Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 9 » Issue 1
Serum cytokine profiling and enrichment analysis reveal the involvement of immunological and inflammatory pathways in stable patients with chronic obstructive pulmonary disease
Authors Bade G, Khan M, Srivastava A, Khare P, Solaiappan K, Guleria R, Palaniyar N, Talwar A
Received 25 January 2014
Accepted for publication 31 March 2014
Published 5 August 2014 Volume 2014:9(1) Pages 759—773
DOI https://doi.org/10.2147/COPD.S61347
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Geetanjali Bade,1 Meraj Alam Khan,2 Akhilesh Kumar Srivastava,1 Parul Khare,1 Krishna Kumar Solaiappan,1 Randeep Guleria,3 Nades Palaniyar,2 Anjana Talwar1
1Department of Physiology, All India Institute of Medical Sciences, New Delhi, India; 2Program in Physiology and Experimental Medicine, The Hospital for Sick Children, Department of Laboratory Medicine and Pathobiology, and Institute of Medical Sciences, Faculty of Medicine, University of Toronto, Toronto, ON, Canada; 3Department of Pulmonary Medicine and Sleep Disorders, All India Institute of Medical Sciences, New Delhi, India
Abstract: Chronic obstructive pulmonary disease (COPD) is a major global health problem. It results from chronic inflammation and causes irreversible airway damage. Levels of different serum cytokines could be surrogate biomarkers for inflammation and lung function in COPD. We aimed to determine the serum levels of different biomarkers in COPD patients, the association between cytokine levels and various prognostic parameters, and the key pathways/networks involved in stable COPD. In this study, serum levels of 48 cytokines were examined by multiplex assays in 30 subjects (control, n=9; COPD, n=21). Relationships between serum biomarkers and forced expiratory volume in 1 second, peak oxygen uptake, body mass index, dyspnea score, and smoking were assessed. Enrichment pathways and networks analyses were implemented, using a list of cytokines showing differential expression between healthy controls and patients with COPD by Cytoscape and GeneGo Metacore™ softwares (Thomson-Reuters Corporation, New York, NY, USA). Concentrations of cutaneous T-cell attracting chemokine, eotaxin, hepatocyte growth factor, interleukin 6 (IL-6), IL-16, and stem cell factor are significantly higher in COPD patients compared with in control patients. Notably, this study identifies stem cell factor as a biomarker for COPD. Multiple regression analysis predicts that cutaneous T-cell-attracting chemokine, eotaxin, IL-6, and stem cell factor are inversely associated with forced expiratory volume in 1 second and peak oxygen uptake change, whereas smoking is related to eotaxin and hepatocyte growth factor changes. Enrichment pathways and network analyses reveal the potential involvement of specific inflammatory and immune process pathways in COPD. Identified network interaction and regulation of different cytokines would pave the way for deeper insight into mechanisms of the disease process.
Keywords: COPD, Bio-Plex assay, biomarkers, pathways, networking
Introduction
Chronic obstructive pulmonary disease (COPD) is a major global health problem. In 2002, it was the fourth worldwide leading cause of death, and it is anticipated to be the third leading cause of death by 2030.1 COPD results from chronic inflammation and eventual irreversible damage to the airways. Several factors, including exposure to environmental pollution and smoking, contribute to the airway damage and obstruction.2 Inflammatory response in COPD is characterized by the increased number of macrophages, neutrophils, and cytotoxic T lymphocytes in the airways and lung parenchyma. Airway inflammation further results in systemic inflammation and other COPD-related manifestations.3–5
Spirometry (forced expiratory volume in 1 second; FEV1), exercise tests (peak oxygen uptake; VO2), and dyspnea scores (a scale to determine the degree of difficulty in breathing) are used for diagnosing and determining the prognostic factors of COPD. These tests are also used for evaluating the treatment efficacy in COPD patients. However, the test results do not fully reflect the burden of COPD.6,7 Aaron et al have studied 19 serum markers and found that only C-reactive protein, myeloperoxidase, and vascular endothelial growth factor show intra- and interpatient reliability and correlation with disease severity.8 Franciosi et al performed a meta-analysis by considering the spirometric, demographic, clinical, cytological, and biochemical variables in stable COPD patients. Their meta-analysis indicates that the concentrations of tumor necrosis factor (TNF) and C-reactive protein have suggestive correlation with disease severity. Further, the existence of high variability among biomarker levels in different studies has also been observed.9 Therefore, additional studies with a well-defined group of patients are needed to identify the sets of potential biomarkers and pathways involved in COPD from different populations.
Cytokines play a critical role in COPD-associated inflammation.10 Interactions among these cytokines are complex, as some cytokines can have different actions under a specific context.11 Thus, studying a panel of cytokine biomarkers, their correlation with pulmonary physiological functions and systemic parameters and the involvement of various pathways and networks would provide a better understanding of the mechanisms underlying COPD. This will also help to identify a nonredundant set of biomarkers for COPD. Measuring cytokine panels from the same samples in multiplex assays will be advantageous for reducing technical variability. Only a limited number of studies have used panels of multiple serum cytokines to determine biomarkers in COPD patients simultaneously.8,12 Furthermore, no studies with multiple biomarkers and pathway analysis have been reported on COPD patients of Indian descent.
In the present study, we measured the serum levels of 48 cytokines in stable COPD and healthy subjects by immunobead Bio-Plex assays (Bio-Rad Laboratories Inc., Hercules, CA, USA) and analyzed their association with FEV1 (percentage predicted), maximum oxygen uptake (peak VO2 predicted), smoking index, dyspnea scores, and body mass index (BMI) by multiple regression analysis. Furthermore, our study identifies that changes in a few key cytokine concentrations explain the changes in lung function related parameters (FEV1 and peak VO2 and smoking index). We also assessed some of the key biomarkers and their common regulatory factors involved in inflammatory and immunological pathways in COPD.
Materials and methods
Ethics statement
This study was approved by the Ethics Committee of All India Institute of Medical Sciences, New Delhi, India. Prior informed written consent was obtained from all participating healthy and COPD volunteers. Blood work and spirometric tests were conducted under the guidelines of the ethics committee.
Patient selection
COPD patients were diagnosed according to the guidelines of Global Initiative for Chronic Obstructive Lung Disease (GOLD) criteria.13 They were either current smokers with COPD or ex-smokers with COPD. Individuals older than 40 years were recruited for this study and were categorized into two main groups: Group 1 included healthy controls with no evidence of COPD (n=9); four and five participants were smokers and nonsmokers, respectively, and group 2 included patients diagnosed with COPD (n=21); eleven of whom were ex-smokers with COPD and ten of them were current smokers with COPD. All the COPD patients selected for the study were in stable condition (no exacerbation for at least 1 month). Participants were excluded specifically when they had a history of COPD exacerbation within the last 1-month period, asthma, or were taking oral steroids or had any conditions that were associated with inflammation such as infection, cancer, congestive heart failure, end-stage renal disease, or connective tissue disorders that could affect the levels of COPD biomarkers.
Measurement of pulmonary functions
FEV1 (percentage predicted) and FVC (forced vital capacity) were measured with a calibrated spirometer (Spiro Air, Medisoft; PK Morgan Ltd., Kent, United Kingdom), using standard methodology at the time of enrollment of the subjects.14 This pulmonary function test value is critical for diagnosing obstructive and restrictive lung diseases.
