Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
Serum Amyloid A in Stable COPD Patients is Associated with the Frequent Exacerbator Phenotype
Authors Zhao D ![]() , Abbasi A, Rossiter HB
, Abbasi A, Rossiter HB ![]() , Su X
, Su X ![]() , Liu H, Pi Y, Sang L, Zhong W, Yang Q, Guo X, Zhou Y
, Liu H, Pi Y, Sang L, Zhong W, Yang Q, Guo X, Zhou Y ![]() , Li T
, Li T ![]() , Casaburi R
, Casaburi R ![]() , Zhang N
, Zhang N ![]()
Received 9 June 2020
Accepted for publication 31 August 2020
Published 30 September 2020 Volume 2020:15 Pages 2379—2388
DOI https://doi.org/10.2147/COPD.S266844
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Dongxing Zhao1,2 ,* Asghar Abbasi2 ,* Harry B Rossiter,2,3 Xiaofen Su,1 Heng Liu,1 Yuhong Pi,1 Li Sang,1 Weiyong Zhong,1 Qifeng Yang,1 Xiongtian Guo,1 Yanyan Zhou,1 Tianyang Li,1 Richard Casaburi,2 Nuofu Zhang1
1State Key Laboratory of Respiratory Disease, National Clinical Research Center for Respiratory Disease, Guangzhou Institute of Respiratory Health, The First Affiliated Hospital of Guangzhou Medical University, Guangzhou, Guangdong 510120, People’s Republic of China; 2Rehabilitation Clinical Trials Center, The Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Center, Torrance, CA, 90502, USA; 3Faculty of Biological Sciences, University of Leeds, Leeds LS2 9JT, UK
*These authors contributed equally to this work
Correspondence: Nuofu Zhang
State Key Laboratory of Respiratory Disease, National Clinical Research Center for Respiratory Disease, Guangzhou Institute of Respiratory Health, The First Affiliated Hospital of Guangzhou Medical University, Guangzhou, Guangdong 510120, People’s Republic of China
Email [email protected]
Background: We sought to determine whether circulating inflammatory biomarkers were associated with the frequent exacerbator phenotype in stable COPD patients ie, those with two or more exacerbations in the previous year.
Methods: Eighty-eight stable, severe, COPD patients (4 females) were assessed for exacerbation frequency, pulmonary function, fraction of expired nitric oxide (FENO); inflammatory variables were measured in venous blood. Logistic regression assessed associations between the frequent exacerbator phenotype and systemic inflammation.
Results: Compared with infrequent exacerbators, frequent exacerbators (n=10; 11.4%) had greater serum concentration (median (25th-75th quartile)) of serum amyloid A (SAA; 134 (84– 178) vs 71 (38– 116) ng/mL; P=0.024), surfactant protein D (SP-D; 15.6 (9.0– 19.3) vs 8.5 (3.6– 14.9) ng/mL; P=0.049) and interleukin-4 (IL-4; 0.12 (0.08– 1.44) vs 0.03 (0.01– 0.10) pg/mL; P=0.001). SAA, SP-D and IL-4 were not significantly correlated with FEV1%predicted or FVC %predicted. After adjusting for sex, age, BMI, FEV1/FVC and smoking pack-years, only SAA remained independently associated with the frequent exacerbator phenotype (OR 1.49[1.09– 2.04]; P=0.012). The odds of being a frequent exacerbator was 18-times greater in the highest SAA quartile (≥ 124.1 ng/mL) than the lowest SAA quartile (≤ 44.1 ng/mL) (OR 18.34[1.30– 258.81]; P=0.031), and there was a significant positive trend of increasing OR with increasing SAA quartile (P=0.008). For SAA, the area under the receiver operating characteristic curve was 0.721 for identification of frequent exacerbators; an SAA cut-off of 87.0 ng/mL yielded an 80% sensitivity and 61.5% specificity.
Conclusion: In stable COPD patients, SAA was independently associated with the frequent exacerbator phenotype, suggesting that SAA may be a useful serum biomarker to inform progression or management in COPD.
Keywords: inflammation, surfactant protein D, interleukin-4
Background
Chronic obstructive pulmonary disease (COPD) is characterized by both an accelerated decline in lung function and periods of acute deterioration in symptoms, termed exacerbations. Exacerbations of respiratory symptoms in COPD are of major importance in terms of their prolonged detrimental effects on patients.1 Frequent exacerbations contribute to long-term decline in lung function as well as to increased morbidity, decreased quality of life, and increased mortality of patients with moderate to severe COPD.2–4 Some patients experience frequent exacerbations, and form a distinct clinical subgroup, the ‘frequent exacerbator’ phenotype, in contrast to patients who experience few exacerbations over time.5 The frequent exacerbator phenotype is recognized as a major phenotype in patients with COPD and occurs across disease severities.5
Exacerbations are typically caused by respiratory tract infections.6 During an acute exacerbation episode, the existing airway and systemic inflammation increase further.7–9 Inflammatory mediators, cytokines and chemokines are released into the circulation and induce recruitment of inflammatory cells into the lung parenchyma. Besides local damage to lung structures, activation of immune cells causes systemic inflammation with increases in acute phase proteins such as fibrinogen and C-reactive protein (CRP).7,10 The airways of frequent exacerbators are more inflamed than those with infrequent exacerbations, and frequent exacerbators have greater concentration of sputum interleukin-6 (IL-6) and −8 (IL-8).11 In addition, during the exacerbation recovery period, sputum IL-6 and serum CRP are greater in the frequent exacerbator phenotype,12 consistent with their greater baseline inflammatory state. In the post-exacerbation recovery period, greater serum concentration of inflammatory mediators, such as CRP, are associated with a shorter time until the next exacerbation.12 As a result, CRP has been proposed as a predictive biomarker for COPD exacerbation.13
Similar to CRP, serum amyloid A (SAA) is an acute phase inflammatory biomarker, and is strongly associated with COPD exacerbations.14 Bozinovski et al14 studied stable and exacerbating COPD patients, and found that SAA was four-fold greater in patients experiencing a severe acute COPD exacerbation. They also identified that the increase in SAA was a more sensitive indicator of acute COPD exacerbation than CRP alone or in combination with dyspnea.14
SAA is a family of proteins that form a major component of the acute-phase inflammatory response and, like CRP, is synthesized mainly in the liver in response to infection, inflammation, injury, or stress.15 Considering that SAA is a sensitive marker of an acute inflammatory state, we aimed to determine the association between the frequent exacerbator phenotype of COPD and SAA concentration in the stable state of COPD. We hypothesized that greater SAA is associated with the frequent exacerbator phenotype in COPD and that the relationship is stronger than for other markers of inflammation.
