Back to Journals » Journal of Hepatocellular Carcinoma » Volume 8
Serum KLKB1 as a Potential Prognostic Biomarker for Hepatocellular Carcinoma Based on Data-Independent Acquisition and Parallel Reaction Monitoring
Authors Che YQ ![]() , Zhang Y
, Zhang Y ![]() , Li HB, Shen D
, Li HB, Shen D ![]() , Cui W
, Cui W
Received 23 June 2021
Accepted for publication 8 September 2021
Published 12 October 2021 Volume 2021:8 Pages 1241—1252
DOI https://doi.org/10.2147/JHC.S325629
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Manal Hassan
Yi-Qun Che,1,2 Yue Zhang,1 Han-Bing Li,1 Di Shen,1 Wei Cui1
1Department of Clinical Laboratory, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100021, People’s Republic of China; 2Center for Clinical Laboratory, Beijing Friendship Hospital, Capital Medical University, Beijing, 100050, People’s Republic of China
Correspondence: Wei Cui
Department of Clinical Laboratory, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, No. 17 Panjiayuan Nanli, Chaoyang District, Beijing, 100021, People’s Republic of China
Tel/Fax +86-10-87788448
Email [email protected]
Purpose: With the advancement of minimally invasive surgery and catheters for hepatocellular carcinoma (HCC), it is becoming more and more inconvenient to get tissues or the tissues gained are insufficient for testing. Screening of blood-derived markers is of great significance for prognosis assessment.
Patients and Methods: Data-independent acquisition (DIA) and parallel reaction monitoring (PRM) were implemented to identify valuable prognostic HCC biomarkers in 48 patients with different prognosis. The potential candidate biomarkers were examined in 205 HCC patients using enzyme-linked immunosorbent assay (ELISA) and then validated in The Cancer Genome Atlas (TCGA) HCC cohort.
Results: DIA screened 86 significantly differentially regulated proteins between patients with poor prognosis and those with good prognosis. Eight proteins from the DIA proteomic analyses were quantified by PRM, and six of them (KLKB1, IGFBP3, SHBG, SAA1, C7, and CD44) presented consistent expression trends between DIA and PRM. Then, the results of ELISA indicated that KLKB1 was abnormally expressed in HCC patients, and the serum level of KLKB1 also exhibited significant changes before and after treatment (P = 0.016). Patients with higher KLKB1 serum levels had significantly superior overall survival (P = 0.035) and progression-free survival (P = 0.027) than those with lower KLKB1 expression. In the TCGA-HCC cohort, Cox regression analysis suggested that KLKB1 was an independent prognostic factor for overall survival (P = 0.032) of HCC patients.
Conclusion: Aberrant expression of KLKB1 was strongly associated with the prognosis of HCC patients. KLKB1 may be used to evaluate the prognosis and guide the treatment for HCC.
Keywords: KLKB1, hepatocellular carcinoma, prognostic, data-independent acquisition, parallel reaction monitoring
Introduction
In China, the estimated age-standardized incidence rate of liver cancer is 343.7 and 122.3 per 100,000 population per year for males and females, ranking 3 and 6, respectively. The mortality rate of liver cancer is 310.6 and 111.5 per 100,000 population per year for males and females, ranking 3 and 4, respectively.1 In addition, it is one of the most lethal primary cancers, with a 5-year relative survival rate of 12.1% in China from 2012 to 2015.2 Trends in the incidence and mortality rates of liver cancer in China and the United States, both sexes combined, were rising.1,3 As the major subtype of liver cancer, hepatocellular carcinoma (HCC) seriously threatens Chinese health and life safety and restricts social and economic development.
Therapeutic modalities for HCC include surgical options (resection or liver transplantation), interventional embolization, radiofrequency ablation (RFA), and chemotherapy. Recurrence and metastasis are important factors that restrict the curative effect of patients with HCC. Accurately predicting recurrence and metastasis of HCC, making the best-individualized treatment in patients, the reasonable choice or combined use of various HCC treatments, avoiding inappropriate or excessive treatment, controlling tumor progression to the maximum extent, improving the overall curative effect and the patients’ quality of life, prolonging the survival period, these strategies mentioned above are important developing directions of the treatment for HCC.4 However, due to the great complexity and heterogeneity of HCC, it is impossible to accurately predict the prognosis and therapeutic response of patients based on the existing clinical staging, imaging diagnosis, and pathological grading.5 In short, there are great limitations in determining the prognosis of patients and guiding the subsequent treatment of HCC.
Therefore, finding stable and efficient prognostic biomarkers for HCC is of great importance. Meanwhile, fine-needle biopsy or tumor tissue biopsy is not required for the diagnosis of HCC in NCCN (National Comprehensive Cancer Network) guidelines. The only clinical biomarker for monitoring HCC recurrence and metastasis is serum alpha-fetoprotein antigen (AFP). However, 30% to 40% of HCC patients may present as AFP-negative.6,7 At present, even though ultrasound (US) plus AFP, either alone or in combination, had a pooled risk ratio for improving survival (US 1.75 vs US plus AFP 1.86), there was no statistical difference between the two strategies.8 Thus, AFP lacks adequate sensitivity and specificity for effective surveillance, necessitating the development of novel effective and reliable non-invasive biomarkers for the prognostic prediction of HCC.
The samples tested in this study were 1–1.5mL serum samples that had been preserved after routine clinical biochemical tests in 2012. Personalized non-invasive analysis methods (cfDNA, ctDNA, NGS, etc.) need 8–10 mL venous blood samples, whereas data-independent acquisition (DIA) only requires 30µl serum to complete the test.9 Therefore, using DIA to screen prognostic markers is both feasible and appropriate.
