Back to Journals » Drug Design, Development and Therapy » Volume 8
Sequential treatment with AT-101 enhances cisplatin chemosensitivity in human non-small cell lung cancer cells through inhibition of apurinic/apyrimidinic endonuclease 1-activated IL-6/STAT3 signaling pathway
Authors Ren T, Shan J, Qing Y, Qian C, Li Q, Liu G, Li M, Li C, Peng Y, Luo H, Zhang S, Zhang W, Wang D, Zhou S
Received 20 July 2014
Accepted for publication 2 September 2014
Published 12 December 2014 Volume 2014:8 Pages 2517—2529
DOI https://doi.org/10.2147/DDDT.S71432
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Tao Ren,1,2,* Jinlu Shan,1,* Yi Qing,1 Chengyuan Qian,1 Qing Li,1 Guoshou Lu,1 Mengxia Li,1 Chongyi Li,1 Yu Peng,1 Hao Luo,1 Shiheng Zhang,1 Weiwei Zhang,1 Dong Wang,1 Shu-Feng Zhou3
1Cancer Center, Daping Hospital and Research Institute of Surgery, Third Military Medical University, Chongqing, People’s Republic of China; 2Oncology Department, The Affiliated Hospital, North Sichuan Medical College, Nanchong, People’s Republic of China; 3Department of Pharmaceutical Sciences, College of Pharmacy, University of South Florida, Tampa, FL, USA
*These authors contributed equally to this work
Abstract: AT-101, known as R-(–)-gossypol, is a potent anticancer agent, but its chemosensitizing effects remain elusive. The present study aimed to examine whether AT-101 could increase the sensitivity of non-small cell lung cancer A549 cells to cisplatin (CDDP) and the underlying mechanisms. We evaluated the efficacy of the sequential treatment with AT-101 and CDDP using both in vitro and in vivo models. Our results showed that as compared to AT-101 or CDDP monotherapy, or AT-101 plus CDDP concurrent treatment, the sequential treatment significantly inhibited cell proliferation and migration and induced tumor cell death. Moreover, the efficacy of the sequential treatment was also confirmed in a mouse A549 xenograft model. Our study revealed that AT-101 inhibited the reduced status of apurinic/apyrimidinic endonuclease 1 (APE1) and attenuated APE1-mediated IL-6/STAT3 signaling activation by decreasing IL-6 protein expression; suppressing the STAT3–DNA binding; and reducing the expression of the downstream antiapoptotic proteins Bcl-2 and Bcl-xL. In conclusion, AT-101 enhances the sensitivity of A549 cells to CDDP in vitro and in vivo through the inhibition of APE1-mediated IL-6/STAT3 signaling activation, providing a rationale for the combined use of AT-101 and CDDP in non-small cell lung cancer chemotherapy.
Keywords: AT101, NSCLC, cisplatin, chemosensitivity, APE1, STAT3, nude mice, apoptosis
Introduction
Lung cancer is the first leading cause of cancer-related death in humans worldwide.1 Lung cancer was the most common cancer worldwide contributing 13% of the total number of new cases diagnosed in 2012. This disease killed 1.59 million patients in 2012. In the People’s Republic of China, lung cancer has become the leading cause of cancer-related death since the 1990s. The crude incidence rate for lung cancer in the People’s Republic of China was 53.57/100,000, accounting for 18.74% of overall new cancer cases diagnosed; and the crude mortality rate for lung cancer was 45.57/100,000, accounting for 25.24% of cancer-related deaths in 2009.2 An estimated 159,260 deaths come from lung cancer (86,930 in men and 72,330 among women) in the US, accounting for approximately 27% of all cancer deaths. Lung cancer includes two major types: small cell lung cancer and non-small cell lung cancer (NSCLC), with the latter accounting for 70%–85%. The primary treatment modalities for lung cancer are surgery, chemotherapy, radiotherapy, and biological therapy. Although current therapeutic strategies for the treatment of NSCLC have made some advancement by the platinum-based standard chemotherapy, the average 5-year survival rate is approximately 17% which has not been significantly improved over the last 40 years.3 Failure of chemotherapy in NSCLC is mainly due to multidrug resistance and dose-limiting adverse reactions. This highlights the urgent need for the discovery of novel therapeutic agents for NSCLC.
Cisplatin (CDDP) is a potent anticancer agent that has been frequently used in the treatment of a broad spectrum of malignancies, including NSCLC, ovarian cancer, and testicular cancer.4 However, development of resistance to CDDP is common during treatment of NSCLC, leading to low overall response rates to CDDP in patients with NSCLC. CDDP-induced adverse effects are dose-dependent and limit the administration of increased dosages, thus compromising its therapeutic efficacy.4 Therefore, for platinum-resistant NSCLC patients, research into new agents or their combinations with currently approved chemotherapeutic agents with different molecular targets is urgently warranted.
Growth factors and cytokines can activate the signal transducer and activator of transcription-3 (STAT3) signaling pathway, which is involved in cell proliferation, differentiation, angiogenesis, and survival.5,6 It has been reported that dysregulation of STAT3 signaling is associated with cancer initiation, growth, development, and metastasis. In particular, aberrant STAT3 signaling pathway contributes to chemoresistance development and enhanced tumor cell migration, and abrogation of STAT3 signaling increases CDDP sensitivity and induces apoptosis in tumor cells.7–10 Furthermore, interleukin-6 (IL-6), an upstream activator of the STAT3 signaling pathway, plays an important role in cancer development and chemoresistance.5,6,11 Thus, the IL-6/STAT3 signaling pathway is considered a potential target for the treatment of CDDP-resistant tumor cells.11 Human apurinic/apyrimidinic endonuclease 1 (APE1)/redox-factor-1 (Ref-1) plays a key role in the repair of oxidized and alkylated bases in mammalian genomes via the base excision repair mechanism.12–15 This important protein was also characterized as a redox activator of a number of additional transcription factors known to be involved in cancer cell signaling and survival, such as nuclear factor-κB, hypoxia-inducible factor-1α, p53, and other proteins. Homozygous deletion of the Ape1 gene in mice leads to embryonic death,16 but heterozygous mice survive and are fertile.17 APE1 is regulated at epigenetic, transcriptional, and posttranscriptional levels and itself can regulate the expression of several genes including STAT3. As a multifunctional protein, dysregulation of APE1 is associated with cancer initiation and development, angiogenesis, progression, and metastasis.12–14,18 Elevated levels of APE1/Ref-1 have been linked to resistance to chemotherapy, poor prognosis, and poor survival. In our recent clinical study, we have found that CDDP-resistant tumors from NSCLC patients had a significantly higher APE1 expression level than CDDP-sensitive tumors, and better overall survival and disease-free survival were noted in NSCLC patients with a low APE1 expression level.19 Inhibition of APE1 by siRNA in A549 cells enhanced the chemosensitivity to CDDP therapy.19
AT-101 (ie, R-(–)-gossypol acetic acid, see Figure 1), a natural BH3-mimetic molecule and pan-Bcl-2 inhibitor, has shown antitumor activity as a single agent and in combination with standard anticancer therapies in a variety of tumor models in mice.20–23 Previous studies have shown that the combination of AT-101 with CDDP treatment significantly inhibited the expression of apoptotic proteins including Bcl-2, BAX, and BAD, as well as regulated the activity of epigenetic proteins, such as DNA methyltransferase and histone deacetylases in ovarian cancer cells.24 This combination therapy overcomes chemoresistance by inducing apoptosis and modulating epigenetics in tumor cells. In addition, a Phase I study of AT-101 and CDDP/etoposide combination therapy in patients with extensive-stage small cell lung cancer also showed promising antitumor effects.25 Furthermore, our recent study has shown that gossypol effectively suppressed the dual-function of APE1.26 However, AT-101 monotherapy does not show remarkable efficacy in clinical trials,27,28 suggesting the necessity for combined use of AT-101 with standard chemotherapeutic agents such as CDDP.