Methods
Patient Population
This cross-sectional study was conducted at the First Affiliated Hospital of Guangzhou Medical University. Patients with severe to very severe COPD (GOLD [Global Initiative for Chronic Obstructive Lung Disease] spirometry stages 3–4),16 were recruited during their stable state. Exclusion criteria included a COPD exacerbation within the previous 4 weeks, lung cancer, bronchiectasis, pneumoconiosis or other restrictive ventilatory dysfunction and chronic systemic inflammatory conditions. Information collected included medical and smoking history, exacerbation frequency, pulmonary function and fraction of expired nitric oxide (FENO). Venous blood was collected and serum was assessed for inflammatory variables. The study protocol was approved by the Medical Ethics Review Committee of the First Affiliated Hospital of Guangzhou Medical University and all subjects provided written informed consent. The study was conducted in accordance with the principles of the Declaration of Helsinki, except for prospective public trial registration.
Subgroups
Moderate and severe exacerbations were captured by a review of medical records. Moderate exacerbations were defined as an acute worsening of respiratory symptoms treated by antibiotics and/or oral steroids and severe exacerbations as a worsening of respiratory symptoms requiring hospitalization or emergency room visit.16 Frequent exacerbations were defined as two or more exacerbations in the previous year.16,17 Subjects were divided into two groups: 1) “frequent exacerbators”, who experienced two or more moderate or severe exacerbations in the 12 months prior to study enrollment; 2) “infrequent exacerbators”, who had one or no exacerbations in the 12 months prior to study enrollment.
Blood Sampling and Measurement of Inflammatory Markers
Venous blood samples were collected from subjects by venipuncture in plain tubes. The samples were centrifuged immediately at 2500 rpm for 15 minutes at room temperature and the serum fraction was collected and frozen at −80°C pending subsequent procedures. The concentration of SAA (R&D, #DY3019-05), SP-D (R&D, #DSFPD0), PRG4 (SAB, #EK2481), LTB4 (Abcam, #ab133040), MMP-8 (R&D, #DMP800), MMP-9 (Bioscience, #85-BMS2016-2) were measured in the serum by an enzyme-linked immunosorbent assay (ELISA). The concentration of GM-CSF, IL-1β, IL-4, IL-6 and IL-8 were measured using human cytokine V-PLEX validated immunoassay kit (Meso Scale Discovery, Gaithersburg, MD). Blood cell counts were made using a hematological analyzer (DxH 800, Beckman Coulter, California, United States). All the measurements were performed in duplicate. The average of each variable is reported.
Pulmonary Function Testing
Spirometry was performed by using a portable spirometer (Carefusion MasterScreen Pneumo, Germany) before, and 20 minutes after, inhalation of 400μg of short-acting β2-agonist (albuterol) in accordance with the ERS/ATS recommendations.18 Spirometric data were reviewed and graded by a quality control software (Sentrysuite V.2.3). Predicted values were calculated according to the use of the reference values from the European Coal and Steel Community (1993) and the application of conversion factors for Chinese adults (male: 0.95, female: 0.93).19
Fractional Exhaled Nitric Oxide (FENO)
FENO was measured by using the NIOX system (NioxMino® ENO analyzer, Aerocrine, Sweden) in accordance with ATS/ERS recommendations.20 FENO was measured before spirometry and measured three times, with differences ≤10% indicating acceptable variance. The mean value of three measurements is reported.
Statistical Analysis
Data were expressed as mean ± standard deviation (SD) or median (25th-75th quartiles) or as percentage. Univariate analyses included independent-samples Student’s t or Mann–Whitney test for continuous variables or chi-square test or Fisher’s exact test for categorical variables between two groups, as appropriate. SAA concentration was divided in quartiles. Relationships among frequent exacerbator phenotype (dependent variable) and clinical data, spirometric values, inflammatory variables and SAA (square root (sqrt) transformed) (independent variables) were assessed by logistic regression. A multivariable logistic regression model was adjusted for age, sex, smoking pack-years, body mass index and FEV1/FVC. The trend for linearity between the odds ratio (OR) and SAA quartiles was estimated by logistic regression including the median of each SAA quartile as a continuous variable. The optimal SAA cut-off point (highest sum of sensitivity and specificity) to identify frequent exacerbators was selected using receiver operating characteristic (ROC) analysis. Correlations between inflammatory variables and pulmonary function were assessed by Spearman correlation. All statistical analyses were performed using SPSS (IBM SPSS V.25.0, Armonk, NY, USA). Statistical significance was set at P<0.05. Figures were created using GraphPad Software (GraphPad Prism V.8.0 for Windows, San Diego, California USA).
Results
Patient Characteristics
A total of 88 patients were enrolled. Patient characteristics are shown in Table 1. Overall, the group was predominantly male (96%), aged 64±7 years with severe or very severe COPD (FEV1 38±9%predicted). Ten (11.4%) patients were frequent exacerbators (3 (2–4) exacerbations in the previous 12 months), while 78 (88.6%) were infrequent exacerbators (0 (0–1) exacerbations in the previous 12 months). Frequent exacerbators had a significantly greater CAT score (13 (9–16) vs 9 (6–12); P=0.028), greater mMRC dyspnea scale (2 (1–3) vs 1 (1–2); P=0.001), a lower post-bronchodilator (BD) FVC (2.01±0.40 vs 2.66±0.60 L; P=0.001), and lower post-BD FVC %predicted (58.3±10.9 vs 75.9±15.8%predicted; P=0.001). There was no significant difference between the two groups in age, sex, BMI, current smoker status, smoking pack-years, post-BD FEV1, post-BD FEV1%predicted or post-BD FEV1/FVC. There was no significant difference between the two groups in total white cell count, or count or percentage of neutrophils and eosinophils (Table 1).