Technological advancements in liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) have greatly expanded our ability to explore proteomes. Currently, three main data acquisition strategies are applied: Data-dependent acquisition (DDA), targeted acquisition by selected or parallel reaction monitoring (SRM or PRM), and DIA.10
In this study, we implemented DIA for discovery and PRM for confirmation of HCC biomarkers in non-fractionated sera. DIA is a recently developed mass spectrometry technology. The advantages of DIA technology over traditional DDA include: 1) collecting all ion information to achieve higher data coverage; 2) reducing the randomness of collection and achieving extremely high detection reproducibility and stability; 3) using fragment ion quantification to greatly improve the quantitative precision, accuracy, and linear range. Based on the advantages described above, DIA technology is especially suitable for high coverage, stable and traceable analysis of large samples.11–13
PRM is an ion monitoring technology based on high resolution and high precision mass spectrometry that can selectively detect target proteins and peptides (such as those undergoing post-translational modification), so as to achieve relative or absolute quantification of target proteins and peptides.14 In terms of application, PRM has higher throughput, specificity, and precision than the traditional Western blot technology to some extent. In addition, PRM-based protein verification is no longer subject to commercial antibodies, and it can be applied to any biological sample to improve the experimental efficiency.15
However, to the best of our knowledge, using DIA and PRM MS-based workflow to research the prognostic biomarkers of HCC has not been reported. In this study, DIA and PRM were utilized to examine the disrupted proteins expressed in HCC sera, and then ELISA and TCGA database were used to confirm whether the selected biomarkers had clinical application value. Our findings revealed several potential prognostic biomarkers, provided a foundation for pathological studies on HCC, and confirmed the DIA-PRM workflow as an efficient analytical tool for proteomic analysis.
Patients and Methods
Patients
The graphic workflow of the present study is shown in Figure 1. A total of 253 newly diagnosed HCC patients were retrospectively enrolled. The participants were divided into two sections: 1) Serum protein expression of 24 patients who survived more than or equal to 5 years, and 24 patients who survived less than 1.5 years were analyzed by DIA. Patients in the two groups were age- and sex-matched. PRM was used to confirm the expression of candidate proteins in HCC patients. 2) The four subgroups, including 32 age- and sex-matched healthy subjects, 35 patients with cirrhosis, 54 patients with liver metastatic cancer, and 205 HCC patients, were applied to verify the candidate proteins using ELISA. Besides, paired serum samples from 30 patients with pre- and post-RFA therapy were collected. Inclusion criteria: 1) patients with definitively diagnosed primary hepatocellular carcinoma; 2) ages 18 to 75; 3) no other major physical diseases or severe liver cancer complications; 4) no prior surgery or interventional therapy; 5) Karnofsky score (KPS) ≥60, and the expected survival time is more than 3 months.16 Exclusion criteria: 1) pregnant or lactating women; 2) patients with severe mental disorders; 3) lost follow-up.
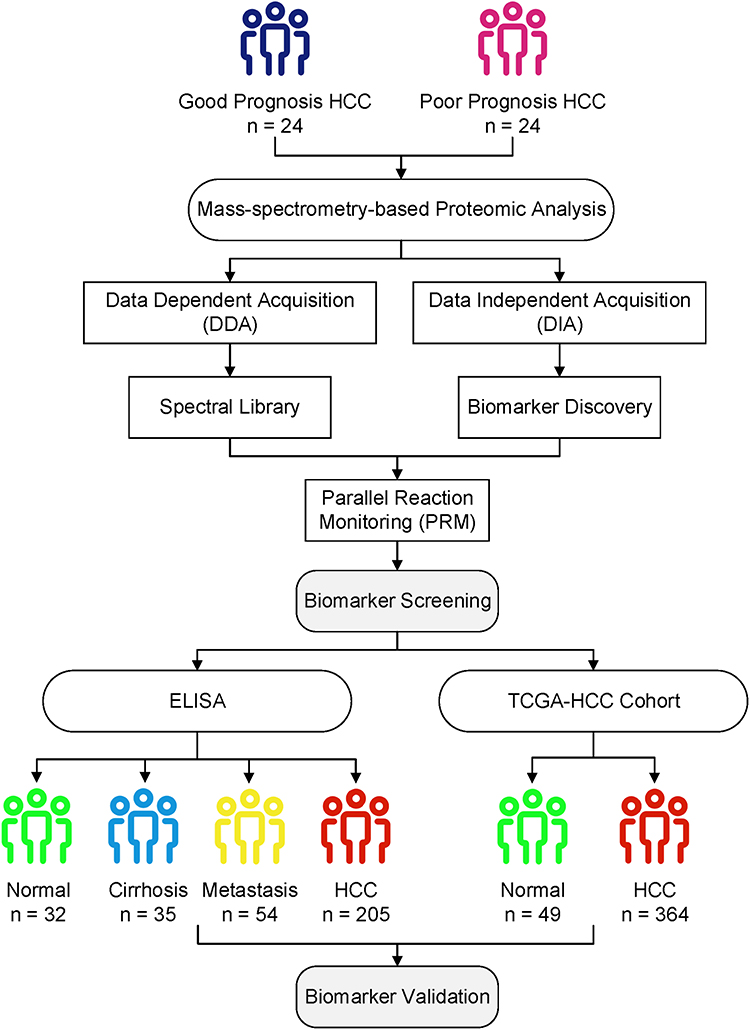 |
Figure 1 Overall experimental design for biomarker screening and validation of the present study. DIA, DDA, and PRM analyses were carried out on 24 good and 24 poor prognosis HCC patients for biomarker screening. Candidate protein biomarkers were validated using the ELISA method and bioinformatics analysis of TCGA transcriptome data. |
All patients and healthy people were from Cancer Hospital, Chinese Academy of Medical Sciences. All samples were collected prior to diagnosis between November 2012 and May 2013. All patients were followed up for an average of 70 months. All of the subjects were selected based on medical and pathology reports. All patients met the pathological or clinical diagnostic criteria.17 Characteristics of HCC patients are summarized in Table 1. Healthy blood samples were obtained from donors in the absence of any disease. The sera of the benign group were provided by non-tumor patients with hepatic cirrhosis. All operations are carried out in the biosafety cabinet, which is calibrated once a year. The trial was conducted in accordance with the Declaration of Helsinki. The study was approved by the Ethics Committee of Cancer Hospital, Chinese Academy of Medical Sciences (Beijing, China), and informed consent was taken from all the patients.