Calculation of BMI
BMI is an independent prognostic factor for disease severity and survival after the diagnosis of COPD.15 BMI is a measure of human body shape/obesity based on an individual’s weight and height. Each body mass index score was calculated by the following formula: BMI = weight (kg)/(height [m])2.
Symptom-limited incremental exercise test
In patients with COPD, a symptom-limited incremental step test elicited the maximum cardiopulmonary and metabolic responses.16 A symptom-limited incremental exercise test was performed on a bicycle ergometer, using 10 W ramp protocol. Exercise was performed while seated on an electronically braked cycle ergometer (Corival; Lode, Groningen, the Netherlands). Heart rate was monitored continuously throughout the protocol using a heart rate monitor (POLAR [Polar Electro Oy, People's Republic of China]). The polar device used in this study had a T31 transmitter and a chest belt (N2965) to transmit heart rate to the system. Breath-by-breath measurements of oxygen consumption (VO2) were made by custom software (vacuumed/vista MX [VacuMed, Ventura, CA, USA]). Standard 12-lead electrocardiograms were obtained at rest, during exercise and in recovery phase. Blood pressure was monitored using a standard cuff sphygmomanometer at rest, every 3 minutes during exercise, and every 2 minutes during recovery. At the end of exercise, the reasons for termination of exercise were obtained from the patient. The VO2 max was the highest VO2 observed during exercise. The exercise was also terminated at exhaustion (intolerable dyspnea, as indicated by the patient); severe desaturation (peripheral capillary oxygen saturation <80%); demand by the patient because of leg cramps, chest pain, or discomfort or request by patients for any other reason.
Evaluation of dyspnea
Dyspnea is one of the common symptoms in COPD either at rest or under conditions of exercise, and this applies to all severities. Dyspnea severity was assessed and scored using the standard Modified Medical Research Council questionnaire.17,18
Measurements of blood markers
A volume of 5 mL of whole blood was collected into a plain tube and allowed to clot for 1 hour. The sera samples were aliquoted after centrifugation at 1,000 × g for 10 minutes and stored at −80°C until further analysis. Concentrations of serum cytokines were analyzed using two complementary Bio-Plex suspension array systems (Bio-Plex Pro Human Cytokine Group 27-Plex Panel and Bio-Plex Pro Human Cytokine Group 21-Plex Panel) to cover the range of all the cytokine biomarkers potentially involved in the pathophysiology of COPD. Forty-eight biomarkers were assessed simultaneously, using the Bio-Plex system. The selection of specific cytokines in the study was based on the previously available reports and the involvement of these cytokine biomarkers in COPD.19 Assays were performed in duplicate by following the standard operating protocol provided by the Bio-Plex Multiplex cytokine assay.
Briefly, anticytokine antibody-conjugated beads were added to individual wells of a 96-well filter plate and adhered using vacuum filtration. After washing, 50 μL prediluted standards and serum samples were added into respective wells, and the filter plates were shaken at 300 rpm for 30 minutes at room temperature. Thereafter, the filter plates were washed and 25 μL prediluted multiplex biotin-conjugated detection antibody was added and incubated for another 30 minutes. After washing, 50 μL prediluted streptavidin-conjugated PE was added for 10 minutes, followed by an additional wash and the addition of 125 μL Bio-Plex assay buffer to each well. Then, filter plates were analyzed using the Bio-Plex Protein Array System, and concentrations of each cytokine were determined using software (Bio-Plex Manager version 6.0). Standard curves were generated for each biomarker. The line of best fit was determined for standard curves by standard recovery methods and by calculating the concentration of each standard.20
Statistical analysis
Cytokines’ levels in sera showed no differences between smokers and nonsmokers within the control group. There were also no statistical differences detected between ex-smokers with COPD and current smokers with COPD. Therefore, we compared the pooled samples (healthy controls versus COPD), using the two-tailed Mann–Whitney test, as outlined in the experimental design (Figure S1). The association of identified serum biomarkers with FEV1 (percentage predicted), peak VO2 (predicted), smoking index, dyspnea scores, and BMI were studied in all subjects, using Spearman correlation analysis. In addition, a step-wise multiple regression analysis with weighted regression equations has also been used for calculating coefficients for FEV1 (percentage predicted), peak VO2 (predicted), smoking index, dyspnea scores, and BMI with serum biomarkers. Multiple regression analysis determines the relationship between a dependent or criterion variable of interest (Y) and a set of K independent variables or potential predictor variables (X1, X2, X3, …, Xk), where the scores on all variables are measured for n number of cases. A multiple regression equation for predicting Y is expressed as Y = A + (B1X1) + (B2X2) + … BkXk. A stepwise multiple regression analysis eliminated the parameters that provided insignificant contribution to the model. The coefficient for each factor was obtained from the final model with the lowest P-value. Calculated and observed values were plotted in a regression line after calculating a common factor for each equation. All the statistical analyses were performed using the Prism version 5 (GraphPad Software, Inc., La Jolla, CA, USA) and SPSS (version 16; SPSS Inc., Chicago, IL, USA) softwares.
Enriched pathways and network analysis
The mean fold changes of the detected biomarkers were calculated between COPD and control subjects. Cytokines showing 1.5-fold changes were used for biological processes, pathways, and network enrichment analyses. The purpose of this analysis was to identify the enriched biological processes and the pathways associated with COPD. Interaction networks among the significantly elevated cytokines in COPD were constructed using Cytoscape and GeneGo Metacore™ software (Thomson Reuters, St Joseph, MI, USA).
Results
Biomarkers CTACK, eotaxin, HGF, IL-6, IL-16, and SCF are elevated in the sera of stable COPD patients
We assessed 48 candidate biomarkers in the serum samples of healthy control and COPD subjects by Bio-Plex assays. Of these, the concentrations, in picograms per milliliter, of 29 cytokines were measurable. The concentrations of the rest of the 19 cytokines were below the detection limit. Mean comparison procedures show that serum levels of six biomarkers (cutaneous T-cell attracting chemokine [CTACK], eotaxin, hepatocyte growth factor [HGF], interleukin 6 [IL-6], IL-16, and stem cell factor [SCF]; P<0.05) are significantly higher in COPD patients compared with in control subjects (Figure 1). The data of cytokines not showing significant differences between the groups and the rest of the biomarkers tested in the study are listed in Table S1.