|
Figure 1 Chemical structures of gossypol and R-(–)-gossypol (AT-101). |
Currently, the mechanisms for better efficacy by the combination therapy of AT-101 and standard chemotherapeutic drugs remain elusive. There is evidence that cellular DNA damage responses can be modulated by the pretreatment with some agents that enhance the sensitivity to DNA damaging agents.29 In the present study, we investigated whether the sequential administration of AT-101 and CDDP increased CDDP sensitivity in NSCLC A549 cells. We further explored the molecular mechanism related to AT-101-enhanced CDDP sensitivity.
Materials and methods
Cell culture
The human pulmonary adenocarcinoma cell line A549 was obtained from Cell Bank of the Chinese Academy of Sciences (Shanghai, People’s Republic of China) and grown in RPMI 1640 medium (Hyclone; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum. The cells were cultured in a humidified incubator at 37°C in a 5% CO2 atmosphere.
Cell proliferation assay
Cell proliferation was measured using the Cell Counting Kit-8 assay (Beyotime Inc., Haimen, Jiangsu, People’s Republic of China), according to the manufacturer’s instructions. Briefly, cells were plated at a density of 8,000 per well on 96-well plates overnight, and then treated with CDDP alone (Jiangsu Hanson Pharmaceutical Co. Ltd., Lianyungang, Jiangsu, People’s Republic of China) or combined with AT-101 (Selleckchem Inc., Shanghai, People’s Republic of China). After culturing for 48 hours, cell viability was quantified by reading the plates at an absorbance of 490 nm on a microplate reader.
Transwell migration assay
The ability of cells to migrate was determined by a Transwell (8 μm pore size; Costar Inc., Costar, NY, USA) assay as described previously.30 In brief, cell suspension was prepared in serum-free medium that contained AT-101, CDDP, or AT-101 plus CDDP (sequential treatment). The cell suspensions were added to the upper chambers, and the lower chambers were filled with RPMI 1640 with 10% fetal bovine serum. After 18 hours incubation at 37°C, cells were fixed using 4% paraformaldehyde solution and migration was visualized by crystal violet staining. The number of migrated cells was counted under an inverted microscope (CKX41; Olympus Corporation, Tokyo, Japan).
Flow cytometric analysis for apoptosis
The apoptosis of cells were determined using flow cytometry. In brief, cells were plated in 6-well plates for 24 hours and then treated with vehicle control (dimethyl sulfoxide [DMSO]), AT-101, CDDP, or AT-101 plus CDDP. After 15 hours’ treatment, cells were harvested and washed once with phosphate-buffered saline. Cell suspensions in 300 μL of phosphate-buffered saline were stained with 50 μL propidium iodide and 50 μL of Annexin V-FITC (BD, Franklin Lakes, NJ, USA) for 15 minutes in the dark at room temperature. Apoptotic cells were measured by flow cytometric analysis.
Enzyme-linked immunosorbent assay
Cells were cultured for 24 hours and then exposed to experimental agents or vehicle control. Supernatant, ie, the tumor-conditioned medium, was collected at indicated time points for IL-6 detection using a human IL-6 enzyme-linked immunosorbent assay (ELISA) kit (NeoBioscience, Shenzhen, People’s Republic of China) according to the manufacturer’s protocol. Tumor-conditioned medium samples were diluted with the sample dilution buffer at a 1:1 ratio. The absorbance was read at an absorbance of 490 nm on a microplate reader (Model 680; Bio-Rad Laboratories Inc., Hercules, CA, USA).
Western blot analysis
The levels of intracellular STAT3, APE1, Bcl-2, and Bcl-xL were determined by Western blot analysis. Briefly, harvested cells were lysed using lysis buffer. Equal amounts of protein was resolved on SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) and then transferred to a polyvinylidene fluoride membrane. The membrane was blocked with 10% milk for 1 hour and then incubated with primary antibody for 1 hour and then secondary antibody for another hour. The following antibodies were used for Western blot assay: anti-APE1 (1:5,000), anti-β-actin (1:5,000), anti-Bcl-2 (1:400), anti-Bcl-xL (1:200), and anti-STAT3 antibodies (1:250) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA).
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) was performed as described previously,31 with some modifications. The biotin-labeled double-stranded oligonucleotide DNA was used as probe that contains the STAT3 direct consensus sequence (5′-GATCCTTCTGGGAATTCCTAGATC-3′) (Beyotime Inc.). Nuclear extracts were prepared from A549 cell samples with different treatments, including DMSO control, 50 ng/mL IL-6 (Sino Biological Inc., Beijing, People’s Republic of China) for 2 hours, and 20 μM AT-101 for 6 hours. To assay APE1/STAT3 interaction, we first prepared a reduced APE1 solution (0.17 ng/μL) with 4 mM dithiothreitol (DTT) at ratio of 9:1 (purified APE1 protein versus DTT) for 10 minutes. Then, the reduced APE1 was added to nuclear extracts for redox reactions, in which the final concentration of DTT was 0.04 mM.
Mouse xenograft models
The experimental protocol in nude mice was approved by the Ethics Committee of the Third Military Medical University, Chongqin, People’s Republic of China. Briefly, 3- to 4-weeks-old BALB/c nude mice were assigned to four groups (n=6 each group). Cultured A549 cells at 90% confluence were harvested to prepare a cell suspension at a concentration of 1.5×106 cells/100 μL. Tumor cells were inoculated subcutaneously in the left anterior axilla of nude mice. When tumor volume reached 200 mm3, mice were treated with the following drugs: 1) vehicle control (sesame oil, China Oil Food Co. [COFCO], Beijing, People’s Republic of China) by oral gavage for 10 consecutive days; 2) AT-101 dissolved in sesame oil (35 mg/kg/day) by oral gavage for 10 consecutive days; 3) CDDP (4 mg/kg/day) by intraperitoneal injection on days 3, 5, 7, and 9; and 4) combination treatment – pretreated with AT-101 for 2 days, followed by administration of CDDP as above.
Statistical analysis
Statistical analysis was performed using the Student’s t-test or analysis of variance. The data are presented as mean ± standard deviation. P<0.05 was considered to be significant statistically.