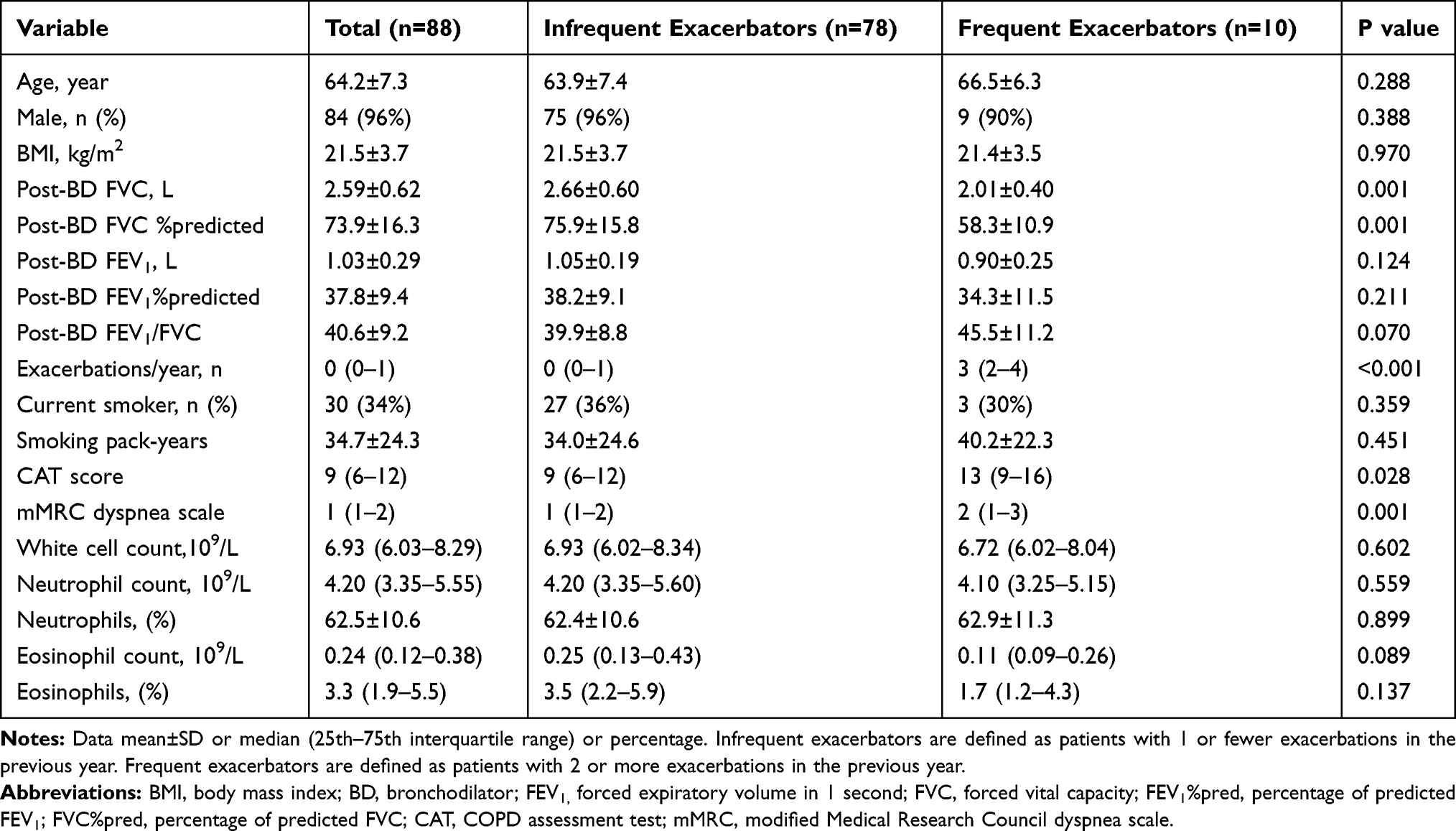 |
Table 1 Patient Characteristics |
Inflammatory Variables in Groups Based on Exacerbation Frequency
SAA was significantly greater in frequent exacerbators compared to infrequent exacerbators (P=0.024) (Table 2; Figure 1A). Similarly, SP-D (P=0.049; Figure 1B) and IL-4 (P=0.001; Figure 1C) concentrations were significantly greater in frequent exacerbators compared to infrequent exacerbators (Table 2). There was no significant difference between the two groups in other inflammatory variables including IL-1β, IL-6, IL-8, CRP, LTB4, GM-CSF, FENO, MMP8, MMP9 and PRG4 (Table 2).
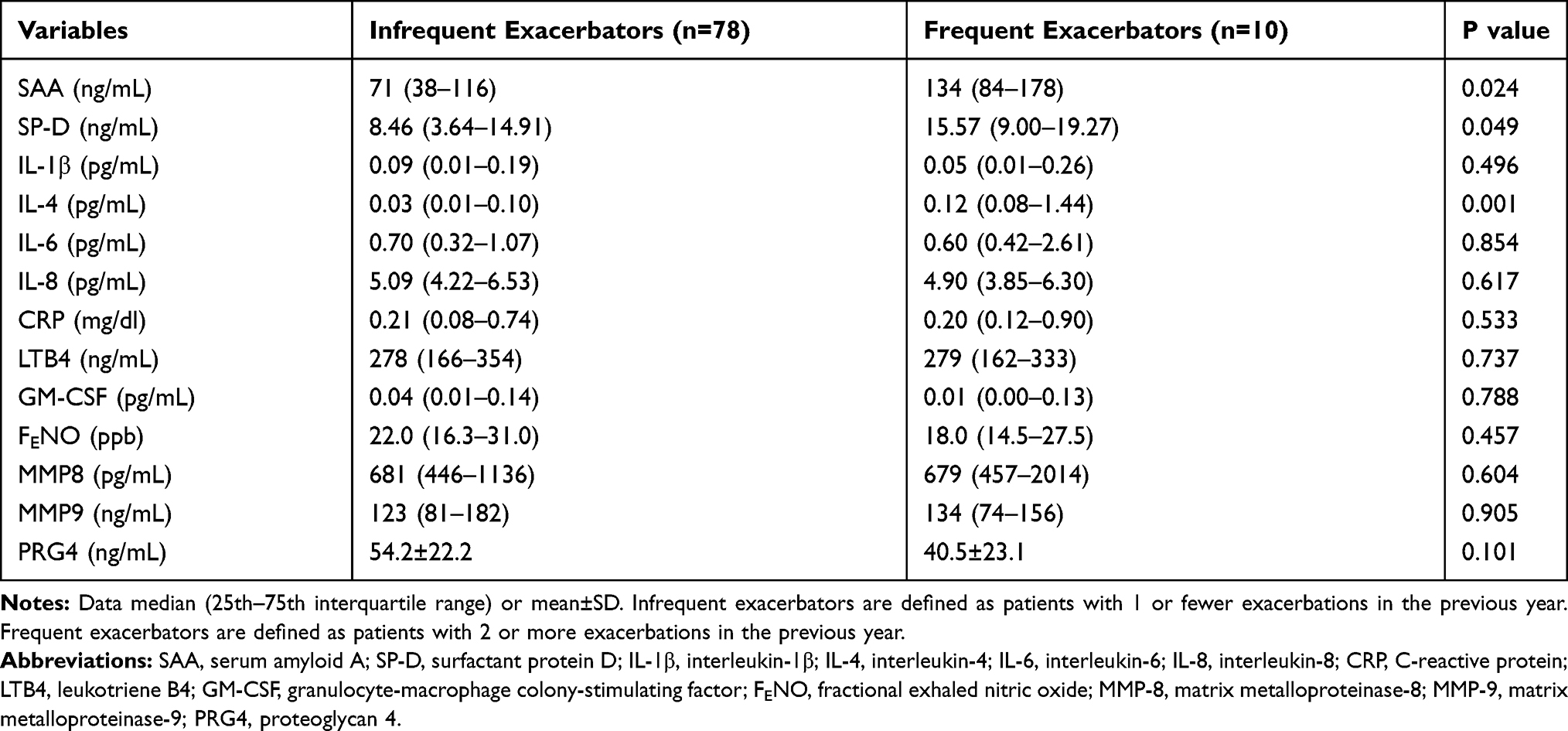 |
Table 2 Inflammatory Variables in Groups Based on Exacerbation Frequency |
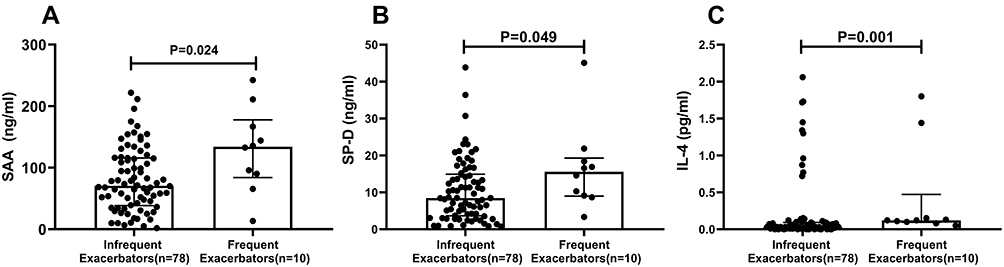 |
Figure 1 Comparison of SAA, SP-D and IL-4 concentration between frequent exacerbators and infrequent exacerbators. Frequent exacerbators (n=10) are defined as patients with 2 or more exacerbations in the previous year. Infrequent exacerbators (n=78) are defined as patients with 1 or none exacerbations in the previous year. (A) SAA concentration. (B) SP-D concentration. (C) IL-4 concentration. Statistical comparisons by Mann–Whitney test. |