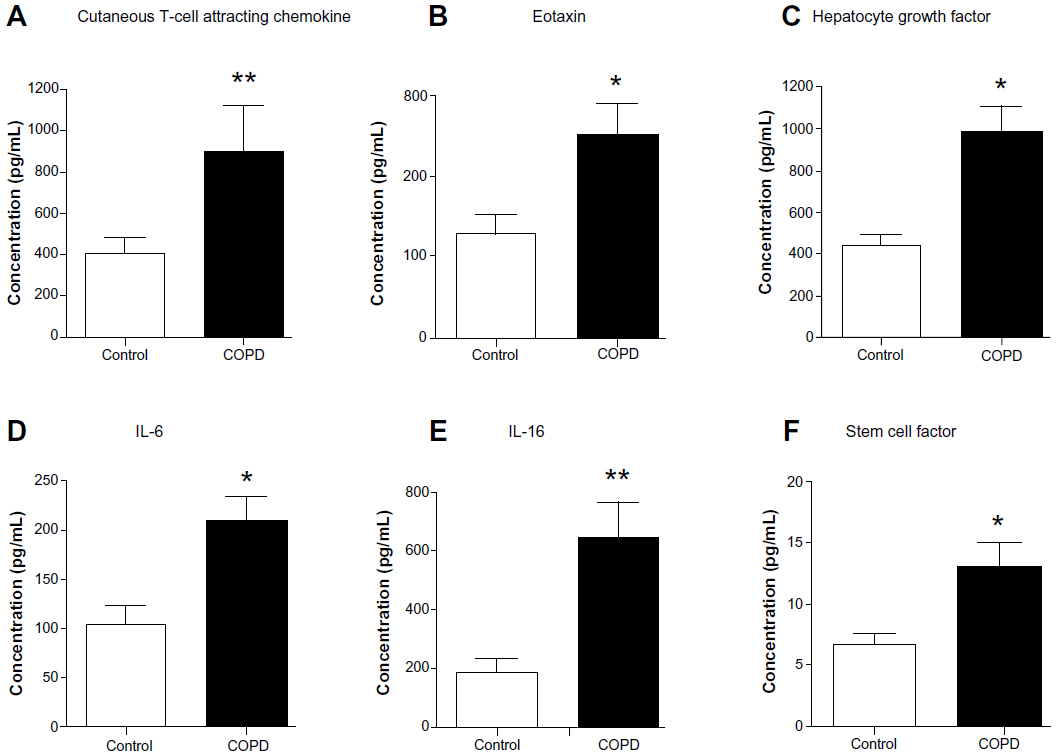 | Figure 1 Serum concentrations of cutaneous T-cell attracting chemokine, eotaxin, hepatocyte growth factor, interleukin 6 (IL-6), IL-16, and stem cell factor are elevated in chronic obstructive pulmonary disease patients compared with control subjects. Serum level of (A) cutaneous T-cell attracting chemokine (P<0.01), (B) eotaxin (P<0.05), (C) hepatocyte growth factor (P<0.05), (D) IL-6 (P<0.05), (E) IL-16 (P<0.01), and (F) stem cell factor (P<0.05) are significantly higher in chronic obstructive pulmonary disease patients (n=21) compared with control subjects (n=9). Significant differences between chronic obstructive pulmonary disease and control subjects are denoted by *P≤0.05 and **P≤0.01, as measured by two-tailed Mann–Whitney test. The data are represented as mean ± standard error. |
CTACK is a T-cell chemokine, whereas eotaxin is a potent chemoattractant for eosinophils and basophils.21,22 HGF is a multifunctional mitogen.23 IL-6 is a permeability-increasing inflammatory cytokine released mainly by macrophages and epithelial cells, and IL-16 is a proinflammatory cytokine released by bronchial epithelium and dendritic cells, cluster of differentiation (CD)8+ and CD4+ T cells.24,25 Hematopoietic factor SCF is secreted by various stroma and inflammatory cells.26 Collectively, all these cytokines are important for recruiting different types of immune cells, cell proliferation, and tissue repair. The cytokines/biomarkers identified by these analyses are highly relevant to COPD and act as putative candidate biomarkers of COPD patients.
Eotaxin is negatively correlated with FEV1, whereas CXCL1 is positively correlated with HGF
To determine the key biomarkers affecting FEV1 (percentage predicted), peak VO2 (predicted), smoking, dyspnea score, and BMI in COPD patients, bivariate correlation analysis was performed. All recruited subjects had complete demographic data, and as expected by the study design, COPD patients have lower FEV1 (percentage predicted) (mean ± standard error: 48.7%±3.9%) compared with age-matched controls (mean ± standard error: 83.6%±1.7%; P<0.001; Table 1). Bivariate analysis of cytokines with each parameter shows that eotaxin concentrations have a strong inverse relationship with FEV1 (percentage predicted; Y=0.106x + 87.08; r=−0.71; P≤0.05; Figure 2A). These data suggest that eotaxin could be one of the important contributors to the decaying lung function. Notably, chemokine (C-X-C motif) ligand 1 (CXCL1) or the growth-related oncogen α (GRO-α) show a strong positive correlation with HGF (Y=8.675x + 124.77; r=0.76; P<0.05; Figure 2B); therefore, CXCL1 and HGF are likely to be dependent variables and/or affected by the same transcription factor. CXCL1 shows statistically significant but low levels of positive correlation with IL-6 (r=0.23; P<0.05) and an inverse relationship with dyspnea (r=−0.53; P<0.05). CXCL1 is a chemotactic factor for monocytes and neutrophils. This protein is produced by a variety of cells including monocytes, endothelial cells, and fibroblasts.27,28 Therefore, CXCL1 may be involved in regulating various components of COPD.
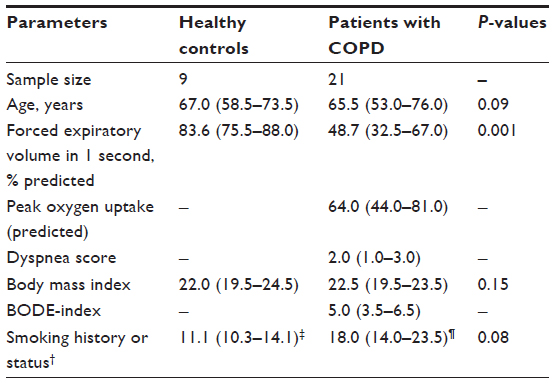 | Table 1 Demographic characteristics of the participants in the study* |
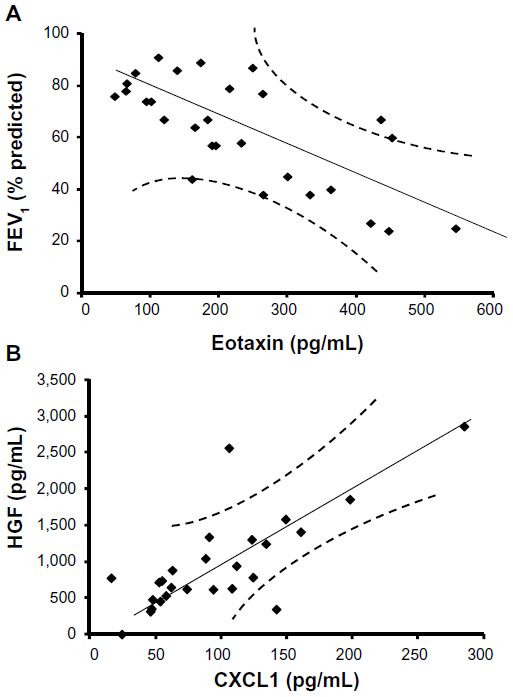 | Figure 2 Increasing severity of airflow limitation is associated with decreasing levels of eotaxin, whereas chemokine (C-X-C motif) ligand 1 (CXCL1) is positively correlated with hepatocyte growth factor. (A) Eotaxin is negatively correlated with forced expiratory volume in 1 second (FEV1; percentage predicted), as represented in the regression line. These data suggest that eotaxin is a good biomarker predicting FEV1 changes in stable chronic obstructive pulmonary disease. (B) The regression line for CXCL1 and hepatocyte growth factor shows a positive correlation, suggesting these two factors are dependent variables or regulated by the same transcription factor. |
Increase in CTACK, eotaxin, IL-6, and SCF significantly contributes to the reduction in FEV1 and peak VO2
To further determine the contribution of all the cytokines to each of these parameters, we conducted a stepwise multiple regression analysis. This systematic analysis identified that eotaxin, IL-6, and SCF are statistically significantly, contributing to the reduction in lung function measured by FEV1 (percentage predicted). Contributions of each parameter are described by the following equation: Y=(−0.02×eotaxin) + (−0.37×IL-6) + (−0.22×SCF) (Table 2; P<0.05). A constant factor of −1.92 converts the Y values equivalent to observed FEV1 (percentage predicted; Figure 3A; r=−0.71; P<0.01). These analyses suggest that an increase in serum concentrations of CTACK, eotaxin, and SCF reflects the decrease in FEV1 observed in the present study, and notably, these three cytokines are sufficient to explain most of the changes seen in FEV1.