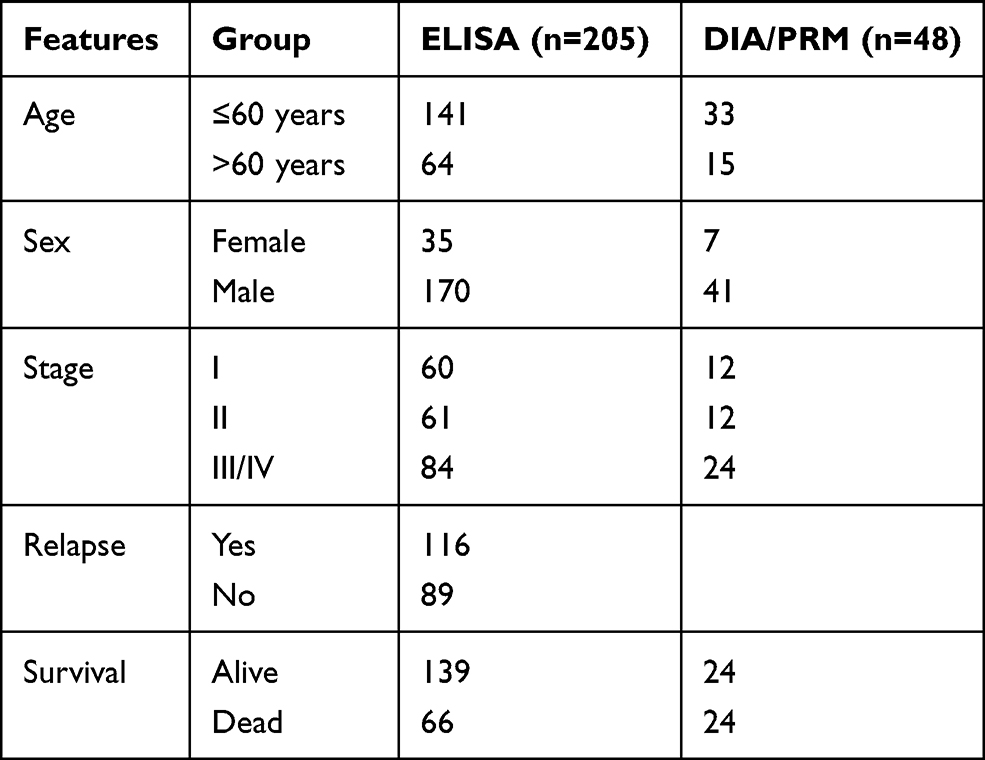 |
Table 1 Basic Clinical Information of HCC Patients |
Serum Collection
Approximately 4mL of venous blood from each patient was collected in SST serum separation tubes (Becton Dickinson) and serum was isolated after centrifugation at 3000g for 10 min at 4°C. After routine biochemical detection, the remaining serum (200μL) was left at room temperature for about 1 h, and centrifuged at 820 g at 4°C for 10 min. The serum was then transferred to the new tube, followed by further centrifugation at 16000g at 4°C for 10min to completely remove the remaining cell debris. Afterward, serum samples were transferred to the new tubes and stored at −80°C.
DIA and PRM Analysis
The protein quantification of serum samples used the BCA method. The filter aided sample preparation (FASP) method was used to process protein lysates as described by Wiśniewski.18 The lysate was depleted from the detergent using 8 M urea and the cysteine residues of denatured proteins were carboxamidomethylated with iodoacetamide. Trypsin buffer was added to each sample and incubated at 37°C overnight. After digestion, the resulting peptides were desalted and lyophilized for storage.
Digested and desalted samples were resuspended in 0.1% formic acid and added with appropriate amount of iRT standard peptides for subsequent analyses. Both types of data acquisition (DDA and DIA) were performed on an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific) coupled with the EASY-nLC 1200 HPLC System. The DDA data were processed by Proteome Discoverer software (Thermo Fisher Scientific) and searched in the human UniProt database appended with the iRT protein sequence. Then, Spectronaut software (Biognosys) generated the spectral library from MS raw files and database search results, and performed the DIA data analysis using default parameters. Gene Ontology (GO) functional annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were performed on the selected proteins using the DAVID database.19 Only terms with FDR < 0.05 and minimum count of 2 were considered significant.
Candidate proteins were selected based on the results of previous proteomic analysis and confirmed by PRM analysis. Initially, the separation and analysis of peptides were performed using the same apparatus as applied in DIA-MS. Then, the obtained PRM raw data were imported into Protein Discoverer software for database search and the human protein dataset “UniProt-organism-9606+reviewed-yes.fasta” was used for analysis. The sub-ion panel of the target peptides was established. Afterward, PRM raw data were imported into SpectroDive software for qualitative and quantitative analysis of the target peptides. The parameters of liquid-phase separation and mass spectrometry are listed in Table 2.
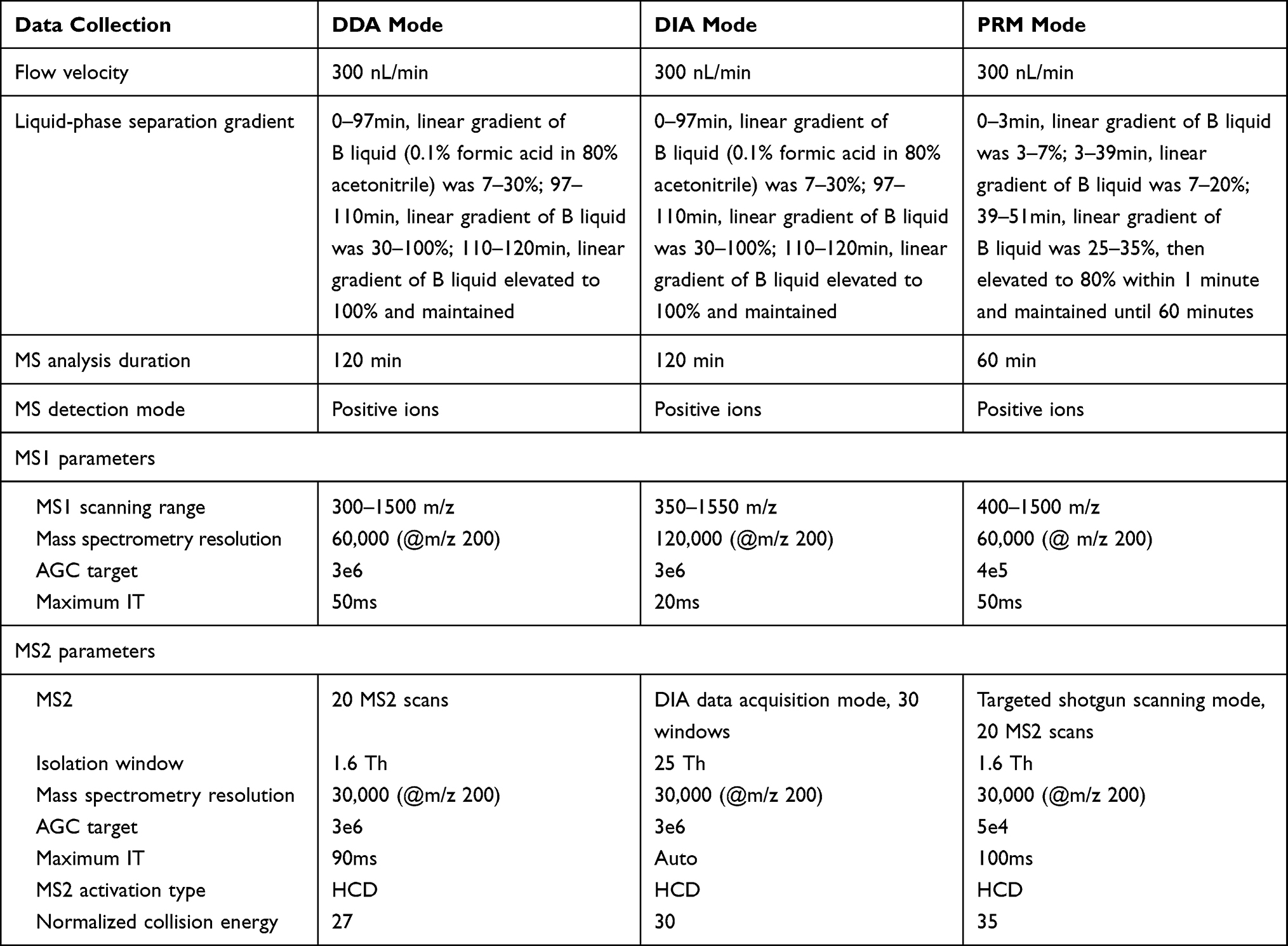 |
Table 2 The Parameters of Liquid-Phase Separation and Mass Spectrometry |
Enzyme‑Linked Immunosorbent Assay (ELISA)