Correlations Between SAA, SP-D, IL-4 and Pulmonary Function
Table 3 shows Spearman correlation between SAA, SP-D, IL-4 and spirometric measures of pulmonary function. For these three inflammatory markers, there was no significant correlation with post-BD FVC %predicted, post-BD FEV1%predicted, and post-BD FEV1/FVC (Table 3).
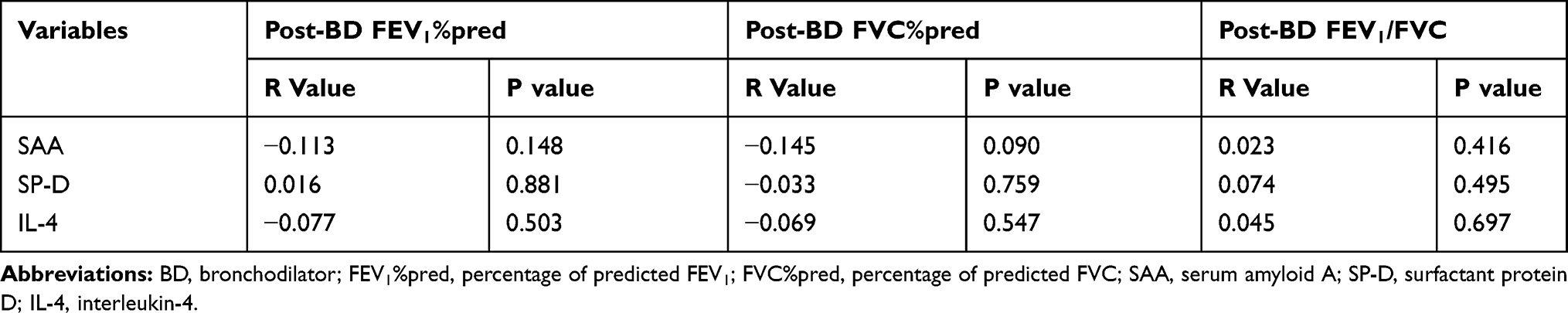 |
Table 3 The Correlation Between SAA, SP-D, IL-4 and Pulmonary Function |
Association Between SAA and Frequent Exacerbations
In the univariate logistic regression models, SAA was significantly associated with frequent exacerbator status (OR 1.33; 95% CI 1.03–1.70; P=0.027). SP-D, IL-4 and other inflammatory variables including IL-1β, IL-6, IL-8, CRP, LTB4, GM-CSF, FENO, MMP8, MMP9, PRG4 were not significantly associated with frequent exacerbator status (Figure 2A). In the multivariable logistic regression model, adjusting for sex, age, BMI, Post-BD FEV1/FVC and smoking pack-years, SAA was independently associated with the frequent exacerbator phenotype (OR 1.49; 95% CI 1.09–2.04; P=0.012) (Figure 2B).