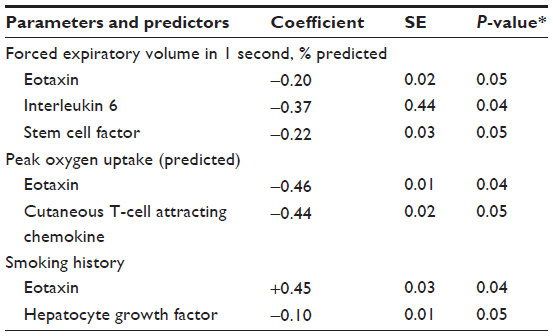 | Table 2 Multiple regression analysis between studied physiological parameters and cytokines showing significant differences between controls and patients with COPD |
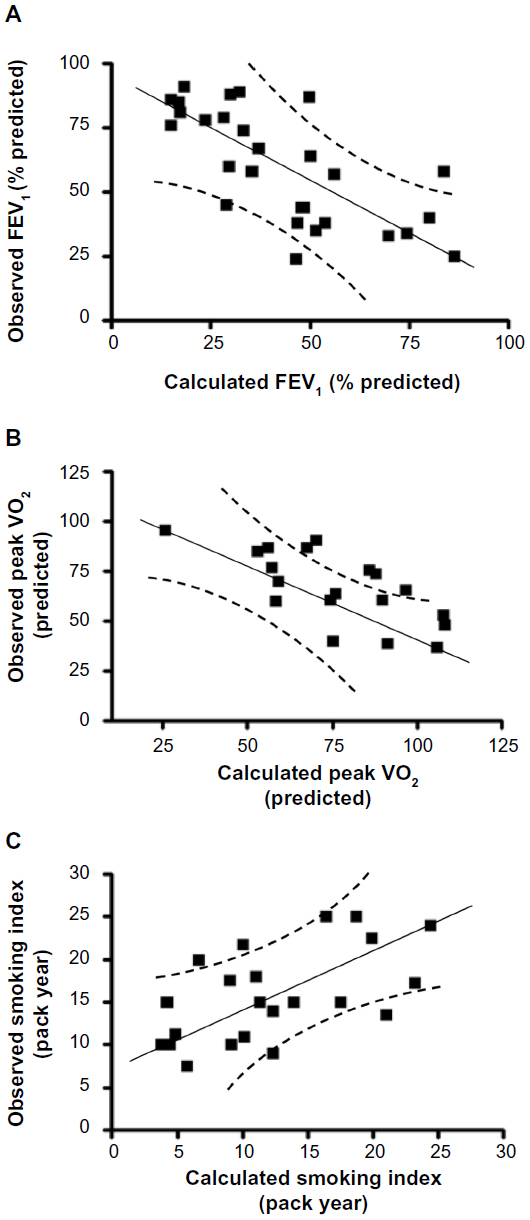 | Figure 3 Cutaneous T-cell attracting chemokine, eotaxin, hepatocyte growth factor, interleukin 6 (IL-6), and stem cell factor concentrations estimate the changes in forced expiratory volume in 1 second (FEV1; percentage predicted), peak oxygen uptake (VO2; predicted), and smoking status by multiple regression analysis. (A) Stepwise multiple regression analysis identified that the variables eotaxin, IL-6, and stem cell factor significantly contribute to the changes in FEV1 (percentage predicted). The values calculated on the basis of these three cytokines correlate well with the changes observed in FEV1 values. (B) Similar analysis shows that cutaneous T-cell attracting chemokine and eotaxin significantly contribute to the changes in the peak VO2. The values calculated based on these two cytokines correlate well with the changes observed in peak VO2. (C) The smoking indices are related to eotaxin and hepatocyte growth factor, and the values correlate well with the predicted and observed smoking indices. |
Furthermore, eotaxin and CTACK significantly contribute to the observed reduction in peak VO2 (predicted). Contributions of these two cytokines to peak VO2 are described by the following equation: Y=(−0.44× CTACK) + (−0.46×eotaxin) (Table 2; P<0.05). A constant factor of −9.1 converts the Y values equivalent to measured peak VO2 (predicted; Figure 3B; r=−0.70; P<0.01). These analyses suggest that increase in CTACK and eotaxin concentrations is related to decrease in peak VO2 observed in these individuals. Once again, these two cytokines are sufficient to explain most of the changes observed in peak VO2. Collectively, these results show that the four key cytokines (CTACK, eotaxin, IL-6, and SCF) explain the significant changes in lung function-related parameters observed in the study population.
Smoking affects the changes in eotaxin and HGF concentrations
To determine the effect of smoking on the changes in cytokine concentrations, we have conducted a stepwise multiple regression analysis of the biomarkers, as explained earlier. This analysis shows that changes in the sera concentrations of eotaxin and HGF are associated with the smoking levels (pack year): Y=(0.45×eotaxin) + (−0.10×HGF) (Table 2; P<0.05). A constant factor of 12.6 converts the Y values equivalent to the observed smoking index (Figure 3C; r=0.58; P<0.05). These data suggest that smoking partially contributes to the increase in eotaxin and decrease in HGF concentrations observed in the sera of smokers in this study population.
Specific immune and inflammatory response pathways are involved in stable COPD
Complex interactions among cytokines, chemokines, and growth factors regulate various immune and inflammatory responses. To determine the pathways that are potentially regulated by these cytokines in COPD, we have conducted enriched pathway analyses. Of the 29 measurable biomarkers, 16 showed higher concentrations (≥1.5 upregulated), whereas two showed lower concentrations (downregulated, ≤1.5) in COPD patients compared with in healthy controls. Pathway enrichment analysis was conducted using these cytokines by Metacore™ platform, with a priori setting of a cutoff threshold (False Discovery Rate (FDR)p, probability value =0.05). Pathways and biological processes regulated by these cytokines include immune responses (mediated by T-helper cell differentiation, riggering receptor expressed on myeloid cells 1 (TREM1) signaling, prostaglandin E2 signaling, and Th17 [T helper 17 cells]-derived cytokines), inflammation mediated by JAK/STAT (Janus kinase/signal transducers and activators of transcription) pathway, histamine signaling), lymphocyte proliferation and regulation, and leukocyte chemotaxis (Figure 4). Therefore, this analysis suggests the involvement of immune response-related pathways in COPD.
 | Figure 4 Pathway enrichment analysis reveals specific immune and inflammatory response pathways in chronic obstructive pulmonary disease. (A) The top most significant enriched biological processes and pathways including immune responses, inflammation, lymphocyte proliferation and regulation, and leukocyte chemotaxis are represented along with the coenrichment P-value (log scale) in the bar graph. (B) The numbers and names of gene sets overlapping with the significant pathways and processes are shown in the table format. |
GCR-α, NF-κβ and C/EBP-β regulate the major cytokine networks in COPD
Specific transcription factors and regulators determine cytokine response.29 To determine the transcription factors that regulate the key cytokines involved in COPD, we have conducted a network analysis. Thirteen of the 18 cytokines (nodes) generated a large single regulatory network. Three main hubs NF-κβ (nuclear factor kappa-light-chain-enhancer of activated B cells), C/EBP-β (CCAAT/enhancer-binding protein beta), and GCR-α (glucocorticoid receptor alpha) regulate these networks (Figure 5). These enriched networks mainly affect mediators involved in immune, inflammatory, and chemotaxis responses. Interconnecting cytokine biomarkers including eotaxin, HGF, IL-6 and SCF are induced either by two or three hubs. Notably, GCR-α directly regulates all four of these cytokines, which are significantly upregulated in stable COPD patients. Therefore, GCR-α is likely a major potential regulator of these cytokine biomarkers in COPD.