Serum kallikrein (KLKB1) concentrations in 32 healthy subjects, 35 cirrhosis patients, 54 liver metastatic carcinoma patients, and 205 HCC patients (including paired samples from 30 HCC patients before and after treatment) were measured in duplicate by ELISA using the commercial kits (MyBioSource, Cat. No: MBS7226286) according to the manufacturer’s instructions. Briefly, the calibration curve was established by diluting the standard solution provided in the kit. The absorbance of each sample was measured against the blank at 450 nm with an ELISA plate reader. The results were plotted against the calibration curve to obtain the amount of the proteins.
TCGA Data Analysis
KLKB1 expression data and clinicopathological features of TCGA-HCC patients were obtained from the Genomic Data Commons (GDC) Data Portal (https://portal.gdc.cancer.gov/).20 The gene expression dataset involved 364 HCC samples and 49 adjacent normal samples.
Gene set enrichment analysis (GSEA) was performed by GSEA 4.1.0 software.21 HCC patients were divided into high- and low-expression groups according to the median expression of KLKB1. “h.all.v7.2.symbols.gmt” was chosen as the reference gene sets. The permutation number was set as 1000. The criteria of significantly enriched gene sets were NOM P < 0.05 and the absolute value of normalized enrichment score (NES) >1.5.
Statistical Analysis
Statistical analyses were performed by SPSS version 22.0 (IBM, Armonk, NY, USA) and GraphPad Prism 7.0 (Graph Pad Software, San Diego, CA, USA). The Kruskal–Wallis test was used to compare serum protein levels in different populations. The diagnostic accuracy of KLKB1 in predicting HCC was assessed by the area under the curve (AUC) identified by the receiver operating characteristic (ROC). Kruskal–Wallis test Wilcoxon matched-pairs signed rank test was employed to compare serum protein levels of patients before and after treatment. In the TCGA-HCC cohort, t-test and one-way ANOVA were applied to analyze the relationship between gene expression and clinicopathological parameters. Survival analysis was conducted using the Kaplan–Meier method and the Cox regression model. P < 0.05 was considered statistically significant.
Results
Protein Identification and Quantification by DIA
The quality control of the data is shown in Supplemental Figure 1. A total of 773 proteins were screened and quantified by DIA, and 441 proteins were present in more than 50% of the patients who received DIA analysis.
The screening criteria for differentially expressed proteins between good (survived more than or equal to 5 years) and poor (survived less than 1.5 years) prognosis groups were as follows: proteins presenting a fold change of more than 1.20 or less than 0.83 and P < 0.05. As depicted in Figure 2A, 86 proteins with significantly different abundances (65 over-abundant and 21 under-abundant) were identified between the two groups. The GO and KEGG pathway analyses of these 86 proteins showed that the functions of differential serum proteins between the poor prognosis group and the good prognosis group were significantly correlated with the complement activation and complement and coagulation cascades (Figure 2B, Supplemental Table 1).