Results
Sequential treatment with AT-101 and CDDP significantly inhibits cell proliferation and migration and induces cell death through suppression of Bcl-2 and Bcl-xL expression
AT-101 was shown to inhibit the proliferation and migration of various types of cancer cells.32–34 Therefore, we examined the effect of the sequential treatment with AT-101 and CDDP on the proliferation of A549 cells. A549 cells were treated with CDDP alone or pretreated with AT-101 for 6 hours, followed by a 48-hour combination treatment. Results showed that the viability of A549 cells was significantly inhibited by the sequential treatment (P<0.05) (Figure 2A). Half maximal inhibitory concentration value of the sequential treatment (1.15 μM) was much smaller than that of CDDP monotherapy (9.5 μM) in A549 cells. Antimigration capacity of the sequential treatment was also assessed by transwell migration assay. As shown in Figure 2B and C, the number of migrated cells was significantly reduced in the combination group, as compared with other groups (P<0.0001).
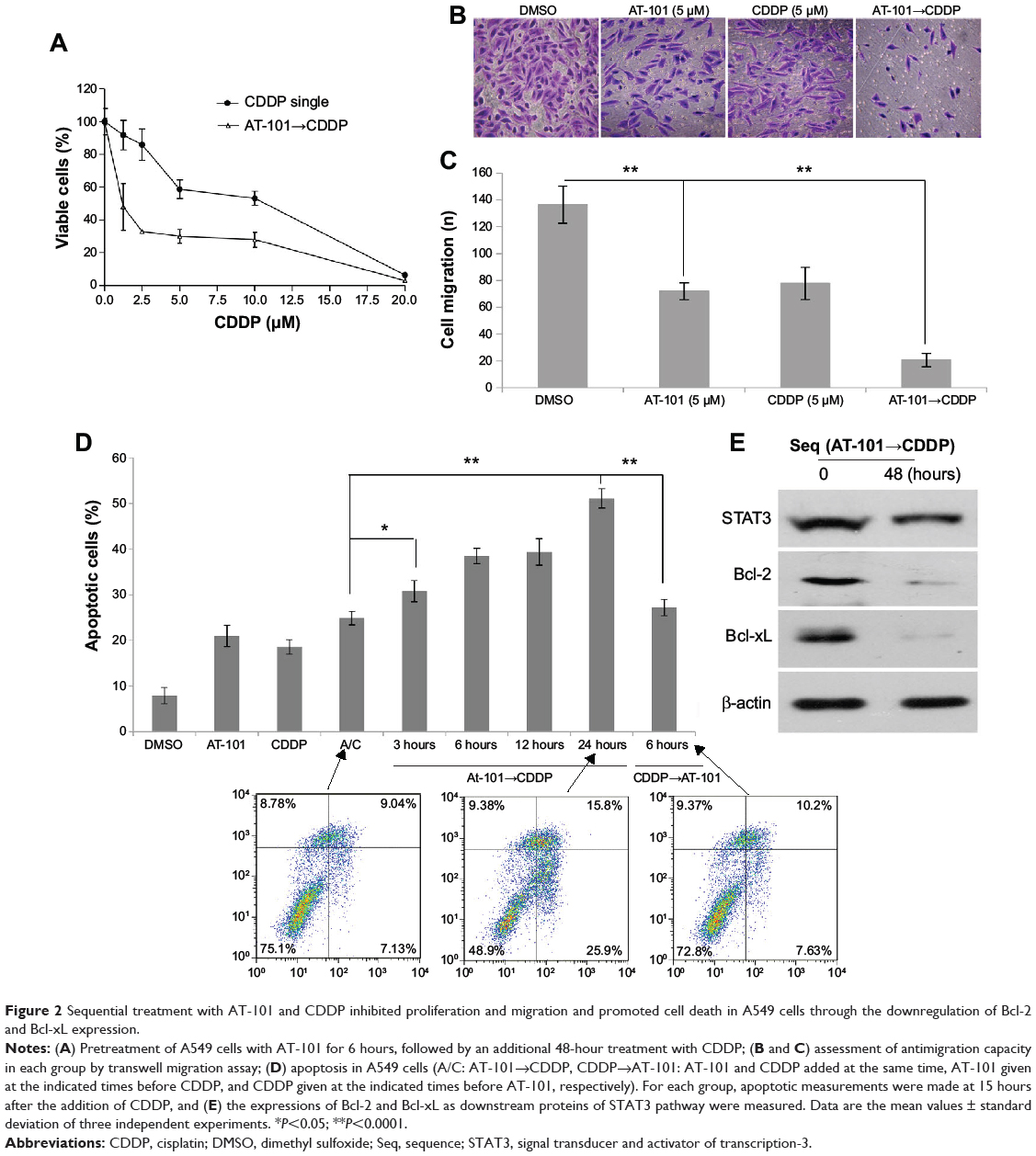
|
Figure 2 Sequential treatment with AT-101 and CDDP inhibited proliferation and migration and promoted cell death in A549 cells through the downregulation of Bcl-2 and Bcl-xL expression. |
AT-101 has been found to induce the apoptosis of various types of cancer cells through mitochondrial and other pathways.22,23,34–45 Since sequential application of anticancer agents can promote cell death by rewiring apoptotic signaling,29 we evaluated the efficacy of the sequential treatment at inducing apoptosis of tumor cells by flow cytometric analysis. Our results showed that pretreatment of AT-101 at least 3 hours prior to CDDP significantly increased the apoptotic rate of tumor cells (P<0.05) (Figure 2D).
Furthermore, the expression of the STAT3 downstream target proteins was also examined by Western blot analysis. As shown in Figure 2E, the expression of Bcl-2 and Bcl-xL were significantly downregulated in A549 cells by the sequential treatment for 48 hours, implicating that the proapoptotic effect of the sequential treatment might be through the inhibition of Bcl-2 and Bcl-xL expression.
CDDP activates IL-6/STAT3 signaling pathway and sequential treatment with AT-101, and CDDP significantly attenuates IL-6/STAT3 signaling activation in A549 cells
CDDP resistance is common in NSCLC patients, and activation of IL-6/STAT3 signaling pathway by CDDP contributes to the occurrence of chemoresistance.46–48 In our study, CDDP-induced IL-6 secretion was examined by ELISA assay. As shown in Figure 3A, an increase in IL-6 secretion was found in A549 cells treated with CDDP in a time-dependent manner (P<0.01).