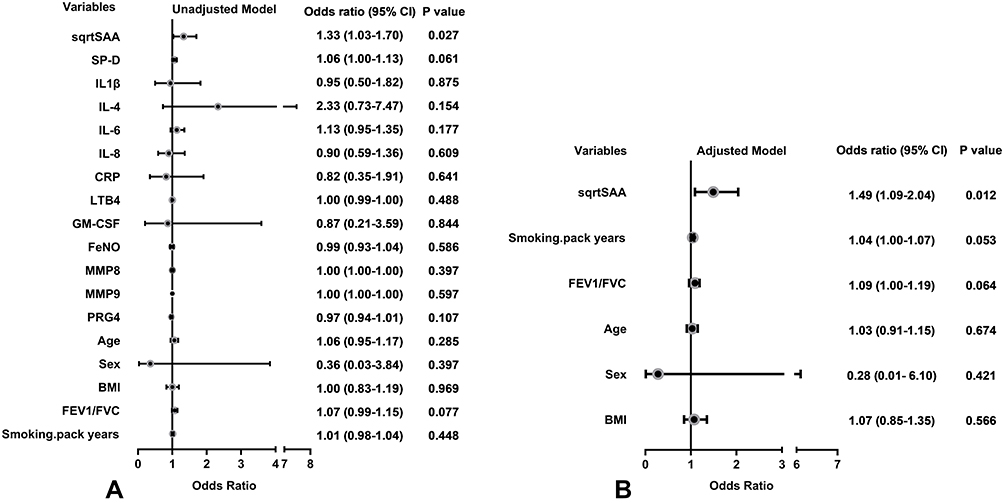 |
Figure 2 Odds ratio for variables associated with the frequent exacerbator phenotype. (A) Univariate logistic regression models. (B) Multivariable logistic regression model adjusting for sex, age, BMI, FEV1/FVC and smoking pack-years. SAA was square root (sqrt) transformed to normalize its distribution. |
Associations Between SAA Quartiles and Frequent Exacerbations
The odds ratio for the association of frequent exacerbator status with quartiles of SAA concentration is presented in Figure 3. After adjusting for sex, age, BMI, Post-BD FEV1/FVC and smoking pack-years, the hazard of frequent exacerbator status in the highest SAA quartile (≥124.1 ng/mL) was 18 times that of the lowest SAA quartile (≤44.1 ng/mL) (OR 18.34; 95% CI 1.30–258.81; P=0.031). In categorical analysis, we found that a significant linear association between the OR for frequent exacerbator status and increasing SAA quartile (P value for trend=0.008) (Figure 3).
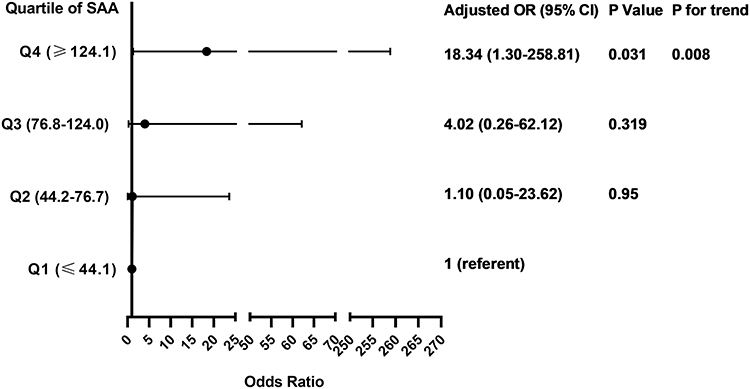 |
Figure 3 Associations between SAA quartiles and the frequent exacerbator phenotype. Multivariable logistic regression model adjusting for sex, age, BMI, FEV1/FVC and smoking pack-years. SAA concentration was divided in quartiles: Q1, ≤44.1 ng/mL; Q2, 44.2–76.7 ng/mL; Q3, 76.8–124.0 ng/mL; Q4, ≤124.1 ng/mL. Linear association between OR and SAA quartile was assessed by P for trend using logistic regression model including the median of each SAA quartile as a continuous variable. |
Sensitivity and Specificity of SAA to Identify the Frequent Exacerbator Phenotype
In ROC analysis, yielded an area under the curve (AUC) of 0.721 (95% CI 0.542–0.899) for the identification of frequent exacerbators. The greatest sum of sensitivity and specificity (1.421) was an SAA of 131.7 ng/mL, corresponding to a sensitivity of 60.0% and specificity of 82.1% (Figure 4). However, choosing to increase test sensitivity (at least 80.0%) did not meaningfully reduce the sum (1.415) and resulted in an SAA of 87.0 ng/mL corresponding to a sensitivity of 80.0% and specificity of 61.5%.
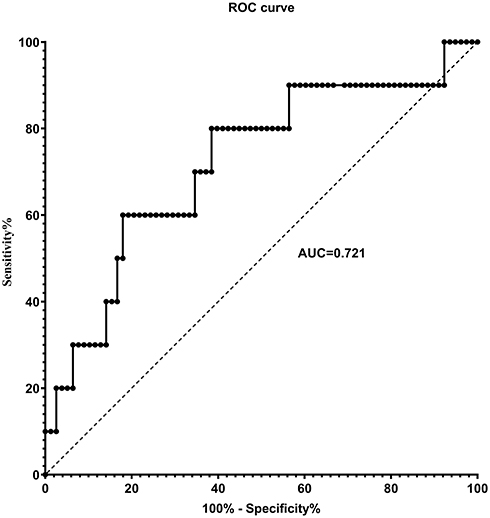 |
Figure 4 Receiver operating characteristic (ROC) for identification of the frequent exacerbator phenotype by serum amyloid A (SAA). ROC analysis yielded an area under the curve (AUC) of 0.721 (95% CI 0.542–0.899) for the identification of frequent exacerbators. The greatest sum of sensitivity and specificity occurred at SAA = 131.7 ng/mL. Selection of a higher specificity (at least 80%) did not meaningfully reduce the sum and resulted in SAA = 87.0 ng/mL to separate frequent and infrequent exacerbators, corresponding to a sensitivity of 80.0% and specificity of 61.5%. |
Discussion
The principal finding of this study was that the serum concentrations of SAA, SP-D and IL-4 were greater in COPD patients who experienced 2 or more exacerbations in the preceding year compared with those who did not. In addition, SAA, but not the other inflammatory biomarkers, was significantly associated with the frequent exacerbator phenotype after adjusting for potential covariates and was independent of spirometric measures of pulmonary impairment and total eosinophil counts or percentage. We also found that the risk of frequent exacerbator status in the highest SAA quartile was 18 times of those in the lowest quartile, suggesting that SAA might be a good indicator of the frequent exacerbator phenotype in COPD. ROC analysis identified SAA = 87.0 ng/mL as providing 80% sensitivity to separate infrequent from frequent exacerbators. Although the inflammatory biomarker SAA is known to be raised during an acute COPD exacerbation,14 this is the first study to our knowledge to demonstrate an association of SAA with the frequent exacerbator phenotype in stable phase of COPD. This observational study cohort contained ~10% frequent exacerbators, which is reflective of the incidence expected in clinical practice. Further research is needed to validate these findings in an independent cohort. Nevertheless, these data show that SAA may be a useful and sensitive biomarker to identify patients at risk of frequent exacerbations, to inform progression and to guide management of COPD.