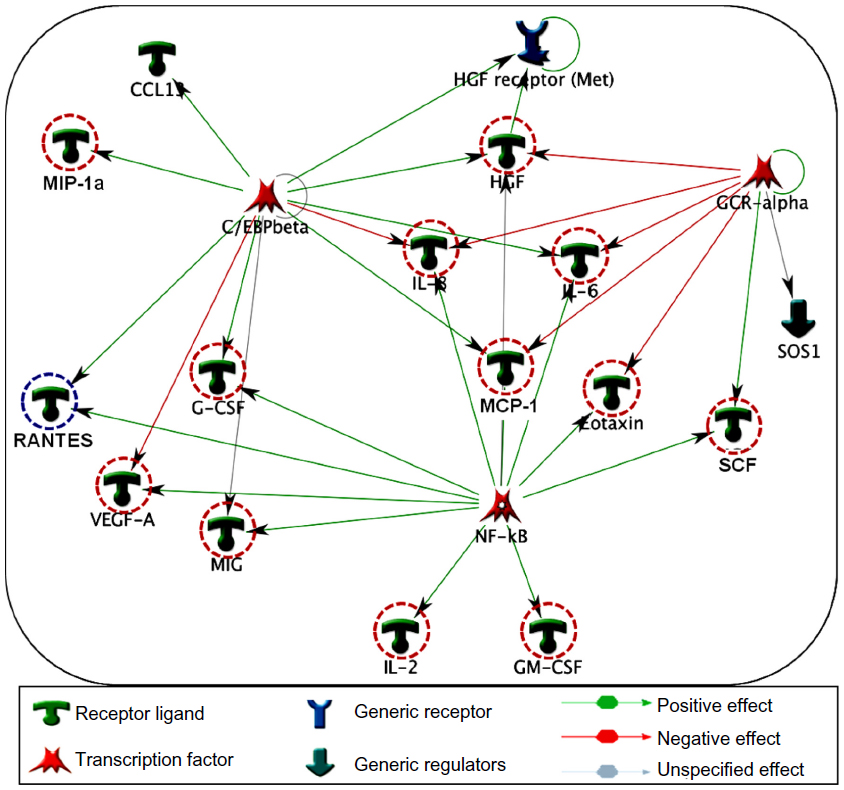 | Figure 5 Interpathway interactions identify cytokine linking network in COPD. Diagram illustrates the network topology, in which individual annotated hubs (GCR-α, nuclear factor kappa-light-chain-enhancer of activated B cells, and C/EBP-β) regulate the thirteen major cytokines. Note that eotaxin, hepatocyte growth factor, interleukin (IL)-6 and stem cell factor interconnecting nodes are induced either by two or three hubs. Red-dotted circles flag the upregulation, whereas blue dotted circles represent the down-regulation of these cytokines in COPD. Red arrows show the positive effects, whereas green arrows represent negative effects. |
Discussion
Serum biomarkers and their effects on COPD-related parameters and pathways that affect COPD have not been fully established. We have assessed the levels of 48 serum biomarkers in COPD patients of Indian descent, using multiplex immunobead-based assays. We have identified some of the key biomarkers involved in pathways associated with stable COPD. In particular, the serum levels of CTACK, eotaxin, HGF, IL-6, IL-16 and SCF are significantly higher in patients with COPD compared with in control subjects (Figure 1). These cytokine levels are expected to reflect long-term effects because our study focuses on stable COPD patients with no exacerbation during the previous 1-month period. To the best of our knowledge, this is the first study that identified an increased serum level of SCF in COPD patients compared with healthy controls. Eotaxin, IL-6, and SCF inversely contribute to the changes in FEV1 (percentage predicted), whereas eotaxin and HGF inversely contribute to the changes in peak VO2 (predicted). Smoking is related to increased eotaxin and decreased HGF concentrations. CXCL1 is positively correlated with IL-6 and HGF, but it is negatively related to dyspnea. Enrichment pathways and network analyses revealed the involvement of inflammatory, chemotaxis, and immune response-related pathways in COPD.
Previously, studies have reported that TNFα can contribute to COPD pathogenesis. Our data show a trend toward increased levels of TNFα in COPD patients (21.1–112.6 pg/mL) compared with in controls (21.1–34.1 pg/mL), but the values are not statistically different (Table S1). This could be a result of a lower level of TNFα in stable COPD patients or because of lower statistical power to detect the change in this cytokine. Regulatory network analysis showed overlapping connectivity among eotaxin, HGF, IL-6, and SCF via C/EBP-β, NF-κβ, and GCR-α transcription factors. In particular, GCR-α is involved in the regulation of all four of these key cytokines in stable COPD patients. The involvement of these cytokines (eotaxin, HGF, IL-6 and SCF) in various pathways and networks indicates their potential regulatory roles in COPD.
CTACK belongs to the C-C motif chemokines family, which plays an important role in the immune–inflammatory processes in many skin diseases.21 CTACK is constitutively produced by epidermal keratinocytes and participates in tissue-specific homing of lymphocytes.30 Increased level of CTACK present in COPD participants compared with controls (Figure 1A) suggests the involvement of CTACK in inflammatory processes related to the disease. In a study involving 48 patients, Pinto-Plata et al have also noted that CTACK levels are elevated in COPD subjects by protein microarray analysis.31 Our study further shows that CTACK negatively affects peak VO2 (Table 2). Therefore, including CTACK in future studies would be useful to confirm the relevance of this marker in other COPD populations.
Eotaxin is a potent chemoattractant for human eosinophils and basophils.22 Our study shows that serum level of eotaxin in stable COPD patients is elevated (Figure 1B). The COPD patients with eosinophil phenotypes show good response to corticosteroids.32 D’Armiento et al have reported that eotaxin levels in plasma and bronchioalveolar lavage can predict rapid deterioration in lung function (FEV1) in COPD patients.33 Similarly, our study shows an inverse relationship between serum eotaxin levels and FEV1 (percentage predicted), as well as peak VO2 in the COPD subjects of Indian descent (Figure 3A and Table 2). Furthermore, smoking is positively correlated with eotaxin, suggesting that smoking potentially contributes to the increase of this cytokine. A previous study reports that smoking increases eotaxin levels in asthmatic subjects,34 but the differences in eotaxin levels are not clearly established in COPD subjects. Pathway enrichment analysis further shows that transcription factors NF-κβ and GCR-α upregulate this cytokine production. Therefore, eotaxin is an important cytokine in COPD.