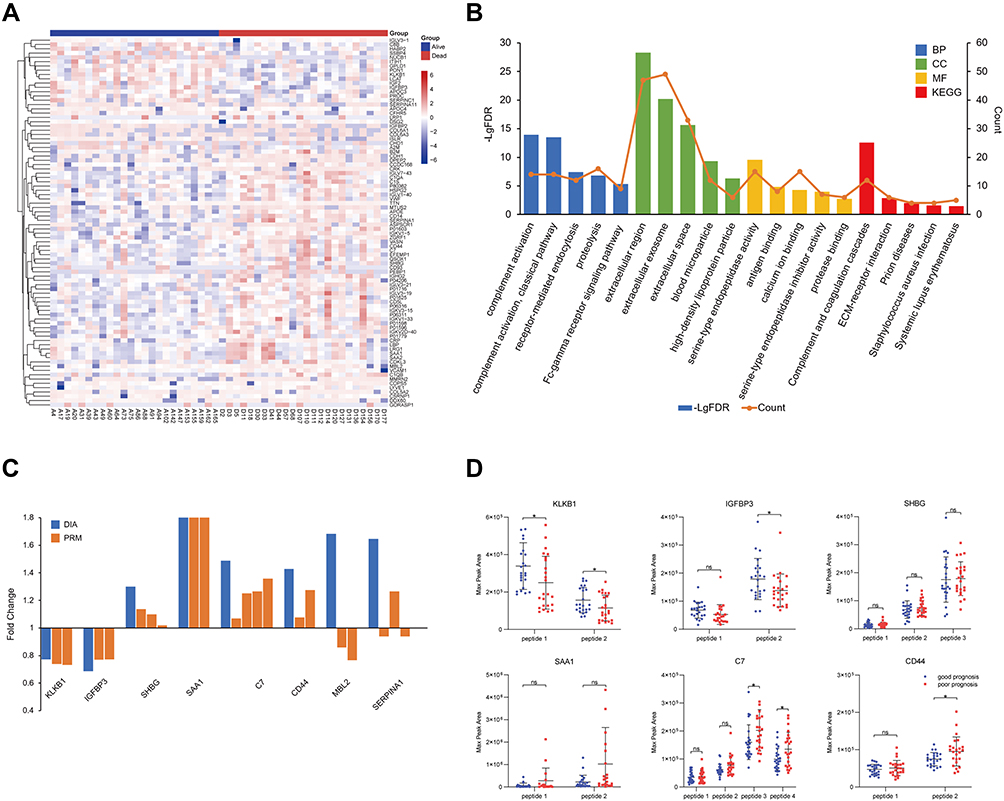 |
Figure 2 Proteomics analyses of HCC patients. Heatmap (A) and enrichment analysis (B) of differentially expressed proteins. (C) The trends of the expression level of eight proteins in patients with different prognosis. The blue bars represent the quantification of proteins using DIA and the brown bars represent the quantification of target peptides using PRM. (D) The quantitative difference (max peak area) in proteins with consistent expression trend between poor prognosis (n = 24) and good prognosis (n = 24) groups. The abscissa represents the target peptides of the proteins. ns p > 0.05, *p < 0.05. |
Confirmation of Key Proteins by PRM
According to the results of protein qualitative and enrichment analyses, 20 target peptides of 8 proteins were selected for PRM confirmation. The quantitative results of the DIA and PRM analyses were consistent for 6 of the 8 proteins (KLKB1, IGFBP3, SHBG, SAA1, C7, and CD44) (Figure 2C). Compared with the good prognosis group, the abundance of two target peptides of KLKB1 and one target peptide of IGFBP3 were significantly reduced (P < 0.05) in the poor prognosis group, while the abundance of two target peptides of C7 and one target peptide of CD44 were significantly increased (Figure 2D). There are significant differences in the abundance of KLKB1 in patients with different prognosis, and there have been few reports about it in HCC so far. As a result, KLKB1 was selected for large sample verification in this study.
Serum KLKB1 Expression in Different Populations
ELISA was performed on 326 participants, including 32 healthy subjects, 35 cirrhosis patients, 54 metastatic carcinoma patients, and 205 HCC patients. Serum protein levels are shown in Figure 3A. KLKB1 expression was significantly lower in HCC patients than in normal subjects (P < 0.0001) and cirrhosis patients (P = 0.046). Besides, serum KLKB1 levels in patients with metastatic carcinoma were also significantly lower than normal subjects (P = 0.002). In addition, KLKB1 discriminated HCC patients from healthy subjects with an AUC of 0.766 and discriminated HCC patients from cirrhosis patients with an AUC of 0.641 (Supplemental Figure 2). The correlation between serum protein levels and clinical characteristics was summarized in Supplemental Table 2. The serum protein levels of KLKB1 were not significantly different in patients of various ages, genders, and stages (P > 0.05). The coefficient of variation (CV) of KLKB1 ELISA microplates was less than 10% (Supplemental Figure 3).
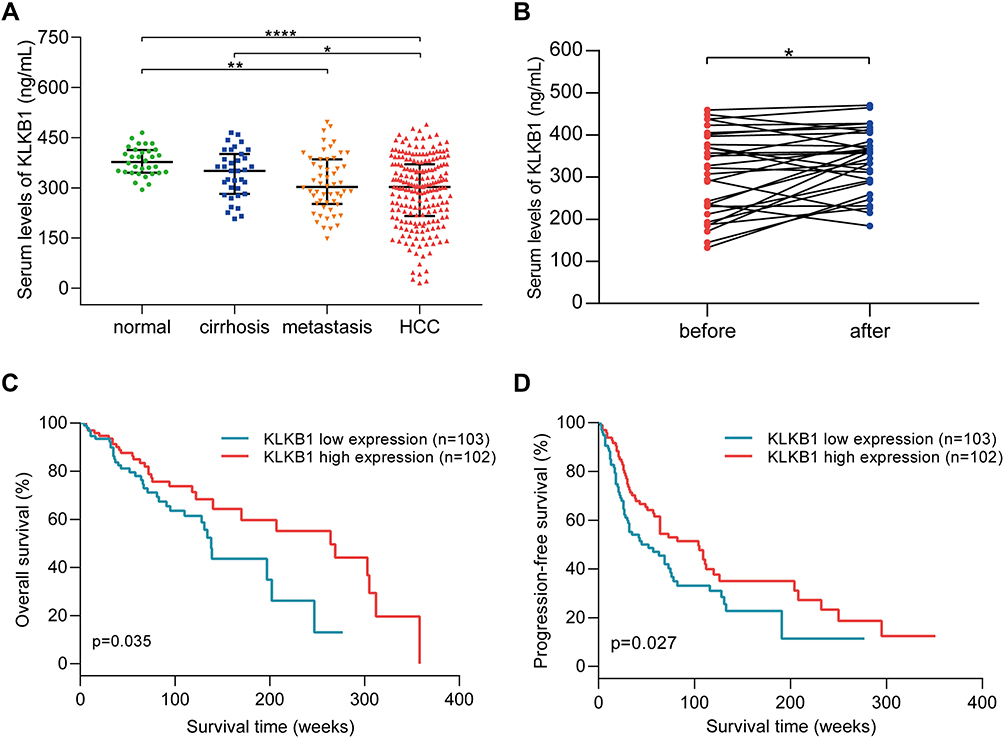 |
Figure 3 The expression of KLKB1 in different populations and its relationship with prognosis of HCC patients. (A) KLKB1 serum levels in healthy subjects, cirrhosis patients, patients with liver metastatic carcinoma, and HCC patients. *p < 0.05, **p < 0.01, and ***p < 0.001. (B) Changes of KLKB1 expression in HCC patients during therapy. *p < 0.05. (C) Overall survival of HCC patients with different expression levels of KLKB1. (D) Progression-free survival of HCC patients with different expression levels of KLKB1. |
Treatment Effect on KLKB1 serum Levels
ELISA was also performed to detect serum levels of KLKB1 in 30 HCC patients before and after treatment. As shown in Figure 3B, the serum KLKB1 level increased significantly (P = 0.016) after the first interventional therapy.