Investigators have long sought a circulating biomarker that separates non-exacerbators from exacerbators. In the ECLIPSE trial, 2138 COPD patients were followed for three years to determine predictors of exacerbations. None of the measured circulating biomarkers were independent predictors of exacerbation frequency. More recently, Gulati and colleagues reported a greater concentration of fibroblast growth factor 23 (FGF23) in frequent exacerbators compared with patients without frequent exacerbations.21 In addition, these investigators showed that FGF23 is independently associated with frequent exacerbations, after adjusting for age, lung function, smoking and oxygen use.21
Previous studies showed that SAA is dramatically increased during acute phase of a COPD exacerbation, and that SAA is a sensitive biomarker for exacerbation severity in COPD patients.14 Other studies showed increased circulating SAA concentration in COPD and other pulmonary diseases such as idiopathic pulmonary fibrosis (IPF).22–24 Formiga et al reported that inspiratory muscle performance was considerably lower in COPD patients with greater SAA concentration.23 In addition, SAA was greater in patients with IPF than in healthy controls.22 Furthermore, serum levels of SAA and CRP together with other inflammatory markers (eg, IL-6, IL-8, TNF-a, IP-10) were significantly greater in COPD patients experiencing an exacerbation than during remission and in healthy control subjects.24
Elevated SAA was reported in patients with cardiovascular disease where COPD was a comorbidity.25 Since half of the deaths in COPD are associated with cardiovascular events, and the incidence of these events increases dramatically after each exacerbation,26,27 and also because SAA is a good predictor of coronary artery disease as well as future cardiovascular events, SAA might be considered as a potential indicator of frequent exacerbation phenotype as well as an indicator of comorbid cardiovascular disease.
As well as acting as a biomarker for the frequent exacerbator phenotype during the stable phase of COPD, evidence suggests that SAA may mediate an acute COPD exacerbation.14 SAA induces lung inflammation in COPD by promoting lung neutrophilia, which is activated by increasing expression of IL-17a in γδT cells.28 In addition, SAA activates the NLRP3 inflammasome and elicits robust TLR2-, MyD88-, and IL-1-dependent pulmonary inflammation.29 Consistent with this, SAA is found in the lungs and bronchoalveolar lavage fluid of smoke exposed COPD patients and endotoxin-challenged mice.30,31
In stable COPD patients, we also showed that the circulating concentration of SP-D was greater in frequent exacerbators compared to infrequent exacerbators, although SP-D was not independently associated with exacerbation. Although no previous study compared the serum concentration of SP-D between frequent and infrequent exacerbators, the ECLIPSE study showed that SP-D significantly, although weakly, predicted exacerbation during the first year of follow-up.17 SP-D is mainly produced by type II alveolar cells in the lung,32 and is involved in regulating pulmonary surfactants, lipid homeostasis and innate immunity in order to protect the lungs from pathogens.33–35 SP-D has many protective properties including anti-inflammatory and anti-oxidant functions34 that associate with protection against the development of COPD.36 SP-D knockout mice have large lungs with enlarged airspaces and activated macrophages; they develop progressive pulmonary emphysema and subpleural fibrosis in association with chronic inflammation.37 In this study, we did not have chest-CT or diffusing capacity measures to identify whether the greater SP-D in frequent exacerbators was associated with greater pulmonary emphysema, although this might be expected.
A major role of SP-D is to regulate antibody-independent immune responses against invading microorganisms.38 Indeed, SP-D deficient mice are more susceptible to respiratory infections with Pneumocystis carinii, influenza, respiratory syncytial virus, and bacteria.39–42 This is consistent with the observation that COPD patients with serum SP-D concentration greater than the 95th percentile of nonsmokers had an increased risk of exacerbation over the following 12 months.43
Our results showed no significant correlation between serum SP-D and spirometric lung function (FEV1%pred, FVC%pred, FEV1/FVC), which is consistent with several other studies.43 The interpretation of association of serum SP-D and lung function is complicated in that serum SP-D is affected not only by lung synthesis, but also by increased leakage of SP-D from the COPD lung into the systemic circulation.36 Nevertheless, in our study, SP-D was not as strongly (or independently) associated with the frequent exacerbator phenotype as SAA.
We also found serum IL-4 was significantly greater in frequent exacerbators, but regression analysis showed no significant independent correlation between IL-4 and exacerbator phenotype in our stable COPD patients. IL-4 is secreted by Th-2 lymphocytes which stimulate the proliferation of B lymphocytes to produce IgG and IgE, and to mediate humoral immunization.44 To our knowledge, this is the first study showing greater concentration of IL-4 in frequent than infrequent exacerbators. Previous studies showed that serum IL-4 is greater in COPD patients during an acute exacerbation compared with stable COPD and control groups.45 Additionally, IL-4 concentration was dramatically increased in Mycoplasma pneumoniae-induced airway diseases, including those occurring in COPD patients.46 These data suggest that Th2 secreted cytokines (eg, IL-4) might play an important role in reducing inflammation. Based on this rationale, greater IL-4 in frequent exacerbators compared with infrequent exacerbators might indicate a protective role of Th2 lymphocytes and their secreted cytokines in balancing immune system function during acute infection and exacerbation. On the other hand, increased concentration of IL-4 due to hyper-activated Th2 cells might increase the concentration of IgE immunoglobulin which leads to airway hyper-responsiveness in exacerbating COPD patients.45 In this regard, the greater IL-4 in frequent exacerbators could reflect increased eosinophilic inflammation, although we found no difference in eosinophilia between frequent and infrequent exacerbators. We also did not find a correlation between IL-4 concentration and spirometric pulmonary function.
This study has limitations. We enrolled fewer frequent exacerbators than infrequent exacerbators. This might affect our ability to detect differences between the two groups. In addition, our study suffers from gender imbalance. The majority of our subjects were men (96%), limiting our ability to assert whether our findings also apply to women with COPD. This observational study was not large enough to provide separate discovery and validation sub-groups. Nevertheless, that we were able to identify the utility of SAA to identify the frequent exacerbator phenotype with a small sample size, suggests that these findings are worth exploring in a larger cohort to validate our findings. In order to increase the clinical utility of SAA to identify frequent exacerbators, we chose to prioritize sensitivity over specificity in identifying an SAA cut-off. Nevertheless, this choice resulted in summed sensitivity and specificity (1.415) that was not meaningfully different from the maximum value (1.421), where specificity was 80%.
Conclusions
This study demonstrated that serum SAA, SP-D and IL-4 concentration were greater in stable COPD patients who had 2 or more exacerbations in the previous year compared with those who experienced 1 or none. Further, SAA was significantly and independently correlated with the frequent exacerbator phenotype but not with spirometric indices of lung function. SAA should be considered as a potential biomarker of the frequent exacerbator phenotype in COPD. These findings provide new insights into the phenotyping COPD and may be useful to inform management of COPD patients.