HGF is a multifunctional heterodimeric protein with mitogenic properties.23 HGF is synthesized by fibroblasts, macrophages, smooth muscle cells, and epithelial cells, and it is an important cytokine for lung development and repair.35–37 HGF concentration is higher in COPD patients compared with in control subjects (Figure 1C). Smoking is negatively correlated with HGF (Figure 3C and Table 2), which could likely affect the repair process. Notably, the human HGF gene has an IL-6 response element, and therefore, HGF is expected to be upregulated during inflammation.38 Collectively increased HGF identified in these patients may reflect the attempt by the host to repair the damaged lung.
IL-6 is a classical marker for inflammation and is associated with many inflammatory disorders.24 An increased concentration of IL-6 detected in stable COPD patients (Figure 1D) is consistent with previous studies showing the increased level of this cytokine in the plasma of COPD patients.39 Monocytes of COPD patients are known to overreact and release more IL-6 than cells from normal subjects.40 IL-6 is negatively related to FEV1 (percentage predicted; Figure 3A and Table 2). Therefore, IL-6 is an important cytokine that could affect systemic inflammation and worsen COPD comorbidity.4
IL-16 is a proinflammatory biomarker released by bronchial epithelium and dendritic cells, CD8+ and CD4+ T cells. It acts as a key element in COPD pathobiology.25 IL-16 secreted by CD8+ cells is involved in regulating the recruitment and activity of CD4+ cells. The present study shows an increase in serum level of IL-16 in the COPD subjects (Figure 1E), reflecting its potential involvement in the inflammatory process of the studied patients. Pinto-Plata et al have studied the serum biomarkers related to inflammation and injury in COPD patients and observed that the levels of the inflammatory markers, including IL-6 and IL-16, were higher in more advanced disease.41 The present finding is consistent with the involvement of these proinflammatory cytokines in the regulation of the disease process.
SCF is a hematopoietic factor.26 Various stroma and inflammatory cells express this factor. SCF stimulates mast cells and induces these cells to adhere to extracellular matrix, and lead to the production of proinflammatory cytokines and chemokines in the tissue.42,43 For the first time, we have identified that COPD patients have higher levels of SCF than control subjects (Figure 1F). This finding suggests that the SCF could directly remodel airway tissues in these patients. Dolgachev et al have shown that SCF, along with IL-31, is involved in remodeling and fibrosis of airways by promoting recruitment and differentiation of bone marrow-derived fibroblast precursors in mice sensitized and chronically challenged with allergens.44 In patients with diffused interstitial lung fibrosis, alveolar fibroblasts secrete high levels of SCF.45 COPD is also characterized by fibrosis and remodeling of airways; hence, SCF is likely to be a key factor involved in permanently altering lung architecture in COPD.
CXCL1, or GRO-α is a chemotactic factor produced by a variety of cells including monocytes, endothelial cells, and fibroblasts. Monocytes and neutrophils from patients with COPD show enhanced chemotactic response to CXCL1.27,28 Induced sputum of COPD patients has increased levels of CXCL1.46 CXCL1 level shows negative correlation with HGF (Figure 3B) and positively correlates with IL-6 and dyspnea scores. The identified key cytokines/biomarkers may be involved in the regulation of the pathophysiology of the disease.
The emerging field of network and pathways analysis can help identify the relationships between systemic inflammatory biomarkers and disease state.47,48 The common distinct functional and regulatory pathways identified in the present study are TH cell differentiation, TREM1 signaling, prostaglandin E2 signaling, Th17-derived cytokines, inflammation (mediated by the JAK-STAT pathway, histamine signaling), and leukocyte chemotaxis (Figure 4). One study shows that prostaglandin E2 is important for the mucosal innate immunity of COPD patients.49 As evident from Figure 5, the network comprises three main seeding nodes (NF-κβ, C/EBP-β and GCR-α), which regulate inflammation and immune regulation. Many of the differentially expressed cytokines are regulated by any of these three nodes, and of them, IL-6, HGF, eotaxin and SCF are significantly higher in COPD subjects. A fuller understanding of these processes and pathways will provide a better panel of COPD serum biomarkers and help us better understand the disease.
One of the limitations of the study is the use of a relatively small sample size. Although 29 stable COPD patients who were selected with stringent exclusion criteria helped us identify key cytokines that are sufficient to explain most of the variability in FEV1 and certain other parameters, and to identify pathways and networks, the cohort is not large enough to identify all of the potential biomarkers. This patient cohort is also not large enough to test comorbidities and to fully determine the effect of smoking on COPD biomarkers. Further studies with large sample size are necessary to validate these biomarkers, and molecular/cell biology-based studies are necessary to determine the mechanisms regulated by these cytokines in COPD.
Conclusion
In the present study, six biomarkers (CTACK, eotaxin, HGF, IL-6, IL-16 and SCF) are significantly elevated in the sera of COPD patients compared with healthy controls. Multiple regression analysis shows that eotaxin, IL-6, and SCF are inversely correlated with FEV1. CTACK and eotaxin are negatively related to peak VO2. Smoking may increase eotaxin and decrease HGF. Investigated enriched pathways and networks suggest involvement of inflammatory JAK-STAT and histamine signaling, immune response medicated by TREM1 and prostaglandin E2 signaling and the regulation of lymphocyte proliferation in the progression and pathogenesis of COPD. Notably, the four cytokines IL-6, HGF, eotaxin and SCF that are higher in COPD showed overlapping interactions with the seeding nodes. The involvement of these cytokines in various pathways and network analyses indicates their potential regulatory roles in the disease process.
Acknowledgment
The authors would like to thank all of the volunteers who took part in this study. The authors are also grateful to Dr Debabrata Ghosh, Department of Physiology, All India Institute of Medical Sciences, New Delhi, for his Molecular Biology laboratory facility support. This work was funded by the All India Institute of Medical Sciences, New Delhi, India (AT). MAK received a postdoctoral fellowship support from the CIHR operating grant MOP-111012 to NP.
