patients with hemoglobin SC disease. Am J Hematol. 2016;91(2):238–242. | ||
MacKenzie LE, Choudhary TR, McNaught AI, Harvey AR. In vivo oximetry of human bulbar conjunctival and episcleral microvasculature using snapshot multispectral imaging. Exp Eye Res. 2016;149:48–58. | ||
Mitropoulos PG, Chatziralli IP, Parikakis EA, Peponis VG, Amariotakis GA, Moschos MM. Intravitreal ranibizumab for stage IV proliferative sickle cell retinopathy: a first case report. Case Rep Ophthalmol Med. 2014;2014:682583. | ||
Moshiri A, Ha NK, Ko FS, Scott AW. Bevacizumab presurgical treatment for proliferative sickle-cell retinopathy-related retinal detachment. Retin Cases Brief Rep. 2013;7(3):204–205. | ||
Babalola OE. Intravitreal bevacizumab (Avastin) associated with secondary hyphaema in a case of proliferative sickle cell retinopathy. BMJ Case Rep. 2010;2010. pii: bcr11.2009.2441. | ||
Walia HS, Yan J. Reversible retinopathy associated with oral deferasirox therapy. BMJ Case Rep. 2013;2013:pii:bcr2013009205. | ||
McKinney CM, Siringo F, Olson JL, Capocelli KE, Ambruso DR, Nuss R. Red cell exchange transfusion halts progressive proliferative sickle cell retinopathy in a teenaged patient with hemoglobin SC disease. Pediatr Blood Cancer. 2015;62(4):721–723. | ||
Gustave BW, Oliver SC, Mathias M, et al. Reversal of paracentral occlusive retinopathy in a case of sickle cell disease using exchange transfusion. Ophthalmic Surg Lasers Imaging Retina. 2013;44(5):505–507. | ||
Canan H, Ulas B, Altan-Yaycioglu R. Hyperbaric oxygen therapy in combination with systemic treatment of sickle cell disease presenting as central retinal artery occlusion: a case report. J Med Case Rep. 2014;8:370. | ||
Myint KT, Sahoo S, Thein AW, Moe S, Ni H. Laser therapy for retinopathy in sickle cell disease. Cochrane Database Syst Rev. 2015;10: CD010790. | ||
Schwartz SG, Flynn HW. Pars plana vitrectomy for primary rhegmatogenous retinal detachment. Clin Ophthalmol. 2008;2(1):57–63. | ||
Chen RW, Flynn HW Jr, Lee WH, et al. Vitreoretinal management and surgical outcomes in proliferative sickle retinopathy: a case series. Am J Ophthalmol. 2014;157(4):870–875. | ||
Odoulami-Yehouessi L, Sounouvou I, Anani L, Tachabi S, Doutentien C, Latoundji S. [Proliferative sickle cell retinopathy revealing Glanzmann thrombasthenia]. J Fr Ophtalmol. 2009;32(10):757. French. | ||
DeWitt MA, Magis W, Bray NL, et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci Transl Med. 2016;8(360):360ra134. | ||
Halperin L. Sickle salmon-patch hemorrhage. ASRS Retina Image Bank. Available from: http://imagebank.asrs.org/file/1789/sickle-salmon-patch-hemorrhage. Accessed August 16, 2017. | ||
Kelly MP. Sickle cell retinopathy. ASRS Retina Image Bank. Available from: http://imagebank.asrs.org/file/721/sickle-cell-retinopathy. Accessed August 16, 2017. | ||
Wu L. Tractional retinal detachment. Medscape Drugs and Diseases. Available from: http://emedicine.medscape.com/article/1224891-overview#a5. Accessed August 16, 2017. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.