Relationship Between Serum KLKB1 and Prognosis of Patient
The patients were divided into high expression and low expression groups according to the median expression of KLKB1. As depicted in Figure 3C and D, patients with lower expression of KLKB1 had observably shorter overall survival (P = 0.035) and progression-free survival (P = 0.027) than those with higher KLKB1 expression.
Exploration of KLKB1 in TCGA Database
Compared with adjacent normal tissues, KLKB1 was significantly down-regulated in tumor tissues (P < 0.0001, Figure 4A). The relationship between KLKB1 expression and clinical characteristics was revealed in Supplemental Table 3. We found that KLKB1 expression was correlated with age (P = 0.005), race (P < 0.001), tumor stage (P = 0.002), tumor grade (P < 0.001), severity of hepatic tissue inflammation (P = 0.002) and Ishak fibrosis score (P = 0.049).22 Cox regression analysis showed that KLKB1 expression was significantly associated with the prognosis of HCC patients. KLKB1 was an independent prognostic factor for overall survival (HR=0.784, P = 0.032), but not for progression-free survival (HR= 0.913, P = 0.242) (Table 3).
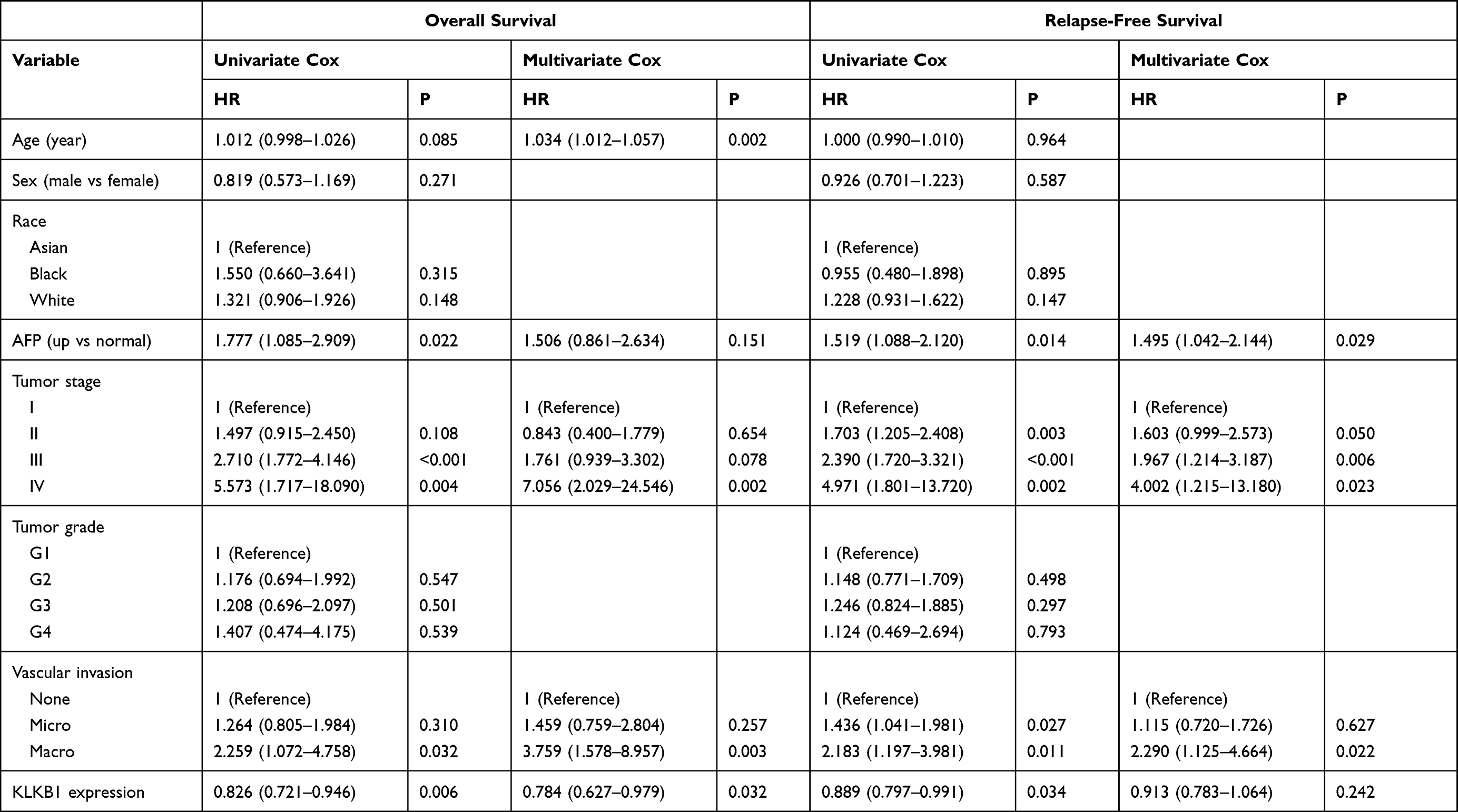 |
Table 3 Cox Regression Analysis of Overall Survival (OS) and Progression-Free Survival (PFS) in HCC Patients from TCGA Database |
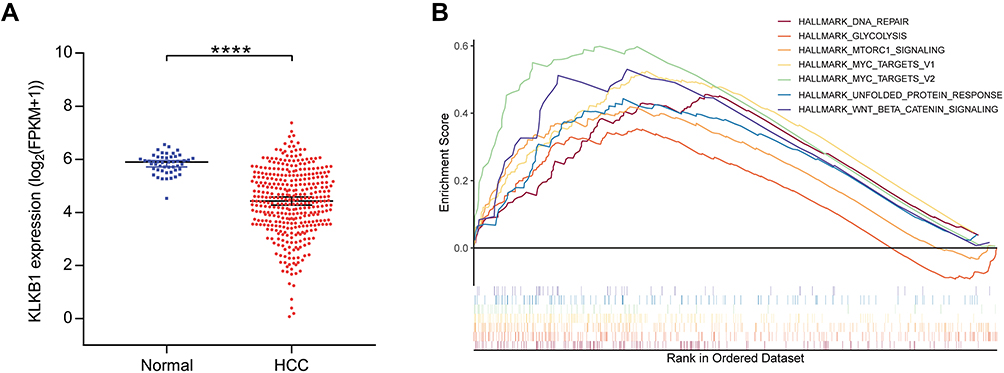 |
Figure 4 KLKB1 expression in TCGA-HCC patients and enrichment plots from gene set enrichment analysis. (A) KLKB1 was obviously downregulated in HCC tissues compared with paracancerous tissues. ****p < 0.0001. (B) The significantly enriched hallmark gene sets in patients with low KLKB1 expression. |
To elucidate the potential mechanisms of the correlation between the KLKB1 expression and biological function, we conducted GSEA to explore the hallmark gene sets enriched in patients with low KLKB1 expression. The enriched gene sets of each gene are shown in Supplemental Table 4. DNA repair, glycolysis, MTORC1 signaling, MYC targets, unfolded protein response, and Wnt/beta-catenin signaling were significantly enriched in the low KLKB1 group (Figure 4B).