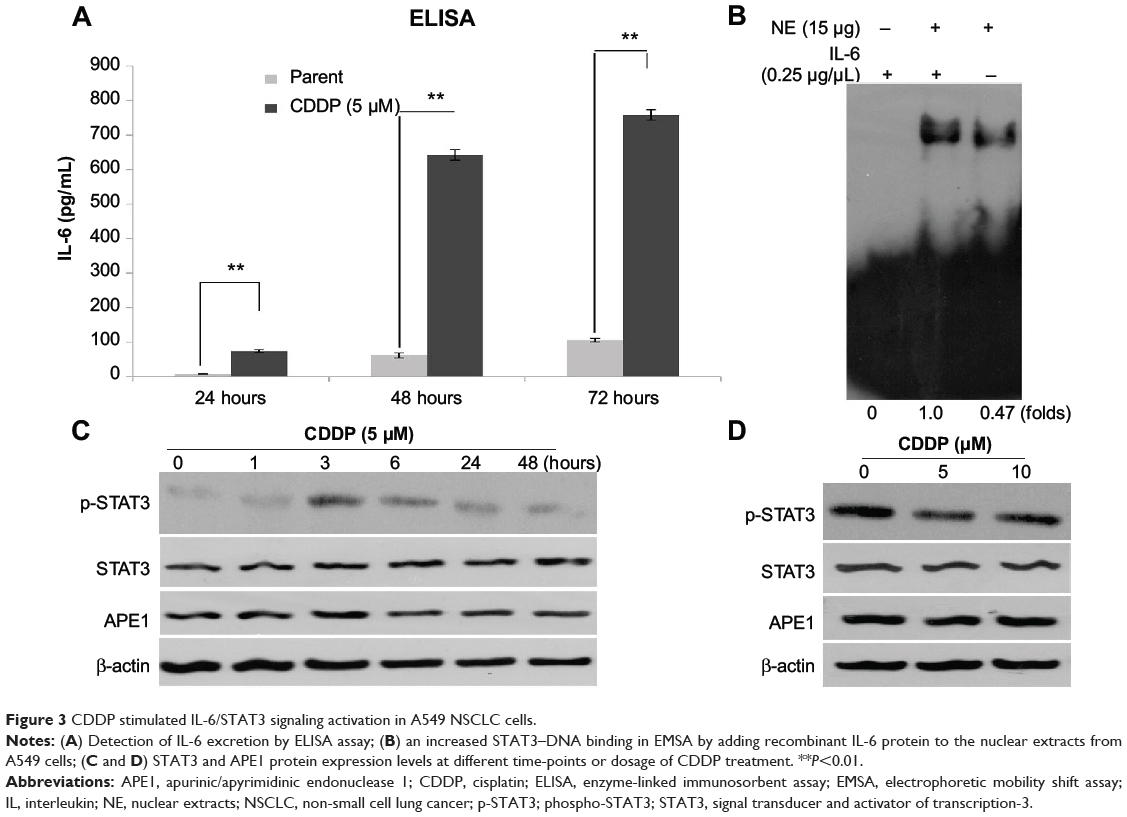
|
Figure 3 CDDP stimulated IL-6/STAT3 signaling activation in A549 NSCLC cells. |
Furthermore, the effect of IL-6 on the binding of STAT3 to DNA was evaluated by EMSA reaction. The EMSA data showed that the ability of STAT3–DNA binding was enhanced in A549 cells treated with recombinant human IL-6 protein (Figure 3B), suggesting that IL-6 promotes STAT3–DNA binding.
Additionally, we investigated whether CDDP upregulated the expression of STAT3 and APE1. As shown in Figure 3C and D, the expression of STAT3 and APE1 in A549 cells was not affected by CDDP treatment. The level of STAT3 or APE1 remained unchanged over time at different concentrations of CDDP. Taken together, CDDP cannot affect STAT3 or APE1 expression but can induce activation of the IL-6/STAT3 signaling pathway.
It has been reported that AT-101 can sensitize tumor cells to chemotherapy24,42 and radiotherapy.37,39,40,49 Furthermore, AT-101 enhances tumor cell killing by EGFR-targeted T-cells through modulating IFN-γ-induced dephosphorylation of STAT3 at phosphorylated Tyr705.45 According to these previous observations, we hypothesized that combined AT-101 and CDDP treatment could inhibit the activation of IL-6/STAT3 signaling pathway. To test this hypothesis, we measured the protein level of IL-6 using ELISA assay. As shown in Figure 4A, the sequential treatment with AT-101 and CDDP significantly attenuated CDDP-induced IL-6 excretion, as compared to the control vehicle-treated or AT-101-treated cells (P<0.0001) in a time-dependent manner (Figure 4B). We further investigated the effect of AT-101 alone or combined with CDDP on STAT3 activity using EMSA. The results demonstrated that the ability of STAT3–DNA binding could be inhibited by treatment with AT-101 (Figure 4C and D). However, the inhibitory extent of combined AT-101 and CDDP treatment was greater than that of AT-101 monotherapy or the vehicle control. Furthermore, using nuclear protein from A549 cells treated with AT-101 for 6 hours, the results showed that addition of CDDP to the EMSA reaction system suppressed the ability of STAT3–DNA binding in a concentration-dependent manner (Figure 4E). Taken together, the sequential treatment was more effective for inhibition of IL-6/STAT3 activation.
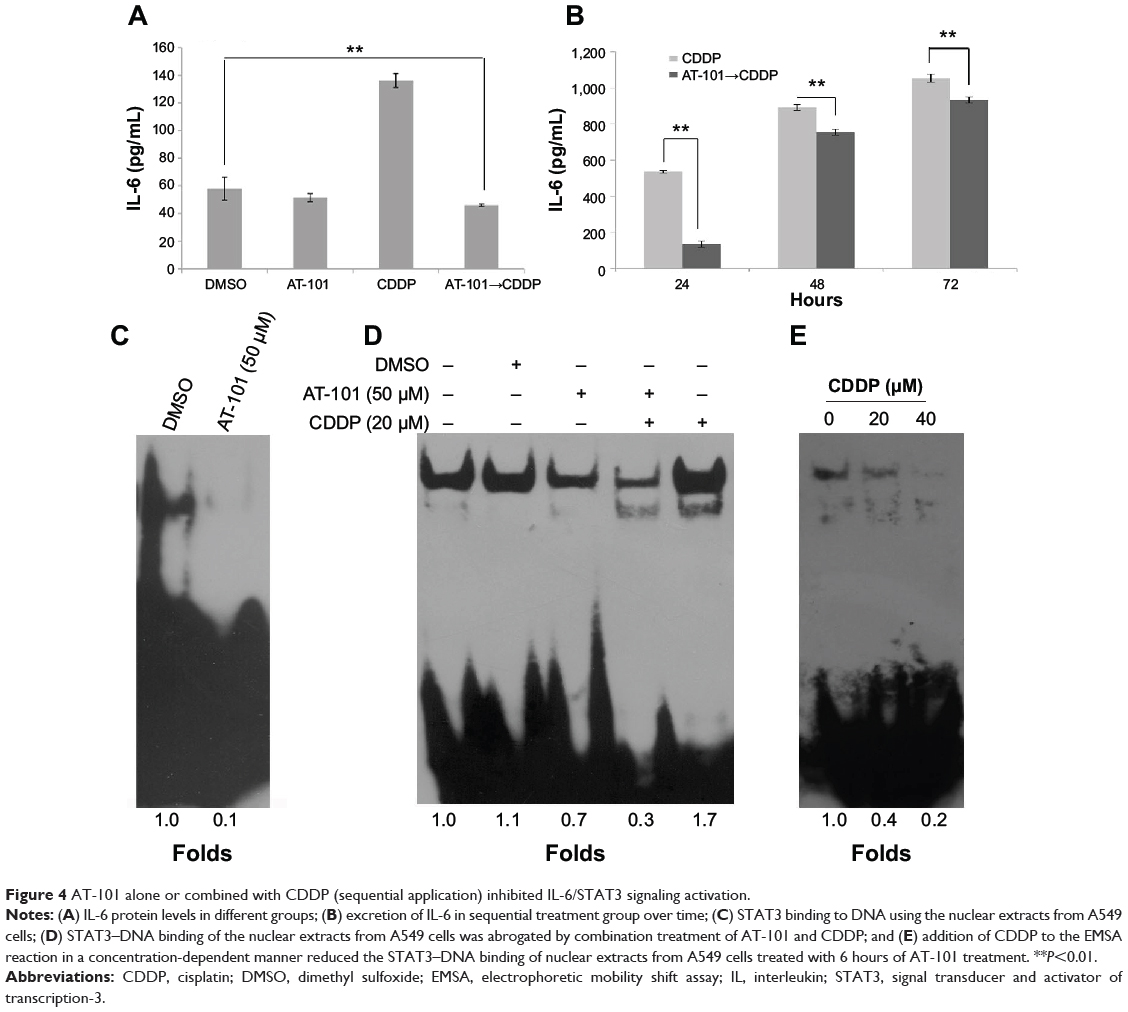
|
Figure 4 AT-101 alone or combined with CDDP (sequential application) inhibited IL-6/STAT3 signaling activation. |
Sequential treatment with AT-101 and CDDP significantly attenuates APE1-enhanced STAT3–DNA binding
Previous studies have revealed that STAT3–DNA binding and transcriptional activity is directly regulated by APE1/Ref-1.31 Gossypol has been identified as a small molecular inhibitor of APE1.26 Therefore, we next determined the effect of AT-101 on either APE1 protein expression or its bioactivity. After 6 hours of AT-101 treatment, APE1 translocated from the nucleus to the cytoplasm (Figure 5A). However, the expression of APE1 was not affected by treatment with AT-101 alone or combined with CDDP over time (Figure 5B–D). We confirmed the regulation of STAT3 transcriptional activity by the redox function of APE1. We found that treatment with AT-101 alone or combined with CDDP might affect the redox function of APE1. In our study, addition of reduced APE1 to nuclear extracts of A549 cells increased STAT3–DNA binding 5.5-fold (Figure 5E), suggesting that APE1 affected STAT3–DNA binding. The EMSA data also showed that in the presence of APE1, AT-101 alone or the sequential treatment (30 minutes pretreatment before CDDP) decreased the ability of STAT3–DNA binding 2.1-fold and 5.4-fold, respectively, as compared to the cells treated with DMSO only (Figure 5E). This data suggests that AT-101 alone and the sequential treatment with AT-101 and CDDP are effective to attenuate APE1-enhanced STAT3–DNA binding.