Abbreviations
BMI, body mass index; BD, bronchodilator; CAT, COPD assessment test; CRP, C-reactive protein; FENO, fractional exhaled nitric oxide; FEV1, forced expiratory volume in one second; FEV1%pred, percentage of predicted FEV1; FVC, forced vital capacity; FVC1%pred, percentage of predicted FVC; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL-1β, interleukin-1β; IL-4, interleukin-4; IL-6, interleukin-6; IL-8, interleukin-8; LTB4, leukotriene B4; MMP-8, matrix metalloproteinases-8; MMP-9, matrix metalloproteinases-9; mMRC, modified Medical Research Council dyspnea scale; NLRP3, NLR family pyrin domain containing 3; PRG4, proteoglycan 4; SAA, serum amyloid A; SP-D, surfactant protein D.
Data Sharing Statement
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
The clinical study was approved by the Medical Ethics Review Committee of the First Affiliated Hospital of Guangzhou Medical University, Guangzhou, China. The reference number for the study is 2016-20. Informed consent about the study procedures were signed and obtained from all the subjects before the study.
Consent for Publication
Not applicable.
Acknowledgments
Results of this study have been presented at the American Thoracic Society 2020 annual conference. The authors would like to thank the investigators and patients at the investigative sites for their support of this study.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This work was funded unconditionally by National Key R&D Program of China (2018YFC1313600 & 2016YFC0901102); Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01S155). The sponsors have no role in design, conduct, data interpretation of the study, and preparation, review or approval of this manuscript.
Disclosure
Professor Richard Casaburi reports grants, personal fees from GlaxoSmithKline, Boehringer Ingelheim, Astra Zeneca, Regeneron, and Genentech, outside the submitted work. The authors declare that they have no other competing interests.
References
1. Celli BR, Barnes PJ. Exacerbations of chronic obstructive pulmonary disease. European Respir J. 2007;29(6):1224–1238. doi:10.1183/09031936.00109906
2. Patel IS, Seemungal TAR, Wilks M, Lloyd-Owen SJ, Donaldson GC, Wedzicha JA. Relationship between bacterial colonisation and the frequency, character, and severity of COPD exacerbations. Thorax. 2002;57(9):759–764. doi:10.1136/thorax.57.9.759
3. Seemungal TAR, Donaldson GC, Paul EA, Bestall JC, Jeffries DJ, Wedzicha JA. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157(5):1418–1422. doi:10.1164/ajrccm.157.5.9709032
4. Seneff MG, Wagner DP, Wagner RP, Zimmerman JE, Knaus WA. Hospital and 1-year survival of patients admitted to intensive care units with acute exacerbation of chronic obstructive pulmonary disease. JAMA. 1995;274(23):1852–1857. doi:10.1001/jama.1995.03530230038027
5. Wedzicha JA, Rabe KF, Martinez FJ, et al. Efficacy of roflumilast in the COPD frequent exacerbator phenotype. Chest. 2013;143(5):1302–1311. doi:10.1378/chest.12-1489
6. Sethi S, Wrona C, Eschberger K, Lobbins P, Cai X, Murphy TF. Inflammatory profile of new bacterial strain exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177(5):491–497. doi:10.1164/rccm.200708-1234OC
7. Gompertz S, O’Brien C, Bayley DL, Hill SL, Stockley RA. Changes in bronchial inflammation during acute exacerbations of chronic bronchitis. European Respir J. 2001;17(6):1112–1119. doi:10.1183/09031936.01.99114901
8. Crooks SW, Bayley DL, Hill SL, Stockley RA. Bronchial inflammation in acute bacterial exacerbations of chronic bronchitis: the role of leukotriene B4. European Respir J. 2000;15(2):274–280. doi:10.1034/j.1399-3003.2000.15b09.x
9. Wedzicha JA, Seemungal TAR, MacCallum PK, et al. Acute exacerbations of chronic obstructive pulmonary disease are accompanied by elevations of plasma fibrinogen and serum IL-6 levels. Thromb Haemost. 2000;84(08):210–215. doi:10.1055/s-0037-1613998
10. Dev D, Wallace E, Sankaran R, Cunniffe J, Emmanuel FXS. Value of C-reactive protein measurements in exacerbations of chronic obstructive pulmonary disease. Respir Med. 1998;92(4):664–667. doi:10.1016/S0954-6111(98)90515-7
11. Bhowmik A, Seemungal TA, Sapsford RJ, Wedzicha JA. Relation of sputum inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax. 2000;55(2):114. doi:10.1136/thorax.55.2.114
12. Perera WR, Hurst JR, Wilkinson TMA, et al. Inflammatory changes, recovery and recurrence at COPD exacerbation. European Respir J. 2007;29(3):527. doi:10.1183/09031936.00092506
13. Hurst JR, Donaldson GC, Perera WR, et al. Use of plasma biomarkers at exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;174(8):867–874. doi:10.1164/rccm.200604-506OC
14. Bozinovski S, Hutchinson A, Thompson M, et al. Serum amyloid a is a biomarker of acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177(3):269–278. doi:10.1164/rccm.200705-678OC
15. Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute‐phase reactant. European j Biochem. 1999;265(2):501–523. doi:10.1046/j.1432-1327.1999.00657.x
16. Global Strategy for the Diagnosis, Management and Prevention of Chronic Obstructive Lung Disease 2020 Report. Global Initiative for Chronic Obstructive Lung Disease (GOLD). Available from: http://www.goldcopd.org.
17. Hurst JR, Vestbo J, Anzueto A, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med. 2010;363(12):1128–1138. doi:10.1056/NEJMoa0909883
18. Pellegrino R, Viegi G, Brusasco V, et al. Interpretative strategies for lung function tests. Eur Respir J. 2005;26(5):948–968. doi:10.1183/09031936.05.00035205