Discussion
Due to the wide application of minimally invasive surgery and catheter in HCC, it is more and more inconvenient to obtain tissue or the acquired tissue is not enough for detection. In that case, it is of great significance to screen a marker in the blood for diagnosis and prognosis assessment, instead of screening the prognostic marker from the tissue chip as usual. The design of this experiment opens up a new way of thinking. This “serum-to-serum” approach evades the heterogeneity of tissue markers. The experiment is aimed at HCC, but this idea is suitable for testing other tumors.
DIA is expected to detect the majority of peptides in most samples, thus reducing the detection challenge mainly to selecting and quantifying the correct peptide fragment signals, also referred to as “peak groups”.23 We established a DIA method for in-depth proteome analysis of HCC. DIA was chosen because it generates a “complete” MS/MS library of peptides from all proteins in the sample, which is particularly useful for mining new biomarkers from irreplaceable and limited clinical samples.
Unlike Western Blot, PRM technology does not require the use of antibody and instead tests the sample by mass spectrum. Thus, it plays an important role in the LC-MS analysis of complex samples by enhancing the analytical selectivity when compared to conventional MRM. Data generated by PRM targeted proteomic measurements are therefore not affected by the same statistical challenges as typical spectrum-centric discovery proteomics experiments.10 Another advantage of PRM is that it uses internal standards, such as stable isotope-labeled synthetic peptides (SIS peptides), for absolute quantification. The PRM target peptides of KLKB1 were VLTPDAFVCR and YSPGGTPTAIK, which are unique peptides derived from trypsin cleavage and could be used for accurate quantitation of serum KLKB1. In addition, we have coupled this approach with ELISA analysis to identify the best prognostic protein biomarkers for HCC that can be detected in nonfractionated sera.
In our study, we detected that KLKB1 protein concentration in peripheral blood was significantly lower in HCC patients. Patients with low KLKB1 expression had a worse prognosis, and the level of KLKB1 in the patients' serum after treatment was significantly higher than before treatment. Excitingly, the transcriptome data and proteome data were in good agreement. Data from the TCGA database showed that KLKB1 expression was significantly reduced in HCC tissues, and KLKB1 expression was correlated with tumor stage, tumor grade, liver tissue inflammation, and Ishak fibrosis score. Moreover, KLKB1 expression is associated with the prognosis of HCC patients and is an independent prognostic factor for the overall survival of HCC patients.
KLKB1 is normally synthesized in hepatocytes and secreted into the bloodstream, where it is implicated in biological processes including surface-dependent activation of blood coagulation, fibrinolysis, kinin generation, and inflammation. Researchers found that the level of KLKB1 in plasma can reflect the severity of liver damage. Plasma KLKB1 was significantly lower in patients with chronic hepatitis B (CHB) compared with normal controls and it was even lower in CHB patients who developed end-stage liver disease, such as acute-on-chronic liver failure (ACLF).24 Li et al reported that KLKB1 was significantly dysregulated in HBV-induced HCC specimens compared with chronic hepatitis B specimens, and KLKB1 is also a hub gene in HBV-induced HCC.25 Besides, KLKB1 is also involved in other cancers. In a previous study, researchers using a spectral library-based proteomics platform found that plasma protein abundance of KLKB1 was significantly lower in pancreatic cancer patients than in diabetics. The biomarker panels containing KLKB1 improved the detection accuracy for the diagnosis of early-stage pancreatic ductal adenocarcinoma in diabetic patients.26 KLKB1 expression was generally elevated in carcinomas of the lung and pleura after treatment with the demethylating agent (5-azacytidine), demonstrating that the KLKB1 gene was down-regulated or silenced by aberrant methylation in lung and pleural cancer, suggesting that KLKB1 may function as a tumor suppressor gene with a critical role in the formation of new blood vessels.27 However, KLKB1 mRNA expression was significantly higher in chronic lymphocytic leukemia (CLL) patients than in healthy blood donors, and it was associated with an increased risk of CLL.28 So far, KLKB1 has been rarely studied in HCC except for one bioinformatics article. Therefore, it is both worthwhile and necessary to look into the clinical significance of serum KLKB1 in HCC.
We conducted GSEA to elucidate the molecular mechanism of poor prognosis in the HCC patients with low KLKB1 expression and found that multiple biological processes associated with poor prognosis were activated. MYC targets was the most significantly enriched gene set. MYC is a proto-oncogene and has a central role in almost every aspect of the oncogenic process, orchestrating proliferation, apoptosis, differentiation, and metabolism.29 We hypothesized that KLKB1 could act as a tumor suppressor via the MYC-related signaling pathway. The results of enrichment analysis provide clues for subsequent mechanism experiments to explore the actual biological effects of KLKB1 in HCC.
Conclusion
The novel DIA/PRM/ELISA workflow implemented in our study can be used to discover and validate the relative efficacies of the protein biomarkers for virtually any other disease. Both protein and mRNA data demonstrated that aberrant expression of KLKB1 was strongly associated with the prognosis of HCC patients. These findings may have implications for predicting the prognosis of HCC.
Data Sharing Statement
Data and materials will be available upon corresponding author approval. Proteomic data of this study has been uploaded to ProteomeXchange and the accession number is PXD027817.