|
Figure 5 AT-101 alone or combined with CDDP targeting APE1 reduced status, leading to the inhibition of STAT3–DNA binding. |
Knockdown of APE1 by siRNA promotes apoptosis in A549 cells that are treated with combined AT-101 and CDDP and blocks APE1-mediated IL-6/STAT3 activation
The results in Figure 4 suggest that the sequential treatment with AT-101 and CDDP significantly attenuated APE1-enhanced STAT3–DNA binding. To further validate this finding, we evaluated the role of APE1 in the regulation of STAT3 activity by knockdown of APE1 in A549 cells. Our study found that APE1 expression was inhibited by transfection of APE1-specific siRNA in A549 cells (Figure 6A). Although knockdown of APE1 did not affect the expression of STAT3 protein, IL-6 level was significantly decreased by knockdown of APE1 in A549 cells (P<0.0001) (Figure 6B). Figure 6C shows that the ability of STAT3–DNA binding was inhibited by knockdown of APE1, but this inhibition could be rescued by adding reduced APE1 into the EMSA reaction system. This data implicated a role of APE1 in the regulation of STAT3 activity. However, cell death rate of APE1-suppressed A549 cell was significantly increased by the sequential treatment with AT-101 and CDDP, as compared to the control cells with APE1 knockdown only (Figure 6D and E) (P<0.05).

|
Figure 6 siRNA–APE1 enhanced cell death in A549 with sequential treatment of AT-101 and CDDP through inhibition of APE1 regulating IL-6/STAT3 signaling. |
Sequential treatment with AT-101 and CDDP inhibited xenograft tumor growth through downregulating APE1/STAT3 signaling enhanced the expressions of Bcl-2, Bcl-xL
To further examine the effects of the combined AT-101 and CDDP treatment on tumor growth in vivo, we established a BALB/c mouse tumor xenograft model by inoculation of human NSCLC A549 cells. Tumor volume and body weight were measured every 4 days from treatment start date. As compared to other groups, the average body weight of mice in the AT-101-alone group was not significantly changed; whereas the body weight of mice in the CDDP-monotherapy group was significantly decreased (P<0.05). In the sequential-treatment group, the average body weight was less compared to the control group (P<0.05), but there was no significant difference between the combination group and AT-101- or CDDP-monotherapy groups (Figure 7A), suggesting that mice could tolerate either AT-101 or the combination therapy. Tumor volume in the combination group was significantly smaller than that in other groups, including vehicle control, AT-101 alone, and CDDP alone (P<0.05) (Figure 7B and C). The weight of tumors in the combination group was significantly lower than that in other groups (P<0.05) (Figure 7D). Furthermore, we tested the similar mechanism for APE1/STAT3 signaling pathway using Western blot. As expected, the expressions of APE1, STAT3, and p-STAT3 were almost not affected when the mice were treated with the vehicle control, AT-101 alone, CDDP alone, or AT-101 plus CDDP (the combination therapy), but Bcl-2 and Bcl-xL expressions were decreased in the combination therapy to some extent. In brief, our data demonstrated that in mouse tumor xenograft model, the sequential treatment with AT-101 and CDDP significantly inhibited tumor growth through inhibition of APE1/STAT3 signaling enhanced expressions of Bcl-2 and Bcl-xL. All of our findings suggest that AT-101 has potential to improve the efficacy of CDDP treatment.