19. Zheng J, Zhong N. Normative values of pulmonary function testing in Chinese adults. Chin Med J. 2002;115(1):50–54.
20. ATS/ERS recommendations for standardized procedures for the online and offline measurement of exhaled lower respiratory nitric oxide and nasal nitric oxide. Am J Respir Crit Care Med. 2005;171(8):912–930. doi:10.1164/rccm.200406-710ST
21. Gulati S, Wells JM, Urdaneta GP, et al. Fibroblast growth factor 23 is associated with a frequent Exacerbator phenotype in COPD: a cross-sectional pilot study. Int J Mol Sci. 2019;20(9):2292. doi:10.3390/ijms20092292
22. Vietri L, Bennett D, Cameli P, et al. Serum amyloid A in patients with idiopathic pulmonary fibrosis. Respir Investig. 2019;57(5):430–434. doi:10.1016/j.resinv.2019.03.010
23. Formiga MF, Vital I, Urdaneta G, et al. Higher serum levels of systemic inflammatory markers are linked to greater inspiratory muscle dysfunction in COPD. Clin Respir J. 2019;13(4):247–255. doi:10.1111/crj.13006
24. Lin TL, Chen WW, Ding ZR, Wei SC, Huang ML, Li CH. Correlations between serum amyloid A, C-reactive protein and clinical indices of patients with acutely exacerbated chronic obstructive pulmonary disease. J Clin Lab Anal. 2019;33(4):e22831. doi:10.1002/jcla.22831
25. Johnson BD, Kip KE, Marroquin OC, et al. Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women: the national heart, lung, and blood institute-sponsored Women’s Ischemia Syndrome Evaluation (WISE). Circulation. 2004;109(6):726–732. doi:10.1161/01.CIR.0000115516.54550.B1
26. Sin DD, Man SF. Why are patients with chronic obstructive pulmonary disease at increased risk of cardiovascular diseases? The potential role of systemic inflammation in chronic obstructive pulmonary disease. Circulation. 2003;107(11):1514–1519. doi:10.1161/01.CIR.0000056767.69054.B3
27. Rodriguez-Roisin R. Is this the beginning of unravelling the puzzle of COPD exacerbations? Eur Respir J. 2005;26(3):376–378. doi:10.1183/09031936.05.00076205
28. Anthony D, Seow HJ, Uddin M, et al. Serum amyloid A promotes lung neutrophilia by increasing IL-17A levels in the mucosa and gammadelta T cells. Am J Respir Crit Care Med. 2013;188(2):179–186. doi:10.1164/rccm.201211-2139OC
29. Ather JL, Ckless K, Martin R, et al. Serum amyloid A activates the NLRP3 inflammasome and promotes Th17 allergic asthma in mice. J Immunol. 2011;187(1):64–73. doi:10.4049/jimmunol.1100500
30. Meng QR, Gideon KM, Harbo SJ, et al. Gene expression profiling in lung tissues from mice exposed to cigarette smoke, lipopolysaccharide, or smoke plus lipopolysaccharide by inhalation. Inhal Toxicol. 2006;18(8):555–568. doi:10.1080/08958370600686226
31. Merkel D, Rist W, Seither P, Weith A, Lenter MC. Proteomic study of human bronchoalveolar lavage fluids from smokers with chronic obstructive pulmonary disease by combining surface-enhanced laser desorption/ionization-mass spectrometry profiling with mass spectrometric protein identification. Proteomics. 2005;5(11):2972–2980. doi:10.1002/pmic.200401180
32. Voorhout WF, Veenendaal T, Kuroki Y, Ogasawara Y, van Golde LM, Geuze HJ. Immunocytochemical localization of surfactant protein D (SP-D) in type II cells, Clara cells, and alveolar macrophages of rat lung. J Histochem Cytochem. 1992;40(10):1589–1597. doi:10.1177/40.10.1527377
33. Fisher JH, Sheftelyevich V, Ho YS, et al. Pulmonary-specific expression of SP-D corrects pulmonary lipid accumulation in SP-D gene-targeted mice. Am J Physiol Lung Cell Mol Physiol. 2000;278(2):L365–373. doi:10.1152/ajplung.2000.278.2.L365
34. Sin DD, Pahlavan PS, Man SFP. Surfactant protein D: a lung specific biomarker in COPD? Ther Adv Respir Dis. 2008;2(2):65–74. doi:10.1177/1753465808088903
35. Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol. 2005;5(1):58–68. doi:10.1038/nri1528
36. Obeidat M, Li X, Burgess S, et al. Surfactant protein D is a causal risk factor for COPD: results of Mendelian randomisation. European Respir J. 2017;50(5):1700657. doi:10.1183/13993003.00657-2017
37. Wert SE, Yoshida M, LeVine AM, et al. Increased metalloproteinase activity, oxidant production, and emphysema in surfactant protein D gene-inactivated mice. Proc Natl Acad Sci U S A. 2000;97(11):5972–5977. doi:10.1073/pnas.100448997
38. Sørensen GL, Hjelmborg JVB, Kyvik KO, Fenger M, Holmskov U. Genetic and environmental influences of surfactant protein D serum levels. Am J Physiol Lung Cell Mol Physiol. 2006;290(5):L1010–1017. doi:10.1152/ajplung.00487.2005
39. Atochina EN, Gow AJ, Beck JM, et al. Delayed clearance of Pneumocystis carinii infection, increased inflammation, and altered nitric oxide metabolism in lungs of surfactant protein-D knockout mice. J Infect Dis. 2004;189(8):1528–1539.
40. LeVine AM, Whitsett JA, Hartshorn KL, Crouch EC, Korfhagen TR. Surfactant protein D enhances clearance of influenza A virus from the lung in vivo. J Immunol. 2001;167(10):5868–5873. doi:10.4049/jimmunol.167.10.5868
41. LeVine AM, Elliott J, Whitsett JA, et al. Surfactant protein-d enhances phagocytosis and pulmonary clearance of respiratory syncytial virus. Am J Respir Cell Mol Biol. 2004;31(2):193–199. doi:10.1165/rcmb.2003-0107OC
42. Wu H, Kuzmenko A, Wan S, et al. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J Clin Invest. 2003;111(10):1589–1602. doi:10.1172/JCI16889
43. Lomas DA, Silverman EK, Edwards LD, et al. Serum surfactant protein D is steroid sensitive and associated with exacerbations of COPD. Eur Respir J. 2009;34(1):95–102. doi:10.1183/09031936.00156508
44. van Oosterhout AJ, Motta AC. Th1/Th2 paradigm: not seeing the forest for the trees? Eur Respir J. 2005;25(4):591–593. doi:10.1183/09031936.05.00014105
45. Wei B, Sheng LC. Changes in Th1/Th2-producing cytokines during acute exacerbation chronic obstructive pulmonary disease. J Int Med Res. 2018;46(9):3890–3902. doi:10.1177/0300060518781642
46. Hao Y, Kuang Z, Jing J, et al. Mycoplasma pneumoniae modulates STAT3-STAT6/EGFR-FOXA2 signaling to induce overexpression of airway mucins. Infect Immun. 2014;82(12):5246–5255. doi:10.1128/IAI.01989-14
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.