Author contributions
The study was conceived and designed by AT, RG, and GB. The experiments were performed by GB, PK, AKS and KKS. The data analysis, pathways analysis, interpretations and manuscript compiling were done by AT, GB, MAK and NP. All authors read and approved the final manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3(11):e442. | |
Pallasaho P, Kainu A, Sovijärvi A, Lindqvist A, Piirilä PL. Combined effect of smoking and occupational exposure to dusts, gases or fumes on the incidence of COPD. COPD. 2014;11(1):88–95. | |
Moermans C, Heinen V, Nguyen M, et al. Local and systemic cellular inflammation and cytokine release in chronic obstructive pulmonary disease. Cytokine. 2011;56(2):298–304. | |
Barnes PJ, Celli BR. Systemic manifestations and comorbidities of COPD. Eur Respir J. 2009;33(5):1165–1185. | |
Agustí A. Systemic effects of chronic obstructive pulmonary disease: what we know and what we don’t know (but should). Proc Am Thorac Soc. 2007;4(7):522–525. | |
Aggarwal AN, Gupta D, Agarwal R, Jindal SK. Comparison of the lower confidence limit to the fixed-percentage method for assessing airway obstruction in routine clinical practice. Respir Care. 2011;56(11):1778–1784. | |
Nishimura K, Izumi T, Tsukino M, Oga T. Dyspnea is a better predictor of 5-year survival than airway obstruction in patients with COPD. Chest. 2002;121(5):1434–1440. | |
Aaron SD, Vandemheen KL, Ramsay T, et al. Multi analyte profiling and variability of inflammatory markers in blood and induced sputum in patients with stable COPD. Respir Res. 2010;11:41. | |
Franciosi LG, Page CP, Celli BR, et al. Markers of disease severity in chronic obstructive pulmonary disease. Pulm Pharmacol Ther. 2006;19(3):189–199. | |
Lu Y, Feng L, Feng L, Nyunt MS, Yap KB, Ng TP. Systemic inflammation, depression and obstructive pulmonary function: a population-based study. Respir Res. 2013;14:53. | |
Barnes PJ. The cytokine network in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2009;41(6):631–638. | |
Lee JS, Rosengart MR, Kondragunta V, et al. Inverse association of plasma IL-13 and inflammatory chemokines with lung function impairment in stable COPD: a cross-sectional cohort study. Respir Res. 2007;8:64. | |
Esteban C, Quintana JM, Egurrola M, et al. Classifying the severity of COPD: are the new severity scales better than the old? Int J Tuberc Lung Dis. 2009;13(6):783–790. | |
Miller MR, Hankinson J, Brusasco V, et al; ATS/ERS Task Force. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–338. | |
Cao C, Wang R, Wang J, Bunjhoo H, Xu Y, Xiong W. Body mass index and mortality in chronic obstructive pulmonary disease: a meta-analysis. PLoS One. 2012;7(8):e43892. | |
Dal Corso S, de Camargo AA, Izbicki M, Malaguti C, Nery LE. A symptom-limited incremental step test determines maximum physiological responses in patients with chronic obstructive pulmonary disease. Respir Med. 2013;107(12):1993–1999. | |
Rabe KF. Improving dyspnea in chronic obstructive pulmonary disease: optimal treatment strategies. Proc Am Thorac Soc. 2006;3(3):270–275. | |
Ferris BG. Epidemiology Standardization Project (American Thoracic Society). Am Rev Respir Dis. 1978;118(6 Pt 2):1–120. | |
Barnes PJ. Mediators of chronic obstructive pulmonary disease. Pharmacol Rev. 2004;56(4):515–548. | |
de Jager W, Rijkers GT. Solid-phase and bead-based cytokine immunoassay: a comparison. Methods. 2006;38(4):294–303. | |
Hayakawa I, Hasegawa M, Matsushita T, et al. Increased cutaneous T-cell-attracting chemokine levels in sera from patients with systemic sclerosis. Rheumatology (Oxford). 2005;44(7):873–878. | |
Menzies-Gow A, Ying S, Sabroe I, et al. Eotaxin (CCL11) and eotaxin-2 (CCL24) induce recruitment of eosinophils, basophils, neutrophils, and macrophages as well as features of early- and late-phase allergic reactions following cutaneous injection in human atopic and nonatopic volunteers. J Immunol. 2002;169(5):2712–2718. | |
Zhang YW, Su Y, Volpert OV, Vande Woude GF. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc Natl Acad Sci U S A. 2003;100(22):12718–12723. | |
Rincon M. Interleukin-6: from an inflammatory marker to a target for inflammatory diseases. Trends Immunol. 2012;33(11):571–577. | |
Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360(23):2445–2454. | |
Al-Muhsen SZ, Shablovsky G, Olivenstein R, Mazer B, Hamid Q. The expression of stem cell factor and c-kit receptor in human asthmatic airways. Clin Exp Allergy. 2004;34(6):911–916. | |
Traves SL, Culpitt SV, Russell RE, Barnes PJ, Donnelly LE. Increased levels of the chemokines GROalpha and MCP-1 in sputum samples from patients with COPD. Thorax. 2002;57(7):590–595. | |
Di Stefano A, Caramori G, Gnemmi I, et al. Association of increased CCL5 and CXCL7 chemokine expression with neutrophil activation in severe stable COPD. Thorax. 2009;64(11):968–975. | |
Hennighausen L, Robinson GW. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev. 2008;22(6):711–721. | |
Kagami S, Sugaya M, Minatani Y, et al. Elevated serum CTACK/CCL27 levels in CTCL. J Invest Dermatol. 2006;126(5):1189–1191. | |
Pinto-Plata V, Toso J, Lee K, et al. Profiling serum biomarkers in patients with COPD: associations with clinical parameters. Thorax. 2007;62(7):595–601. | |
Barbu C, Iordache M, Man MG. Inflammation in COPD: pathogenesis, local and systemic effects. Rom J Morphol Embryol. 2011;52(1):21–27. | |
D’Armiento JM, Scharf SM, Roth MD, et al. Eosinophil and T cell markers predict functional decline in COPD patients. Respir Res. 2009;10:113. | |
Krisiukeniene A, Babusyte A, Stravinskaite K, Lotvall J, Sakalauskas R, Sitkauskiene B. Smoking affects eotaxin levels in asthma patients. J Asthma. 2009;46(5):470–476. | |
Crestani B, Marchand-Adam S, Quesnel C, et al. Hepatocyte growth factor and lung fibrosis. Proc Am Thorac Soc. 2012;9(3):158–163. | |
Ware LB, Matthay MA. Keratinocyte and hepatocyte growth factors in the lung: roles in lung development, inflammation, and repair. Am J Physiol Lung Cell Mol Physiol. 2002;282(5):L924–L940. | |
Mason RJ. Hepatocyte growth factor: the key to alveolar septation? Am J Respir Cell Mol Biol. 2002;26(5):517–520. | |
Miyazawa K, Kitamura A, Kitamura N. Structural organization and the transcription initiation site of the human hepatocyte growth factor gene. Biochemistry. 1991;30(38):9170–9176. | |
Debigaré R, Marquis K, Côté CH, et al. Catabolic/anabolic balance and muscle wasting in patients with COPD. Chest. 2003;124(1):83–89. | |
Aldonyte R, Jansson L, Piitulainen E, Janciauskiene S. Circulating monocytes from healthy individuals and COPD patients. Respir Res. 2003;4:11. | |
Pinto-Plata V, Casanova C, Müllerova H, et al. Inflammatory and repair serum biomarker pattern: association to clinical outcomes in COPD. Respir Res. 2012;13:71. | |
Berlin AA, Lincoln P, Tomkinson A, Lukacs NW. Inhibition of stem cell factor reduces pulmonary cytokine levels during allergic airway responses. Clin Exp Immunol. 2004;136(1):15–20. | |
Gagari E, Rand MK, Tayari L, et al. Expression of stem cell factor and its receptor, c-kit, in human oral mesenchymal cells. Eur J Oral Sci. 2006;114(5):409–415. | |
Dolgachev VA, Ullenbruch MR, Lukacs NW, Phan SH. Role of stem cell factor and bone marrow-derived fibroblasts in airway remodeling. Am J Pathol. 2009;174(2):390–400. | |
Ding L, Dolgachev V, Wu Z, et al. Essential role of stem cell factor-c-Kit signalling pathway in bleomycin-induced pulmonary fibrosis. J Pathol. 2013;230(2):205–214. | |
Costa C, Rufino R, Traves SL, et al. CXCR3 and CCR5 chemokines in induced sputum from patients with COPD. Chest. 2008;133(1):26–33. | |
Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 2008;118(11):3546–3556. | |
Agustí A, Edwards LD, Rennard SI, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS One. 2012;7(5): e37483. | |
Zhang W, Case S, Bowler RP, Martin RJ, Jiang D, Chu HW. Cigarette smoke modulates PGE(2) and host defence against Moraxella catarrhalis infection in human airway epithelial cells. Respirology. 2011;16(3):508–516. |
Supplementary materials
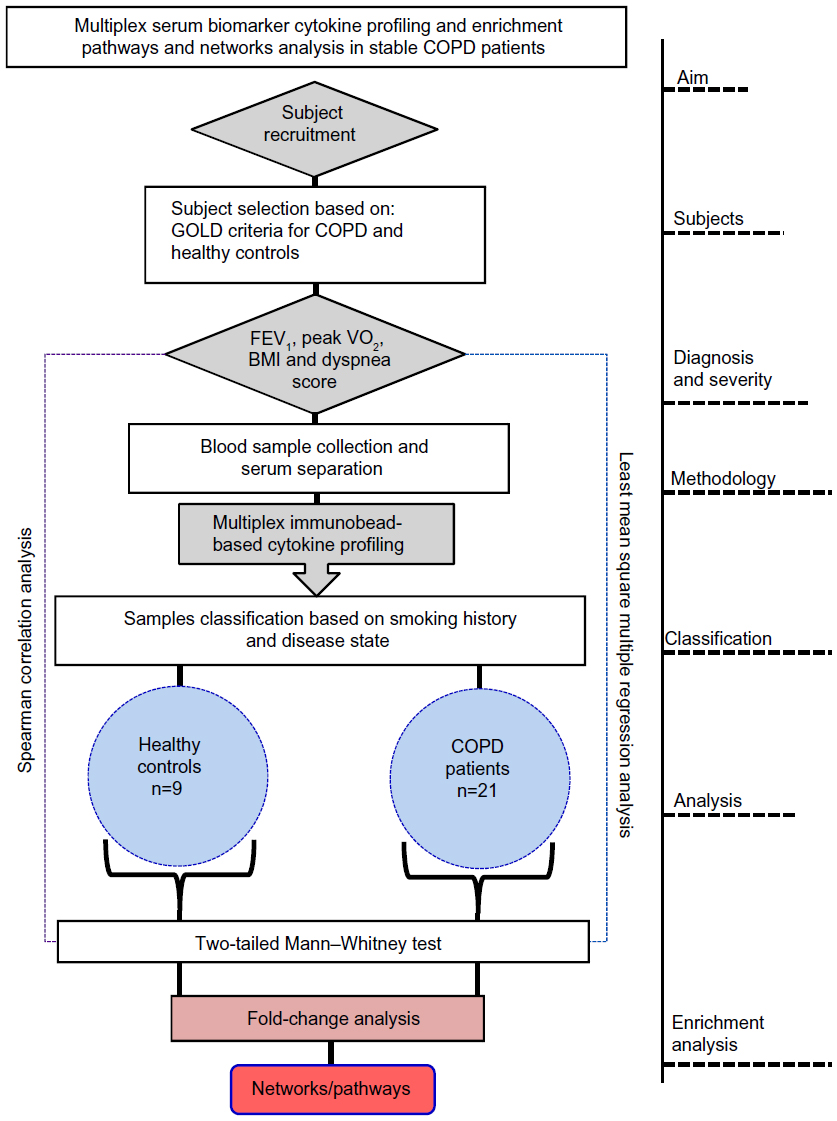 | Figure S1 Flow diagram of the experimental design showing overall aim and analysis steps involved in the present study. |
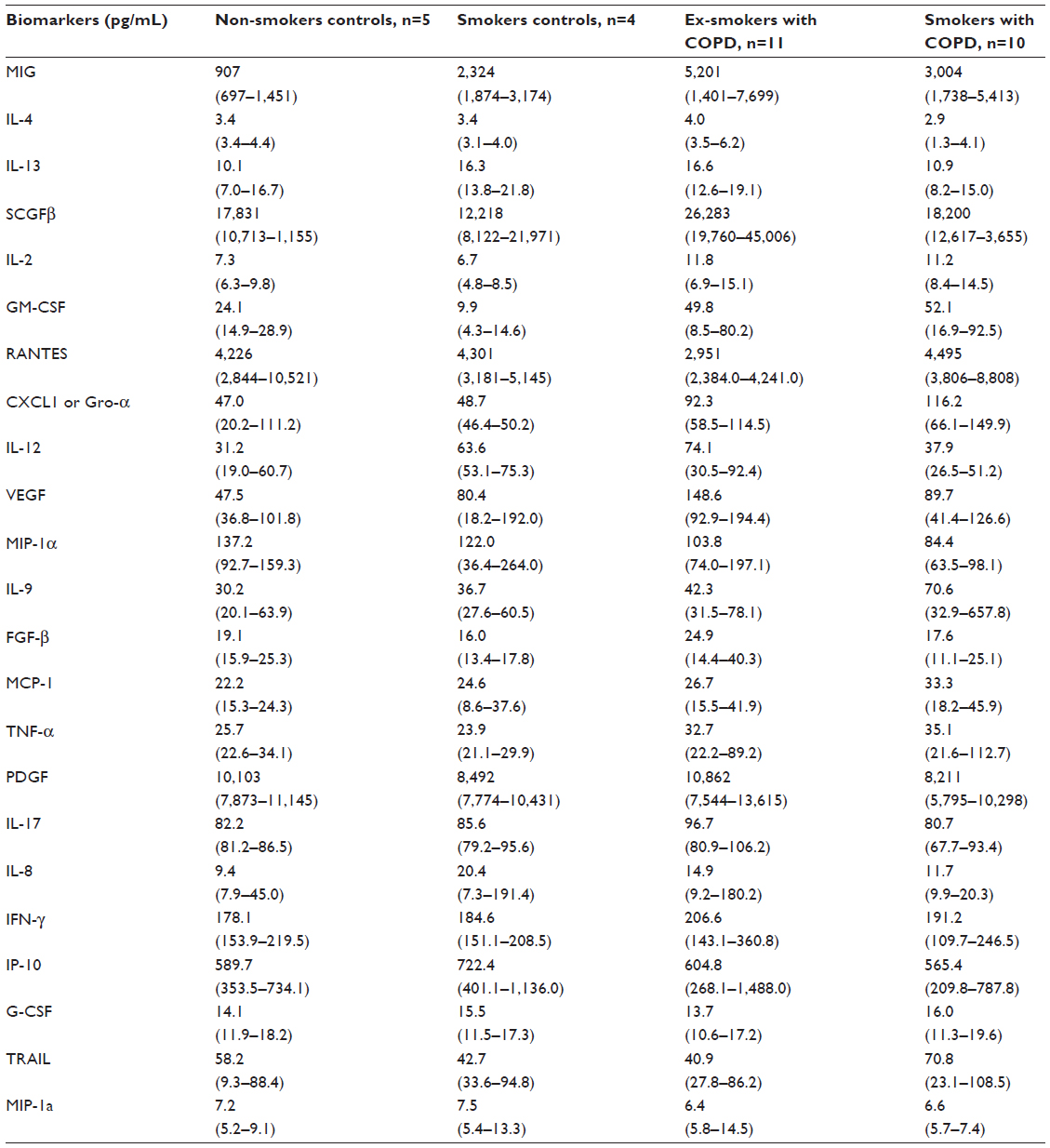 | Table S1 Concentrations (pg/mL) of biomarkers detected in the sera samples among groups1* |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.