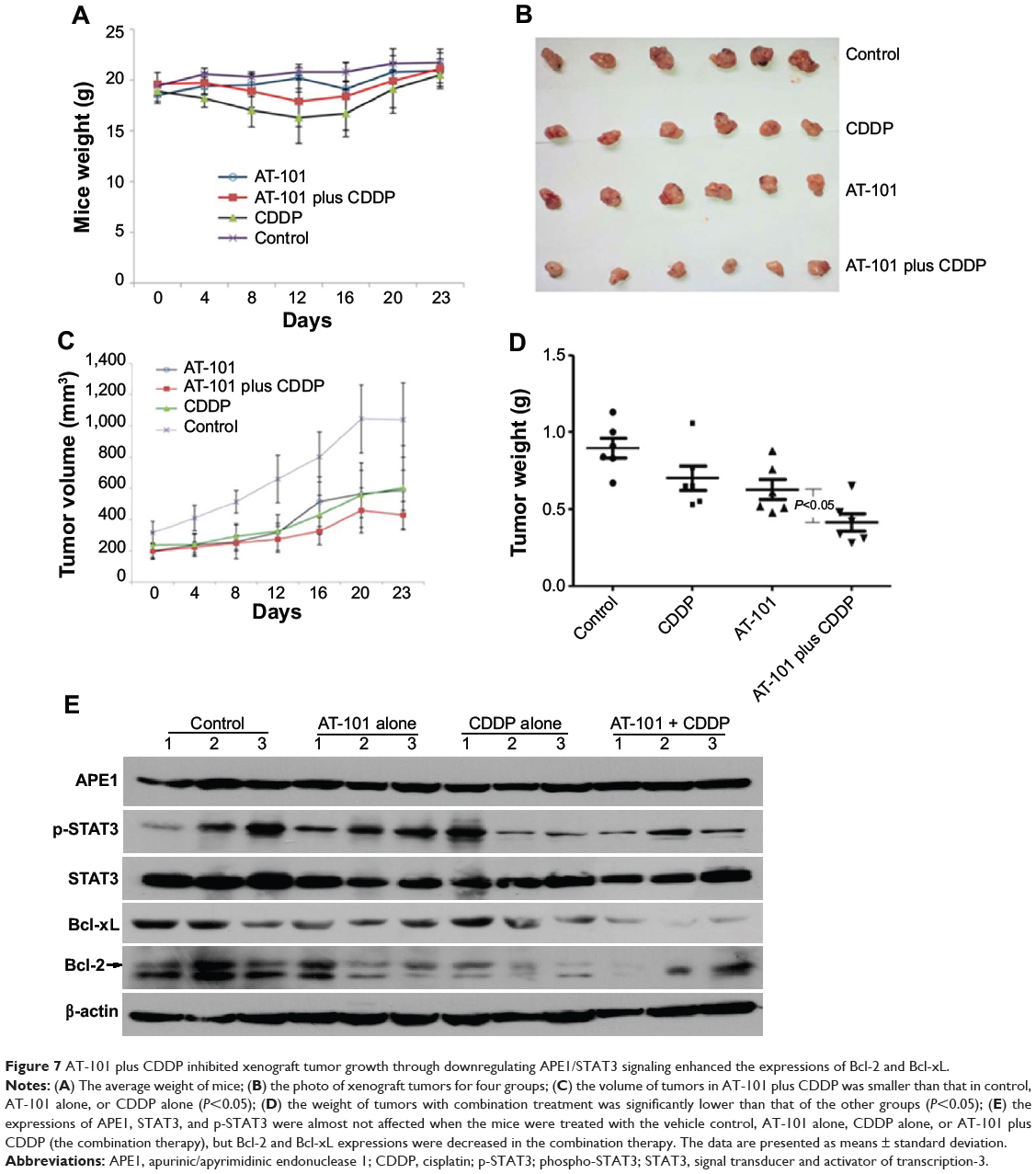
|
Figure 7 AT-101 plus CDDP inhibited xenograft tumor growth through downregulating APE1/STAT3 signaling enhanced the expressions of Bcl-2 and Bcl-xL. |
Discussion
In the present study, we demonstrated for the first time that AT-101 is capable of enhancing CDDP’s antitumor efficacy in NSCLC cells in vitro and in vivo through sequential treatment. We found that AT-101-induced chemosensitivity was associated with the suppression of the expression of antiapoptotic protein: Bcl-2 and Bcl-xL. Importantly, we revealed that the mechanisms underlying the enhanced antitumor effects of the sequential treatment with AT-101 and CDDP involve the inhibition of APE1-mediated IL-6/STAT3 signaling activation.
CDDP is one of the most frequently used chemotherapeutic agents for NSCLC treatment. The efficacy of CDDP for NSCLC treatment is limited, due to either intrinsic or acquired chemoresistance following CDDP administration.50–54 The mechanisms related to CDDP resistance found in many NSCLC cell lines appear to be multifactorial, including enhancement of drug detoxification, alteration of drug transport system, disruption of apoptotic and autophagic cell death pathways, and change of DNA damage tolerance mechanisms.51–54 Among them, antiapoptosis is a critical factor for CDDP-induced chemoresistance. STAT3 is a key player in signal transduction of growth factors, such as epidermal growth factor and the entire IL-6 family of cytokines. Downstream target genes of STAT3 signaling pathway are involved in cell proliferation, differentiation, angiogenesis, and survival.6,11 Dysregulation of IL-6/STAT3 activity in cancer cells contributes to chemoresistance, and inhibition of IL-6/STAT3 signaling activation can make tumor cells sensitive to CDDP.8,55 Our study demonstrated similar results (Figure 3), which are consistent with reports regarding CDDP-mediated activation of IL-6/STAT3 signaling pathway. Taken together, activation of IL-6/STAT3 signaling pathway could be a risk factor in inducing CDDP chemoresistance. Therefore, suppressing IL-6/STAT3 activation may be a favorable therapeutic strategy to overcome CDDP-induced chemoresistance.
AT-101 has shown antitumor activity as a single agent and in combination with standard anticancer therapies.20–23 AT-101 also reverses chemoresistance by promoting tumor cell apoptosis through different pathways.24,42,56,57 Furthermore, AT-101 affects the phosphorylation of STAT3.45 Several studies suggest that sequential treatment may be a valuable approach in clinical practice to overcome chemotherapeutic resistance and toxicity.29 Therefore, we reasoned that the sequential treatment with AT-101 and CDDP might promote A549 cell death through the inhibition of IL-6/STAT3 signaling activation.
Our study showed that sequential treatment with AT-101 and CDDP significantly inhibited cell proliferation and migration, as well as promoted apoptosis of cultured A549 cells. Further, our mouse xenograft model demonstrated that tumor-growth inhibition in the combination group was more effective compared to the control, AT-101, or CDDP monotherapy. To explore the mechanisms involved in the combination therapy, we investigated the inhibitory effect of combination therapy on the IL-6/STAT3 signaling pathway. Results showed that IL-6 protein expression was downregulated in the combination group, as compared to CDDP alone group, suggesting that sequential treatment can attenuate CDDP-induced IL-6 overexpression. Sahin et al58 reported that targeted therapies in a sequential manner can effectively overcome drug resistance by targeting and reprogramming the signaling pathways in highly aggressive breast cancer. Saiyin et al59 also showed that the autophagy inhibitor LY294002-loaded hydrophilic and pH-responsive hyperbranched poly(acylhydrazone)-doxorubicin micelles could rapidly enter tumor cells, and then sequentially release LY294002 and doxorubicin in response to an intracellular acidic microenvironment, resulting in the inhibition of cell proliferation. Interestingly, there was a synergistic effect between LY294002 and doxorubicin, such that the preferentially liberated LY294002 suppressed the autophagic process in tumor cells and made them more sensitive to the subsequent release of doxorubicin. Our EMSA data showed that STAT3–DNA binding was inhibited by the treatment with AT-101 alone or combined with CDDP, but that the latter was more effective. In brief, the combination therapy significantly induces cell death and inhibits activation of the IL-6/STAT3 signaling pathway in A549 cells. However, the mechanisms for the regulation of IL-6/STAT3 signaling activity remains elusive.