Ethics Approval and Informed Consent
Informed consent was obtained from all patients and the protocol was approved by the Ethics Committee of Cancer Hospital, Chinese Academy of Medical Sciences.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (2017-I2M-3-005 & 2017-I2M-1-013) and National Natural Science Foundation of China (81972016).
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
2. Zeng H, Chen W, Zheng R, et al. Changing cancer survival in China during 2003–15: a pooled analysis of 17 population-based cancer registries. Lancet Glob Health. 2018;6(5):e555–e567. doi:10.1016/S2214-109X(18)30127-X
3. Islami F, Miller KD, Siegel RL, Fedewa SA, Ward EM, Jemal A. Disparities in liver cancer occurrence in the United States by race/ethnicity and state. CA Cancer J Clin. 2017;67(4):273–289. doi:10.3322/caac.21402
4. Lurje I, Czigany Z, Bednarsch J, et al. Treatment strategies for hepatocellular carcinoma - a multidisciplinary approach. Int J Mol Sci. 2019;20(6):1465. doi:10.3390/ijms20061465
5. Liu PH, Hsu CY, Hsia CY, et al. Prognosis of hepatocellular carcinoma: assessment of eleven staging systems. J Hepatol. 2016;64(3):601–608. doi:10.1016/j.jhep.2015.10.029
6. Chen S, Chen H, Gao S, et al. Differential expression of plasma microRNA-125b in hepatitis B virus-related liver diseases and diagnostic potential for hepatitis B virus-induced hepatocellular carcinoma. Hepatol Res. 2017;47(4):312–320. doi:10.1111/hepr.12739
7. Han LL, Lv Y, Guo H, Ruan ZP, Nan KJ. Implications of biomarkers in human hepatocellular carcinoma pathogenesis and therapy. World J Gastroenterol. 2014;20(30):10249–10261. doi:10.3748/wjg.v20.i30.10249
8. Heimbach JK, Kulik LM, Finn RS, et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology. 2018;67(1):358–380. doi:10.1002/hep.29086
9. Adalsteinsson VA, Ha G, Freeman SS, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun. 2017;8(1):1324. doi:10.1038/s41467-017-00965-y
10. Rosenberger G, Bludau I, Schmitt U, et al. Statistical control of peptide and protein error rates in large-scale targeted data-independent acquisition analyses. Nat Methods. 2017;14(9):921–927. doi:10.1038/nmeth.4398
11. Selevsek N, Chang CY, Gillet LC, et al. Reproducible and consistent quantification of the Saccharomyces cerevisiae proteome by SWATH-mass spectrometry. Mol Cell Proteomics. 2015;14(3):739–749. doi:10.1074/mcp.M113.035550
12. Huang Q, Yang L, Luo J, et al. SWATH enables precise label-free quantification on proteome scale. Proteomics. 2015;15(7):1215–1223. doi:10.1002/pmic.201400270
13. Gillet LC, Leitner A, Aebersold R. Mass spectrometry applied to bottom-up proteomics: entering the high-throughput era for hypothesis testing. Annu Rev Anal Chem. 2016;9(1):449–472. doi:10.1146/annurev-anchem-071015-041535
14. Bourmaud A, Gallien S, Domon B. Parallel reaction monitoring using quadrupole-orbitrap mass spectrometer: principle and applications. Proteomics. 2016;16(15–16):2146–2159. doi:10.1002/pmic.201500543
15. Canessa EH, Goswami MV, Alayi TD, Hoffman EP, Hathout Y. Absolute quantification of dystrophin protein in human muscle biopsies using parallel reaction monitoring (PRM). J Mass Spectrom. 2020;55(2):e4437. doi:10.1002/jms.4437
16. Karnofsky DA, Burchanot JH. The Clinical Evaluation of Chemotherapeutic Agents. New York: New York Columpia Univ. Press; 1949.
17. Fu J, Wang H. Precision diagnosis and treatment of liver cancer in China. Cancer Lett. 2018;412:283–288. doi:10.1016/j.canlet.2017.10.008
18. Wiśniewski JR. Filter-aided sample preparation for proteome analysis. Methods Mol Biol. 2018;1841:3–10.
19. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi:10.1038/nprot.2008.211
20. Weinstein JN, Collisson EA, Davis C, Donehower L; Cancer Genome Atlas Research Network. The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45(10):1113–1120. doi:10.1038/ng.2764.
21. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. doi:10.1073/pnas.0506580102
22. Ishak K, Baptista A, Bianchi L, et al. Histological grading and staging of chronic hepatitis. J Hepatol. 1995;22(6):696–699. doi:10.1016/0168-8278(95)80226-6
23. Reiter L, Rinner O, Picotti P, et al. mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nat Methods. 2011;8(5):430–435. doi:10.1038/nmeth.1584
24. Sun Z, Liu X, Wu D, et al. Circulating proteomic panels for diagnosis and risk stratification of acute-on-chronic liver failure in patients with viral hepatitis B. Theranostics. 2019;9(4):1200–1214. doi:10.7150/thno.31991
25. Li Z, Xu J, Cui H, Song J, Chen J, Wei J. Bioinformatics analysis of key biomarkers and potential molecular mechanisms in hepatocellular carcinoma induced by hepatitis B virus. Medicine. 2020;99(20):e20302. doi:10.1097/MD.0000000000020302
26. Peng H, Pan S, Yan Y, et al. Systemic proteome alterations linked to early stage pancreatic cancer in diabetic patients. Cancers. 2020;12(6):1534. doi:10.3390/cancers12061534
27. Wong J, Sia YY, Misso NL, Aggarwal S, Ng A, Bhoola KD. Effects of the demethylating agent, 5-azacytidine, on expression of the kallikrein-kinin genes in carcinoma cells of the lung and pleura. Patholog Res Int. 2011;2011:167046.
28. Adamopoulos PG, Kontos CK, Papageorgiou SG, Pappa V, Scorilas A. KLKB1 mRNA overexpression: a novel molecular biomarker for the diagnosis of chronic lymphocytic leukemia. Clin Biochem. 2015;48(13–14):849–854. doi:10.1016/j.clinbiochem.2015.04.007
29. Chen H, Liu H, Qing G. Targeting oncogenic myc as a strategy for cancer treatment. Signal Transduct Target Ther. 2018;3:5. doi:10.1038/s41392-018-0008-7
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.