APE1 is the primary enzyme responsible for recognition and incision of noncoding apurinic/apyrimidinic sites in DNA resulting from spontaneous, chemical, or DNA glycosylase-mediated hydrolysis of the N-glycosyl bond initiated by the DNA base excision repair pathway.12–14 APE1 mainly shows 5′-endonuclease activity, but also exhibits minor 3′-phosphodiesterase, 3′-phosphatase, and 3′→5′ exonuclease activities. Previous reports revealed that the DNA damage response pathways can be dynamically reprogrammed through prior exposure to a drug that can modulate cell signaling pathways to increase the cell sensitivity to DNA damaging agents in a time-dependent manner.29,60 Emerging evidence supports the opinion that APE1 can regulate STAT3 transcriptional activity through its reduced status.31 Gossypol, a potential anticancer agent, has been identified as an APE1 inhibitor in our previous study.26 Therefore, we hypothesized that the inhibitory effect of the sequential treatment with AT-101 and CDDP on the IL-6/STAT3 signaling pathway might be associated with the inhibition of the redox function of their upstream protein, APE1. To this end, we performed an EMSA assay to examine the effect of the reduced APE1 on STAT3–DNA binding in A549 cells. We found that sequential treatment significantly attenuates APE1-enhanced STAT3–DNA binding, as compared with AT-101 alone. Moreover, the expression of Bcl-2 and Bcl-xL, two downstream proteins of STAT3, were downregulated by the combination therapy. The above data demonstrated that AT-101 might be an ideal agent to sensitize NSCLC A549 cells to CDDP via inhibition of APE1-mediated IL-6/STAT3 signaling activation.
Conclusion
In conclusion, we revealed for the first time that the sequential treatment with AT-101 and CDDP promotes apoptosis of A549 cells death in vitro and in vivo. More importantly, our findings showed that APE1 redox functions could be inhibited by the AT-101 and CDDP combination therapy, leading to suppression of IL-6/STAT3 signaling. Notably, downregulation of antiapoptotic protein Bcl-2 and Bcl-xL expression, and induction of cell apoptosis, are also related to the mechanisms of action of this combination therapy. Taken together, these data suggest that the sequential application of AT-101 and CDDP is an attractive novel approach for overcoming CDDP chemoresistance in NSCLC treatment. Further studies are warranted to demonstrate the efficacy and safety of AT-101 combined with CDDP in the clinical setting.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (No. 30872975).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
Chen W, Zheng R, Zhang S, et al. The incidences and mortalities of major cancers in China, 2009. Chin J Cancer. 2013;32(3):106–112. | ||
DeSantis CE, Lin CC, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin. 2014;64(4):252–271. | ||
Maccio A, Madeddu C. Cisplatin: an old drug with a newfound efficacy – from mechanisms of action to cytotoxicity. Expert Opin Pharmacother. 2013;14(13):1839–1857. | ||
O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med. 2013;368(2):161–170. | ||
Yu H, Jove R. The STATs of cancer – new molecular targets come of age. Nat Rev Cancer. 2004;4(2):97–105. | ||
Huang S, Chen M, Shen Y, et al. Inhibition of activated Stat3 reverses drug resistance to chemotherapeutic agents in gastric cancer cells. Cancer Lett. 2012;315(2):198–205. | ||
Han Z, Feng J, Hong Z, et al. Silencing of the STAT3 signaling pathway reverses the inherent and induced chemoresistance of human ovarian cancer cells. Biochem Biophys Res Commun. 2013;435(2):188–194. | ||
Lo HW, Cao X, Zhu H, Ali-Osman F. Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin Cancer Res. 2008;14(19):6042–6054. | ||
Yue P, Zhang X, Paladino D, et al. Hyperactive EGF receptor, Jaks and Stat3 signaling promote enhanced colony-forming ability, motility and migration of cisplatin-resistant ovarian cancer cells. Oncogene. 2012;31(18):2309–2322. | ||
Siveen KS, Sikka S, Surana R, et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta. 2014;1845(2):136–154. | ||
Bhakat KK, Mantha AK, Mitra S. Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid Redox Signal. 2009;11(3):621–638. | ||
Abbotts R, Madhusudan S. Human AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancer. Cancer Treat Rev. 2010;36(5):425–435. | ||
Busso CS, Lake MW, Izumi T. Posttranslational modification of mammalian AP endonuclease (APE1). Cell Mol Life Sci. 2010;67(21):3609–3620. | ||
Kelley MR, Georgiadis MM, Fishel ML. APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Curr Mol Pharmacol. 2012;5(1):36–53. | ||
Xanthoudakis S, Smeyne RJ, Wallace JD, Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sci U S A. 1996;93(17):8919–8923. | ||
Meira LB, Devaraj S, Kisby GE, et al. Heterozygosity for the mouse Apex gene results in phenotypes associated with oxidative stress. Cancer Res. 2001;61(14):5552–5557. | ||
Al-Safi RI, Odde S, Shabaik Y, Neamati N. Small-molecule inhibitors of APE1 DNA repair function: an overview. Curr Mol Pharmacol. 2012;5(1):14–35. | ||
Wang D, Xiang DB, Yang XQ, et al. APE1 overexpression is associated with cisplatin resistance in non-small cell lung cancer and targeted inhibition of APE1 enhances the activity of cisplatin in A549 cells. Lung Cancer. 2009;66(3):298–304. | ||
Imai A, Zeitlin BD, Visioli F, et al. Metronomic dosing of BH3 mimetic small molecule yields robust antiangiogenic and antitumor effects. Cancer Res. 2012;72(3):716–725. | ||
Loberg RD, McGregor N, Ying C, Sargent E, Pienta KJ. In vivo evaluation of AT-101 (R-(-)-gossypol acetic acid) in androgen-independent growth of VCaP prostate cancer cells in combination with surgical castration. Neoplasia. 2007;9(12):1030–1037. | ||
Ko CH, Shen SC, Yang LY, Lin CW, Chen YC. Gossypol reduction of tumor growth through ROS-dependent mitochondria pathway in human colorectal carcinoma cells. Int J Cancer. 2007;121(8):1670–1679. | ||
Pang X, Wu Y, Wu Y, et al. (-)-Gossypol suppresses the growth of human prostate cancer xenografts via modulating VEGF signaling-mediated angiogenesis. Mol Cancer Ther. 2011;10(5):795–805. | ||
Karaca B, Atmaca H, Bozkurt E, et al. Combination of AT-101/cisplatin overcomes chemoresistance by inducing apoptosis and modulating epigenetics in human ovarian cancer cells. Mol Biol Rep. 2013;40(6):3925–3933. | ||
Schelman WR, Mohammed TA, Traynor AM, et al. A phase I study of AT-101 with cisplatin and etoposide in patients with advanced solid tumors with an expanded cohort in extensive-stage small cell lung cancer. Invest New Drugs. 2014;32(2):295–302. | ||
Qian C, Li M, Sui J, et al. Identification of a novel potential antitumor activity of gossypol as an APE1/Ref-1 inhibitor. Drug Des Devel Ther. 2014;8:485–496. | ||
Baggstrom MQ, Qi Y, Koczywas M, et al. A phase II study of AT-101 (Gossypol) in chemotherapy-sensitive recurrent extensive-stage small cell lung cancer. J Thorac Oncol. 2011;6(10):1757–1760. | ||
Sonpavde G, Matveev V, Burke JM, et al. Randomized phase II trial of docetaxel plus prednisone in combination with placebo or AT-101, an oral small molecule Bcl-2 family antagonist, as first-line therapy for metastatic castration-resistant prostate cancer. Ann Oncol. 2012;23(7):1803–1808. | ||
Lee MJ, Ye AS, Gardino AK, et al. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149(4):780–794. | ||
Ren T, Qing Y, Dai N, et al. Apurinic/apyrimidinic endonuclease 1 induced upregulation of fibroblast growth factor 2 and its receptor 3 induces angiogenesis in human osteosarcoma cells. Cancer Sci. 2014;105(2):186–194. | ||
Cardoso AA, Jiang Y, Luo M, et al. APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival. PLoS One. 2012;7(10):e47462. | ||
Huang YW, Wang LS, Dowd MK, Wan PJ, Lin YC. (–)-Gossypol reduces invasiveness in metastatic prostate cancer cells. Anticancer Res. 2009;29(6):2179–2188. | ||
Huang YW, Wang LS, Chang HL, et al. Molecular mechanisms of (–)-gossypol-induced apoptosis in human prostate cancer cells. Anticancer Res. 2006;26(3A):1925–1933. | ||
Jiang J, Slivova V, Jedinak A, Sliva D. Gossypol inhibits growth, invasiveness, and angiogenesis in human prostate cancer cells by modulating NF-κB/AP-1 dependent- and independent-signaling. Clin Exp Metastasis. 2012;29(2):165–178. | ||
Hsiao WT, Tsai MD, Jow GM, Tien LT, Lee YJ. Involvement of Smac, p53, and caspase pathways in induction of apoptosis by gossypol in human retinoblastoma cells. Mol Vis. 2012;18:2033–2042. | ||
Volate SR, Kawasaki BT, Hurt EM, et al. Gossypol induces apoptosis by activating p53 in prostate cancer cells and prostate tumor-initiating cells. Mol Cancer Ther. 2010;9(2):461–470. | ||
Xu L, Yang D, Wang S, et al. (–)-Gossypol enhances response to radiation therapy and results in tumor regression of human prostate cancer. Mol Cancer Ther. 2005;4(2):197–205. | ||
Balakrishnan K, Burger JA, Wierda WG, Gandhi V. AT-101 induces apoptosis in CLL B cells and overcomes stromal cell-mediated Mcl-1 induction and drug resistance. Blood. 2009;113(1):149–153. | ||
Zerp SF, Stoter R, Kuipers G, et al. AT-101, a small molecule inhibitor of anti-apoptotic Bcl-2 family members, activates the SAPK/JNK pathway and enhances radiation-induced apoptosis. Radiat Oncol. 2009;4:47. | ||
Moretti L, Li B, Kim KW, Chen H, Lu B. AT-101, a pan-Bcl-2 inhibitor, leads to radiosensitization of non-small cell lung cancer. J Thorac Oncol. 2010;5(5):680–687. | ||
McGregor N, Patel L, Craig M, Weidner S, Wang S, Pienta KJ. AT-101 (R-(–)-gossypol acetic acid) enhances the effectiveness of androgen deprivation therapy in the VCaP prostate cancer model. J Cell Biochem. 2010;110(5):1187–1194. | ||
Hu W, Wang F, Tang J, et al. Proapoptotic protein Smac mediates apoptosis in cisplatin-resistant ovarian cancer cells when treated with the anti-tumor agent AT101. J Biol Chem. 2012;287(1):68–80. | ||
Kisim A, Atmaca H, Cakar B, et al. Pretreatment with AT-101 enhances tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis of breast cancer cells by inducing death receptors 4 and 5 protein levels. J Cancer Res Clin Oncol. 2012;138(7):1155–1163. | ||
Billard C. Development of Noxa-like BH3 mimetics for apoptosis-based therapeutic strategy in chronic lymphocytic leukemia. Mol Cancer Res. 2012;10(6):673–676. | ||
Thakur A, Lum LG, Schalk D, et al. Pan-Bcl-2 inhibitor AT-101 enhances tumor cell killing by EGFR targeted T cells. PLoS One. 2012;7(11):e47520. | ||
Ikuta K, Takemura K, Kihara M, et al. Overexpression of constitutive signal transducer and activator of transcription 3 mRNA in cisplatin-resistant human non-small cell lung cancer cells. Oncol Rep. 2005;13(2):217–222. | ||
Kulesza DW, Carré T, Chouaib S, Kaminska B. Silencing of the transcription factor STAT3 sensitizes lung cancer cells to DNA damaging drugs, but not to TNFα- and NK cytotoxicity. Exp Cell Res. 2013;319(4):506–516. | ||
Tezcanli Kaymaz B, Bozok Cetintas V, Eroglu Z, Kosova B. Suppression of STAT3 by chemically modified siRNAs increases the chemotherapeutic sensitivity of parental and cisplatin-resistant non-small cell lung cancer cells. J BUON. 2014;19(1):145–152. | ||
Akagunduz O, Karaca B, Atmaca H, et al. Radiosensitization of hormone-refractory prostate cancer cells by gossypol treatment. J BUON. 2010;15(4):763–767. | ||
Galluzzi L, Senovilla L, Vitale I, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31(15):1869–1883. | ||
Koberle B, Tomicic MT, Usanova S, Kaina B. Cisplatin resistance: preclinical findings and clinical implications. Biochim Biophys Acta. 2010;1806(2):172–182. | ||
Galluzzi L, Vitale I, Michels J, et al. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5:e1257. | ||
Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev. 2007;33(1):9–23. | ||
Martin LP, Hamilton TC, Schilder RJ. Platinum resistance: the role of DNA repair pathways. Clin Cancer Res. 2008;14(5):1291–1295. | ||
Sugimura K, Miyata H, Tanaka K, et al. Let-7 expression is a significant determinant of response to chemotherapy through the regulation of IL-6/STAT3 pathway in esophageal squamous cell carcinoma. Clin Cancer Res. 2012;18(18):5144–5153. | ||
Deng S, Yuan H, Yi J, et al. Gossypol acetic acid induces apoptosis in RAW264.7 cells via a caspase-dependent mitochondrial signaling pathway. J Vet Sci. 2013;14(3):281–289. | ||
Li G, Liu L, Shan C, et al. RhoA/ROCK/PTEN signaling is involved in AT-101-mediated apoptosis in human leukemia cells in vitro and in vivo. Cell Death Dis. 2014;5:e998. | ||
Sahin O, Wang Q, Brady SW, et al. Biomarker-guided sequential targeted therapies to overcome therapy resistance in rapidly evolving highly aggressive mammary tumors. Cell Res. 2014;24(5):542–559. | ||
Saiyin W, Wang D, Li L, et al. Sequential release of autophagy inhibitor and chemotherapeutic drug with polymeric delivery system for oral squamous cell carcinoma therapy. Mol Pharm. 2014;11(5):1662–1675. | ||
Janes KA, Reinhardt HC, Yaffe MB. Cytokine-induced signaling networks prioritize dynamic range over signal strength. Cell. 2008;135(2):343–354. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.